

ABSTRACT
The Gram-positive actinomycete Amycolatopsis sp. strain ATCC 39116 is used for the industrial production of natural vanillin. Previously, the only gene deletion performed in this strain targeted the gene vdh, coding for a vanillin dehydrogenase. The generation of this mutant suffered from a high number of illegitimate recombinations and a low rate of homologous recombination. To alleviate this, we constructed an optimized deletion system based on a modified suicide vector. Thereby, we were able to increase the rate of homologous integration from less than 1% of the analyzed clones to 20% or 50%, depending on the targeted gene. We were furthermore able to reduce the screening effort needed to identify homogenotes through the use of the rpsL gene from Saccharopolyspora erythraea, which confers streptomycin sensitivity on clones still carrying the suicide vector. The new suicide vector is p6SUI5ERPSL, and its applicability was demonstrated by the deletion of three Amycolatopsis gene clusters. The deletion of the first of the gene clusters, coding for an aldehyde oxidase (yagRST), led to no altered phenotype compared to the parent strain; deletion of the second, coding for a vanillic acid decarboxylase (vdcBCD), led to a phenotype that was strongly impaired in its growth with vanillic acid as the sole carbon source and also unable to form guaiacol; and deletion of the third, coding for a vanillate demethylase (vanAB), led to only a negligible impact in comparison. Therefore, we showed that decarboxylation of vanillic acid is the main degradation pathway in Amycolatopsis sp. ATCC 39116 while the demethylation plays only a minor role and does not compensate the deletion of vdcBCD.
IMPORTANCE Amycolatopsis sp. ATCC 39116 is an important microorganism used for the production of natural vanillin from ferulic acid. In contrast to this importance, it has previously been shown that this strain is hard to manipulate on a genetic level. We therefore generated an optimized system to facilitate the deletion of genes in this strain. This allowed us to greatly reduce the time and work requirements for generating deletions. This could allow the improvement of vanillin production in the future and also the elucidation of metabolic pathways. To test our deletion system, we deleted three gene clusters in Amycolatopsis sp. ATCC 39116. One showed no involvement in the metabolism of vanillin, while the second proved to be the main pathway of vanillic acid degradation and completely stopped the formation of the off-flavor guaiacol. The third appeared to have only a negligible impact on the degradation of vanillic acid.
KEYWORDS: Actinomycetes, Amycolatopsis, genetic modification, metabolic engineering, vanillin, vanillic acid, guaiacol, gene deletion
INTRODUCTION
The actinomycete Amycolatopsis sp. strain ATCC 39116 is an important organism for the production of natural vanillin from ferulic acid at an industrial level. The annual vanillin market exceeded 16 kilotons in 2010, but most of it is produced from petrochemical sources (1). Many other strains, such as Saccharomyces cerevisiae (2), Escherichia coli (3), or Pycnoporus cinnabarinus (4), have been analyzed and adapted for the production of vanillin, but Amycolatopsis still remains the strain with the highest concentrations and yields (5). Even though this strain is an important vanillin producer, there are only two publications that performed a genetic optimization of this strain (5, 6). The deletion performed in this strain aimed at the vanillin dehydrogenase gene vdh and reduced the formation of vanillic acid by 90% (strain F33, Amycolatopsis sp. ATCC 39116 Δvdh). Furthermore, the genes ech and fcs, coding for enoyl coenzyme A (enoyl-CoA) hydratase/aldolase and feruloyl-CoA synthetase, respectively, were expressed under the control of the strong constitutive erythromycin promoter. The resulting mutant reached the currently highest described vanillin concentration obtained from ferulic acid without extraction of the fermentation broth (5). The other publications focused on the optimization of the fermentation conditions (7–10) or on the use of adsorbing materials (11). The transformation of this strain has been established in 2002, the genome sequence has been available since 2012, and usable expression vectors with different promoters have existed since 2014 (12–14). However, there is still a lack of efficient methods for the genetic modification of Amycolatopsis sp. ATCC 39116. This is a strong contrast to other strains that have been modified for vanillin production through means of genetic modification (1–3, 15).
While generating the first deletion mutant of Amycolatopsis sp. ATCC 39116, it became obvious that nonhomologous integration of DNA is one of the major problems in genetically modifying this strain (5, 6). This problem is also well known for other actinomycetes like Mycobacterium spp. (16). Furthermore, there are a large number of different methods that have been developed to generate genetic modifications in actinomycetes, which also shows the difficulties of genetic manipulation with these organisms (17). Sometimes, it is even necessary to optimize these methods for one specific strain, as was the case for Amycolatopsis sp. ATCC 39116 (13), and even PCR can pose a problem in these GC-rich strains (18).
Another problem was the low rate of homologous recombination when trying to achieve the second recombination event, leading to either the deletion of the target gene or the restoration of the genotype of the wild type. Large amounts of clones had to be screened to identify potential homogenotes, and the cultivation times necessary to generate these clones were quite long (5). Several different methods have been applied to facilitate the identification and isolation of homogenotes. These were, for example, based on the sensitivity toward 5-fluoroorotic acid (19), sucrose (20), or streptomycin (21).
Streptomycin resistance can be conferred by mutations in the small ribosomal protein S12, which is encoded by the rpsL gene (22, 23). It was shown elsewhere that the resistance is recessive in strains possessing both resistant and sensitive copies of the rpsL gene (22). This makes it an interesting candidate to establish counterselection systems in a wide variety of strains since the only prerequisite is a streptomycin-resistant rpsL gene in the targeted strain (21, 24–26). Alternatively, it is possible to make the homogenotes differ visibly from heterogenotes. This was achieved with enzymes such as a β-glucuronidase (27), the green fluorescent protein (28), or luciferase (29).
The aim of this study was to develop suicide vectors suitable for the deletion of genes in Amycolatopsis sp. ATCC 39116 in a faster and less error-prone way. To accomplish this, we analyzed the previously used vector p6sui (5) to determine and remove problematic sequence areas that lead to nonhomologous integrations. To test the generated suicide plasmids, we performed knockouts of an aldehyde oxidase cluster consisting of three subunits (yagRST); the vdcBCD cluster, coding for a vanillic acid decarboxylase; and the vanAB cluster, coding for a vanillate demethylase.
Aldehyde oxidases are large enzyme complexes, and some are known to be able to oxidize vanillin to vanillic acid without requiring NAD(P) as a cofactor. Structurally similar compounds like protocatechuic aldehyde are metabolized as well, albeit with widely differing reaction rates (30). Since the vdh deletion mutant is still capable of producing reduced amounts of vanillic acid, it was previously speculated that an aldehyde oxidase might be responsible for this property (5, 6). The vanillic acid decarboxylase was targeted because it is known that Amycolatopsis sp. ATCC 39116 produces guaiacol from vanillic acid (6). Similar clusters have already been analyzed in strains like Bacillus subtilis or Streptomyces sp. strain D7, although no deletions or disruptions have been performed so far to our knowledge (31, 32). The formation of guaiacol has been a major problem in the spoilage of food and beverages and might also pose a problem as an off-flavor in vanillin (33).
In addition to the degradation of vanillic acid to guaiacol, Amycolatopsis sp. ATCC 39116 is also known to produce protocatechuic acid as another degradation product (6, 34). This reaction is catalyzed by vanillic acid demethylases and produces formaldehyde as a by-product (35, 36). The vanAB cluster, coding for this enzyme, and the vdcBCD cluster are interesting candidates for deletion as they might allow the identification of the main degradation pathway of vanillic acid in Amycolatopsis sp. ATCC 39116. The deletion could also allow easier quantification of the remaining vanillic acid formation and might also have a positive effect on the viability of the cells by preventing the formation of toxic formaldehyde. A similar effect was demonstrated in Burkholderia cepacia TM1 while growing on vanillic acid. Heterologously expressing enzymes of the RuMP cycle, which assimilate formaldehyde, led to an increased growth of this strain (37).
With the constructed suicide vector, we were able to generate the three mutants described above and achieved homologous integration rates of between 20% and 50% of the screened clones. Furthermore, we were able to significantly speed up the time required to identify homogenotes that lost the suicide plasmid, again leading to either a restoration of the genetic organization of the wild type or the deletion of the target gene. These vectors are now available to generate further genetic modifications in Amycolatopsis. Especially, genes involved in the formation of vanillyl alcohol might be interesting targets for future research because this compound poses a major problem in Amycolatopsis sp. ATCC 39116 (5) as well as in other microorganisms like Saccharomyces cerevisiae (2).
RESULTS
Construction of the deletion fragments.
The target gene clusters yagRST and vdcBCD were chosen for two different reasons. Previous results showed that F33 still produces small amounts of vanillic acid, and it was speculated that a vanillin oxidase might be responsible for this (5, 6). Amycolatopsis sp. ATCC 39116 possesses several gene clusters that could possibly code for aryl aldehyde oxidase complexes. One of them was chosen for a deletion since it exhibited homologies toward putative xanthine oxidase complexes that have been shown to also utilize vanillin as the substrate (30).
The vdcBCD and vanAB gene clusters were chosen to elucidate the main degradation pathway of vanillic acid in Amycolatopsis. Both protocatechuic acid and guaiacol were identified as degradation products in this strain previously (6). The vdcBCD cluster exhibited a high homology to a similar cluster in Streptomyces sp. D7 that was shown to be capable of decarboxylating vanillic acid to guaiacol (31). The vanAB cluster shows high homologies to a characterized vanillate demethylase cluster from Pseudomonas sp. strain HR199 (35) and other confirmed or putative vanillate demethylases. The flanking regions of vdcBCD, vanAB, and yagRST were then amplified, fused, and sequenced to generate the deletion fragments.
Construction of the suicide plasmids.
Previous studies done with Amycolatopsis sp. ATCC 39116 and with members of the genus Amycolatopsis in general have shown that it is quite difficult to generate deletion mutants (5, 6, 38). Therefore, our first goal was to improve the homologous integration of the generated suicide plasmids. The generation of the first and only Amycolatopsis sp. ATCC 39116 deletion mutant required a very long time period, and hundreds of mutant candidates had to be screened (5, 6).
The first step was to analyze the sequence of the plasmid p6SUI2 for problematic sequences. These analyses identified an open reading frame (ORF) that exhibited a high identity to the putative integrase gene int3 (GenBank accession no. ADE34474.1). This part of the plasmid was cloned from Tn5096 as part of the apramycin resistance gene (12, 39). Since this enzyme could be responsible for the nonhomologous integration of the suicide vector, it was a prime target for removal. The removal of this sequence led to the construction of plasmid p6SUI3.
After cloning of the flanking region of the yagRST cluster into vector p6SUI3, we transformed strain F33 with the finished construct and checked the resulting mutants for homologous integration events. Since we could not identify any such event, we then tried to identify which part of the vector facilitated the nonhomologous recombination into the chromosome. We used the primer combinations p6sui_for4 (5′-CAGCATCGCCAGTCACTATGGC-3′) and p6sui_rev1 (5′-CGGTTCATGTGCAGCTCCATCAGC-3′) and p6sui_for2 (5′-GCTGATGGAGCTGCACATGAACCG-3′) and p6sui_rev2 (5′-CACGTAGATCACATAAGCACCAAGCG-3′), as well as p6sui_for3 (5′-CGCTTGGTGCTTATGTGATCTACGTG-3′) and p6sui_rev3 (5′-GCCATAGTGACTGGCGATGCTG-3′), to amplify all regions of the integrated plasmid and to determine the locus for the nonhomologous integration. The first combination amplified the apramycin resistance gene, the second amplified the E. coli and partial Amycolatopsis origin of replication, and the third one amplified the yagRST flanking region, including 78 bp of the upstream and 14 bp of the downstream region. PCR results showed that the origins of replication oriV and pArep as well as the resistance cassette were intact. Only primers p6sui_for3 and p6sui_rev3 resulted in no amplification, which indicated that the plasmid integrated over either the fused flanking region or the short sequences adjacent to it. We generated two plasmids based on these findings that were missing either 78 bp upstream (p6SUI4::ΔyagRST) or 14 bp downstream (p6SUI3RF::ΔyagRST) of the flanking regions, and we tested these for homologous integration. Plasmid p6SUI4::ΔyagRST showed 2 homologous insertions for 20 clones that were checked into the left flank of the yagRST cluster, while p6SUI3RF::ΔyagRST showed no homologous integration at all.
Since removing these 78 bp through means of restriction enzymes would have also removed the EcoRV site and since we still observed nonhomologous integrations, we chose to further reduce the size of the plasmid and to introduce a new EcoRV site by PCR, thus generating plasmid p6SUI5. Thereby, a total of 787 bp was removed between the apramycin resistance cassette and oriV. Vector maps of the suicide vectors are shown in Fig. 1. The obtained vectors, together with their unique restriction sites that are not present in annotated genes or other important regions, are shown in Fig. 2.
FIG 1.

Vector maps of the suicide vectors that were used and generated in this study. All single cutter enzymes that do not cut inside any of the annotated genes and other elements are shown on the vector maps. apraR, apramycin resistance gene; oriV, pBR origin of replication for E. coli; pArep, partly deleted, nonfunctional origin of replication for Amycolatopsis from plasmid pA387; int3, putative integrase sequence; permE, erythromycin promoter from Saccharopolyspora erythraea; rpsL, gene coding for 30S ribosomal subunit protein S12 from S. erythraea; aacC1, part of a putative acetyltransferase gene.
FIG 2.

Schematic illustration of the construction of the vectors used in this study. Primers used in the construction are depicted as blue arrows. apraR, apramycin resistance gene; oriV, pBR origin of replication for E. coli; pArep, remaining nonfunctional part of the origin of replication for Amycolatopsis from plasmid pA387; int3, putative integrase sequence; permE, erythromycin promoter from Saccharopolyspora erythraea; rpsL, gene coding for 30S ribosomal subunit protein S12 from S. erythraea; aacC1, part of a putative acetyltransferase sequence; ΔyagRST, fused flanking region of the yagRST cluster.
Generation of the yagRST deletion mutant.
The two heterogenotes that were generated using plasmid p6SUI4::ΔyagRST were then cultivated to generate the second recombination event leading to either the deletion of the yagRST cluster or the restoration of the wild type. For this, we cultivated both mutants for 3 weeks while plating out the culture broth every 2 to 3 days on glucose-yeast extract medium (GYM) agar. Growing colonies were then transferred to plates containing 50 μg/ml apramycin or no antibiotic to check for the loss of the suicide plasmid. After approximately 2,000 screened clones, we were able to identify eight clones which showed a loss of the apramycin resistance. From these, only one showed the loss of the yagRST cluster while the others showed a restoration of the wild type. We then checked for the reintegration of the yagRST cluster using PCR, which showed no reintegration of the cluster into another locus.
Counterselection system.
The second problem in generating Amycolatopsis sp. ATCC 39116 deletion mutants was the low rate of homologous recombination after the suicide vector had integrated into the genome and heterogenotes were generated (5). To alleviate this, a counterselection system was used that allowed a first separation of clones that still carry the plasmid from those that do not, without requiring a thorough analysis of each one. We decided to use the rpsL system (21). This system is based on the dominant phenotype of streptomycin sensitivity but requires a strain that is resistant to streptomycin due to mutations of the rpsL gene to begin with (22). An inspection of the Amycolatopsis rpsL sequence showed that the strain should be sensitive to streptomycin. To verify this, we did a growth experiment of the cells in liquid GYM supplemented with 0, 25, 50, and 100 μg/ml of streptomycin over 24 h. None of the cultures containing streptomycin showed any growth under these conditions (Fig. 3). Several streptomycin-resistant mutants were then generated as described in Materials and Methods. One of them was further characterized and named S33. Its growth was tested in GYM with 0, 100, and 500 μg/ml of streptomycin, where it grew to an optical density at 600 nm (OD600) of 9.2, 9.1, and 7.3, respectively, after 9.5 h. Sequencing of the rpsL gene of this mutant showed the exchange of lysine for arginine at position 42. To confer streptomycin sensitivity to the now-resistant clones, the rpsL gene from Saccharopolyspora erythraea NRRL 2338 (40) was chosen and first cloned into the vectors p6pkan and p6permE containing the kanamycin and erythromycin promoters, respectively (12). To test the functionality of the counterselection system, strain S33 was transformed with these plasmids. A single clone of each transformation was then cultivated on GYM agar plates containing either 50 μg/ml apramycin or 50 μg/ml apramycin and 100 μg/ml streptomycin. The strain containing p6permE::rpsL was unable to grow on medium containing both antibiotics, except for a few small single colonies. The mutant S33 p6pkan::rpsL showed no impaired growth under these conditions (data not shown).
FIG 3.
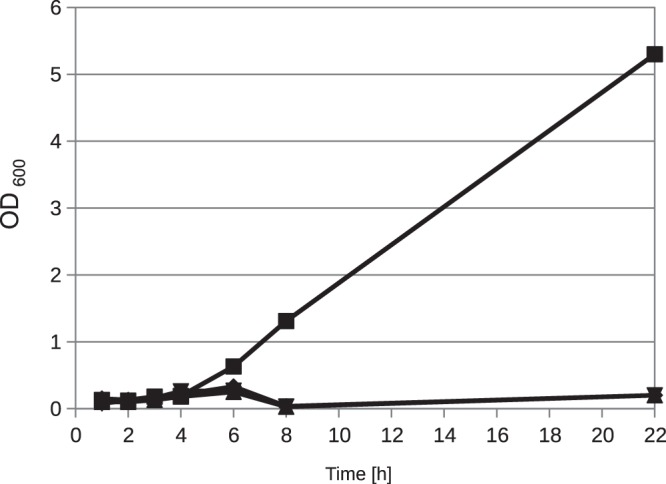
Growth experiment with Amycolatopsis sp. ATCC 39116 in the presence of streptomycin. The cells were grown for 14 h in liquid GYM; then, 1% (vol/vol) of the cells was transferred to fresh GYM supplemented with 0 (■), 25 (◆), 50 (▲), or 150 (▼) μg/ml of streptomycin. Cultures were cultivated at 42°C, and the optical density was measured at a wavelength of 600 nm.
Since the rpsL gene under the control of the kanamycin promoter was obviously not suitable for our purpose and the one under the control of the erythromycin promoter showed the expected streptomycin sensitivity, we only used this one for further experiments. This was then cloned into the suicide vector p6SUI5 as described above, resulting in the vector p6SUI5ERPSL.
Generation of the vdcBCD and vanAB deletion mutants.
To generate the suicide plasmids for vdcBCD, the ΔvdcBCD deletion construct was ligated into the EcoRV-digested vectors as described above. Afterwards, transformation of S33 was carried out as described before, and the clones were analyzed for the homologous integration of the suicide plasmids. For each strain, 100 clones were checked via PCR. We observed homologous integration for 50% of the mutants based on p6SUI5::ΔvdcBCD and p6SUI5ERPSL::ΔvdcBCD, while none could be identified for p6SUI2::ΔvdcBCD and p6SUI3::ΔvdcBCD.
We then tried to generate the second recombination event for one of the correct S33 (p6SUI5ERPSL::ΔvdcBCD) clones as described above. After 24 h of incubation, we plated 100 μl of 10−6-diluted culture broth onto GYM agar plates containing either 250 μg/ml streptomycin or no antibiotic at all. The same was done after 48 h. More than 100 clones could be identified on each plate, with the streptomycin plates generally showing smaller colonies. Sixty clones were transferred from each plate onto GYM agar plates containing 50 μg/ml apramycin or no antibiotic to screen for the loss of the suicide plasmid (see Fig. S1 in the supplemental material). Twenty-six of 60 clones showed a loss of apramycin resistance after 24 h, when the culture was plated first onto agar plates containing streptomycin. Otherwise, no loss of apramycin resistance could be observed. Sixteen colonies showed growth on streptomycin as well as on apramycin. After 48 h, all of the clones treated with streptomycin showed a loss of the apramycin resistance, while no loss occurred for the untreated clones (data not shown). The resulting apramycin-sensitive clones were checked for the deletion of vdcBCD and showed a deletion of the cluster in about half of the cases. One of these mutants was sequenced and checked for reintegration of the deleted gene cluster and then referred to as Amycolatopsis sp. ATCC 39116 Δvdh ΔvdcBCD Strr.
The vanAB deletion mutant was directly generated using vector p6SUI5ERPSL. Otherwise, the procedure was identical to the generation of the vdcBCD deletion mutant described above. Four clones were checked after the transformation, and each one showed the homologous integration of the suicide plasmid into the flanking region of the vanAB cluster. The resulting clones were then cultivated without selection pressure and analyzed with regard to their streptomycin and apramycin resistance as described previously. Through this, we were able to generate mutant Amycolatopsis sp. ATCC 39116 Δvdh ΔvanAB Strr.
Characterization of the deletion mutants.
Since the effects of these deletions on Amycolatopsis sp. ATCC 39116 were unknown, we characterized the generated mutants. For the ΔvdcBCD and ΔvanAB mutants, we used flask experiments to analyze the growth on and biotransformation of vanillic acid. For the ΔyagRST mutant, we verified the phenotype during 2-liter-scale fermentations. The biotransformation of vanillic acid for the ΔvdcBCD mutant showed that the mutant is no longer capable of forming guaiacol but still consumes vanillic acid, albeit at a significantly lower rate than S33 (Fig. 4). Protocatechuic acid was detectable in only trace amounts in both cases (data not shown). S33 completely consumed the vanillic acid during the first 6 h of the experiment, while the ΔvdcBCD mutant required between 10 and 26 h to fully consume this compound. The guaiacol concentration for S33 rose sharply until 2.5 h and started to decrease only after 4.5 h, when vanillic acid was depleted.
FIG 4.

Biotransformation of 0.3% (wt/vol) vanillic acid by resting cells of Amycolatopsis sp. ATCC 39116 Δvdh Strr and Amycolatopsis sp. ATCC 39116 Δvdh ΔvdcBCD Strr. Vanillic acid concentrations are represented by ■ for ΔvdcBCD and ▲ for Δvdh. Guaiacol concentrations are represented by ● for ΔvdcBCD and ▶ for Δvdh.
With vanillic acid as the only carbon source, we were able to observe a strongly reduced growth rate in the vdcBCD mutant starting between 8 and 24 h and reaching its maximum after 50 h (Fig. 5). After 24 h, it had just reached the same optical density as both S33 and the ΔvanAB mutant reached after 8 h. S33 and the ΔvanAB mutant showed growth after 6 h and reached their maxima after 24 and 32 h, respectively. The growth rate for the ΔvanAB mutant was comparable to that of S33, being only slightly lower, but the cultures showed a higher final optical density, albeit with a lot of variance between the triplicates. After 32 h, both S33 and the ΔvanAB mutant showed a decline in optical density, while the ΔvdcBCD mutant showed an ongoing increase.
FIG 5.

Growth of strains S33, S33 ΔvdcBCD, and S33 ΔvanAB in MSM with vanillic acid (0.1%, wt/vol) as sole carbon source. Cells were grown at 42°C in 250-ml flasks with baffles. All growth experiments were performed as triplicates, and the mean value is shown together with the standard deviation for S33 (■), S33 ΔvanAB (▲), and S33 ΔvdcBCD (◆). The left panel shows the change of the optical density at 400 nm, and the right panel shows the concentrations of vanillic acid (black lines) and guaiacol (gray lines). Protocatechuic acid was not detectable in any sample.
The metabolite concentrations showed a similar picture. S33 depleted the vanillic acid slightly faster than the ΔvanAB mutant and also reached slightly higher guaiacol concentrations. Small amounts of vanillic acid were still remaining in the ΔvanAB cultures after 24 h. The guaiacol concentration increased steadily for both strains and started to fall only after the depletion of vanillic acid. The ΔvdcBCD mutant showed no decrease in vanillic acid until after 10 h into the cultivation and also no detectable guaiacol formation. Protocatechuic acid was not detectable at any point during the cultivation for any of the mutants. Interestingly, after 2 h the vanillic acid concentration had already decreased by 0.2 g/liter for the ΔvanAB mutant and S33 without the strains showing growth. After 4 h, between 40 and 50% of the supplied vanillic acid was already degraded, with both strains just entering the exponential phase. The bulk of the detectable growth occurred only after the vanillic acid was depleted.
During the fermentations of the ΔyagRST mutant, we observed a phenotype that was similar to that of the S33 mutant. The only differences were a slight decrease in vanillin and vanillic acid concentrations in comparison to S33. The consumption of ferulic acid remained unchanged under these conditions (Fig. 6).
FIG 6.

Two-liter fermentations of mutants F33 (left) and F33 ΔyagRST (right) with ferulic acid as the substrate. The cells were grown for 10 h until they reached the stationary phase before feeding was started. Metabolite concentrations were determined every hour by HPLC: ferulic acid (■), vanillin (◆), and vanillic acid (▼).
DISCUSSION
Amycolatopsis sp. ATCC 39116 is an important organism for the production of natural vanilla flavor (6, 9, 41). Several laboratories have studied this strain, but most of them focused on the optimization of fermentation parameters (7, 8, 10, 11). So far, only one knockout and one genomic integration have been described for this strain, which highlights the difficulties in genetically manipulating it (5, 6). Therefore, in this study we focused on the optimization of our knockout vectors to ease future studies with this strain. The target gene clusters coding for an aldehyde oxidase, a vanillate decarboxylase, and a vanillate demethylase were chosen to identify their role in vanillic acid formation or vanillic acid degradation, respectively.
Since we observed a large number of nonhomologous integrations during the generation of the abovementioned knockout mutant F33 (5), we first tried to reduce the size of our suicide plasmid and to determine the cause of these illegitimate recombinations. A first sequence analysis showed the presence of a putative integrase int3 homologue on our suicide plasmid, which was part of the previously cloned apramycin resistance cassette (12). The removal of this sequence resulted in vector p6SUI3 but showed no change in the rate of homologous integration.
We then tried to identify intact areas of the vector inside our transformants, to determine the locus of nonhomologous integration. We were able to pinpoint this to an area 78 bp upstream of the yagRST fused flanking region, which when removed dramatically increased the rate of homologous insertion from <1% as shown for p6SUI3::ΔyagRST and in our previous study (5) to 10%. The removed sequence shows only short areas with less than 20 bp of homology with the Amycolatopsis genome. It has been shown in E. coli that homologous overlaps of 5 to 14 bp can be enough for recombination, but even on p6SUI5, there remain homologous areas of 17 to 19 bp (42). These remaining homologous regions could be the reason for the remaining illegitimate recombinations that still occur with these vectors.
The 78-bp-long sequence which was removed also shows some repeats and inverted repeats with a maximum length of 6 bp, which is shorter than most of the inverted repeats that were found flanking other integrating elements, for example, in the phage λ or ϕC31 (43, 44). Especially for the actinophage ϕC31, the conserved attachment site TTG is very short and the inverted repeats possess lengths of only 4, 7, and 9 bp (44). Additionally, this sequence shows 100% identity with several class 1 integron sequences from various organisms (Proteus mirabilis, Serratia marcescens, and Pseudomonas putida) (e.g., NCBI accession number KJ620481.1). The sequence homology comprises a total of 169 bp upstream of the EcoRV site that was used for integration of the various flanking regions and downstream of oriV and shows 100% sequence identity except for a 26-bp gap. However, the annotation of the class 1 integron sequences shows that the homologous sequence contains part of the gene aacC1, coding for the protein 3-N-aminoglycoside acetyltransferase, which probably confers resistance to aminoglycoside antibiotics (45). Important sequences for the functioning of an integrase, like the attachment site attB or the integrase itself, are not present on this plasmid. If they are present, then they exhibit no homologies to other already-known ones (46).
Since Amycolatopsis sp. ATCC 39116 possesses 26 genes encoding enzymes annotated as integrases or site-specific integrases (e.g., accession numbers WP_027936153.1 and WP_039791203.1) in its genome, it is also possible that a part of the suicide vector is recognized by one of these enzymes as a pseudo-attP site, thus facilitating recombination into the genome. It has been shown in other organisms that the well-known Streptomyces phage ϕC31 integrates at sites that show only between 18 and 31% sequence identity to the normal attP site (47). Since most of these integrases are uncharacterized, it cannot be concluded with certainty if this is also the case here. Another question would then be why the replicating expression plasmids generated previously in our laboratory are replicating without being modified (12), especially since all our newly generated plasmids presented in this study derive from plasmid pRLE6, which is a fragment of vector pRLE60 that is not further modified in Amycolatopsis sp. ATCC 39116 (13, 48). The cause of the drastic difference in the rate of homologous integration between p6SUI3 and p6SUI5 is therefore currently unknown.
The deletion of an additional 787 bp of the vector backbone between the apramycin resistance cassette and the E. coli origin of replication oriV, including the previously mentioned 78 bp, led to the construction of vector p6SUI5 and showed a further increase in homologous integrations to up to 100% in the case of the vanAB deletion and 50% in the case of the vdcBCD mutant. The rare but still occurring nonhomologous integrations are most likely due to random double-strand breaks of the vector DNA that induce the cellular DNA repair systems that then introduce the suicide vector at loci containing nonhomologous or only short homologous areas into the genome (49, 50).
Another problem with the generation of knockouts in Amycolatopsis sp. ATCC 39116 is the low rate of homologous recombination, leading to the removal of the integrated suicide plasmid. We previously applied a system based on the gene gusA, coding for a β-glucuronidase (5). Colonies of clones which still carry the suicide plasmid would occur in blue when overlaid with 5-bromo-4-chloro-1H-indol-3-yl β-d-glucopyranosiduronic acid (X-Gluc), while clones which lost it would keep their natural yellow color (5, 27). The main problem with this approach was the low homologous recombination rate, since about 100 colonies had to be screened to identify a single one that lost the suicide plasmid, as was demonstrated in this study with the deletion of the yagRST cluster. Another problem stems from the inherent yellow color of the cells, which makes it hard to differentiate between colored and noncolored cells during screening since the blue coloring often ends up as a weak green hue. We also speculated that the gusA gene might lead to more nonhomologous recombination events in Amycolatopsis sp. ATCC 39116 (5).
To overcome these difficulties, we tried to establish a counterselection system that allowed a reduction of the number of clones still carrying the suicide plasmid before performing a more thorough analysis. To accomplish this goal, we chose the rpsL-based streptomycin sensitivity system which was successfully applied in Streptomyces roseosporus and other Gram-positive bacteria (21). To use this system, we first generated a spontaneous streptomycin-resistant mutant that showed a mutation in the rpsL gene consistent with mutations observed in streptomycin-resistant mutants of Mycobacterium smegmatis (51). The streptomycin-sensitive rpsL gene from Saccharopolyspora was chosen to obtain a copy of the gene that is homologous enough to Amycolatopsis to prevent problems during expression but is also not completely homologous, to prevent recombination via this gene.
During tests with the rpsL gene under the control of the kanamycin and erythromycin promoter, we observed no streptomycin-sensitive clones with the former. Most likely, the expression of the sensitive rpsL gene under the control of the kanamycin promoter is not strong enough to confer its normally dominant phenotype (12, 22). This is consistent with results from Neisseria gonorrhoeae, where a streptomycin-sensitive rpsL gene from E. coli did not result in change of the phenotype, likely due to its poor expression (52).
While generating the second homologous recombination, we observed a loss of the apramycin resistance in 26 clones after only 24 h while after 48 h all of the clones plated onto streptomycin had lost their apramycin resistance. The fact that not all streptomycin-resistant clones are also apramycin sensitive is most likely due to mutations in the sensitive rpsL gene or its promoter region, since any mutation that inhibits its function will lead to a streptomycin-resistant phenotype. Over time, the number of clones that lose the vector through homologous recombination increases, thus leading to more clones that are streptomycin resistant as well as apramycin sensitive. Clones that were not subjected to streptomycin showed no loss of apramycin resistance after 24 h or 48 h. This is a huge improvement over the 5 to 21 days which were previously required to generate homogenotes and also over the rates at which these appeared (5).
The analysis of the generated homogenotes showed that the deletion of the yagRST cluster using vector p6SUI4::ΔyagRST in 8.5% of the analyzed clones. For the clusters vanAB and vdcBCD, we observed the deletion of the targeted gene in about 50% of the clones, as would be expected for a nonessential gene.
The characterization of the ΔyagRST mutant showed a phenotype that was nearly identical to that of mutant S33. Previously, it was shown that these enzymes are capable of oxidizing vanillin to vanillic acid (30). The reduced formation of vanillic acid was most likely indirectly caused by the reduced vanillin concentration. It can be assumed that this enzyme either is not capable of utilizing vanillin or is at least not active under these conditions. The other three genes that are clustered together with the three subunits of yagRST are a transcription regulator, a putative epoxide hydrolase, and a conserved hypothetical protein and allow no conclusions to be drawn in regard to their function.
The deletion of the vdcBCD cluster had a more noticeable effect. The generated mutant shows a strong reduction in growth rate and an increase in lag phase while growing on vanillic acid as the sole carbon source. This indicates that the decarboxylation to guaiacol is indeed the main degradation pathway of vanillic acid while the demethylation pathway plays only a minor role under these conditions (6). This is further supported by the limited impact of the deletion of the ΔvanAB cluster. While the deletion mutant showed a slight reduction in growth rate and vanillic acid depletion, the final optical density of the deletion mutant was higher than that of parent strain S33, although with a large variance between the three growth experiments. Since no further putative vanAB homologues exist in Amycolatopsis sp. ATCC 39116, it is unlikely that the vanAB deletion is complemented by another enzyme complex catalyzing the same reaction. The theoretical possibility that the deletion has a positive effect on the growth of Amycolatopsis sp. ATCC 39116 due to the reduced formation of formaldehyde can now also be excluded. This might have various reasons. The nearly imperceptible impact of the deletion shows that this reaction contributes only relatively little to the vanillic acid degradation, thus also limiting the formation of formaldehyde from this step. Additionally, further degradation most likely involves demethylation to catechol which is then cleaved. This step will probably also produce formaldehyde, which the strain then has to detoxify (34).
Under growth conditions, the decarboxylation seems to be the main pathway utilized by Amycolatopsis sp. ATCC 39116, which is also supported by the strong smell caused by guaiacol that is noticeable in strain S33 immediately after supplementation with vanillic acid. To our knowledge, this is the first deletion of a vdcBCD cluster in any strain to date, while the deletion of vanAB has already been performed and studied in multiple strains. It always led to a vanillic acid-negative phenotype, for example, in Corynebacterium glutamicum (53), Rhodococcus jostii RHA1 (54), or Agrobacterium fabrum (55). This is in strong contrast to the phenotype observed with the deletion mutant presented here. The vanillate decarboxylase VdcBCD has been characterized in multiple strains, e.g., Streptomyces sp. D7 (31), Bacillus subtilis (32), Nocardia sp. (56), or Rhodotorula rubra (57), but has so far not been deleted or disrupted in these strains. A homology search for the VanA sequence from Amycolatopsis sp. ATCC 39116 resulted in a large number of hits with sequence identities of more than 75% in strains like Nocardia, Streptomyces, and Amycolatopsis. As an example, we were able to identify a likely VanA (79% amino acid identity) as well as VdcC (76% identity) homologue in Streptomyces sp. strain NRRL S-813. Thus, it seems probable that at least other Streptomyces strains possess both pathways to degrade vanillic acid either to protocatechuate or to guaiacol, even though this has not been studied so far.
In conclusion, we were able to develop an improved knockout system for Amycolatopsis sp. ATCC 39116, which shows a drastically reduced rate of nonhomologous integrations and which thereby allows for a faster generation of hetero- and homogenotes. The yagRST cluster does not seem to have an impact on the vanillin metabolism of our strain. The vdcBCD cluster is responsible for decarboxylation of vanillic acid to guaiacol and is also the main way for our strain to utilize vanillic acid as sole carbon source. The deletion of the vanAB cluster has only a negligible effect during growth on vanillic acid and is not required for growth on this compound to any degree.
MATERIALS AND METHODS
Bacterial strains, plasmids, and cultivation conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. Cells of Escherichia coli were grown in lysogeny broth (LB) medium at 30°C (58), and cells of Amycolatopsis sp. ATCC 39116 or Saccharopolyspora erythraea were grown in liquid GYM (4 g/liter glucose, 4 g/liter yeast extract, 10 g/liter malt extract) or on GYM agar plates (additionally 2 g/liter CaCO3, 18 g/liter agar) (modified from the work of Shima et al. [59]) at 42°C and 37°C, respectively. For transformation purposes, cells of Amycolatopsis were grown in tryptic soy broth (TSB) liquid medium which was prepared according to manufacturer specifications. Additionally, cells of Amycolatopsis could be grown in liquid modified H16 mineral salts medium (MSM) (60) [Na2HPO2·2H2O, 4.5 g; KH2PO4, 1.5 g; NH4Cl, 1 g; MgSO4·7H2O, 0.2 g; CaCl2·2H2O, 0.02 g; Fe(III)NH4-citrate, 1.2 mg; trace element solution SL6 (61), 0.1 ml; demineralized water to 1,000 ml]. Suitable carbon sources were added as filter-sterilized stock solutions to the mineral medium. For the selection of transformants, the following antibiotics were used: apramycin at 50 μg/ml and streptomycin at 250 μg/ml. The optical density of cultures was measured at 600 or 400 nm.
TABLE 1.
List of strains and plasmids used in this study
| Bacterial strain or plasmid | Description | Reference or source |
|---|---|---|
| Strains | ||
| Amycolatopsis sp. ATCC 39116 Δvdh (F33) | vdh deletion mutant of Amycolatopsis sp. ATCC 39116 | 5 |
| Amycolatopsis sp. ATCC 39116 Δvdh Strr (S33) | Spontaneously streptomycin-resistant mutant of F33 | This study |
| Amycolatopsis sp. ATCC 39116 Δvdh ΔyagRST (FMALDOX1) | vdh and yagRST cluster deletion mutant of Amycolatopsis sp. ATCC 39116 | This study |
| Amycolatopsis sp. ATCC 39116 Δvdh ΔvdcBCD Strr (FMVDC1) | vdh and vdcBCD deletion mutant of Amycolatopsis sp. ATCC 39116, based on S33 and thus streptomycin resistant | This study |
| Amycolatopsis sp. ATCC 39116 Δvdh ΔvanAB Strr (HPVANAB1) | vdh and vanAB deletion mutant of Amycolatopsis sp. ATCC 39116, based on S33 and thus streptomycin resistant | This study |
| Saccharopolyspora erythraea NRRL 2338 | Wild type | 40 |
| E. coli K-12 ER2925 | ara-14 leuB6 fhuA31 lacY1 tsx-78 glnV44 galK2 galT22 mcrA dcm-6 hisG4 rfbD1 R(zgb-210::Tn10)Tets endA1 rpsL136 dam13::Tn9 xylA5 mtl-1 thi-1 mcrB1 hsdR2 | New England BioLabs, Inc. (Ipswich, MA) |
| Plasmids | ||
| p6pkan | E. coli-Amycolatopsis shuttle vector carrying an apramycin resistance gene and the kanamycin promoter | 12 |
| p6pkan::rpsL | p6pkan with rpsL gene from Saccharopolyspora erythraea cloned into the NheI/HindIII site | This study |
| p6permE | E. coli-Amycolatopsis shuttle vector carrying an apramycin resistance gene and the erythromycin promoter from Saccharopolyspora erythraea | 12 |
| p6permE::rpsL | p6permE with rpsL gene from Saccharopolyspora erythraea cloned into the NheI/HindIII site | This study |
| p6SUI2 | Amycolatopsis suicide plasmid, replicates in E. coli, carries an apramycin resistance gene, internally named p6SUI2, designated p6sui in the referenced paper | 5 |
| p6SUI3 | PmlI/HindIII fragment of p6SUI2, blunted using T4 polymerase and religated afterward, removing int3 sequence | This study |
| p6SUI5 | Amplification with the primers pBBR_Ori_Rev_EcoRV and ApraR_Rev_EcoRV with p6SUI3 as the template, removes more of the vector backbone and adds an EcoRV restriction site | This study |
| p6SUI5::permE::rpsL (p6SUI5ERPSL) | p6SUI5 with rpsL gene under the control of the erythromycin promoter cloned into the NruI site | This study |
| p6SUI2::ΔvdcBCD | p6SUI2 with flanking regions of vdcBCD cluster inserted into the EcoRV site | This study |
| p6SUI3::ΔvdcBCD | p6SUI3 with flanking regions of vdcBCD cluster inserted into the EcoRV site | This study |
| p6SUI5::ΔvdcBCD | p6SUI5 with flanking regions of vdcBCD cluster inserted into the EcoRV site | This study |
| p6SUI5::permE::rpsL::ΔvdcBCD (p6SUI5ERPSL::ΔvdcBCD) | p6SUI5::permE::rpsL with flanking regions of vdcBCD cluster inserted into the EcoRV site | This study |
| p6SUI5::permE::rpsL::ΔvanAB (p6SUI5ERPSL::ΔvanAB) | p6SUI5::permE::rpsL with flanking regions of vanAB cluster inserted into the EcoRV site | This study |
| p6SUI2::ΔyagRST | p6SUI2 with flanking regions of yagRST cluster cloned into the EcoRV restriction site | This study |
| p6SUI3::ΔyagRST | p6SUI3 with flanking regions of yagRST cluster cloned into the EcoRV restriction site | This study |
| p6SUI4::ΔyagRST | p6SUI3::ΔyagRST where 78 bp upstream of the flanking regions has been removed via PCR | This study |
| p6SUI3RF::ΔyagRST | p6SUI3::ΔyagRST where 14 bp downstream of the flanking regions has been removed via PCR | This study |
Identification of target genes.
To identify potentially interesting target genes, a literature search was performed. BLAST (62) was used to search for homologous proteins in Amycolatopsis sp. ATCC 39116. The basis for these sequence searches was the genome sequence published by Davis et al. in 2012 (14). Through this, we identified the gene clusters vdcBCD, vanAB, and yagRST, coding for the vanillate decarboxylase complex, vanillate demethylase complex, and a putative aldehyde oxidase complex, respectively.
DNA isolation and manipulation.
Plasmid DNA from E. coli was isolated using the peqGOLD plasmid miniprep kit I (PeqLab GmbH, Erlangen, Germany) according to the manual. Genomic DNA from Amycolatopsis sp. ATCC 39116 and Saccharopolyspora erythraea was isolated using the method described by Ausubel et al. (63). DNA digestion was performed with restriction enzymes from Thermo Fisher Scientific (Thermo Fisher Scientific GmbH, Schwerte, Germany) using buffers and conditions as described by the supplier. Isolation of DNA from agarose gels was performed using the peqGOLD gel extraction kit (PeqLab GmbH, Erlangen, Germany). Phosphorylation and dephosphorylation of DNA were performed using T4 polynucleotide kinase or FastAP thermosensitive alkaline phosphatase (Thermo Fisher Scientific GmbH, Schwerte, Germany), respectively. PCR was performed using Phusion Hot-Start II high-fidelity polymerase (Thermo Fisher Scientific GmbH, Schwerte, Germany) with a modified combination of the Touchdown and Slowdown PCR protocols described by Frey et al. (18). The GC buffer supplied by the manufacturer was used, and 10% (vol/vol) dimethyl sulfoxide (DMSO) was added to each PCR mixture. All heating and cooling rates were limited to 1.5°C/s and 2.5°C/s, respectively. The annealing temperature was calculated using the Phusion Tm calculator supplied by Thermo Fisher Scientific. The first two cycles started with the calculated annealing temperature +5°C, which was then decreased every two cycles by 1°C for a total of 20 cycles. Afterward, 15 cycles were performed with the calculated annealing temperature. Thirty seconds at 98°C ensured complete denaturing of the template during initial denaturation as well as in the following cycles. All other concentrations and parameters were chosen according to manufacturer specifications. The template for PCR was always genomic DNA isolated from cells of Amycolatopsis sp. ATCC 39116.
Transfer of DNA.
Plasmids were transferred into E. coli K-12 ER2925 using the rubidium chloride method (58) and into Amycolatopsis sp. ATCC 39116 using the direct mycelium transformation technique previously described (13).
Construction of the suicide plasmids.
To generate the different generations of the suicide plasmid presented in this study, we used the previously described vector p6sui as a basis (5). Internally, this vector is designated p6SUI2, thus the numbering of the derivative vectors as 3, 4, and 5, respectively. A schematic overview of the vector construction can be seen in Fig. 1. p6SUI3 was generated by digesting p6SUI2 with the enzymes PmlI and HindIII, removing part of the vector backbone containing a sequence coding for an integrase (int3) gene. The restriction fragment was blunted afterward using the T4 polymerase enzyme and religated, and E. coli ER2925 was transformed with the resulting plasmid. The vectors p6SUI4 and p6SUI3RF were constructed specifically for the deletion of the yagRST cluster on the basis of plasmid p6SUI3::ΔyagRST, the construction of which is detailed further below. Plasmid p6SUI4::ΔyagRST was generated by PCR using the primers p6sui_rev_2 (5′-CACGTAGATCACATAAGCACCAAGCG-3′) and aldox_LF_For (5′-GTGCGGGAGGCGGTGC-3′). Plasmid p6SUI3RF::ΔyagRST was generated using the primers aldox_RF_Rev (5′-CTCTGCCGATCAGCGCACC-3′) and p6sui_For_4 (5′-CAGCATCGCCAGTCACTATGGC-3′). The resulting fragment was phosphorylated and religated, and E. coli ER2925 was transformed with it. Plasmid p6SUI5 was generated by PCR using the primers ApraR_Rev_EcoRV (5′-ATCCTCAGCCAATCGACTGGCG-3′) and pBBR_Ori_Rev_EcoRV (5′-ATCGGCTAGGTGAAGATCC-3′) with p6SUI3 as the template. The resulting amplicon was phosphorylated and religated, and E. coli ER2925 was transformed with the resulting plasmid. To generate the plasmids containing the streptomycin-sensitive rpsL gene from Saccharopolyspora erythraea under the control of the pkan or permE promoter, it was first amplified from isolated S. erythraea genomic DNA using the primers rspL_For_NheI_SD (5′-ATATGCTAGCAGAAGGGCCGGTCCATGCCC-3′) and rpsL_Rev_HindIII (5′-ATATAAGCTTGGGCATCAGCTCTTCTCCTTC-3′). The resulting fragment was purified, digested with NheI and HindIII, and then ligated into the NheI/HindIII-digested and dephosphorylated p6pkan or p6permE plasmid (12). This led to the construction of the vectors p6pkan::rpsL and p6permE::rpsL. The rpsL gene under the control of the erythromycin promoter was then amplified from p6permE::rpsL using the primers pErmE_For (5′-CAGCCCGACCCGAGCAC-3′) and rpsL_Rev_HindIII. The resulting amplicons were then phosphorylated and cloned into the dephosphorylated NruI site of the p6SUI5 plasmid, resulting in the construction of the plasmid p6SUI5ERPSL.
To generate the knockout vectors for specific genes, the fused flanking regions (∼1.5 kb each) of the targeted gene had to be cloned into the EcoRV site of p6SUI, p6SUI3, p6SUI5, and p6SUI5ERPSL. The flanking regions were amplified using the PCR method described above with isolated genomic DNA of Amycolatopsis sp. ATCC 39116 as the template. The left flanking region of the vdcBCD cluster was amplified using the primers vdcBCD_LF_For (5′-GCAGAACGACGCGGAGGAGG-3′) and vdcBCD_LF_Rev_RF (5′-CACATCAGCGTGTCCGCCCGCGCCCTCCCTGTTAGTTC-3′). The right flanking region was amplified using the primers vdcBCD_RF_For_LF (5′-GAACTAACAGGGAGGGCGCGGGCGGACACGCTGATGTGG-3′) and vdcBCD_RF_rev (5′-GTGTCGCGCAGGCGGC-3′).
The procedure was identical for the yagRST deletion construct with the exception of the primers used. For the left flank, these were aldox_LF_For (5′-GTGCGGGAGGCGGTGC-3′) and aldox_LF_Rev_RF (5′-CGACCATAACGAGACCCACCGACGTACCCGGCGGGACAGCC-3′). The right flank was amplified using the primers aldox_RF_For_LF (5′-GGCTGTCCCGCCGGGTACGTCGGTGGGTCTCGTTATGGTCG-3′) and aldox_RF_Rev (5′-CTCTGCCGATCAGCGCACC-3′).
The same steps were performed for the vanAB deletion construct. The left flank was amplified using primers vanALF_For (5′-CGCCCAGGCGGCCGG-3′) and VanA_lF_rev_rF (5′-CTGCCCCGCGCCGGCCTGCAGCTCCTTCGGTGTAGATG-3′). The right flank was amplified using the primers VanB_rF_for_lF (5′-CATCTACACCGAAGGAGCTGCAGGCCGGCGCGGGGCAG-3′) and VanB_reFla_r (5′-CGCCCAGGCGGCCGG-3′). The resulting amplicons of each deletion target were purified and combined in a following fusion PCR. The fused construct was then phosphorylated and cloned into the dephosphorylated EcoRV-linearized suicide vector.
Generation of a streptomycin-resistant Amycolatopsis sp. ATCC 39116 mutant.
To make use of the rpsL gene in the suicide plasmids, a resistant mutant with a mutated rpsL gene of Amycolatopsis sp. ATCC 39116 was generated. This study was based on the Δvdh mutant since it is our current production strain. The strain was grown in liquid GYM in the presence of increasing concentrations of streptomycin starting with 10 μg/ml for 24 h and further steps of 25 μg/ml, 50 μg/ml, and 100 μg/ml for 24 h each. After 24 h, 0.1% (vol/vol) of the culture was transferred to fresh GYM with the increased streptomycin concentration. Finally, the cells were streaked out on GYM agar plates with 250 μg/ml streptomycin. Total DNA was isolated from multiple colonies, and the rpsL gene was sequenced to identify mutations.
Generation and identification of deletion mutants.
The strains F33 and S33 were transformed with the different plasmids as described above. After at least 3 days of incubation at 37°C, clones were transferred to fresh GYM agar plates and grown for another 24 h. Genomic DNA of these putative heterogenotes was isolated, and a PCR was done using the primer combinations Aldox_LF_Diag (5′-GCTCTTGAGCTCGTCGAGG-3′) and Aldox_RF_Diag (5′-GCACACCGCCAAGCGACAC-3′), vdcBCD_LF_For_Diag (5′-GAAGCGGCAGGTGCGGG-3′) and vdcBCD_RF_Rev_Diag (5′-GCGGGAAGTCGTCGGTCACC-3′), and VanA_lF_for_diag (5′-GGAGGACCACGACCTGTCC-3′) and VanB_rFrev_diag2 (5′-GGCGTGCCTGTTGTCGG-3′), respectively. If the presence of the suicide plasmid at the desired integration locus could be shown, the clones were transferred into liquid GYM. An 0.1% (vol/vol) amount of the cultures was transferred to fresh GYM once or twice a day, depending on their growth, and then cultivated for up to 5 days in total. Afterward, 200 μl of the culture was diluted to 10−6 with 0.9% sterile saline and then plated onto GYM agar plates. If a plasmid containing the rpsL gene was used, the culture was plated onto GYM agar plates containing 250 μg/ml streptomycin. Growing colonies were then plated onto GYM agar plates with and without apramycin to screen for the loss of the suicide plasmid. Genomic DNA from clones sensitive to apramycin was then isolated and checked for the loss of the target gene using the same primer combinations as above. To determine if the genes were reinserted in another locus, we also tried to directly amplify the deleted genes using the primers Aldox2_For_SD_NheI (5′-ATATGCTAGCAGGAGGCCACGTCATGGACGCGGAG-3′) and Aldox2_Rev_HindIII (5′-ATATAAGCTTCACGGCAACGCGGGC-3′) for the yagRST cluster, vdcB_For (5′-ATATCATATGCGGCTGATCGTCGGC-3′) and vdcD_Rev (5′-ATATCTCGAGCATCAGCGCACCCGCAG-3′) for the vdcBCD cluster, and vanA_For_NheI (5′-ATATGCTAGCATGACTGCGTTCGTGCGCG-3′) and vanB2_Rev_DraI (5′-ATATTTTAAACGCCGGCCTACAGATCCAGC-3′).
Characterization of deletion mutants.
To identify the phenotypic effects of the deleted genes on Amycolatopsis sp. ATCC 39116, two methods were used. Mutant FMVDC1 (Δvdh ΔvdcBCD Strr) was analyzed in both 2-liter fermentations and flask experiments to determine changes in metabolite production and growth on vanillic acid, respectively. The vanAB deletion mutant (Δvdh ΔvanAB Strr) was characterized only regarding its growth on vanillic acid. Mutant FMALDOX1 (Δvdh ΔyagRST) was characterized only in 2-liter fermentations. This deletion was expected to impact only the formation of vanillic acid from vanillin or ferulic acid. However, mutant F33 (Δvdh), i.e., the parent strain, is already incapable of growing on ferulic acid or vanillin when used as the sole carbon source, thus making growth experiments with these compounds impossible (5, 6). To determine growth on vanillic acid, cells were grown in 50 ml of GYM for 14 h at 42°C. Afterward, the cells were washed twice with MSM and then used to inoculate 50 ml of MSM with 0.1% (wt/vol) vanillic acid added as sole carbon source. The remaining washed cells of strain FMVDC1 were then supplied with 0.3% (wt/vol) vanillic acid, too, to observe the formation of guaiacol.
The 2-liter fermentations were done as described by Fleige et al. (5).
Measurement of aromatic compounds in the supernatant.
To determine the concentration of aromatic compounds in the supernatant, we used a Dionex UltiMate 3000 high-performance liquid chromatography (HPLC) system (Dionex, Idstein, Germany). Samples of the supernatant were taken, centrifuged for 3 min at 16,000 × g, diluted 1:100 with double-distilled water, and then measured. The separation of the different compounds was done using an Acclaim 120 C8 column (particle size, 5 μm; column size, 250 mm by 2.1 mm) with a gradient of 0.1% (vol/vol) formic acid and acetonitrile in a range from 15% to 80% with a flow rate of 0.3 ml/min. The gradient used is shown in Table 2. Quantification was done using external standards of the compounds of interest. Measurements were performed using the MWD-3000 multiple-wavelength detector (Dionex, Idstein, Germany) at the wavelengths 259, 280, 285, and 340 nm. Data analysis was done using the software Chromeleon 6.8 (Dionex, Idstein, Germany).
TABLE 2.
HPLC gradient used to separate aromatic compounds in the culture supernatantsa
| Retention time (min) | % acetonitrile | % 0.1% (vol/vol) formic acid |
|---|---|---|
| 0 | 15 | 85 |
| 6 | 15 | 85 |
| 11 | 50 | 50 |
| 18 | 75 | 25 |
| 19 | 80 | 20 |
| 23 | 80 | 20 |
| 26 | 15 | 85 |
| 30 | 15 | 85 |
Flow rate was set to 0.3 ml/min. All changes in concentration were performed as linear gradients.
Accession number(s).
Vector sequences have been made available under the following NCBI accession numbers: p6SUI2, KX863719; p6SUI3, KX863720; p6SUI5, KX863721; p6SUI5ERPSL, KX863722. The accession numbers of the deleted gene clusters are as follows: yagT, WP_020416700.1; yagS, WP_020416699.1; yagR, WP_020416698.1; vanAB, WP_020422153.1 and WP_020422151.1; vdcBCD, WP_020421911.1, WP_020421910.1, and WP_020421909.1.
Supplementary Material
ACKNOWLEDGMENTS
We thank Isabelle Plugge for excellent technical assistance.
This study, including the efforts of Florian Meyer and Alexander Steinbüchel, was financially supported by Symrise AG. The funders had no role in study design, data collection and interpretation, or the decision to submit the study for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02660-16.
REFERENCES
- 1.Brochado AR, Matos C, Møller BL, Hansen J, Mortensen UH, Patil KR. 2010. Improved vanillin production in baker's yeast through in silico design. Microb Cell Fact 9:84. doi: 10.1186/1475-2859-9-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hansen EH, Møller BL, Kock GR, Bünner CM, Kristensen C, Jensen OR, Okkels FT, Olsen CE, Motawia MS, Hansen J. 2009. De novo biosynthesis of vanillin in fission yeast (Schizosaccharomyces pombe) and baker's yeast (Saccharomyces cerevisiae). Appl Environ Microbiol 75:2765–2774. doi: 10.1128/AEM.02681-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li K, Frost JW. 1998. Synthesis of vanillin from glucose. J Am Chem Soc 120:10545–10546. doi: 10.1021/ja9817747. [DOI] [Google Scholar]
- 4.Falconnier B, Lapierre C, Lesage-Meessen L, Yonnet G, Brunerie P, Colonna-Ceccaldi B, Corrieu G, Asther M. 1994. Vanillin as a product of ferulic acid biotransformation by the white-rot fungus Pycnoporus cinnabarinus I-937: identification of metabolic pathways. J Biotechnol 37:123–132. doi: 10.1016/0168-1656(94)90003-5. [DOI] [Google Scholar]
- 5.Fleige C, Meyer F, Steinbüchel A. 2016. Metabolic engineering of the actinomycete Amycolatopsis sp. strain ATCC 39116 towards enhanced production of natural vanillin. Appl Environ Microbiol 82:3410–3419. doi: 10.1128/AEM.00802-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fleige C, Hansen G, Kroll J, Steinbüchel A. 2013. Investigation of the Amycolatopsis sp. strain ATCC 39116 vanillin dehydrogenase and its impact on the biotechnical production of vanillin. Appl Environ Microbiol 79:81–90. doi: 10.1128/AEM.02358-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma X, Daugulis AJ. 2014. Effect of bioconversion conditions on vanillin production by Amycolatopsis sp. ATCC 39116 through an analysis of competing by-product formation. Bioprocess Biosyst Eng 37:891–899. [DOI] [PubMed] [Google Scholar]
- 8.Max B, Carballo J, Cortés S, Domínguez JM. 2012. Decarboxylation of ferulic acid to 4-vinyl guaiacol by Streptomyces setonii. Appl Biochem Biotechnol 166:289–299. doi: 10.1007/s12010-011-9424-7. [DOI] [PubMed] [Google Scholar]
- 9.Muheim A, Lerch K. 1999. Towards a high-yield bioconversion of ferulic acid to vanillin. Appl Microbiol Biotechnol 51:456–461. doi: 10.1007/s002530051416. [DOI] [Google Scholar]
- 10.Pérez-Rodríguez N, Oliveira RP, Agrasar AM, Domínguez JM. 2016. Ferulic acid transformation into the main vanilla aroma compounds by Amycolatopsis sp. ATCC 39116. Appl Microbiol Biotechnol 100:1677–1689. doi: 10.1007/s00253-015-7005-3. [DOI] [PubMed] [Google Scholar]
- 11.Ma X, Daugulis AJ. 2014. Transformation of ferulic acid to vanillin using a fed-batch solid-liquid two-phase partitioning bioreactor. Biotechnol Prog 30:207–214. doi: 10.1002/btpr.1830. [DOI] [PubMed] [Google Scholar]
- 12.Fleige C, Steinbüchel A. 2014. Construction of expression vectors for metabolic engineering of the vanillin-producing actinomycete Amycolatopsis sp. ATCC 39116. Appl Microbiol Biotechnol 98:6387–6395. doi: 10.1007/s00253-014-5724-5. [DOI] [PubMed] [Google Scholar]
- 13.Priefert H, Achterholt S, Steinbüchel A. 2002. Transformation of the Pseudonocardiaceae Amycolatopsis sp. strain HR167 is highly dependent on the physiological state of the cells. Appl Microbiol Biotechnol 58:454–460. doi: 10.1007/s00253-001-0920-5. [DOI] [PubMed] [Google Scholar]
- 14.Davis JR, Goodwin LA, Woyke T, Teshima H, Bruce D, Detter C, Tapia R, Han S, Han J, Pitluck S, Nolan M, Mikhailova N, Land ML, Sello JK. 2012. Genome sequence of Amycolatopsis sp. strain ATCC 39116, a plant biomass-degrading actinomycete. J Bacteriol 194:2396–2397. doi: 10.1128/JB.00186-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plaggenborg R, Overhage J, Steinbüchel A, Priefert H. 2003. Functional analyses of genes involved in the metabolism of ferulic acid in Pseudomonas putida KT2440. Appl Microbiol Biotechnol 61:528–535. doi: 10.1007/s00253-003-1260-4. [DOI] [PubMed] [Google Scholar]
- 16.Kalpana GV, Bloom BR, Jacobs WR. 1991. Insertional mutagenesis and illegitimate recombination in mycobacteria. Proc Natl Acad Sci U S A 88:5433–5437. doi: 10.1073/pnas.88.12.5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siegl T, Luzhetskyy A. 2012. Actinomycetes genome engineering approaches. Antonie Van Leeuwenhoek 102:503–516. doi: 10.1007/s10482-012-9795-y. [DOI] [PubMed] [Google Scholar]
- 18.Frey UH, Bachmann HS, Peters J, Siffert W. 2008. PCR-amplification of GC-rich regions: “slowdown PCR”. Nat Protoc 3:1312–1317. doi: 10.1038/nprot.2008.112. [DOI] [PubMed] [Google Scholar]
- 19.Boeke JD, Lacroute F, Fink GR. 1984. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet 197:345–346. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]
- 20.Pelicic V, Reyrat J-M, Gicquel B. 1996. Generation of unmarked directed mutations in mycobacteria, using sucrose counter-selectable suicide vectors. Mol Microbiol 20:919–925. doi: 10.1111/j.1365-2958.1996.tb02533.x. [DOI] [PubMed] [Google Scholar]
- 21.Hosted TJ, Baltz RH. 1997. Use of rpsL for dominance selection and gene replacement in Streptomyces roseosporus. J Bacteriol 179:180–186. doi: 10.1128/jb.179.1.180-186.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lederberg J. 1951. Streptomycin resistance: a genetically recessive mutation. J Bacteriol 61:549–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ozaki M, Mizushima S, Nomura M. 1969. Identification and functional characterization of the protein controlled by the streptomycin-resistant locus in E. coli. Nature 222:333–339. doi: 10.1038/222333a0. [DOI] [PubMed] [Google Scholar]
- 24.Dean D. 1981. A plasmid cloning vector for the direct selection of strains carrying recombinant plasmids. Gene 15:99–102. doi: 10.1016/0378-1119(81)90108-6. [DOI] [PubMed] [Google Scholar]
- 25.Stibitz S, Black W, Falkow S. 1986. The construction of a cloning vector designed for gene replacement in Bordetella pertussis. Gene 50:133–140. doi: 10.1016/0378-1119(86)90318-5. [DOI] [PubMed] [Google Scholar]
- 26.Sander P, Meier A, Böttger EC. 1995. rpsL+: a dominant selectable marker for gene replacement in mycobacteria. Mol Microbiol 16:991–1000. doi: 10.1111/j.1365-2958.1995.tb02324.x. [DOI] [PubMed] [Google Scholar]
- 27.Myronovskyi M, Welle E, Fedorenko V, Luzhetskyy A. 2011. β-Glucuronidase as a sensitive and versatile reporter in actinomycetes. Appl Environ Microbiol 77:5370–5383. doi: 10.1128/AEM.00434-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Craney A, Hohenauer T, Xu Y, Navani NK, Li Y, Nodwell J. 2007. A synthetic luxCDABE gene cluster optimized for expression in high-GC bacteria. Nucleic Acids Res 35:e46. doi: 10.1093/nar/gkm086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nordeen SK. 1988. Luciferase reporter gene vectors for analysis of promoters and enhancers. Biotechniques 6:454–458. [PubMed] [Google Scholar]
- 30.Panoutsopoulos GI, Kouretas D, Beedham C. 2004. Contribution of aldehyde oxidase, xanthine oxidase, and aldehyde dehydrogenase on the oxidation of aromatic aldehydes. Chem Res Toxicol 17:1368–1376. doi: 10.1021/tx030059u. [DOI] [PubMed] [Google Scholar]
- 31.Chow KT, Pope MK, Davies J. 1999. Characterization of a vanillic acid non-oxidative decarboxylation gene cluster from Streptomyces sp. D7. Microbiology 145:2393–2403. doi: 10.1099/00221287-145-9-2393. [DOI] [PubMed] [Google Scholar]
- 32.Lupa B, Lyon D, Shaw LN, Sieprawska-Lupa M, Wiegel J. 2008. Properties of the reversible nonoxidative vanillate/4-hydroxybenzoate decarboxylase from Bacillus subtilis. Can J Microbiol 54:75–81. doi: 10.1139/W07-113. [DOI] [PubMed] [Google Scholar]
- 33.Chang S-S, Kang D-H. 2004. Alicyclobacillus spp. in the fruit juice industry: history, characteristics, and current isolation/detection procedures. Crit Rev Microbiol 30:55–74. doi: 10.1080/10408410490435089. [DOI] [PubMed] [Google Scholar]
- 34.Sutherland JB, Crawford DL, Pometto AL III. 1983. Metabolism of cinnamic, p-coumaric, and ferulic acids by Streptomyces setonii. Can J Microbiol 29:1253–1257. doi: 10.1139/m83-195. [DOI] [PubMed] [Google Scholar]
- 35.Priefert H, Rabenhorst J, Steinbüchel A. 1997. Molecular characterization of genes of Pseudomonas sp. strain HR199 involved in bioconversion of vanillin to protocatechuate. J Bacteriol 179:2595–2607. doi: 10.1128/jb.179.8.2595-2607.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brunel F, Davison J. 1988. Cloning and sequencing of Pseudomonas genes encoding vanillate demethylase. J Bacteriol 170:4924–4930. doi: 10.1128/jb.170.10.4924-4930.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitsui R, Kusano Y, Yurimoto H, Sakai Y, Kato N, Tanaka M. 2003. Formaldehyde fixation contributes to detoxification for growth of a nonmethylotroph, Burkholderia cepacia TM1, on vanillic acid. Appl Environ Microbiol 69:6128–6132. doi: 10.1128/AEM.69.10.6128-6132.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dhingra G, Kumari R, Bala S, Majumdar S, Malhotra S, Sharma P, Lal S, Cullum J, Lal R. 2003. Development of cloning vectors and transformation methods for Amycolatopsis. J Ind Microbiol Biotechnol 30:195–204. doi: 10.1007/s10295-003-0040-6. [DOI] [PubMed] [Google Scholar]
- 39.Banh Q, Arenskötter M, Steinbüchel A. 2005. Establishment of Tn5096-based transposon mutagenesis in Gordonia polyisoprenivorans. Appl Environ Microbiol 71:5077–5084. doi: 10.1128/AEM.71.9.5077-5084.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Labeda DP. 1987. Transfer of the type strain of Streptomyces erythraeus (Waksman 1923) Waksman and Henrici 1948 to the genus Saccharopolyspora Lacey and Goodfellow 1975 as Saccharopolyspora erythraea sp. nov., and designation of a neotype strain for Streptomyces erythraeus. Int J Syst Bacteriol 37:19–22. doi: 10.1099/00207713-37-1-19. [DOI] [Google Scholar]
- 41.Priefert H, Rabenhorst J, Steinbüchel A. 2001. Biotechnological production of vanillin. Appl Microbiol Biotechnol 56:296–314. doi: 10.1007/s002530100687. [DOI] [PubMed] [Google Scholar]
- 42.Kumagai M, Ikeda H. 1991. Molecular analysis of the recombination junctions of lambda bio transducing phages. Mol Gen Genet 230:60–64. doi: 10.1007/BF00290651. [DOI] [PubMed] [Google Scholar]
- 43.Berg DE, Davies J, Allet B, Rochaix J-D. 1975. Transposition of R factor genes to bacteriophage lambda. Proc Natl Acad Sci U S A 72:3628–3632. doi: 10.1073/pnas.72.9.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rausch H, Lehmann M. 1991. Structural analysis of the actinophage phiC31 attachment site. Nucleic Acids Res 19:5187–5189. doi: 10.1093/nar/19.19.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davies J, O'Connor S. 1978. Enzymatic modification of aminoglycoside antibiotics: 3-N-acetyltransferase with broad specificity that determines resistance to the novel aminoglycoside apramycin. Antimicrob Agents Chemother 14:69–72. doi: 10.1128/AAC.14.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Novais A, Baquero F, Machado E, Canton R, Peixe L, Coque TM. 2010. International spread and persistence of TEM-24 is caused by the confluence of highly penetrating Enterobacteriaceae clones and an IncA/C2 plasmid containing Tn1696::Tn1 and IS5075-Tn21. Antimicrob Agents Chemother 54:825–834. doi: 10.1128/AAC.00959-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bi Y, Liu X, Zhang L, Shao C, Ma Z, Hua Z, Zhang L, Li L, Hua W, Xiao H, Wei Q, Zheng X. 2013. Pseudo attP sites in favor of transgene integration and expression in cultured porcine cells identified by Streptomyces phage phiC31 integrase. BMC Mol Biol 14:20. doi: 10.1186/1471-2199-14-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lal R, Khanna R, Dhingra N, Khanna M, Lal S. 1998. Development of an improved cloning vector and transformation system in Amycolatopsis mediterranei (Nocardia mediterranei). J Antibiot (Tokyo) 51:161–169. doi: 10.7164/antibiotics.51.161. [DOI] [PubMed] [Google Scholar]
- 49.Ashizawa Y, Yokochi T, Ogata Y, Shobuike Y, Kato J, Ikeda H. 1999. Mechanism of DNA gyrase-mediated illegitimate recombination: characterization of Escherichia coli gyrA mutations that confer hyper-recombination phenotype. J Mol Biol 289:447–458. doi: 10.1006/jmbi.1999.2758. [DOI] [PubMed] [Google Scholar]
- 50.Ikeda H. 1995. A novel assay for illegitimate recombination in Escherichia coli: stimulation of lambda bio transducing phage formation by ultra-violet light and its independence from RecA function. Adv Biophys 31:197–208. doi: 10.1016/0065-227X(95)99392-3. [DOI] [PubMed] [Google Scholar]
- 51.Springer B, Kidan YG, Prammananan T, Ellrott K, Böttger EC, Sander P. 2001. Mechanisms of streptomycin resistance: selection of mutations in the 16S rRNA gene conferring resistance. Antimicrob Agents Chemother 45:2877–2884. doi: 10.1128/AAC.45.10.2877-2884.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnston DM, Cannon JG. 1999. Construction of mutant strains of Neisseria gonorrhoeae lacking new antibiotic resistance markers using a two gene cassette with positive and negative selection. Gene 236:179–184. doi: 10.1016/S0378-1119(99)00238-3. [DOI] [PubMed] [Google Scholar]
- 53.Merkens H, Beckers G, Wirtz A, Burkovski A. 2005. Vanillate metabolism in Corynebacterium glutamicum. Curr Microbiol 51:59–65. doi: 10.1007/s00284-005-4531-8. [DOI] [PubMed] [Google Scholar]
- 54.Chen H-P, Chow M, Liu C-C, Lau A, Liu J, Eltis LD. 2012. Vanillin catabolism in Rhodococcus jostii RHA1. Appl Environ Microbiol 78:586–588. doi: 10.1128/AEM.06876-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Campillo T, Renoud S, Kerzaon I, Vial L, Baude J, Gaillard V, Bellvert F, Chamignon C, Comte G, Nesme X, Lavire C, Hommais F. 2014. Analysis of hydroxycinnamic acid degradation in Agrobacterium fabrum reveals a coenzyme A-dependent, beta-oxidative deacetylation pathway. Appl Environ Microbiol 80:3341–3349. doi: 10.1128/AEM.00475-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dhar A, Lee K-S, Dhar K, Rosazza JPN. 2007. Nocardia sp. vanillic acid decarboxylase. Enzyme Microb Technol 41:271–277. doi: 10.1016/j.enzmictec.2007.02.002. [DOI] [Google Scholar]
- 57.Huang Z, Dostal L, Rosazza JP. 1993. Mechanisms of ferulic acid conversions to vanillic acid and guaiacol by Rhodotorula rubra. J Biol Chem 32:23954–23958. [PubMed] [Google Scholar]
- 58.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 59.Shima J, Hesketh A, Okamoto S, Kawamoto S, Ochi K. 1996. Induction of actinorhodin production by rpsL (encoding ribosomal protein S12) mutations that confer streptomycin resistance in Streptomyces lividans and Streptomyces coelicolor A3(2). J Bacteriol 178:7276–7284. doi: 10.1128/jb.178.24.7276-7284.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schlegel HG, Kaltwasser H, Gottschalk G. 1961. Ein Submersverfahren zur Kultur wasserstoffoxydierender Bakterien: Wachstumsphysiologische Untersuchungen. Arch Mikrobiol 38:209–222. doi: 10.1007/BF00422356. [DOI] [PubMed] [Google Scholar]
- 61.Pfennig N. 1974. Rhodopseudomonas globiformis, sp. n., a new species of the Rhodospirillaceae. Arch Microbiol 100:197–206. doi: 10.1007/BF00446317. [DOI] [Google Scholar]
- 62.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 63.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. 2001. Current protocols in molecular biology. John Wiley & Sons, New York, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.