

ABSTRACT
Microdiversification of a planktonic freshwater bacterium was studied by comparing 37 Polynucleobacter asymbioticus strains obtained from three geographically separated sites in the Austrian Alps. Genome comparison of nine strains revealed a core genome of 1.8 Mb, representing 81% of the average genome size. Seventy-five percent of the remaining flexible genome is clustered in genomic islands (GIs). Twenty-four genomic positions could be identified where GIs are potentially located. These positions are occupied strain specifically from a set of 28 GI variants, classified according to similarities in their gene content. One variant, present in 62% of the isolates, encodes a pathway for the degradation of aromatic compounds, and another, found in 78% of the strains, contains an operon for nitrate assimilation. Both variants were shown in ecophysiological tests to be functional, thus providing the potential for microniche partitioning. In addition, detected interspecific horizontal exchange of GIs indicates a large gene pool accessible to Polynucleobacter species. In contrast to core genes, GIs are spread more successfully across spatially separated freshwater habitats. The mobility and functional diversity of GIs allow for rapid evolution, which may be a key aspect for the ubiquitous occurrence of Polynucleobacter bacteria.
IMPORTANCE Assessing the ecological relevance of bacterial diversity is a key challenge for current microbial ecology. The polyphasic approach which was applied in this study, including targeted isolation of strains, genome analysis, and ecophysiological tests, is crucial for the linkage of genetic and ecological knowledge. Particularly great importance is attached to the high number of closely related strains which were investigated, represented by genome-wide average nucleotide identities (ANI) larger than 97%. The extent of functional diversification found on this narrow phylogenetic scale is compelling. Moreover, the transfer of metabolically relevant genomic islands between more distant members of the Polynucleobacter community provides important insights toward a better understanding of the evolution of these globally abundant freshwater bacteria.
KEYWORDS: Polynucleobacter, ecophysiology, environmental genomics, functional diversity
INTRODUCTION
Genetic analysis of environmental bacteria indicates an extremely high diversity compared to what is known from macroscopic organisms (1, 2). Data from whole-genome sequencing in particular have revealed that differences between individuals can be extensive even on the species level or beyond (microdiversity) (3–5). Often, multiple closely related genotypes coexist in a particular habitat (6–8), which seems to contradict the concept of competitive exclusion (9, 10). However, the physiological relevance of observed genetic differences is widely unclear, making the drawing of ecological conclusions difficult. Multiple explanations for the discordance between the high numbers of coexisting closely related genotypes and ecological theory are conceivable. Either (i) the observed genetic variation represents to a large extent neutral diversification (11–13), (ii) closely related genotypes indeed occupy distinct ecological niches, implicating a vast number of species-like units (14, 15), or (iii) concepts of evolution, which were originally developed for macroorganisms, are not readily applicable to microbes, and novel models have to be developed (16–19).
Genetic variation between individuals includes both the presence of differing gene content (nonhomologous genes) and the variation in alleles (differences between homologous genes). Substantial intraspecies genomic diversity in bacteria derives from the nonhomologous genes (20, 21). Often, the largest part of differential gene content is clustered in genomic islands (GIs), genome regions at which horizontal gene transfer and gene loss occur at high rates (20–22). GIs were initially associated with virulence in pathogenic bacteria, termed pathogenicity islands (23, 24), but are also frequently found in nonpathogenic bacteria (25–28). Corresponding to the mechanisms of gene acquisition and loss, two types of GIs can be distinguished (29). Additive GIs stem from the legacy of mobile genetic elements and can harbor a varied number of gene cassettes. On the other hand, replacement GIs are suggested to originate from homologous recombination of the whole GI fragment (30). They are usually found at conserved genomic locations and harbor genes with similar assigned functions, which can in many cases be related to cell surface properties (31) potentially linked to phage predation (32–34).
Concerning pathogenic bacteria, there are plenty of examples where crucial physiological differences between closely related strains are provided by the differential presence of genomic islands (35, 36). However, evolution in pathogenic and not host-associated bacteria usually differs essentially due to contrasting population structures, i.e., physically highly constrained populations in pathogens (e.g., compare references 37 and 6). For Prochlorococcus bacteria, it was demonstrated by means of single-cell genomics that the flexible genes contribute to niche adaptation (6). This genus was described as a federation of diversified cells, which provide an extensive pool of traits that different environmental conditions can select for (38). Substrate-specific adaptations to distinct environments were attributed to flexible genes in metagenomics comparisons between geographically separated populations (39). Yet, Luo and Konstantinidis (40) pointed out that better knowledge of genomic variability between putative ecotypes is needed to draw robust conclusions. In addition, ecophysiological experiments linked to genome analysis are fundamental for assessing the relevance of genomic variability (41). In studies comparing coastal and open-ocean bacteria, it was shown that genes involved in the copper stress response are found in GIs and are preferentially present in coastal strains (42). Expression of these genes was upregulated under increased copper levels (43), and respective knockout mutants showed higher sensitivity to copper stress (42). In actinobacteria from marine sediments, biosynthetic gene clusters linked to antibiotic production gave rise to different competitive strategies of two closely related species (44). The aforementioned references were all investigating diversity on a genus-like level, i.e., comparing groups including individuals with less than 95 to 96% ANI, which is a suggested threshold for species delineation (45). Experimental evidence for ecophysiological differences above that threshold, as given for soil bacteria by Kopac et al. (46), was rarely demonstrated in marine or freshwater bacteria.
Bacterial diversification cannot a priori be assumed to be similar in marine and freshwater ecosystems. Compared to marine systems, conditions in freshwaters are generally more variable, and drastic fluctuations of biotic and abiotic factors often appear on shorter time scales (47). Genome reduction in Prochlorococcus was found to be related to specialization to narrow ecological niches at the expense of versatility and competitiveness under changing conditions (48). Marine bacteria experience rather limited changes in temperature (49) compared to freshwater bacteria in smaller water bodies (47). Niche partitioning between layers of stratified water columns (49, 50) is not expected for freshwater bacteria (e.g., Polynucleobacter asymbioticus) preferentially dwelling in shallow freshwater systems, which represent thermally highly dynamic habitats (47). Moreover, the spatial separation of inland waters crucially influences the population structure of microorganisms (51, 52), in contrast to a more continuous horizontal transition between habitats in the ocean (53). It could be hypothesized that stronger gene flow barriers and the more variable environment, which may provide more microniches for potential niche partitioning, lead to higher microbial diversification in freshwater ecosystems.
A particularly interesting taxon for studying diversification in freshwater is the Polynucleobacter cluster PnecC (54), due to its cosmopolitan distribution (55) and high global abundance (56). Its ecological success has been shown to be the result of diversification rather than a generalist adaptation, as lineages within the PnecC cluster reveal distinct ecological characteristics (55, 57, 58). A closer investigation of isolated strains illustrated substantial differences regarding ecophysiological and genomic traits among strains exhibiting more than 99% 16S rRNA gene sequence similarity (59). The PnecC cluster represents a cryptic species complex, including a few described species and an unknown number of undescribed species (60). Polynucleobacter asymbioticus, formerly termed the F10 lineage (47, 57), has so far been detected only in the Austrian Alps. The species is particularly abundant in dystrophic ponds, where it accounted for up to 46% of total bacterioplankton (61). Investigating differences between individuals on narrow phylogenetic scales, i.e., within P. asymbioticus, is fundamental for enlightening the ecological significance of the diversity within the PnecC cluster. Here, we studied variation between 37 P. asymbioticus strains, isolated from small ponds at three different sites and altitudes. Nine selected strains were subjected to genome analysis. Focusing on gene content differences between these strains, the ecophysiological relevance of two additive GIs was investigated by growth experiments in the laboratory.
RESULTS
Isolation and characterization of P. asymbioticus strains.
Thirty-seven strains affiliated with the species P. asymbioticus were obtained from nine habitats at three different sites in the Austrian Alps by using nonstandard isolation protocols (Fig. 1 and Tables S1 and S2). All isolates were collected between 2003 and 2007, with one exception collected in 2014. Affiliation with the species P. asymbioticus was verified by multilocus sequence analysis (MLSA) of 11 loci. Phylogeny based on 16S-23S internal transcribed spacer (ITS) sequences shows clear separation of P. asymbioticus strains from other members of the genus (Fig. 2). Regarding ITS sequences (515 bp, including two tRNA genes), only two genotypes can be distinguished among the 37 P. asymbioticus strains. According to genome content (see below), one ITS genotype is termed lineage simplex (Latin for “simple, pure, plain”) and the other lineage amplus (Latin for “large, great, ample”) (Fig. 1). Lineage simplex was found at all three collection sites, whereas lineage amplus was found exclusively at the Rauriser Urwald site. Nine genotypes can be distinguished by MLSA, two from the Loibersbacher Höhe site, five from the Trög site, and three from the Rauriser Urwald site. Interestingly, the predominant MLSA genotype from Loibersbacher Höhe is also found at Rauriser Urwald. Although the core genome phylogeny reveals that the respective strain, MWH-Recht1, diverged significantly from the Loibersbacher Höhe isolates, its close position to these isolates in the phylogenetic tree is conspicuous (Fig. 2). The 37 strains were further characterized in detail for three GIs (see below). These characterizations combined with MLSA revealed 17 different P. asymbioticus genotypes in total.
FIG 1.

Isolated P. asymbioticus strains and their diversity regarding MLSA and three GI variants. Upper left, maps depicting geographic locations of the sites of origin of the 37 cultivated P. asymbioticus strains. Air line distances between the sites are given in kilometers, and maximum distances between the habitats within the sites are given in meters. Satellite pictures were adopted from Google Earth. Bottom left, the three sites, with a photo of one habitat from each site. Right table, characterization of the 37 strains sorted by site of origin (color code), habitat of origin, sampling date, and MLSA sequence type. The left boxes are colored according to lineage affiliation (yellow for simplex, and blue for amplus), while boxes on the right represent the GI variants ACD, NIT, and CSC, respectively. The GI variant CSC is characterized for each strain concerning presence/absence patterns of selected genes (A to I) and sequence types of one gene present in 27 strains (1 to 6, and − for strains lacking the gene). For each strain, the presence of the GI variant ACD (catechol 1,2-dioxygenase gene [catA]) is indicated by a cyan box. The presence of NIT (assimilatory nitrate reductase catalytic subunit gene [nasA]) is displayed by red boxes. Sequence types of the genes are given by numbers in the boxes. In cases where no number is shown, the respective gene was not subjected to DNA sequencing. The black brackets on the right enclose strains obtained from single samples.
FIG 2.

Phylogeny of P. asymbioticus. (A) Unrooted phylogenetic tree (RAxML) based on a genome alignment of the nine genome-sequenced P. asymbioticus strains. Bootstrap support is given for both RAxML (left) and NJ (right) tree calculations. (B) Phylogenetic tree (NJ) based on 16S-23S ITS sequences of P. asymbioticus and related strains. Bootstrap values are depicted at the nodes. The tree is rooted on the sequence of Cupriavidus metallidurans strain CH34.
Genome characteristics of P. asymbioticus.
Nine genomes, three from each collection site, were subjected to genome analysis (Table 1). The genome sizes vary from 2.14 to 2.38 Mbp. On average, 93.4% of the DNA is coding sequence, and the gene numbers range from 2,180 to 2,413 genes. All nine genomes share the signatures of the previously characterized passive lifestyle of the type strain of P. asymbioticus (47). This includes the lack of genes encoding flagella, the lack of chemotaxis genes, and a quite low number of genes putatively involved in the transduction of environmental signals. None of the nine sequenced strains encode systems putatively involved in light harvesting which were found in other Polynucleobacter species (59).
TABLE 1.
General features of the nine P. asymbioticus genomesa
| Strain | DNA contigsb | N50 (Mbp) | Genome size (Mbp) | Gene count | GC content (%) | Coding DNA (%) | tRNA count |
|---|---|---|---|---|---|---|---|
| QLW-P1DMWA-1 | 1 | Closed | 2.16 | 2,180 | 44.8 | 93.7 | 38 |
| P1-4-10KL | 2 | 1.11 | 2.16 | 2,207 | 44.8 | 93.7 | 38 |
| P1-Kol8 | 8 | 0.76 | 2.17 | 2,217 | 44.8 | 93.5 | 38 |
| MWH-Tro-7-1-4 | 4 | 0.67 | 2.23 | 2,275 | 44.8 | 93.6 | 39 |
| MWH-Tro-8-2-9 | 3 | 1.27 | 2.27 | 2,305 | 44.8 | 93.3 | 38 |
| Tro8F10W22 | 1 | Closed | 2.22 | 2,235 | 44.8 | 93.8 | 39 |
| MWH-Recht1 | 8 | 0.81 | 2.14 | 2,189 | 44.8 | 93.5 | 38 |
| MWH-RechtKolB | 1 | Closed | 2.36 | 2,393 | 44.7 | 92.6 | 41 |
| MWH-RechtKol4 | 1 | Closed | 2.38 | 2,413 | 44.7 | 92.6 | 40 |
Accession numbers are given in Table S1.
Only contigs >3,000 bp are considered.
The two lineage amplus genomes are considerably larger than those assigned to lineage simplex. ANI values for pairwise comparisons within the lineages are all higher than 98.8%, while the values for comparisons between lineages simplex and amplus range from 97.7 to 98.0% (Table 2). The ANI values correlate with the fraction of homologous genes in pairwise comparisons. Values for the percentage of homologous genes are all higher than 90.0% within lineages and ranged from 82.0 to 83.6% between lineages. The core genome of the nine investigated strains contains 1,820 genes. Accordingly, 18% of genes are contained in the auxiliary genome. Two pairs of strains show exceptionally high similarities, respectively, with ANI values of 100.0% (QLW-P1DMWA-1 and P1-4-10KL) and 99.9% (MWH-RechtKolB and MWH-RechtKol4). The differences between the genomes of QLW-P1DMWA-1 and P1-4-10KL are constituted by only 22 single nucleotide polymorphisms (SNPs) and differences in a giant gene (see below), which cannot be exactly assessed due to sequence assembly ambiguities in P1-4-10KL. These two almost-clonal strains were obtained from the same habitat but from different samplings that were 4 years apart (Fig. 1).
TABLE 2.
ANI values and percent fractions of homologous genes
| Strain | % ANI value (below the diagonal) or % fraction of homologous genes (above the diagonal) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| QLW-P1DMWA-1 | P1-4-10KL | P1-Kol8 | MWH-Tro-7-1-4 | MWH-Tro-8-2-9 | Tro8F10W22 | MWH-Recht1 | MWH-RechtKolB | MWH-RechtKol4 | |
| QLW-P1DMWA-1 | 97.2 | 93.0 | 90.1 | 91.1 | 90.2 | 92.6 | 81.7 | 81.3 | |
| P1-4-10KL | 100.0 | 92.5 | 89.6 | 90.6 | 89.6 | 92.0 | 81.3 | 80.9 | |
| P1-Kol8 | 99.6 | 99.6 | 89.4 | 89.8 | 88.6 | 92.4 | 81.1 | 81.0 | |
| MWH-Tro-7-1-4 | 99.1 | 99.1 | 99.1 | 90.8 | 91.4 | 88.0 | 81.1 | 80.7 | |
| MWH-Tro-8-2-9 | 99.4 | 99.4 | 99.5 | 99.1 | 91.2 | 89.9 | 80.7 | 80.2 | |
| Tro8F10W22 | 98.8 | 98.9 | 99.0 | 99.0 | 99.1 | 89.2 | 81.3 | 80.7 | |
| MWH-Recht1 | 99.4 | 99.4 | 99.5 | 99.1 | 99.4 | 98.9 | 81.6 | 81.3 | |
| MWH-RechtKolB | 98.0 | 98.0 | 98.0 | 97.9 | 97.9 | 97.7 | 97.9 | 96.5 | |
| MWH-RechtKol4 | 97.9 | 97.9 | 97.9 | 97.9 | 97.9 | 97.7 | 97.8 | 99.9 | |
Core genome phylogeny.
Whole-genome alignment of the nine strains produced a consensus sequence of 1.81 Mbp. A phylogenetic tree based on that alignment (Fig. 2) reveals a weak trend toward higher relatedness of strains originating from the same habitat. However, there is one clear exception, strain MWH-Recht1, which is more closely related to the strains isolated from Loibersbacher Höhe than to any other genome-sequenced strain. Its conspicuous position in the phylogenetic tree is also represented in the MLSA data (Fig. 1). Furthermore, MWH-Recht1 represents the only simplex strain isolated from Rauriser Urwald.
Genomic islands.
Seventy-six percent of the auxiliary genes are clustered in GIs. The gene synteny in the core genome is highly conserved among the nine investigated genomes, and the position of GIs can therefore be compared between different genomes according to gene context. There are in total 24 positions where GIs are located (Fig. 3C). These positions are strain-specifically occupied by different GIs. The genomes of lineage simplex contain 8 to 10 GIs, and the two genomes of lineage amplus contain 16 GIs. There are only two positions (positions 2 and 16) which are occupied by a GI in all genomes.
FIG 3.

Genomic islands in P. asymbioticus. (A) The nine P. asymbioticus genomes are shown in a linear representation. The genome position with respect to the origin of replication is shown on the horizontal axis at the bottom. GIs are depicted as colored boxes. The 28 GI variants are represented by different colors. The six variants for which a putative metabolic function was inferred from gene annotation are framed in black and named (ACD, aromatic compound degradation; NIT, assimilatory nitrate reduction; LIP, lipid metabolism; COD, carbon monoxide dehydrogenation; SUL, sulfate transport; HMR, heavy metal resistance). Two further variants which are discussed in the text are named as well (CSC, cell surface composition; GGR, giant gene region). The small bars above and below the genomes depict tRNAs. Those tRNAs which are supposed to be potential target sites for integrases, as inferred from the localizations of GIs, are highlighted in red, and the respective genome positions in the nine genomes are connected. The positions of the 16S rRNA and glutamine synthetase (glnA) genes, of which sequences are publicly available from various Polynucleobacter strains, are indicated by black ovals. (B) The number of SNPs from a pairwise comparison of the QLW-P1DMWA-1 and the MWH-RechtKol4 genomes is shown in a sliding window (window size, 10 kbp; step size, 1 kbp) along the genome of QLW-P1DMWA-1. The dashed line indicates the average number of SNPs in the aligned sequences. Gaps in the graph result from alignment gaps larger than 5 kbp. GIs of QLW-P1DMWA-1 are depicted by colored bars. GIs which are suspected to stem from homologous recombination events, suggested by a SNP peak in flanking regions, are framed in black. (C) The 24 genome positions where GIs can be found among the nine genomes are numbered and displayed at the respective QLW-P1DMWA-1 genome positions. The presence of GIs at these positions is indicated by crosses, colored as in panel A. The nine genomes are arranged one below the other in the same order as in panel A.
The GIs were grouped into 28 different variants, indicated by different colors in Fig. 3 (see also Table S5). A GI variant is characterized by the presence of homologous genes, or genes with similar annotations concerning gene function, in GIs of different genomes. For six out of the 28 variants, a putative metabolic function was inferred from gene annotations (Fig. 3). For two such GI variants (aromatic compound degradation [ACD] and assimilatory nitrate reduction [NIT]), the metabolic potential was confirmed (Fig. 4), and their distribution among all 37 P. asymbioticus strains (Fig. 1) and some other strains affiliated with the PnecC cluster (Fig. 5) was assessed. There is one GI variant (cell surface composition [CSC]) for which gene annotation suggests that it is involved in the synthesis of exposed structures of the cell surface. Homology between the GIs of different strains is low for this variant, except between strains QLW-P1DMWA-1 and P1-4-10KL (identical) and between MWH-RechtKolB and MWH-RechtKol4 (99.996% similarity). The diversity concerning CSC was further characterized (Fig. 1 and Table S6). Another conspicuous GI variant (giant gene region [GGR]) is present in all simplex genomes but not in MWH-Tro8-2-9 and the two amplus genomes. It includes a giant gene of obscure function (see references 62 and 29), which is annotated as “uncharacterized conserved protein, contains a C-terminal beta-barrel porin domain.” This gene contains repetitive elements that precluded DNA sequence assembly for all but two strains, QLW-P1DMWA-1 and Tro8F10W22, for which it exhibits DNA sequence lengths of 31.3 and 42.3 kbp, respectively. The other 20 GI variants were not further characterized, as they contain either a large fraction of genes annotated as hypothetical proteins or at least not a set of genes that suggested assignment to one specific function. In total, 37% of the GI genes are annotated as hypothetical proteins, compared to 11% of the core genes.
FIG 4.

Metabolic potential of two GI variants. (A) Extra growth in medium amended with benzoate compared to benzoate-free medium as a percentage of the growth in benzoate-free medium. Error bars depict standard deviations of the results from two replicates. (B) Extra growth in medium amended with nitrate compared to nitrogen-depleted medium as a percentage of average extra growth in media amended with ammonia and urea, respectively, compared to nitrogen-depleted medium. Error bars depict standard deviations of the results from two replicates.
FIG 5.

Horizontal gene transfer (HGT) of GIs. (A) A phylogenetic tree (RAxML) based on 344 conserved genes among the nine genome-sequenced P. asymbioticus strains and four genome-sequenced Polynucleobacter strains not affiliated to the same species is shown on the left. The tree is rooted on Polynucleobacter rarus strain MT-CBb6A5, which is not shown in the tree. Numbers at the nodes display bootstrap support from 100 rapid bootstrap inferences. Next to the strains, the presence of three different DNA fragments corresponding to three GI variants (ACD, NIT, and HMR) is indicated by colored segments. Homologies are illustrated with respect to reference fragments indicated by stars. Sequence similarities are represented by the transparencies of the colors and written on the top next to the segments in small boldface numbers (%). Alignment lengths to the reference fragments are represented by lengths of the segments and written below the similarity values (in kilobase pairs). ANI values between the respective genomes and the reference genomes are written next to the similarity values after the slash. (B) The three reference DNA fragments from panel A are shown in detail. Genes are colored according to COG categories. The gene symbols of those genes associated with the presumed functions of the three GI variants are written above or below the genes.
Evidence for physiological function of two GIs.
The GI variant ACD contains an operon for the degradation of benzoate to succinyl-coenzyme A (CoA) (Fig. 5). Six strains, three of which possess the GI variant and three which lack it, were tested for their growth potential on benzoate as an additional carbon and energy source. The growth of strains harboring ACD was significantly enhanced by the addition of benzoate, whereas the growth of strains lacking ACD was not (Fig. 4A).
NIT contains an operon for assimilatory nitrate reduction (Fig. 5). The same six strains from before, four possessing NIT and two not, were tested for their potential to use nitrate as a nitrogen source in media depleted of other nitrogen sources. Strains harboring NIT were profiting from the presence of nitrate in the growth media, whereas the other strains were not (Fig. 4B).
Diversity of ACD and NIT.
In order to gain broader insight into the potential for functional diversification provided by GIs, the presence/absence pattern of ACD and NIT was investigated for all 37 P. asymbioticus isolates (Fig. 1). The presence was inferred from PCR targeting a key gene of each operon, the catechol 1,2-dioxygenase gene (catA) and the assimilatory nitrate reductase catalytic subunit gene (nasA), respectively. Selected PCR fragments were subsequently sequenced, providing sequences of 672 bp and 771 bp in length, respectively. All isolates from the Loibersbacher Höhe site lack catA, in contrast to the Trög site, all isolates from which contain the gene. Among the isolates from Rauriser Urwald, all amplus strains contain catA, whereas the one simplex strain lacks it. Regarding respective gene sequences, no variation was found within either the Trög site or the Rauriser Urwald site. Between the sites, the sequences exhibit 96.9% similarity. The nasA gene was detected in all sites and again in all strains from Trög. Four different nasA sequence types with similarities ranging from 94.9% (sequence types 1 and 3 in Fig. 1) to 99.5% (sequence types 2 and 3 in Fig. 1) were obtained. The sequence type found in Loibersbacher Höhe was also found in Trög, which is the only site from which different nasA sequence types were obtained.
Mechanisms of recombination: replacement and additive GIs.
Flexible genes show elevated fractions of best BLAST hits assigned to classes other than Betaproteobacteria and families other than Burkholderiaceae (Fig. S1). Furthermore, these genes exhibit atypical GC content (Fig. S2). This suggests that recombination plays a larger role for flexible genes than for core genes. The underlying transfer mechanisms for specific GIs can be evaluated by different hallmarks of recombination.
tRNAs are known target sites for integrases, and hence, the presence of tRNAs points to an integrase-mediated transfer of the respective GIs. tRNAs can be found at half of the 24 GI positions, whereas genes annotated as integrases are also frequently present in the respective GIs. In most cases, the tRNAs flank the GIs on the posterior end (counted from the origin of replication). One exception, where the tRNA is located at the anterior end, is the GI NIT in strain MWH-Tro8-2-9. Interestingly, this is the only GI found in position 9 and is orientated as a reverse complement to the other GIs of the NIT variant. There are other cases (positions 16 and 21) where tRNAs are located within GIs. Possibly, the respective GIs stem from the legacy of two integration events at both sides of the tRNAs. The GIs at position 21 (ACD in MWH-RechtKolB and MWH-RechtKol4, Fig. 3), for example, contain the operon for the degradation of aromatic compounds posterior to the tRNA, whereas the gene cassette anterior to the tRNA contains functionally unrelated genes. Furthermore, at those GI positions where tRNAs are located, different GI variants are often present in different genomes (positions 5, 12, 15, 16, and 24) (Fig. 3C). All the aforementioned characteristics suggest assignment of the respective GIs into the additive type, according to the definition by López-Pérez et al. (29), which applies to 54% of the 24 GI positions.
On the other hand, a conspicuous accumulation of SNPs in the flanking regions of some GIs suggests homologous recombination events as origins of the GIs at positions 2, 7, 10, 14, 17, and 18 (Fig. 3B). For replacement GIs, as defined by López-Pérez et al. (29), homologous recombination is the most likely transfer mechanism (30). Another characteristic for replacement GIs is the presence of genes with similar assigned function but completely different genes in the GIs of different genomes. This is the case for the GI variant CSC at position 2. This kind of GI was found in various taxa, including archaea, and was suggested to be transferred by a single homologous recombination event of the whole island (30).
Diversity of CSC.
An attempt was made to assess the diversity of CSC, which is coding for the synthesis of exposed structures of the cell surface and is assumed to play a crucial role in phage recognition, for all 37 P. asymbioticus strains. Regarding the presence/absence pattern of six tested genes affiliated with CSC, nine different genotypes were detected (Fig. 1 and Table S6). By sequencing 842 bp of one of the six genes, peptidoglycan/lipopolysaccharide (LPS) O-acetylase, six sequence types were found among the 27 strains that possess it. All together, 13 different CSC genotypes were detected among the 37 strains (Fig. 1).
Horizontal transfer of GIs across species boundaries.
The sequences of a few GIs with exceptionally high similarity were found in genome-sequenced strains not affiliated with P. asymbioticus, exhibiting ANI values of around 80% with P. asymbioticus genomes (Fig. 5). The most conspicuous example is a fragment of 43.5 kbp (heavy metal resistance [HMR] at GI position 20 in MWH-Tro8-2-9) found with 99.6% similarity in a strain with 79.9% ANI. This points to interspecific horizontal transfer of whole GIs in Polynucleobacter. The GI may be transferred by integrases, as indicated by the presence of a gene annotated as integrase/recombinase XerD. Flanking tRNAs are not present on either side of the GI, but tRNAs are located at position 20 in the two P. asymbioticus genomes that do not harbor a GI at this genomic location (Fig. 3A). Other examples for interspecifically shared GIs are the variants ACD and NIT (Fig. 5), which were shown to potentially provide additional metabolic functions to the strains possessing them (Fig. 4).
The legacy of an ancient event of horizontal gene transfer may be found in a sulfur-oxidizing (SOX) gene cluster, for which BLAST analyses suggest an alphaproteobacterial origin (see Fig. 7 in reference 47). The cluster is located between GI positions 15 and 16 and is present in all nine P. asymbioticus genomes with >94% homology. There is a second SOX gene cluster located directly after the GI at position 7. SNP peaks in the flanking regions, especially at the posterior end of the GI (Fig. 3B), suggest that this gene cluster is the result of a homologous recombination. The SOX gene cluster is located exactly at this position. It is identical in all seven simplex genomes, but only a part of it is present in the two amplus genomes and with only 70% DNA sequence similarity, which generates the SNP peaks in Fig. 3B.
Mobility of GIs.
The various lines of evidence shown above suggest that the GIs of P. asymbioticus are predominantly the result of horizontal gene transfer (intra- and interspecific). This assumption is further supported by the fact that homologous GI genes of different genomes excessively exhibit either identical sequences or relatively low (<97%) sequence similarities (Fig. 6A). This disproportionality between GI and non-GI genes becomes more apparent when the proportion of identical genes is plotted against the average gene similarity in pairwise comparisons of strains (Fig. 6B). The fraction of identical gene pairs among homologous genes is particularly high when GI genes from strains originating from the same site are compared (Fig. 6A, left). Yet, even if comparisons between strains originating from different sites are considered, 58% of the homologous genes contained in GIs are identical. In comparison, among the non-GI genes, only 44% of the respective genes are identical (Fig. 6A, right). The evidence mentioned above suggests that the distribution of GIs is uncoupled from the core genome to some extent, which can enhance the spreading of GIs across distant sites.
FIG 6.
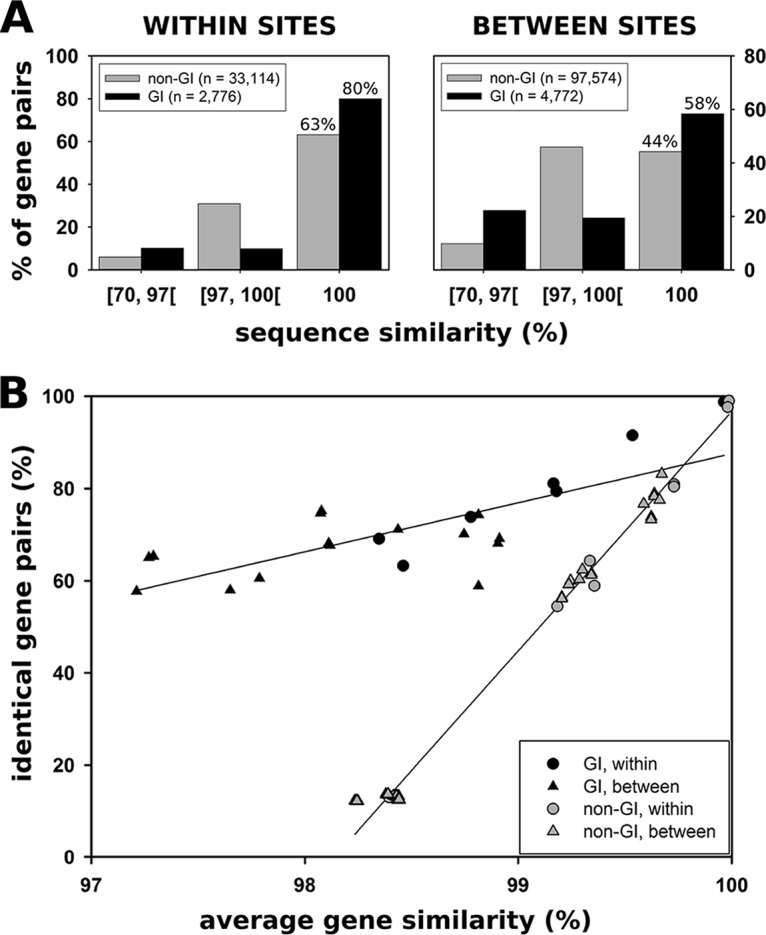
Sequence similarities of GI and non-GI genes. (A) Proportion of gene pairs exhibiting similarities in the range of ≥70% to <97%, in the range of ≥97% to <100%, and 100% sequence identity. The results are shown for GI and non-GI genes. The data were classified into comparisons between strains isolated from the same site (left) and comparisons between strains isolated from different sites (right). (B) For pairwise comparisons between strains, the proportion of genes exhibiting 100% sequence identity is plotted against the average gene similarity in the respective data set. GI genes exhibit an elevated number of identical genes at similar average gene similarities compared to non-GI genes. Linear regressions are shown for all data with respect to GI genes (y = 136 + 11x, r2 = 0.58) and for all data with respect to non-GI genes (y = 54 + 52x, r2 = 0.99).
Concurrent diversity.
When collectively viewing different sampling time points, the possibility that the observed diversity is generated due to changes in the respective populations through time cannot be excluded. On the other hand, the diversity of genotypes found within a single sample from a single habitat at a given time point (concurrent diversity) can definitely be attributed to the coexistence of different genotypes. When data from MLSA and characterization of the three GI variants CSC, ACD, and NIT are taken into account (Fig. 1), there are seven samples that exhibit concurrent diversity: (i) Pond-1, July 2004; (ii) Pond-1, October 2005; (iii) Trög-6, October 2007; (iv) Trög-7, October 2007; (v) Trög-8, October 2007; (vi) Rechteckteich, October 2006, and (vii) Unterer Teich, October 2007. Concerning the two Pond-1 samples from 2004 and 2005, it is noticeable that strains harboring the potential for nitrate assimilation coexist with strains that do not exhibit this potential. In comparison to the concurrent diversity, it should be noted that an almost-clonal (22 genome-wide SNPs) isolate to strain QLW-P1DMWA-1, namely, strain P1-4-10KL, was isolated 4 years later from the same habitat.
Distribution of GI and non-GI genes among COG categories.
The distribution among the different COG categories of the P. asymbioticus GI and non-GI genes is plotted in Fig. 7. The following categories are clearly overrepresented in the GIs: cell wall/membrane/envelope biogenesis, inorganic ion transport and metabolism, transcription, defense mechanism, and mobilome: prophages, transposons.
FIG 7.

COG distribution of GI and non-GI genes. The number of genes assigned to a certain COG category as a percentage of all genes assigned to COG categories is shown for the genes within GIs and the remaining genes of all nine P. asymbioticus genomes.
Flexible non-GI genes.
Twenty-five percent of the flexible genes are located outside GIs. Just like the flexible genes found in GIs, these are assumed to stem from the legacy of recombination (Fig. S1 and S2). A conspicuously high fraction of the respective genes can be attributed to the COG category “mobilome: prophages, transposons” (Fig. S3). The flexible non-GI genes are rather scattered, i.e., 62% are found in clusters of three or fewer adjacent genes (Fig. S4). In total, 40% of the flexible non-GI genes are annotated as hypothetical proteins. The flexible gene content outside GIs may represent, at least to some extent, remnants of former GIs which are diminishing in the course of genome streamlining.
Gene content variability in various taxa.
The extent of gene content variation within various bacterial and archaeal taxa was assessed by plotting ANI values versus bidirectional best BLAST hits (BBHs), a proxy for the number of homologous genes (Fig. 8). BBHs are plotted as percentage values of the average gene count of the respective genome pairs. BBH percentage values markedly below the average curve are observed for the archaea Sulfolobus and Haloquadratum. P. asymbioticus shows no noticeable abnormalities with respect to this characteristic.
FIG 8.
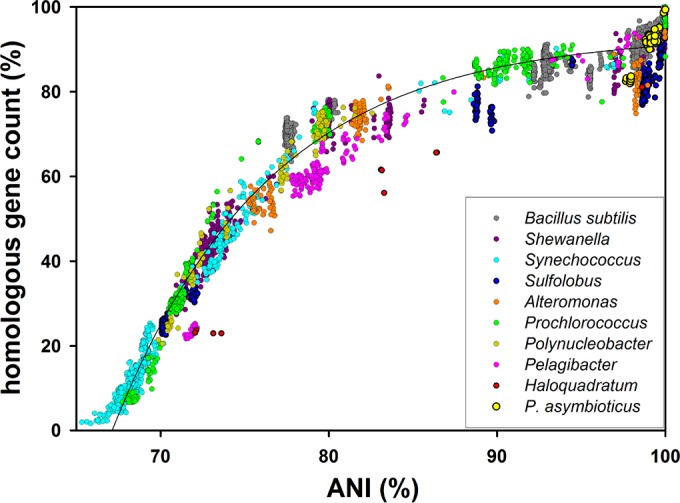
Relative proportion of homologous genes within various taxa. The number of homologous genes as a percentage of the average gene count of the respective pair of genomes is plotted against the ANI values of the same pair. Genomes within different taxa of nonendosymbiotic bacteria and archaea are compared. The black line shows an exponential curve fit for all data in the plot.
DISCUSSION
The vast extent of intraspecific gene content diversity found in many bacteria is challenging the ecological relevance of the current species concept for bacteria (14, 17). The ecophysiological versatility within species and its consequences for the inference of ecosystem functions are widely unclear (38, 41, 63). Moreover, evolutionary mechanisms for the generation and maintenance of microdiversity are only partially resolved (19, 33, 64). The work presented here, comprehending DNA sequencing data on 37 conspecific isolates, nine being whole-genome sequenced (ANI, >97.6%), in combination with ecophysiological tests on selected strains, should contribute to elucidating this matter.
Ecological significance of conspecific differences in GI content.
The results of the assimilation experiments present clear evidence for the functionality of the two tested GI variants, ACD and NIT. Consequently, these GIs potentially provide a fitness advantage to the encoding strains. The investigated P. asymbioticus strains all originate from habitats rich in humic matter and hence rich in aromatic compounds. For strain QLW-P1DMWA-1, comprehensive tests concerning the assimilation of humic substances and certain aromatic compounds were conducted (47). Interestingly, no potential for the direct assimilation of humic substances was found. The strain was merely indirectly benefiting from humic substances by utilizing photooxidation products generated by the impact of UV and visible light on humic substances (65). In this context, the potential for degrading aromatic compounds may be a crucial benefit for the strains harboring ACD if more easily utilizable carbon sources are scarce (66). As well, the potential for assimilating nitrate can be advantageous when nitrogen is limiting (67). According to gene annotation, four more GIs of P. asymbioticus are also likely to provide accessory metabolic capabilities, in addition to the metabolic functions encoded in the core genome. If only these six metabolically functional GIs are considered and differences in the core genome are neglected, 64 metabolically different phenotypes are theoretically realizable. Importantly, seven such phenotypes are found among the nine genome-sequenced strains. Genome sequencing of further P. asymbioticus strains would presumably increase the number of metabolically functional GIs (= x) found within the species, which would exponentially increase the number of possible metabolically different phenotypes (= 2x). It is clear from the above that the number of functionally distinguishable phenotypes within P. asymbioticus is quite high. The fitness advantage gained through the additional metabolic capability of an acquired GI can be reduced by the fitness deficit of harboring additional genetic material in the cell (68). Generally, in Polynucleobacter, there seems to be strong selection pressure toward small genomes, indicated by a high fraction of coding bases and small genome sizes compared to its closest related genera (47). Diversification by GIs, i.e., the partitioning of certain functions among closely related strains, may be an elegant way to reduce competition and minimize the amount of genetic material for a single cell (69). As a consequence of such microniche partitioning, the niche space of a species can be expanded and exploited more efficiently (70). Assuming that partitioning of functional capabilities between different genotypes within taxa is a suitable way to minimize the required genetic material per cell, it could be expected that the fraction of differing gene content between pairs of genomes would be particularly high for taxa with small genome sizes. However, such a trend is not evident from the data presented in Fig. 8, where smaller values of %BBH indicate higher variability in gene content. Comparisons within members of the genus “Candidatus Pelagibacter,” which harbor the smallest genomes in the data set (1.27 to 1.46 Mbp), exhibit %BBH values slightly below the trendline, yet a similar correlation is not seen for the taxa next in genome size, namely, Prochlorococcus (1.48 to 2.68 Mbp) and Polynucleobacter (1.61 to 3.16 Mbp). On the other hand, BBH counts markedly below the trend line are observed for the archaea Sulfolobus (2.06 to 2.99 Mbp) and Haloquadratum (3.02 to 3.59 Mbp). For Haloquadratum walsbyi, the large extent of accessory genes was attributed to the diverse nutrient pool exploited by the single numerically dominant species in a saltern crystallizer (70). While Polynucleobacter is in trend with marine bacteria concerning the extent of gene content variation, differences are evident if the distribution of functional categories assigned to the genes contained in GIs is examined (Fig. 7). Compared to “Candidatus Pelagibacter ubique” and Prochlorococcus marinus (supplemental material S3 in reference 33), it is noticeable that genes affiliated with the categories “energy production and conversion” and “lipid metabolism and transport” are overrepresented in the GIs of P. asymbioticus. High relative frequencies in the category “inorganic ion transport and metabolism” are evidenced for both P. asymbioticus and “Candidatus Pelagibacter ubique” but not Prochlorococcus marinus. Genes assigned to the category “cell wall/membrane/envelope biogenesis” are overrepresented in all three taxa. The properties of the cell surface may essentially be shaped by these genes and are decisive for phage recognition. Besides grazing by phagotrophic protists, phage predation is a principal cause of mortality for pelagic bacteria (71) and is assumed to play a crucial role in maintaining the diversity of prokaryotic populations as explained by the evolutionary model of constant diversity (33, 72). The diversity found in the GI variant CSC (Fig. 1 and Table S6), which is involved in synthesis of cell surface components, conforms to the requirements for this model. In view of the concept of periodic selection on the other hand (64, 73), the revealed metabolic differences between strains would probably not allow for the concurrent diversity found. In addition, considering the competitive exclusion principle (9), it can be doubted that the microniche partitioning realized via the acquisition of distinct GIs reduces competition between different P. asymbioticus strains effectively enough to maintain genotype richness within the species.
Mobility of GIs.
The hints for homologous recombination at six GI positions (Fig. 3B), as well as the presence of tRNAs (Fig. 3A) and/or integrase genes in/adjacent to additive GIs, suggest that they were acquired by horizontal gene transfer. Thus, the distribution of GIs can be uncoupled from the core genome to a certain extent (Fig. 6). This may be crucial for the effective spread of genetic material among geographically separated Polynucleobacter populations. While a genotype introduced into a foreign habitat may stay rare or go extinct in the resident population, the GIs of the immigrated genotype potentially rise to high abundance in the Polynucleobacter community. In this context, it has to be considered that recombination is not limited to conspecific individuals but that the possibility for interspecific transfer (Fig. 5) enhances the probability of an effective distribution of GIs. Where phages mediate the transfer of GIs, which is suggested by the presence of phage integrase genes in certain GIs, it is conceivable that phages are serving as a vector for dispersal across sites. The action of broad-host-range phages (74) might allow for interspecific exchange of GIs. Enhanced gene flow with respect to certain genomic regions (i.e., GIs) may be particularly important in freshwater bacteria, for which dispersal is limited due to the spatial separation between habitats. Considering MLSA and genome sequencing data, it is assumed that no strains originating from different sites and exhibiting an ANI of >99.7% are found among the 37 P. asymbioticus isolates. This could be a result of undersampling or local adaptation, but it is also likely that it indicates dispersal limitations. The position of MWH-Recht1 in the core-genome phylogeny (Fig. 2), as well as its similarity to strains from Loibersbacher Höhe concerning MLSA (Fig. 1), suggests a past dispersal event. However, ANI values which in all comparisons are <99.5% show that the strain had already diverged considerably from other isolates. In comparison, López-Pérez and colleagues give an example of isolates, exhibiting an ANI of 99.97%, which were isolated from marine samples taken more than 1,000 km away and 5 years apart (29). On the other hand, for thermoacidophilic archaea isolated from distant geothermal regions, it was shown that geographic barriers isolate core genomes and partition the flexible gene pool to a greater extent than assumed here for Polynucleobacter (75).
Horizontal gene transfer across species boundaries indicates that a very large interspecific gene pool is accessible for a single Polynucleobacter species. Comparable examples were found in free-living marine bacteria (5). Thus, it is reasonable to discount the auxiliary genome for bacterial species delineation at least in certain taxa. Compared to eukaryotic species, in addition to the extent of functional diversification found within species, the appearance of genetic similarities between distinct species is also a basal difference between the two domains. If different environments are compared, for distinct species, it is conceivable that certain adaptations in bacterial populations are habitat specific rather than species specific. An example from the data presented in this work is the GI variant NIT, encoding an operon for the assimilation of nitrate. This GI is present in all isolates from the Trög site, even when different species (P. asymbioticus and MWH-Tro8-2-5gr, ANI 80%) are considered, but absent from several isolates from the Loibersbacher Höhe site, affiliated with the same two species (QLW-P1DATA-2 and MWH-Tro8-2-5gr, considered to be conspecific strains, ANI 99%). Thus, in addition to niche partitioning between concurrent cells, GIs may also provide the potential for rapid local adaptation.
In conclusion, we have shown that the flexible genome and especially GIs provide the basis for the physiological, and consequently, ecological breadth of P. asymbioticus. This species cannot be considered an enclosed assemblage of individuals with identical functionalities. The notion of a federation of functionally diversified cells, similar to that described for marine Prochlorococcus (38), is likely applicable even on the species level. Yet, in contrast to marine ecosystems, the spatially separated and shallow habitats of P. asymbioticus impose contrasting principles for population structuring. The mobility of GIs across distant sites in combination with the large accessible gene pool evidenced by horizontal transfer across species boundaries may be a crucial aspect for the evolution of Polynucleobacter bacteria.
MATERIALS AND METHODS
Isolation and characterization of strains.
Bacterial strains were isolated from several dystrophic ponds in the Austrian Alps, mainly by using acclimatization methods (76) (Tables S1 and S2). The majority of strains were isolated according to the dilution acclimatization protocol (DAM) (61), and all strains isolated in October 2007 (Fig. 1) were obtained by targeted isolation employing DAM and screening of cultures for P. asymbioticus strains by using species-specific PCR primers. All P. asymbioticus strains were characterized by multilocus sequence analysis (MLSA) of 11 loci (47).
Genome sequencing, assembly, and annotation.
Genome sequencing and assembly of the P. asymbioticus type strain QLW-P1DMWA-1 have been described elsewhere (77). The genomes of the eight other P. asymbioticus strains were obtained as follows. DNA used for genome sequencing was extracted from biomass of the strains grown in liquid nutrient broth-soyotone-yeast extract (NSY) medium (76), as described previously (77). Illumina paired-end sequencing was performed by Eurofins Genomics using a MiSeq instrument with 2 × 300-bp sequencing mode for the Tro8F10W22 genome and 2 × 150-bp for the other genomes. Reads that did not pass the default Illumina filter procedure (chastity filter) were discarded. The numbers of reads obtained and respective quality statistics are given in Table S3. Reads were assembled using Velvet 1.2.10 (78), in which default settings were used, except for read category (shortPaired1), coverage cutoff (auto), and expected coverage (auto). Multiple hash lengths were tested, and for each genome, the hash length producing the best-performing assembly according to total assembly length, N50, and number of Ns was selected (Table S4). The obtained contigs for each genome were ordered according to the synteny of the closed QLW-P1DMWA-1 genome by using progressiveMauve (79). Subsequently, Ns were removed from assembled sequences, and contigs were split at these positions. Small contigs representing sequences present as an identical copy at a few different loci in the genome could be identified by clearly elevated coverage and were used for gap closure. Furthermore, some gaps could be manually closed after the elimination of low-quality sequences at the ends of contigs. The genome sequences were annotated by using the Integrated Microbial Genomes-Expert Review (IMG/ER) annotation pipeline (80).
Genome analysis.
ANI values were calculated using the IMG/ER comparative analysis system (80). Homologous genes were identified using the same analysis system, with a threshold of >70% gene sequence identity as the homology criterion. This analysis yielded 1,820 core genes among the nine P. asymbioticus genomes. The residual genes were designated flexible genes. Classification into Clusters of Orthologous Groups (COG) categories was adopted from the IMG/ER annotation.
Phylogenetic trees.
A multiple whole-genome alignment of the nine P. asymbioticus genomes was performed by using progressiveMauve (79). The alignment was refined with Gblocks version 0.91b (81) using default parameters in order to omit poorly aligned positions. The resulting alignment of 1.81 Mbp was used to calculate a maximum likelihood-based RAxML tree (82) by using the CIPRES Science Gateway version.3.3 (83), and a neighbor-joining (NJ) tree (84) was made by using the software MEGA7 (85). The 16S-23S internal transcribed spacer (ITS) sequences of the nine P. asymbioticus strains and 13 related strains were aligned using MUSCLE (86). An NJ tree was calculated using MEGA7. RAxML bootstrap values were calculated by 100 rapid bootstrap inferences, and NJ bootstrap values were calculated from 1,000 replications.
Genomic islands.
GIs were defined for each genome as a consecutive sequence of flexible genes longer than 10 kbp. The borders of each such sequence were extended until the cluster of flexible genes was interrupted by a consecutive sequence of core genes longer than 5 kbp. It should be noted that as a consequence of this definition, a few core genes are included in GIs. Genes contained in GIs were designated GI genes, and genes located outside were designated non-GI genes. Due to the highly conserved synteny among the nine P. asymbioticus genomes, the genomic localizations of the GIs were comparable between the strains. Each GI was assigned to a genomic position (1 to 24) according to the intergenomic gene homology of the adjacent genes. The classification of GIs into different variants was based on gene homology or similar functional annotation between GIs of different genomes.
Benzoate and nitrate assimilation experiments.
The potential for the assimilation of benzoate and nitrate was tested for six selected P. asymbioticus strains in batch culture experiments analogous to the substrate assimilation tests described by Hahn et al. (47). Due to the potentially toxic effect of benzoate, the substrate was added to a final concentration of 500 mg · liter−1 in three steps, i.e., 50 mg · liter−1 initial concentration, 150 mg · liter−1 after 5 days of growth, and 300 mg · liter−1 after 10 days of growth. The nitrate assimilation experiment was performed with four specifically modified growth media. The medium contained either nitrate (medium 1), ammonia (medium 2), or urea (medium 3) as the major nitrogen source, or it was nitrogen depleted (medium 4). All four media were modifications of NSY medium (87), i.e., Ca(NO3)2·4H2O was omitted from the inorganic basal medium (IBM), and organic supplements were added in reduced amounts (10 mg · liter−1 each nutrient broth, soyotone, and yeast extract). As an organic carbon supply, 100 mg · liter−1 each Mg-acetate, Na-propionate, succinic acid, pyruvic acid·Na2·6H2O, levulinate, d-galacturonic acid sodium salt, and phosphoenolpyruvic acid were added. All four media were further supplemented with 0.7 mg · liter−1 vitamins (biotin, cobalamin, folic acid, nicotinic acid, pantothenic acid, pyridoxine, riboflavin, thiamine, and vitamin K). Finally, media 1 to 3 were supplemented with the different nitrogen sources in equimolar concentrations, i.e., 43 mg · liter−1 Ca(NO3)2·4H2O (medium 1), 19 mg · liter−1 NH4Cl plus 27 mg · liter−1 CaCl2·2H2O (medium 2), and 11 mg · liter−1 urea plus 27 mg · liter−1 CaCl2·2H2O (medium 3). Medium 4 was supplemented with 27 mg · liter−1 CaCl2·2H2O and no extra nitrogen source. Changes in optical density at 575 nm (ΔOD575) values were determined in reference to the nitrogen-depleted medium 4. For representation of the results as presented in Fig. 4B, the ΔOD575 value for medium 1 (nitrate) is given as a percentage of the mean ΔOD575 from medium 2 (ammonia) and medium 3 (urea).
Diversity of ACD and NIT.
The GI variants ACD and NIT contain an operon associated with aromatic compound degradation and nitrate assimilation, respectively. For each operon, one key gene, the catechol 1,2-dioxygenase gene (catA) or the assimilatory nitrate reductase catalytic subunit gene (nasA), respectively, was selected as a proxy for testing the presence/absence of the respective GI variant in the 37 P. asymbioticus strains. PCR primers were designed for the amplification of partial gene sequences (758 bp and 1,310 bp, respectively). The GI variant was assumed to be absent in the tested strain if the PCR yielded no product. Selected PCR products were sequenced, and a number was assigned to the respective sequence type (1 to 2 for catA and 1 to 4 for nasA; Fig. 1).
Diversity of CSC.
Six genes contained in the GI variant CSC of at least some P. asymbioticus genomes and putatively involved in cell surface structuring were selected for testing their presence/absence in the 37 P. asymbioticus strains (Table S6). PCR primers were designed for amplification of partial gene sequences. PCR products from amplification of the partial peptidoglycan/LPS O-acetylase gene were sequenced. Each strain is assigned a letter according to the presence/absence pattern concerning the six PCR products (A to I, Fig. 1), followed by a number specifying the peptidoglycan/LPS O-acetylase sequence type (1 to 6, Fig. 1). Absence of the peptidoglycan/LPS O-acetylase gene is depicted by a minus sign.
Similarities between homologous GI and non-GI genes.
A BLASTn of the GI genes from all nine P. asymbioticus genomes was performed using the software BLAST+ (88). The same BLASTn was executed for all non-GI genes. The BLASTn hits were separated into comparisons between strains isolated from the same site and comparisons between strains isolated from different sites. Hits with <70% sequence similarity and hits with <70% sequence coverage were removed from further analysis.
Best BLAST hit analysis.
The genes of the nine genomes were divided into four groups: (I) core non-GI genes, (II) core GI genes, (III) flexible non-GI genes, and (IV) flexible GI genes. Only the genes of strain QLW-P1DMWA-1 were used in the groups containing core genes (I and II) in order to omit redundancy stemming from homologous genes and to reduce computational expenses. For all protein sequences in each of the four groups, a BLASTp against the nonredundant protein sequences (nr) database from NCBI (89) was performed using the BLAST+ software (88). BLAST hits against Polynucleobacter gene sequences in the database were excluded. The BLAST results were analyzed using the software MEGAN6 Community Edition (90) with the lowest common ancestor (LCA) parameters min. score, 50; max. expected, 0.1; min. percent identity, 0.0; top percent, 1.0E-10; min. support percent, 0.0 (off); min. support, 1; and min. complexity, 0.0. These settings result in only the best BLAST hit being used for taxonomic assignment.
Homologous gene content within different taxa.
The IMG/ER public genomes database was screened for the following keywords in genome name: Alteromonas, Bacillus subtilis, Haloquadratum, Pelagibacter, SAR11, Polynucleobacter, Prochlorococcus, Shewanella, Sulfolobus, and Synechococcus. All single-cell genomes and genomes with >50 contigs were removed from the obtained data set, resulting in 257 genomes in the final data set. The ANI and number of bidirectional-best BLAST hits (BBH) were calculated for each genome pair within the taxa by using the IMG/ER comparative analysis system (80). The BBH count includes hits of genes having 70% or more identity and at least 70% coverage of the shorter gene and was used as a proxy for the number of homologous genes shared between the respective genome pair. Percentage values were calculated with respect to the average of the protein-coding gene count from the two compared genomes.
Accession number(s).
The genome sequences were deposited in GenBank under the accession numbers CP000655, CP015016 to CP015018, CP015922, LVLD00000000, LVLE00000000, LVLF00000000, LVLG00000000, LVJO00000000, LZFI00000000, LZMQ00000000, and LZMR00000000. The GenBank/EMBL accession numbers for further nucleotide sequences used in this study are AJ879778, AJ879783, AJ879799 to AJ879801, FN429654, FN429668, FN429705, FN429706, FN429710 to FN429713, FN429735, FN556009, FN823098, FN823099, FN823102, FN823104, FN823145, FN823146, FN823182, FN823183, FN823185 to FN823187, FN823190, FN823208, HE646440 to HE647131, LN908882, and LT622179 to LT622237. All strains and respective accession numbers are provided in Table S1.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for the contribution to isolation of strains by Jan Jezbera, who prematurely passed away in 2013.
This study was supported by the Austrian Science Fund (FWF) project I482-B09 (Ecological diversification in Polynucleobacter), the European Science Foundation (ESF) project FREDI, and the Austrian Academy of Sciences project 23791 (DOC fellowship).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02266-16.
REFERENCES
- 1.Pace NR. 2009. Mapping the tree of life: progress and prospects. Microbiol Mol Biol Rev 73:565–576. doi: 10.1128/MMBR.00033-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Handelsman J. 2004. Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev 68:669–685. doi: 10.1128/MMBR.68.4.669-685.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welch RA, Burland V, Plunkett G III, Redford P, Roesch P, Rasko D, Buckles EL, Liou S-R, Boutin A, Hackett J, Stroud D, Mayhew GF, Rose DJ, Zhou S, Schwartz DC, Perna NT, Mobley HLT, Donnenberg MS, Blattner FR. 2002. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci U S A 99:17020–17024. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schloter M, Lebuhn M, Heulin T, Hartmann A. 2000. Ecology and evolution of bacterial microdiversity. FEMS Microbiol Rev 24:647–660. doi: 10.1111/j.1574-6976.2000.tb00564.x. [DOI] [PubMed] [Google Scholar]
- 5.López-Pérez M, Rodriguez-Valera F. 2016. Pangenome evolution in the marine bacterium Alteromonas. Genome Biol Evol 8:1556–1570. doi: 10.1093/gbe/evw098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kashtan N, Roggensack SE, Rodrigue S, Thompson JW, Biller SJ, Coe A, Ding H, Marttinen P, Malmstrom RR, Stocker R, Follows MJ, Stepanauskas R, Chisholm SW. 2014. Single-cell genomics reveals hundreds of coexisting subpopulations in wild Prochlorococcus. Science 344:416–420. doi: 10.1126/science.1248575. [DOI] [PubMed] [Google Scholar]
- 7.Gonzaga A, Martin-Cuadrado AB, Lopez-Perez M, Megumi Mizuno C, Garcia-Heredia I, Kimes NE, Lopez-Garcia P, Moreira D, Ussery D, Zaballos M, Ghai R, Rodriguez-Valera F. 2012. Polyclonality of concurrent natural populations of Alteromonas macleodii. Genome Biol Evol 4:1360–1374. doi: 10.1093/gbe/evs112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hunt DE, David LA, Gevers D, Preheim SP, Alm EJ, Polz MF. 2008. Resource partitioning and sympatric differentiation among closely related bacterioplankton. Science 320:1081–1085. doi: 10.1126/science.1157890. [DOI] [PubMed] [Google Scholar]
- 9.Hardin G. 1960. The competitive exclusion principle. Science 131:1292–1297. doi: 10.1126/science.131.3409.1292. [DOI] [PubMed] [Google Scholar]
- 10.Koeppel AF, Wu M. 2014. Species matter: the role of competition in the assembly of congeneric bacteria. ISME J 8:531–540. doi: 10.1038/ismej.2013.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kimura M. 1983. The neutral theory of molecular evolution. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 12.Thompson JR, Pacocha S, Pharino C, Klepac-Ceraj V, Hunt DE, Benoit J, Sarma-Rupavtarm R, Distel DL, Polz MF. 2005. Genotypic diversity within a natural coastal bacterioplankton population. Science 307:1311–1313. doi: 10.1126/science.1106028. [DOI] [PubMed] [Google Scholar]
- 13.Haegeman B, Weitz JS. 2012. A neutral theory of genome evolution and the frequency distribution of genes. BMC Genomics 13:196. doi: 10.1186/1471-2164-13-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doolittle WF, Zhaxybayeva O. 2009. On the origin of prokaryotic species. Genome Res 19:744–756. doi: 10.1101/gr.086645.108. [DOI] [PubMed] [Google Scholar]
- 15.Koeppel AF, Wertheim JO, Barone L, Gentile N, Krizanc D, Cohan FM. 2013. Speedy speciation in a bacterial microcosm: new species can arise as frequently as adaptations within a species. ISME J 7:1080–1091. doi: 10.1038/ismej.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barberán A, Casamayor EO, Fierer N. 2014. The microbial contribution to macroecology. Front Microbiol 5:203. doi: 10.3389/fmicb.2014.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fraser C, Alm EJ, Polz MF, Spratt BG, Hanage WP. 2009. The bacterial species challenge: making sense of genetic and ecological diversity. Science 323:741–746. doi: 10.1126/science.1159388. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez-Valera F, Ussery DW. 2012. Is the pan-genome also a pan-selectome? F1000Res 1:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cordero OX, Polz MF. 2014. Explaining microbial genomic diversity in light of evolutionary ecology. Nat Rev Microbiol 12:263–273. doi: 10.1038/nrmicro3218. [DOI] [PubMed] [Google Scholar]
- 20.Mira A, Martin-Cuadrado AB, D'Auria G, Rodriguez-Valera F. 2010. The bacterial pan-genome: a new paradigm in microbiology. Int Microbiol 13:45–57. [DOI] [PubMed] [Google Scholar]
- 21.Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. 2005. The microbial pan-genome. Curr Opin Genet Dev 15:589–594. doi: 10.1016/j.gde.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 22.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, Margarit y Ros I, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N, Khouri H, Radune D, Dimitrov G, Watkins K, O'Connor KJB, Smith S, Utterback TR, White O, Rubens CE, Grandi G, Madoff LC, Kasper DL, Telford JL, Wessels MR, Rappuoli R, Fraser CM. 2005. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A 102:13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hacker J, Bender L, Ott M, Wingender J, Lund B, Marre R, Goebel W. 1990. Deletions of chromosomal regions coding for fimbriae and hemolysins occur in vitro and in vivo in various extraintestinal Escherichia coli isolates. Microb Pathog 8:213–225. doi: 10.1016/0882-4010(90)90048-U. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt H, Hensel M. 2004. Pathogenicity islands in bacterial pathogenesis. Clin Microbiol Rev 17:14–56. doi: 10.1128/CMR.17.1.14-56.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palenik B, Brahamsha B, Larimer FW, Land M, Hauser L, Chain P, Lamerdin J, Regala W, Allen EE, McCarren J, Paulsen I, Dufresne A, Partensky F, Webb EA, Waterbury J. 2003. The genome of a motile marine Synechococcus. Nature 424:1037–1042. doi: 10.1038/nature01943. [DOI] [PubMed] [Google Scholar]
- 26.Coleman ML, Sullivan MB, Martiny AC, Steglich C, Barry K, DeLong EF, Chisholm SW. 2006. Genomic islands and the ecology and evolution of Prochlorococcus. Science 311:1768–1770. doi: 10.1126/science.1122050. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez-Valera F, Martin-Cuadrado AB, Lopez-Perez M. 2016. Flexible genomic islands as drivers of genome evolution. Curr Opin Microbiol 31:154–160. doi: 10.1016/j.mib.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 28.Grote J, Thrash JC, Huggett MJ, Landry ZC, Carini P, Giovannoni SJ, Rappe MS. 2012. Streamlining and core genome conservation among highly divergent members of the SAR11 clade. mBio 3(5):e00252-12. doi: 10.1128/mBio.00252-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.López-Pérez M, Gonzaga A, Rodriguez-Valera F. 2013. Genomic diversity of “deep ecotype” Alteromonas macleodii isolates: evidence for pan-Mediterranean clonal frames. Genome Biol Evol 5:1220–1232. doi: 10.1093/gbe/evt089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.López-Pérez M, Martin-Cuadrado AB, Rodriguez-Valera F. 2014. Homologous recombination is involved in the diversity of replacement flexible genomic islands in aquatic prokaryotes. Front Genet 5:147. doi: 10.3389/fgene.2014.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nazarenko EL, Crawford RJ, Ivanova EP. 2011. The structural diversity of carbohydrate antigens of selected Gram-negative marine bacteria. Marine Drugs 9:1914–1954. doi: 10.3390/md9101914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shimada M, Kadowaki T, Taniguchi Y, Inagawa H, Okazaki K, Soma G-I. 2012. The involvement of O-antigen polysaccharide in lipopolysaccharide in macrophage activation. Anticancer Res 32:2337–2341. [PubMed] [Google Scholar]
- 33.Rodriguez-Valera F, Martin-Cuadrado AB, Rodriguez-Brito B, Pasic L, Thingstad TF, Rohwer F, Mira A. 2009. Explaining microbial population genomics through phage predation. Nat Rev Microbiol 7:828–836. doi: 10.1038/nrmicro2235. [DOI] [PubMed] [Google Scholar]
- 34.Avrani S, Wurtzel O, Sharon I, Sorek R, Lindell D. 2011. Genomic island variability facilitates Prochlorococcus-virus coexistence. Nature 474:604–608. doi: 10.1038/nature10172. [DOI] [PubMed] [Google Scholar]
- 35.Lloyd AL, Henderson TA, Vigil PD, Mobley HLT. 2009. Genomic islands of uropathogenic Escherichia coli contribute to virulence. J Bacteriol 191:3469–3481. doi: 10.1128/JB.01717-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Juhas M, Power PM, Harding RM, Ferguson DJP, Dimopoulou ID, Elamin ARE, Mohd-Zain Z, Hood DW, Adegbola R, Erwin A, Smith A, Munson RS, Harrison A, Mansfield L, Bentley S, Crook DW. 2007. Sequence and functional analyses of Haemophilus spp. genomic islands. Genome Biol 8:R237. doi: 10.1186/gb-2007-8-11-r237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Achtman M. 2012. Insights from genomic comparisons of genetically monomorphic bacterial pathogens. Philos Trans R Soc Lond B Biol Sci 367:860–867. doi: 10.1098/rstb.2011.0303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biller SJ, Berube PM, Lindell D, Chisholm SW. 2015. Prochlorococcus: the structure and function of collective diversity. Nat Rev Microbiol 13:13–27. doi: 10.1038/nrmicro3378. [DOI] [PubMed] [Google Scholar]
- 39.Coleman ML, Chisholm SW. 2010. Ecosystem-specific selection pressures revealed through comparative population genomics. Proc Natl Acad Sci U S A 107:18634–18639. doi: 10.1073/pnas.1009480107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo C, Konstantinidis KT. 2011. Phosphorus-related gene content is similar in Prochlorococcus populations from the North Pacific and North Atlantic Oceans. Proc Natl Acad Sci U S A 108:E62–E63. doi: 10.1073/pnas.1018662108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwalbach MS, Tripp HJ, Steindler L, Smith DP, Giovannoni SJ. 2010. The presence of the glycolysis operon in SAR11 genomes is positively correlated with ocean productivity. Environ Microbiol 12:490–500. doi: 10.1111/j.1462-2920.2009.02092.x. [DOI] [PubMed] [Google Scholar]
- 42.Stuart RK, Brahamsha B, Busby K, Palenik B. 2013. Genomic island genes in a coastal marine Synechococcus strain confer enhanced tolerance to copper and oxidative stress. ISME J 7:1139–1149. doi: 10.1038/ismej.2012.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stuart RK, Dupont CL, Johnson DA, Paulsen IT, Palenik B. 2009. Coastal strains of marine Synechococcus species exhibit increased tolerance to copper shock and a distinctive transcriptional response relative to those of open-ocean strains. Appl Environ Microbiol 75:5047–5057. doi: 10.1128/AEM.00271-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patin NV, Duncan KR, Dorrestein PC, Jensen PR. 2016. Competitive strategies differentiate closely related species of marine actinobacteria. ISME J 10:478–490. doi: 10.1038/ismej.2015.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim M, Oh HS, Park SC, Chun J. 2014. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol 64:346–351. doi: 10.1099/ijs.0.059774-0. [DOI] [PubMed] [Google Scholar]
- 46.Kopac S, Wang Z, Wiedenbeck J, Sherry J, Wu M, Cohan FM. 2014. Genomic heterogeneity and ecological speciation within one subspecies of Bacillus subtilis. Appl Environ Microbiol 80:4842–4853. doi: 10.1128/AEM.00576-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hahn MW, Scheuerl T, Jezberova J, Koll U, Jezbera J, Simek K, Vannini C, Petroni G, Wu QL. 2012. The passive yet successful way of planktonic life: genomic and experimental analysis of the ecology of a free-living Polynucleobacter population. PLoS One 7:e32772. doi: 10.1371/journal.pone.0032772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dufresne A, Garczarek L, Partensky F. 2005. Accelerated evolution associated with genome reduction in a free-living prokaryote. Genome Biol 6:R14. doi: 10.1186/gb-2005-6-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.West NJ, Scanlan DJ. 1999. Niche-partitioning of Prochlorococcus populations in a stratified water column in the eastern North Atlantic Ocean. Appl Environ Microbiol 65:2585–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rocap G, Larimer FW, Lamerdin J, Malfatti S, Chain P, Ahlgren NA, Arellano A, Coleman M, Hauser L, Hess WR, Johnson ZI, Land M, Lindell D, Post AF, Regala W, Shah M, Shaw SL, Steglich C, Sullivan MB, Ting CS, Tolonen A, Webb EA, Zinser ER, Chisholm SW. 2003. Genome divergence in two Prochlorococcus ecotypes reflects oceanic niche differentiation. Nature 424:1042–1047. doi: 10.1038/nature01947. [DOI] [PubMed] [Google Scholar]
- 51.Whitaker RJ, Grogan DW, Taylor JW. 2003. Geographic barriers isolate endemic populations of hyperthermophilic archaea. Science 301:976–978. doi: 10.1126/science.1086909. [DOI] [PubMed] [Google Scholar]
- 52.Lindström ES, Langenheder S. 2012. Local and regional factors influencing bacterial community assembly. Environ Microbiol Rep 4:1–9. doi: 10.1111/j.1758-2229.2011.00257.x. [DOI] [PubMed] [Google Scholar]
- 53.Müller AL, de Rezende JR, Hubert CR, Kjeldsen KU, Lagkouvardos I, Berry D, Jorgensen BB, Loy A. 2014. Endospores of thermophilic bacteria as tracers of microbial dispersal by ocean currents. ISME J 8:1153–1165. doi: 10.1038/ismej.2013.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hahn MW. 2003. Isolation of strains belonging to the cosmopolitan Polynucleobacter necessarius cluster from freshwater habitats located in three climatic zones. Appl Environ Microbiol 69:5248–5254. doi: 10.1128/AEM.69.9.5248-5254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hahn MW, Koll U, Jezberova J, Camacho A. 2015. Global phylogeography of pelagic Polynucleobacter bacteria: restricted geographic distribution of subgroups, isolation by distance and influence of climate. Environ Microbiol 17:829–840. doi: 10.1111/1462-2920.12532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jezberová J, Jezbera J, Brandt U, Lindstrom ES, Langenheder S, Hahn MW. 2010. Ubiquity of Polynucleobacter necessarius ssp. asymbioticus in lentic freshwater habitats of a heterogeneous 2000 km area. Environ Microbiol 12:658–669. doi: 10.1111/j.1462-2920.2009.02106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jezbera J, Jezberova J, Brandt U, Hahn MW. 2011. Ubiquity of Polynucleobacter necessarius subspecies asymbioticus results from ecological diversification. Environ Microbiol 13:922–931. doi: 10.1111/j.1462-2920.2010.02396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jezbera J, Jezberova J, Koll U, Hornak K, Simek K, Hahn MW. 2012. Contrasting trends in distribution of four major planktonic betaproteobacterial groups along a pH gradient of epilimnia of 72 freshwater habitats. FEMS Microbiol Ecol 81:467–479. doi: 10.1111/j.1574-6941.2012.01372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hahn MW, Jezberová J, Koll U, Saueressig-Beck T, Schmidt J. 2016. Complete ecological isolation and cryptic diversity in Polynucleobacter bacteria not resolved by 16S rRNA gene sequences. ISME J 10:1642–1655. doi: 10.1038/ismej.2015.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hahn MW, Schmidt J, Pitt A, Taipale SJ, Lang E. 2016. Reclassification of four Polynucleobacter necessarius strains as representatives of Polynucleobacter asymbioticus comb. nov., Polynucleobacter duraquae sp. nov., Polynucleobacter yangtzensis sp. nov. and Polynucleobacter sinensis sp. nov., and emended description of Polynucleobacter necessarius. Int J Syst Evol Microbiol 66:2883–2892. doi: 10.1099/ijsem.0.001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hahn MW, Pockl M, Wu QL. 2005. Low intraspecific diversity in a Polynucleobacter subcluster population numerically dominating bacterioplankton of a freshwater pond. Appl Environ Microbiol 71:4539–4547. doi: 10.1128/AEM.71.8.4539-4547.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reva O, Tummler B. 2008. Think big–giant genes in bacteria. Environ Microbiol 10:768–777. doi: 10.1111/j.1462-2920.2007.01500.x. [DOI] [PubMed] [Google Scholar]
- 63.Cardinale BJ. 2011. Biodiversity improves water quality through niche partitioning. Nature 472:86–89. doi: 10.1038/nature09904. [DOI] [PubMed] [Google Scholar]
- 64.Cohan FM, Perry EB. 2007. A systematics for discovering the fundamental units of bacterial diversity. Curr Biol 17:R373–R386. doi: 10.1016/j.cub.2007.03.032. [DOI] [PubMed] [Google Scholar]
- 65.Anesio AM, Graneli W, Aiken GR, Kieber DJ, Mopper K. 2005. Effect of humic substance photodegradation on bacterial growth and respiration in lake water. Appl Environ Microbiol 71:6267–6275. doi: 10.1128/AEM.71.10.6267-6275.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Steinberg CEW, Kamara S, Prokhotskaya VY, Manusadzianas L, Karasyova TA, Timofeyev MA, Jie Z, Paul A, Meinelt T, Farjalla VF, Matsuo AYO, Kent Burnison B, Menzel R. 2006. Dissolved humic substances–ecological driving forces from the individual to the ecosystem level? Freshwat Biol 51:1189–1210. doi: 10.1111/j.1365-2427.2006.01571.x. [DOI] [Google Scholar]
- 67.Elser JJ, Bracken ME, Cleland EE, Gruner DS, Harpole WS, Hillebrand H, Ngai JT, Seabloom EW, Shurin JB, Smith JE. 2007. Global analysis of nitrogen and phosphorus limitation of primary producers in freshwater, marine and terrestrial ecosystems. Ecol Lett 10:1135–1142. doi: 10.1111/j.1461-0248.2007.01113.x. [DOI] [PubMed] [Google Scholar]
- 68.Giovannoni SJ, Cameron Thrash J, Temperton B. 2014. Implications of streamlining theory for microbial ecology. ISME J 8:1553–1565. doi: 10.1038/ismej.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kempes CP, Wang L, Amend JP, Doyle J, Hoehler T. 2016. Evolutionary tradeoffs in cellular composition across diverse bacteria. ISME J 10:2145–2157. doi: 10.1038/ismej.2016.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cuadros-Orellana S, Martin-Cuadrado A-B, Legault B, D'Auria G, Zhaxybayeva O, Papke RT, Rodriguez-Valera F. 2007. Genomic plasticity in prokaryotes: the case of the square haloarchaeon. ISME J 1:235–245. doi: 10.1038/ismej.2007.35. [DOI] [PubMed] [Google Scholar]
- 71.Pernthaler J. 2005. Predation on prokaryotes in the water column and its ecological implications. Nat Rev Microbiol 3:537–546. doi: 10.1038/nrmicro1180. [DOI] [PubMed] [Google Scholar]
- 72.Thingstad TF. 2000. Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol Oceanogr 45:1320–1328. doi: 10.4319/lo.2000.45.6.1320. [DOI] [Google Scholar]
- 73.Atwood KC, Schneider LK, Ryan FJ. 1951. Periodic selection in Escherichia coli. Proc Natl Acad Sci U S A 37:146–155. doi: 10.1073/pnas.37.3.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jensen EC, Schrader HS, Rieland B, Thompson TL, Lee KW, Nickerson KW, Kokjohn TA. 1998. Prevalence of broad-host-range lytic bacteriophages of Sphaerotilus natans, Escherichia coli, and Pseudomonas aeruginosa. Appl Environ Microbiol 64:575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reno ML, Held NL, Fields CJ, Burke PV, Whitaker RJ. 2009. Biogeography of the Sulfolobus islandicus pan-genome. Proc Natl Acad Sci U S A 106:8605–8610. doi: 10.1073/pnas.0808945106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hahn MW, Stadler P, Wu QL, Pockl M. 2004. The filtration-acclimatization method for isolation of an important fraction of the not readily cultivable bacteria. J Microbiol Methods 57:379–390. doi: 10.1016/j.mimet.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 77.Meincke L, Copeland A, Lapidus A, Lucas S, Berry KW, Del Rio TG, Hammon N, Dalin E, Tice H, Pitluck S, Richardson P, Bruce D, Goodwin L, Han C, Tapia R, Detter JC, Schmutz J, Brettin T, Larimer F, Land M, Hauser L, Kyrpides NC, Ivanova N, Goker M, Woyke T, Wu QL, Pockl M, Hahn MW, Klenk HP. 2012. Complete genome sequence of Polynucleobacter necessarius subsp. asymbioticus type strain (QLW-P1DMWA-1T). Stand Genomic Sci 6:74–83. doi: 10.4056/sigs.2395367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang J, Williams P, Huntemann M, Anderson I, Mavromatis K, Ivanova NN, Kyrpides NC. 2012. IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res 40:D115–D122. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 82.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 83.Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees, p 1–8. Proceedings of the Gateway Computing Environments Workshop (GCE), 14 November 2010, New Orleans, LA. [Google Scholar]
- 84.Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. [DOI] [PubMed] [Google Scholar]
- 85.Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hahn MW, Lunsdorf H, Wu Q, Schauer M, Hofle MG, Boenigk J, Stadler P. 2003. Isolation of novel ultramicrobacteria classified as Actinobacteria from five freshwater habitats in Europe and Asia. Appl Environ Microbiol 69:1442–1451. doi: 10.1128/AEM.69.3.1442-1451.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.NCBI Resource Coordinators. 2013. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 41:D8–D20. doi: 10.1093/nar/gks1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huson DH, Beier S, Flade I, Górska A, El-Hadidi M, Mitra S, Ruscheweyh H-J, Tappu R. 2016. MEGAN Community Edition—interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput Biol 12:e1004957. doi: 10.1371/journal.pcbi.1004957. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.