

Abstract
Voltage-gated Kv1.1/Kvβ1.1 A-type channels, as a natural complex, can switch from fast to slow inactivation under oxidation/reduction conditions. The mode-switching of inactivation, which is mediated by a cysteine residue in the inactivation ball domain of the Kvβ1.1 N terminus, can regulate membrane electrical excitability. In the present study, we identified a mechanism whereby inactivation in Kv1.1/Kvβ1.1 channels is regulated by calcium influx. The rise in intracellular calcium, due to either influx from extracellular space or release from intracellular stores, eliminates fast inactivation induced by Kvβ1.1, resulting in slower inactivation and increased steady-state current. This oxidation-independent calcium effect is mediated through the Kvβ1.1 N terminus, not the C terminus. We propose that a coupling between calcium influx and inactivation of voltage-gated A-type K+ channels occurs as a result of membrane depolarization and may contribute to afterhyperpolarization as negative feedback to control neuronal excitability.
Keywords: calcium entry, afterhyperpolarization, Xenopus oocytes, two-electrode voltage clamp
Calcium entry is essential for a number of cellular functions, including secretion, contraction, and hormone actions. Upon membrane depolarization, changes in intracellular calcium occur through the opening of voltage-activated calcium channels or depletion of stores by the second messenger inositol trisphosphate (IP3) (1, 2). In neurons, an increase in the free intracellular calcium causes neuronal excitability and triggers neurotransmitter release during the action potential (3). Consequently, influx of calcium during action potentials can lead to increases in cytosolic calcium that can activate potassium currents, which are largely known as calcium-activated potassium channels that regulate cell excitability (4). It has been shown that elevation of intracellular calcium mainly activates two classes of calcium-dependent K+ channels, large-conductance (BK) and small-conductance (SK), both of which reduce membrane excitability (5, 6). Opening of BK channels contributes to spike repolarization and early spike fast afterhyperpolarization (fAHP), whereas SK channels are responsible for medium afterhyperpolarization (mAHP) (7, 8). However, there is also a slow component of afterhyperpolarization (sAHP) that acts as a negative-feedback inhibitor of repetitive firing mediated by non-BK, non-SK channels and remains unidentified (9).
The Kvβ1.1 subunit associates with the noninactivating Kv1.1 α subunit to form rapidly inactivating A-type channels (Kv1.1/Kvβ1.1). The conversion of the delayed rectifier Kv1.1 current to A-type current is mediated by an inactivation ball domain (first 34 residues) in the N terminus of Kvβ1.1 that binds initially to the cytoplasmic channel surface and then enters the pore as an extended peptide. As a native complex, Kv1.1/Kvβ1.1 A-type K+ channels exhibit characteristics of rapid inactivation right after activation in response to membrane depolarization. The time course of inactivation of A-type currents primarily determines action potential waveform and duration and repolarization in neurons (10-13). Therefore, modulation of Kv1.1/Kvβ1.1 channel inactivation can greatly affect neuronal firing.
Studies on Kvβ1.1 knockout mice demonstrated that disruption of Kvβ1.1 inactivation resulted in reduced spike broadening and sAHP, indicating that fast inactivation of Kvβ1.1 is involved in the regulation of action potential broadening and afterhyperpolarization (AHP) (14). It has been proposed that action potential broadening contributes to an increase in intracellular calcium and that calcium influx can lead to a slow activation of sAHP current (IsAHP) (4, 9, 15). A question arose as to whether calcium influx could directly affect the inactivation of Kvβ1.1-dependent voltage-gated A-type K+ channels that are known to regulate neuronal firing. Here, we show that calcium influx, induced by either ionophores or IP3 signaling, directly affects Kv1.1 inactivation induced by Kvβ1.1, resulting in slower inactivation and increased steady-state Kv1.1 current. The calcium effect is Kvβ1.1 N-terminus-dependent. We propose that a coupling between calcium influx and inactivation of voltage-gated A-type K+ channels occurs as a result of membrane depolarization. Such a coupling may provide a physiological mechanism whereby slowed inactivation of A-type K+ channels by calcium contributes to sAHP that serves as a negative feedback to regulate neuronal excitability.
Materials and Methods
Molecular Biology and Channel Expression. Human Kv1.1, Kv1.4 channels, and Kvβ1.1 were subcloned after double digestion with EcoRI and BglII into the pBluescript vector pBJ/KSM (16). Chimera constructs and mutations were generated by PCR with the QuikChange mutagenesis method (Stratagene). The Kv1.1βN/Kv1.4 chimera was constructed on the backbone of the Kv1.4 α subunit by equivalently replacing its proximal N-terminal residues with N-terminal residues (1-70) of Kvβ1.1. The Kv1.4N/Kvβ1.1C chimera was constructed on the backbone of Kvβ1.1 by replacing its N-terminal residues 1-70 with Kv1.4 N-terminal residues 1-70. In Kvβ1.1C7S, the cysteine residue was mutated to serine. For oocyte expression, cDNAs were transcribed in vitro with the T3 mMESSAGE mMACHINE kit (Ambion, Austin, TX) after linearization of the cDNA construct with NotI. Entire inserts of all clones were verified by in-house automated sequencing.
Two-Electrode Voltage-Clamp Recording in Xenopus Oocytes. For channel expression, defolliculated Xenopus oocytes (stage V-VI) were selected and injected with 46 nl of solution containing 0.05-40 ng of cRNAs by using a Drummond microinjector (Drummond Scientific, Broomall, PA). One to 4 days after injection of cRNA, individual oocytes were impaled with two microelectrodes (0.5-1.0 MΩ) filled with 3 M KCl in a 40-μl recording chamber. The chamber was constantly perfused with ND-96 recording solution containing 96 mM NaMeSO3, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, and 5 mM Hepes (pH 7.6); chloride was replaced with 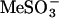 to eliminate chloride-activated current. Currents were recorded by using a Gene-Clamp 500 amplifier (Axon Instruments, Union City, CA) or an OC-725C amplifier (Warner Instruments, Hamden, CT). All recordings were conducted at room temperature (22 ± 1°C). Calcium ionophores ionomycin and A23187; IP3; and kinase inhibitors 19-36, HA-1077, and genistein were from Sigma. KN93 was from Calbiochem and RP-adenosine cyclic 3′,5′-phosphorothioate (RP-cAMPS) was from Research Biochemicals (Natick, MA). HA-1077 (10 mM) and RP-cAMPS (10 mM) for protein kinase A and 19-36 (1 mM) for PKC were injected into oocytes for a period of 10-60 min before application of ionophore A23817. KN-93 (50 μM) for calmodulin-dependent protein kinase II was incubated with oocytes for a period of 10-30 min before application of the ionophore.
to eliminate chloride-activated current. Currents were recorded by using a Gene-Clamp 500 amplifier (Axon Instruments, Union City, CA) or an OC-725C amplifier (Warner Instruments, Hamden, CT). All recordings were conducted at room temperature (22 ± 1°C). Calcium ionophores ionomycin and A23187; IP3; and kinase inhibitors 19-36, HA-1077, and genistein were from Sigma. KN93 was from Calbiochem and RP-adenosine cyclic 3′,5′-phosphorothioate (RP-cAMPS) was from Research Biochemicals (Natick, MA). HA-1077 (10 mM) and RP-cAMPS (10 mM) for protein kinase A and 19-36 (1 mM) for PKC were injected into oocytes for a period of 10-60 min before application of ionophore A23817. KN-93 (50 μM) for calmodulin-dependent protein kinase II was incubated with oocytes for a period of 10-30 min before application of the ionophore.
Macro Patch Recording of Kv1.1/Kvβ1.1 Channels. Recordings from macro inside-out patches excised from Xenopus oocytes were conducted by using an EPC9 amplifier (HEKA Electronics, Lembrecht/Pfalz, Germany), low-pass filtered at 2 kHz and sampled at 10 kHz. Patch pipettes were fabricated from borosilicate thin-wall glass (WI 30-0035, Harvard Apparatus) and had a resistance of ≈1.0 MΩ when filled with the extracellular ND-96 solution. The intracellular (bath) solution consisted of 120 mM KCl, 2 mM MgCl2, 10 mM Hepes, 1 mM EGTA, 0.137 mM CaCl2, and 4 mM glucose (pH 7.2). CaCl2 was added to the intracellular solution in an amount to yield the desired concentration of free Ca2+, as calculated with the cabuf program (17).
Data Acquisition and Analysis. Data were acquired and analyzed with pulse software (HEKA Electronics, Lambrecht/Pfalz, Germany) and a Pentium III PC. Current was low-pass filtered at 200 Hz and digitized at 1.0 kHz by using an ITC-16 interface (Instrutech, Mineola, NY). Data were analyzed with pulsefit, igor, or microcal origin 5.0. Inactivation time constants (τ) were obtained by fitting a single-exponential function to the decaying phases of Kv currents. All data are given as mean ± SEM for 4-24 cells, and statistical analysis was performed by using Student's t test.
Results
Calcium Ionophores Eliminate Fast Inactivation of Kv1.1/Kvβ1. We started by replicating the effects of Kvβ1.1 on Kv1.1 channels coexpressed in Xenopus oocytes. As expected, the noninactivating Kv1.1 current is converted to an inactivating A-type current induced by Kvβ1.1 with fast (τf = 8.9 ± 0.9 ms) and slow (τs = 240 ± 27 ms) inactivation time constants during a 200-ms pulse at +50 mV (Fig. 1 A-C). When the calcium ionophore ionomycin (1 μM) was perfused to raise the level of intracellular calcium in oocytes, the fast inactivation by Kvβ1.1 was eliminated without affecting the amplitude of initial peak current. As a result, the slow inactivating current (τ = 150 ± 34 ms) remained, and the noninactivating steady-state current, measured at the end of a 200-ms pulse at +50 mV, was increased by ≈65% (Fig. 1D). Increasing the concentration of ionomycin to 10 μM resulted in even slower inactivating current, with a time constant of 325 ± 105 ms, and increased steady-state current ≈3.0-fold (Fig. 1E). Fig. 1F shows the dose-dependent effects of ionomycin on inactivation. To confirm the calcium effect observed with ionomycin, we also used another calcium ionophore, A23187. A23187 (10 μM) ablated the fast inactivation induced by Kvβ1.1, resulting in a noninactivating Kv1.1 current with a time constant of 316 ± 39 ms (Fig. 2 A-C). A23187 also increased the steady-state current ≈3.0-fold (Fig. 2D). To determine whether the calcium effects were mediated through activation of kinases, we tested several kinase inhibitors on the inactivation: genestein for tyrosine kinase, HA-1077 (10 mM) and RP-cAMPS (10 mM) for protein kinase A, 19-36 (1 mM) for PKC, and KN-93 (50 μM) for calmodulin-dependent protein kinase II. None of them, from concentrations of 25 μM to 10 mM, had any effect on inactivation (data not shown).
Fig. 1.

Calcium ionophore ionomycin eliminates fast inactivation of A-type Kv1.1/Kvβ1.1 channels induced by Kvβ1.1. (A) Kv1.1 outward current traces were recorded in Xenopus oocytes by a family of depolarizing potentials from -60 to +50 mV with a 10-mV increment for 200 ms from a holding potential at -80 mV. (B) A-type current traces of Kv1.1 coexpressed with Kvβ1.1 in oocytes with the same protocol as in A. (C) Comparison of inactivation kinetics of Kv1.1 alone and Kv1.1 plus Kvβ1.1 at +50 mV from A and B. (D) A family of current traces recorded before and after applications of 1 μM ionomycin. Voltage protocols used are the same as in A. (E) Current traces recorded before and after application of 10 μM ionomycin with the same voltage protocol as in A. (F) Comparisons of inactivation and steady-state current from D and E at 50 mV.
Fig. 2.

Calcium ionophore A-23187 also eliminates fast inactivation of Kv1.1/Kvβ1.1 K+ channels. Xenopus oocytes injected with Kv1.1/Kvβ1.1 were recorded in Cl--free solution (1 mM Ca2+/NaMeSO3), held at -80 mV, and pulsed from -60 to +50 mV in 10-mV steps for 200 ms. A family of current traces was recorded from oocytes before (A) and after (B) applications of 10 μM A-23187. (C) Comparison of inactivation kinetics in the absence and presence of A-23187 (10 μM). Current was recorded with a single pulse from a holding potential at -80 mV and stepped to +50 mV for 200 ms. (D) Current-voltage relationship of steady-state current in A-23187 (▪) and control (○) from A and B.
IP3 Signaling also Ablates the Fast Inactivation of Kv1.1/Kvβ1. Previous results with calcium ionophores suggested to us that inactivation of voltage-gated Kv1.1/Kvβ1.1 A-type channels was affected by calcium influx. To investigate whether calcium released from intracellular stores by IP3 signaling can mimic the effect of calcium ionophores, we injected 46 nl of 2 mM IP3 into oocytes expressing Kv1.1/Kvβ1.1 channels. IP3 release of intracellular Ca2+ can activate Ca2+-dependent Cl- current. To avoid any contamination of this calcium-activated chloride current, a Cl--free solution was used. Injection of IP3 changed the fast inactivating Kv1.1/Kvβ1.1 current (τ = 9.9 ± 0.3 ms) to a slow inactivating current with a time constant of 245 ± 14 ms, and the steady-state current was doubled (Fig. 3A). The IP3 effect on inactivation is consistent with that of calcium ionophores. It is also noticeable that the IP3 effect gradually disappeared over a period of 15 min when inactivation was partially restored (Fig. 3A).
Fig. 3.

Time courses of IP3 effects on inactivation and steady-state current of Kv1.1/Kvβ1.1 channels. Representative current traces were recorded in oocytes in Cl--free recording solution with a single pulse from a holding potential at -80 mV and stepped to +50 mV for 200 ms with a 10-s interval. (A) Extracellular recording solution contains 1.0 mM calcium. Arrow indicates the injection of 46 nl of 2 mM IP3. Current sample traces showing inactivation and steady-state current were taken at different time points, indicated by 1-3. (B) IP3 effects on inactivation and steady-state currents in either Ca2+-free or 1 mM Ca2+ external Cl--free solution. The voltage protocol used is the same as in A. Arrow, time point when IP3 was injected. Current traces showing the changes of inactivation and steady-state current at different time points are indicated by 1-5.
We thought that the relatively long-lasting IP3 effect was due to an entry of extracellular Ca2+ through the opening of voltagegated calcium channels in response to membrane depolarization, because our recording ND-96 solution contained a standard 1 mM calcium. We then switched to a calcium-free recording solution. In the absence of calcium in the bath solution, IP3 had a similar effect on Kv1.1/Kvβ1.1 inactivation. In contrast, the effect lasted <2 min before returning to a baseline level. When 1 mM calcium was added back to the bath solution, the IP3 effect resulting in a slower inactivation was restored (Fig. 3B), and then it decreased gradually over a period of 15 min, a long-lasting effect similar to that seen in Fig. 3A.
Effect of Free Calcium on Inactivation of Kv1.1/Kvβ1.1. To further determine whether calcium directly affects inactivation, we used inside-out macro patches excised from Xenopus oocytes and investigated the effect of free calcium on inactivation of Kv1.1/Kvβ1.1. To obtain specific free Ca2+ concentrations, 1 mM EGTA was added to the bath solution, and different concentrations of CaCl2 were added as calculated with the cabuf program. With normal free calcium (25 nM), Kv1.1/Kvβ1.1 current inactivates rapidly, with an inactivation time constant of 7.0 ± 0.7 ms (n = 18 patches) at 50 mV. Upon application of calcium (930 nM) to the cytoplasmic side of macro inside-out patches, the fast inactivation was eliminated, resulting in a markedly slowed inactivation time constant of 59 ± 4 ms (n = 7 patches). Fast inactivation was returned upon washout of calcium (Fig. 4A). Calcium (930 nM) also increased both peak current and the steady-state currents ≈1-fold and 13-fold (n = 10), respectively (Fig. 4B). Application of different concentrations of calcium ranging from 25 to 930 nM resulted in a linear increase of the inactivation time constant, and increased peak and sustained currents were recorded from inside-out patches in a concentration-dependent manner (Fig. 4 C and D). Extremely high concentrations of calcium (550 μM) applied to the insideout patches (n = 4) abolished inactivation, leaving noninactivating currents like Kv1.1 alone, with activation slightly slowed (data not shown). Activation of Kv1.1/Kvβ1.1 current was unaffected by calcium concentrations up to 930 nM, as indicated by the similar kinetics. These results suggest that calcium directly affects Kv1.1/Kvβ1.1 current.
Fig. 4.

The concentration-dependent effect of intracellular free calcium on inactivation and sustained currents of Kv1.1/Kvβ1.1 currents. (A) Currents were recorded from macro inside-out patches in response to a voltage pulse of +50 mV from -80 mV for 200 ms before and after application of 930 nM free calcium to the bath solution (as calculated with the cabuf program) and washout. (B) Effects of different concentrations of calcium, ranging from 25 to 930 nM, added to the cytoplasmic side of inside-out patches on inactivation and peak current and sustained current. Inset shows the normalized traces according to the peak current and compares the inactivation before and after 930 nM calcium was added to the bath. (C) Inactivation time constant changes with the increasing concentrations of calcium added to the bath. (D) A summary of changes in ratios of steady-state current (Iss) and peak current (Imax) in different concentrations of free calcium. Data are presented in mean ± SEM of 7-18 patches.
The Calcium Effect Is Kvβ1.1 N-Terminal-Dependent. To be certain that calcium specifically affects the inactivation of Kv1.1/Kvβ1.1, we investigated effects of ionomycin on another A-type channel, Kv1.4. In oocytes, Kv1.4 currents activate instantaneously and inactivate with a time constant of 36 ± 2.0 ms. With application of 10 μM ionomycin, inactivation of Kv1.4 channels was not altered, indicating that calcium has no effect on inactivation of Kv1.4 A-type channels (Fig. 5A). This result also suggests that the calcium effect induced by ionophores on inactivation is specific to Kv1.1/Kvβ1.1. To further dissect whether Kvβ1.1 is responsible for the calcium effect, we then made a chimera, Kvβ1.1N/Kv1.4, in which the Kv1.4 N terminus was truncated and replaced with an equal number of N-terminal residues, 1-70, of Kvβ1.1. Like the WT Kv1.4, the chimera shows a fast inactivation, with a time constant of 23 ± 0.9 ms. With application of 10 μM ionomycin, fast inactivation of the chimera (Kvβ1.1N/Kv1.4) was eliminated, leaving a slow inactivating current with a time constant of 464 ± 115 ms. The steady-state current also increased ≈2.0-fold (Fig. 5B). This result suggested that the calcium effect is mediated through the Kvβ1.1 N terminus.
Fig. 5.

Effect of calcium ionophore ionomycin on inactivation is Kvβ1.1 N-terminus-dependent. (A) Representative Kv1.4 current traces were recorded in oocytes at +50 mV before and after application of ionomycin (10 μM) in Cl--free NaMeSO3 recording solution with 1 mM Ca2+. The inactivation time constant of Kv1.4 is 36 ± 2 ms. (B) Representative current traces of a Kvβ1.1N/Kv1.4 chimera in which the Kv1.4 N terminus was truncated and replaced with an equal number of N-terminal residues, 1-70, of Kvβ1.1 in the presence and absence of ionomycin (10 μM), using the same voltage protocol as in A. (C) Current traces of Kv1.4N/Kvβ1.1C chimera coexpressed with Kv1.1 before and after application of ionomycin (10 μM). The Kv1.4N/Kvβ1.1C chimera was constructed on the backbone of Kvβ1.1 by replacing its N-terminal residues (1-70) with Kv1.4 N-terminal residues 1-70. (D) Current traces of Kvβ1.1 C7S mutant (cysteine at position 7 was mutated to serine) and Kv1.1 coexpressed before and after application of ionomycin (10 μM).
In a similar way, we also constructed a Kv1.4N/Kvβ1.1C chimera in which the proximal N terminus of Kvβ1.1 was truncated and replaced with N-terminal residues of Kv1.4. When the Kv1.4N/Kvβ1.1C chimera coexpressed with Kv1.1, this channel complex gave rise to a typical A-type current with fast inactivation of 43 ± 9 ms. However, ionomycin (10 μM) had no effect on the fast inactivation of the Kv1.4N/Kvβ1.1C chimera coexpressed with Kv1.1 (Fig. 5C). Because redox-sensitive inactivation is also conferred by Kvβ1.1 and fast inactivation of Kvβ1.1 can be abolished when the cysteine residue at position 7 (C7) is oxidized, mutating the cysteine into serine (C7S) prevents oxidation from removing inactivation in Kv1.1/Kvβ1.1 channels (18). To determine whether calcium acted by oxidizing Kvβ1.1, we coexpressed Kvβ1.1C7S with Kv1.1, which gave rise to a typical A-type current with an inactivation time constant of 14 ± 1.0 ms. Ionomycin at 10 μM still was able to ablate the fast inactivation, resulting in a noninactivating current with a time constant of 1,229 ± 413 ms and increasing steady-state current ≈2-fold (Fig. 5D). Therefore, these results demonstrated that the calcium effect on inactivation is oxidation-independent and Kvβ1 N-terminal-specific.
Discussion
The present study describes the functional coupling of intracellular calcium with inactivation of voltage-gated Kv1.1/Kvβ1.1 A-type K+ channels that are prominent for neuronal functions. The experiment illustrates that calcium can affect the kinetics of Kvβ1.1 inactivation, resulting in the switch of inactivation of A-type K+ channels from “fast” to “slow.” The mode-switching of inactivation by calcium is specific to the N terminus of the Kvβ1.1 subunit. Calcium does not affect inactivation of Kv1.4 channels. We hypothesize that the slowed inactivation of voltagegated Kv1.1/Kvβ1.1 K+ channels by calcium serves as a negative feedback mechanism to increase potassium conductance and may contribute to the regulation of repolarization and AHP during the action potentials in neurons.
Specific Modulation of Kvβ1.1 Inactivation Gate by Calcium. Intracellular injection of calcium activates potassium conductance in snail neurons, and the conductance is due to the opening of potassium channels gated by intracellular calcium (19). The fact that the increase in potassium conductance in response to calcium can be partially altered by charybdotoxin also suggests that other voltage-gated potassium channels might be modulated by the rise of intracellular calcium (20).
In this study, our experimental results show that intracellular calcium, because of either influx from the extracellular space or an intracellular store, can directly regulate the inactivating activity of voltage-gated Kv1.1/Kvβ1.1 current, resulting in slowly inactivating channels. In the absence of extracellular calcium, the calcium effect on inactivation is short (less than ≈2 min). When calcium (1 mM) is added back to the extracellular solution, the calcium effect can last ≈15 min, indicating that the increased time course of the calcium effect on inactivation depends on extracellular calcium. In addition, direct application of calcium ranging from 25 to 930 nM to the cytoplasmic side of macro inside-out patches dose-dependently removes fast inactivation of Kv1.1/Kvβ1.1 current, indicating that calcium can directly affect inactivation of the Kvβ1.1 ball domain. Yet, calcium does not affect inactivation of Kv1.4, another A-type K+ channel. It has been reported that Kvβ1.1 can further potentiate the fast inactivation of Kv1.4 (18, 21). When Kv1.4 and Kvβ1.1 coexpressed in oocytes, we indeed observed the faster inactivation (8 ms) of Kv1.4/Kvβ1.1 current. Interestingly, when either ionomycin (10 μM) or A23187 (10 μM) was perfused, the induced fast inactivation by Kvβ1.1 (8 ms) was only partially reversed, with an inactivation time constant of 17 ms, compared with 40-ms inactivation of Kv1.4 alone (Fig. 6). We speculate that the accelerated inactivation of Kv1.4/Kvβ1.1 was not fully reversed by calcium ionophores because of the following reasons. (i) Accelerated inactivation of Kv1.4 induced by Kvβ1.1 is caused by concerted action of both N termini of Kv1.4 and Kvβ1.1, because Kvβ1.1 indeed binds to the N terminus (such as the NAB motif) of Kv1.4 to inactivate (21, 22). (ii) The Kv1.4/Kvβ1.1 complex inactivates faster (inactivation time constant τ = 8 ms) than the Kvβ1.1N/Kv1.4 chimera, in which the N terminus of Kv1.4 is deleted (τ = 23 ms), indicating that the Kv1.4 N terminus significantly contributed to the accelerated inactivation of Kv1.4/Kvβ1.1. (iii) Calcium can affect only inactivation induced by Kvβ1.1, not the N terminus of Kv1.4. This claim is also supported by the evidence of our experiments that calcium affects the Kvβ1.1N/Kv1.4 chimera, not WT Kv1.4 alone, indicating that calcium affects Kvβ1.1 only. To fully understand how calcium regulates the inactivation of the Kv1.4/Kvβ1.1 complex, such questions as where and how Kv1.4 and Kvβ1.1 N termini interact and how much the Kv1.4 N terminus contributes to the accelerated inactivation of Kv1.4/Kvβ1.1 should be further investigated.
Fig. 6.

Effects of calcium ionophores on inactivation of Kv1.4/Kvβ1.1. Current traces of Kv1.4 alone or Kv1.4/Kvβ1.1 were recorded in oocytes at +50 mV from -80 mV. Inactivation time constants of Kv1.4 alone and Kv1.4/Kvβ1.1 are 40 ± 6 and 8 ± 0.8 ms, respectively. The inactivation time constant of Kv1.4/Kvβ1.1 in the presence of ionomycin (10 μM), identical to that of A23187 (10 μM), is 17 ± 2.6 ms.
By constructing chimeras in which the Kvβ1.1 N terminus was either attached to Kv1.4 (Kvβ1.1N1-70/Kv1.4 chimera) or removed from Kvβ1.1 (Kv1.4N/Kvβ1.1 chimera), we further demonstrated that the calcium effect on inactivation of Kv1.1/Kvβ1.1 is Kvβ1.1 N-terminus-specific. Although sequence alignment does not show obvious calcium-binding domains in the N terminus of Kvβ1.1, calcium can still specifically affect the inactivation gate of the Kvβ1.1. It was recently shown that calcium can activate BK potassium channels that lack the calcium bowl and RCK domains (23).
It is known that the rapid inactivation of voltage-gated Kv1.1/Kvβ1.1 A-type channels is mediated by an inactivating ball domain in the N terminus of Kvβ1.1 through binding to the inner pore of the open Kv1.1 channels (24). In response to the cellular environment, the fast inactivating A-type Kv1.1/Kvβ1.1 can be converted to a delayed rectifier by oxidation of cysteine residue C7 in the inactivation ball of the Kvβ1.1 N terminus. The oxidation effect on mode-switching of inactivation can be reversed when the cellular reducing condition is resumed (18, 25). Mutating C7 to serine removes the oxidation effect. We showed that the C7S mutant of Kvβ1.1 still retained the calcium effect on inactivation, indicating that the calcium effect is oxidation-independent. Given that calcium can induce slowed inactivation, we hypothesize that the calcium effect on Kvβ1.1 provides a negative feedback mechanism during the membrane depolarization and that tuning of the inactivation time constant is critical in controlling neuronal excitability.
Slowed Inactivation of A-Type K+ Channels May Contribute to sAHP. Voltage-gated K+ channels are activated by membrane depolarization and serve to repolarize the membrane through conduction of an outward K+ current. Specifically, K+ channels typically either repolarize action potentials and synaptic potentials or limit the rise of these potentials. K+ currents are critical in determining action-potential duration. During a spike train, the inactivation of K+ currents can produce progressive broadening of the spikes. Neurons use inactivating K+ channels to modulate firing frequency and shape the electrical signaling properties. Slow inactivation, however, is involved in repolarization of action potential and attenuation of cell excitability. The Kvβ1.1 knockout mice demonstrated that disruption of Kvβ1.1 inactivation caused reduced spike broadening and AHP (14), suggesting that inactivation of Kvβ1.1 may contribute to the regulation of AHP during action potentials.
AHP has three components: fAHP, mAHP, and sAHP. fAHP is activated immediately during action potential and lasts several tens of milliseconds. It has been suggested that current underlying fAHP is through Ca2+-activated BK (Maxi K)-type K+ channels because of their blockade by low concentrations of tetraethylammonium, iberiotoxin, and paxilline (26-28), although exact identity of these channels has not been determined. mAHP is also activated rapidly after an action potential but decays with a time course of 50 to several hundred milliseconds (29, 30).
The slow activation of sAHP current (IsAHP) underlying sAHP was originally described in neurons in the myenteric plexus (31), and shaker-like A-type current was also identified in the myenteric neurons (32). The molecular identity of sAHP, the third component, remains so far unknown. sAHP controls cell excitability by hyperpolarizing the membrane, thus preventing the membrane potential from reaching threshold for further firing. After calcium influx, the IsAHP has time to peak on the order of hundreds of milliseconds and decays with a time constant of 1-2 s. Our data demonstrated that either calcium ionophores or IP3 signaling through elevation of intracellular calcium can dramatically alter the fast inactivation of Kvβ1.1-dependent A-type current, resulting from a mode-switching of the channel inactivation from fast to slow. The calcium-induced transition of inactivation mode-switching during the action potential is likely to contribute to the sAHP. As a result, the inactivation time constant of Kvβ1.1 was changed from <10 ms to a slow inactivation of hundreds of milliseconds. Oliver et al. (33) recently also reported that membrane lipids can regulate inactivation of Kv1.1/Kvβ1.1 by converting A-type channels into delayed rectifiers, and vice versa. The bidirectional control of Kv1.1/Kvβ1.1 inactivation by lipids is mediated through immobilizing/mobilizing the β ball domain, suggesting a common mechanism for a dynamic regulation of neuronal excitability (33). We, therefore, propose that a coupling between calcium influx and inactivation of voltage-gated A-type K+ channels occurs as a result of membrane depolarization. Such a coupling may provide a physiological mechanism that slowed inactivation of A-type K+ channels by calcium can contribute to the sAHP that serves as a negative feedback in regulation of neuronal excitability.
Acknowledgments
We thank Drs. Ken Rhodes and Jim Barrett for their consistent support of this project and Dr. Stephen Lin for critical discussion and comments on the manuscript. KW.W. thanks Connie Wang for her inspiration and discussion.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: AHP, afterhyperpolarization; BK, large-conductance calcium-dependent K+ channels; RP-cAMPS, RP-adenosine cyclic 3′,5′-phosphorothioate; fAHP, fast AHP; IP3, inositol trisphosphate; mAHP, medium AHP; sAHP, slow AHP.
References
- 1.Berridge, M. J. (1993) Nature 361, 315-325. [DOI] [PubMed] [Google Scholar]
- 2.Collet, C. & Jacquemond, V. (2002) Biophys. J. 82, 1509-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borst, J. G. & Sakmann, B. (1996) Nature 383, 431-434. [DOI] [PubMed] [Google Scholar]
- 4.Sah, P. & Davies, P. (2000) Clin. Exp. Pharmacol. Physiol. 27, 657-663. [DOI] [PubMed] [Google Scholar]
- 5.Cui, J., Cox, D. H. & Aldrich, R. W. (1997) J. Gen. Physiol. 109, 647-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kohler, M., Hirschberg, B., Bond, C. T., Kinzie, J. M., Marrion, N. V., Maylie, J. & Adelman, J. P. (1996) Science 273, 1709-1714. [DOI] [PubMed] [Google Scholar]
- 7.Sah, P. & Faber, E. S. (2002) Prog. Neurobiol. 66, 345-353. [DOI] [PubMed] [Google Scholar]
- 8.Adelman, J. P., Shen, K. Z., Kavanaugh, M. P., Warren, R. A., Wu, Y. N., Lagrutta, A., Bond, C. T. & North, R. A. (1992) Neuron 9, 209-216. [DOI] [PubMed] [Google Scholar]
- 9.Abel, H. J., Lee, J. C., Callaway, J. C. & Foehring, R. C. (2004) J. Neurophysiol. 91, 324-335. [DOI] [PubMed] [Google Scholar]
- 10.Connor, J. A. & Stevens, C. F. (1971) J. Physiol. 213, 21-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rudy, B. (1988) Neuroscience 25, 729-749. [DOI] [PubMed] [Google Scholar]
- 12.Bardoni, R. & Belluzzi, O. (1993) J. Neurophysiol. 69, 2222-2231. [DOI] [PubMed] [Google Scholar]
- 13.Storm, J. F. (1987) Brain Res. 435, 387-392. [DOI] [PubMed] [Google Scholar]
- 14.Giese, K. P., Storm, J. F., Reuter, D., Fedorov, N. B., Shao, L. R., Leicher, T., Pongs, O. & Silva, A. J. (1998) Learn. Mem. 5, 257-273. [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson, M. B., Konnerth, A. & Augustine, G. J. (1991) Proc. Natl. Acad. Sci. USA 88, 380-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jow, F. & Wang, K. (2000) Brain Res. Mol. Brain Res. 80, 269-278. [DOI] [PubMed] [Google Scholar]
- 17.Ishii, T. M., Silvia, C., Hirschberg, B., Bond, C. T., Adelman, J. P. & Maylie, J. (1997) Proc. Natl. Acad. Sci. USA 94, 11651-11656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rettig, J., Heinemann, S. H., Wunder, F., Lorra, C., Parcej, D. N., Dolly, J. O. & Pongs, O. (1994) Nature 369, 289-294. [DOI] [PubMed] [Google Scholar]
- 19.Meech, R. W. (1972) Comp. Biochem. Physiol. A Physiol. 42, 493-499. [DOI] [PubMed] [Google Scholar]
- 20.Greffrath, W., Martin, E., Reuss, S. & Boehmer, G. (1998) J. Physiol. (London) 513, 493-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu, W., Xu, J. & Li, M. (1996) Neuron 16, 441-453. [DOI] [PubMed] [Google Scholar]
- 22.Xu, J., Yu, W., Jan, Y. N., Jan, L. Y. & Li, M. (1995) J. Biol. Chem. 270, 24761-24768. [DOI] [PubMed] [Google Scholar]
- 23.Piskorowski, R. & Aldrich, R. W. (2002) Nature 420, 499-502. [DOI] [PubMed] [Google Scholar]
- 24.Zhou, M., Morais-Cabral, J. H., Mann, S. & MacKinnon, R. (2001) Nature 411, 657-661. [DOI] [PubMed] [Google Scholar]
- 25.Jing, J., Chikvashvili, D., Singer-Lahat, D., Thornhill, W. B., Reuveny, E. & Lotan, I. (1999) EMBO J. 18, 1245-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams, P. R., Constanti, A., Brown, D. A. & Clark, R. B. (1982) Nature 296, 746-749. [DOI] [PubMed] [Google Scholar]
- 27.Lancaster, B. & Nicoll, R. A. (1987) J. Physiol. 389, 187-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shao, L. R., Halvorsrud, R., Borg-Graham, L. & Storm, J. F. (1999) J. Physiol. (London) 521, 135-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goh, J. W. & Pennefather, P. S. (1987) J. Physiol. (London) 394, 315-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sah, P. & McLachlan, E. M. (1992) J. Neurophysiol. 68, 1834-1841. [DOI] [PubMed] [Google Scholar]
- 31.Hirst, G. D., Johnson, S. M. & van Helden, D. F. (1985) J. Physiol. (London) 361, 315-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Starodub, A. M. & Wood, J. D. (2000) Neuroscience 99, 389-396. [DOI] [PubMed] [Google Scholar]
- 33.Oliver, D., Lien, C. C., Soom, M., Baukrowitz, T., Jonas, P. & Fakler, B. (2004) Science 304, 265-270. [DOI] [PubMed] [Google Scholar]