

ABSTRACT
The genome of influenza virus (viral RNA [vRNA]) is associated with the nucleoprotein (NP) and viral RNA-dependent RNA polymerases and forms helical viral ribonucleoprotein (vRNP) complexes. The NP-vRNA complex is the biologically active template for RNA synthesis by the viral polymerase. Previously, we identified human pre-mRNA processing factor 18 (Prp18) as a stimulatory factor for viral RNA synthesis using a Saccharomyces cerevisiae replicon system and a single-gene deletion library of Saccharomyces cerevisiae (T. Naito, Y. Kiyasu, K. Sugiyama, A. Kimura, R. Nakano, A. Matsukage, and K. Nagata, Proc Natl Acad Sci USA, 104:18235–18240, 2007, https://doi.org/10.1073/pnas.0705856104). In infected Prp18 knockdown (KD) cells, the synthesis of vRNA, cRNA, and viral mRNAs was reduced. Prp18 was found to stimulate in vitro viral RNA synthesis through its interaction with NP. Analyses using in vitro RNA synthesis reactions revealed that Prp18 dissociates newly synthesized RNA from the template after the early elongation step to stimulate the elongation reaction. We found that Prp18 functions as a chaperone for NP to facilitate the formation of NP-RNA complexes. Based on these results, it is suggested that Prp18 accelerates influenza virus RNA synthesis as an NP chaperone for the processive elongation reaction.
IMPORTANCE Templates for viral RNA synthesis of negative-stranded RNA viruses are not naked RNA but rather RNA encapsidated by viral nucleocapsid proteins forming vRNP complexes. However, viral basic proteins tend to aggregate under physiological ionic strength without chaperones. We identified the pre-mRNA processing factor Prp18 as a stimulatory factor for influenza virus RNA synthesis. We found that one of the targets of Prp18 is NP. Prp18 facilitates the elongation reaction of viral polymerases by preventing the deleterious annealing of newly synthesized RNA to the template. Prp18 functions as a chaperone for NP to stimulate the formation of NP-RNA complexes. Based on these results, we propose that Prp18 may be required to maintain the structural integrity of vRNP for processive template reading.
KEYWORDS: host factor, protein chaperones, ribonucleoprotein, viral RNA synthesis
INTRODUCTION
The genome of influenza A virus is composed of eight-segmented and single-stranded RNAs of negative polarity and exists as a ribonucleoprotein (RNP) (termed viral RNP [vRNP]) complex with nucleoprotein (NP) and viral RNA-dependent RNA polymerases (RdR Pol) consisting of three subunits, PB1, PB2, and PA. NP binds to the virus genome (viral RNA [vRNA]) through a phosphate backbone without sequence specificity (1, 2). vRNP forms a helical structure, and RdR Pol is located at the open end of the RNP hairpin structure formed by the interaction between the 5′ and 3′ ends of vRNA (3–6). The vRNP complex is essential for the transcription and replication of the influenza virus genome as an enzyme-template complex, and some host factors have been found to be involved in these reactions (reviewed in reference 7). Transcription from vRNA initiates with a capped oligoribonucleotide derived from cellular pre-mRNAs as a primer, whereas viral genome replication is primer independent and host factor dependent and generates full-length vRNA through cRNA synthesized from vRNA (7).
It has been postulated that NP encapsidates de novo RNA and regulates the RdR Pol function through the interaction with PB1 and PB2 (8–12). It has been reported that encapsidation is coupled with replication processes (11) and initiated by successive targeting of the exogenous NP monomer to RdR Pol, which is distinct from the replicative polymerase and binds to the 5′ end of nascent RNA (13), and additional NPs are then subsequently recruited by NP-NP oligomerization (14). Encapsidation is also important for the stabilization of nascent cRNA to protect the virus from degradation by cellular nucleases (15).
It is known that the maximal level of replication and transcription of the influenza virus genome requires not only viral components associated with virions but also some host factors present in infected cells (11, 16–22). We reconstituted a cell-free viral RNA synthesis system with virion-associated vRNP and nuclear extracts prepared from uninfected HeLa cells (19, 23). By reconstitution and dissection of the cell-free system, we have identified RAF-1/Hsp90, IREF-1/MCM, and RAF-2p48/UAP56/BAT1 as host factors that stimulate viral RNA synthesis (24–26). In addition, we also established an influenza virus replicon system in Saccharomyces cerevisiae and identified Tat-SF1 as a host factor using a yeast single-gene deletion library (27). RAF-1/Hsp90 regulates the assembly of the viral RNA polymerase complex and is also involved in its stabilization during transfer between templates (28). IREF-1/MCM stabilizes replicating polymerase complexes by stabilizing the interaction between nascent cRNA and PA (24). RAF-2p48 and Tat-SF1 facilitate viral RNA synthesis as NP chaperones (11, 26, 27).
Here, we have identified another host factor, Prp18, as a stimulatory factor for influenza virus RNA synthesis using the yeast single-gene deletion library screening system (27). Prp18 is associated with U5 snRNP and plays an important role in catalytic step II in pre-mRNA splicing (29–31) to stabilize the interaction of the ends of exons with loop 1 of U5 snRNA (32). We found that Prp18 stimulates the RNA synthesis of the influenza virus genome and interacts with NP directly in vitro and in vivo. Biochemical analyses indicated that Prp18 is involved in the formation of NP-RNA complexes. Based on these results, we propose that Prp18 is one of the NP chaperones.
RESULTS
Prp18 stimulates viral RNA synthesis in vitro.
Previously, we established an influenza virus replicon system in yeast and a screening system using a single-gene deletion yeast library, both of which enabled us to identify candidate host factors involved in influenza virus RNA synthesis (27). Using this system, we have identified Prp18 as one of the candidate host factors. The RNA synthesis level was decreased by 40% in Prp18 deletion yeast cells compared to that in wild-type cells (27). Yeast Prp18 shares 39% identity with human Prp18. This factor is associated with U5 snRNP and functions in catalytic step II in pre-mRNA splicing as a splicing factor (29–32). Prp18 contains a splicing factor motif (SFM) in the N-terminal region, which is a Prp4-like motif with two weakly folded helices, and a Prp18 motif in the C-terminal region (33). The C-terminal Prp18 domain associates with U5 snRNP and the Slu7 protein during splicing (29, 34). The N-terminal Prp18 SFM domain interacts with the basic protein hCypH (35).
To confirm that Prp18 has stimulatory activity for viral RNA synthesis, we carried out cell-free influenza virus RNA synthesis assays. We purified recombinant glutathione S-transferase (GST)-Prp18 and GST proteins using an Escherichia coli expression system (Fig. 1A). vRNP, as an enzyme source, was incubated with an exogenously added 53-nucleotide (nt)-long model vRNA (termed v53) in the presence of GST-Prp18 or GST, and synthesized RNAs were analyzed by electrophoresis through a denaturing gel. GST-Prp18 stimulated viral RNA synthesis from exogenous v53 as well as endogenous RNAs in a Prp18 dose-dependent manner (Fig. 1B), suggesting that Prp18 enhances the viral RNA synthesis activity, possibly by interacting with RdR Pol, NP, and/or the virus genome.
FIG 1.
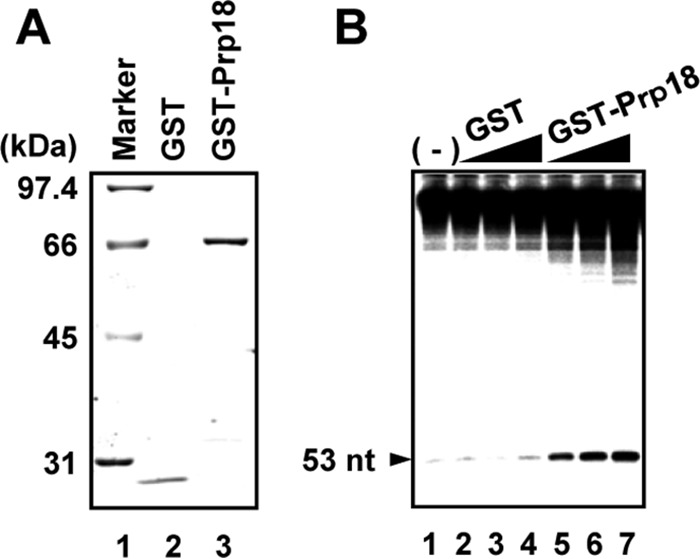
Stimulation of viral RNA synthesis by Prp18 in vitro. (A) Purified GST and GST-Prp18 were separated by SDS-PAGE and visualized by Coomassie brilliant blue staining. (B) Effect of Prp18 on cell-free RNA synthesis. Cell-free RNA synthesis was carried out by using vRNP (3 ng of NP equivalents) as an enzyme source and v53 in the presence of increasing amounts of GST (lane 2, 30 ng; lane 3, 100 ng; lane 4, 300 ng) and GST-Prp18 (lane 5, 30 ng; lane 6, 100 ng; lane 7, 300 ng). RNA products from endogenous viral RNA present in vRNP are shown at the top of a 10% PAGE gel in the presence of 8 M urea, and RNA products from the 53-mer model vRNA are indicated by an arrowhead.
Effect of Prp18 knockdown on influenza virus-infected cells.
Next, we examined the function of Prp18 in influenza virus-infected cells using small interfering RNA (siRNA) against Prp18 (siPrp18). The amount of Prp18 was reduced to <10% in cells transfected with Prp18 siRNA compared to that in control siRNA-transfected cells (Fig. 2A). The transfection of Prp18 siRNA showed negligible cytotoxicity compared to that of control siRNA (Fig. 2B). The virus titer in Prp18 knockdown (KD) cells decreased to <10% of that in control cells (Fig. 2C). Although the amount of NP was saturated at 7 h postinfection (hpi), the protein synthesis levels of PB1, PB2, PA, and NP were reduced by ∼40% in siPrp18-transfected cells compared with those in control siRNA-transfected cells (Fig. 2D) (the signal intensities were measured by using Image J software). Based on these results, it is possible that Prp18 is involved in viral protein synthesis and/or viral RNA synthesis. Thus, we investigated the levels of vRNA, cRNA, and viral mRNA in infected cells by quantitative reverse transcription-PCR (RT-PCR). The levels of segment 3, 5, and 7 vRNAs, cRNAs, and their mRNAs were decreased in Prp18 KD cells (Fig. 2E). Because Prp18 is one of the splicing factors, it is possible that Prp18 KD may have some effects on the splicing of viral pre-mRNAs and therefore on viral RNA synthesis. To examine this, we determined the ratio of the level of M1 mRNA to that of M2 mRNA, the latter of which is generated from M1 mRNA by splicing. In Prp18 KD cells, the amounts of both M1 and M2 mRNAs were reduced by >40% compared to those in control cells, but the ratio of the M2 mRNA level to the M1 mRNA level was decreased by Prp18 KD only slightly (Fig. 2F), indicating that Prp18 KD has little effect on splicing under the conditions employed here.
FIG 2.

Effect of Prp18 knockdown on viral RNA synthesis. (A) HeLa cells were transfected with control siRNA or siPrp18. At 72 h posttransfection, cell lysates were subjected to Western blotting using anti-Prp18 and anti-β-actin antibodies. (B) Seventy-two hours after transfection of siRNA, the viability of control and Prp18 KD cells was determined by trypan blue staining. The averages and standard deviations determined from three independent experiments are shown. (C) Seventy-two hours after transfection of siRNA, control and Prp18 KD cells were infected with influenza virus at an MOI of 0.01. The culture supernatants collected at 12, 24, 36, 48, 60, and 72 hpi were subjected to plaque assays to examine the production of infectious virions. The average titers and standard deviations determined from three independent experiments are shown. (D) Seventy-two hours after transfection of siRNA, control (lanes 1, 3, 5, and 7) and Prp18 KD (lanes 2, 4, 6, and 8) cells were infected with influenza virus at an MOI of 3. At 3, 5, and 7 hpi, the cell lysates were subjected to Western blotting with anti-PB1, anti-PB2, anti-PA, anti-NP, and anti-β-actin antibodies. (E) Control and Prp18 KD cells were infected with influenza virus at an MOI of 3 and incubated for 3, 5, and 7 h. Total RNA was prepared from cells, and quantitative RT-PCR was carried out with primer sets specific for segment 3 vRNA, segment 3 cRNA, segment 3 mRNA, segment 5 vRNA, segment 5 cRNA, segment 5 mRNA, segment 7 vRNA, segment 7 cRNA, and segment 7 mRNA. The results were normalized to the level of 18S rRNA. The averages and standard deviations determined in three independent experiments are shown. The level of significance was determined by Student's t test (*, P < 0.01; **, P < 0.05; ***, P < 0.5). (F) HeLa cells were transfected with control siRNA or siPrp18. At 72 h posttransfection, cells were infected with influenza virus at an MOI of 3 and incubated for 5 h. Total RNA was prepared from cells, and quantitative RT-PCR was carried out with primer sets specific for M1 mRNA and M2 mRNA. The results were normalized to the level of 18S rRNA. The ratio of the amount of M2 mRNA to that of M1 mRNA was also determined in three independent experiments, with standard deviations. The level of significance was determined by Student's t test (*, P < 0.01; **, P < 0.05).
Prp18 is involved in primary transcription of incoming vRNP.
Because the newly synthesized viral genome served as a template for viral mRNA synthesis, we investigated the effect of Prp18 on primary transcription from incoming vRNP in Prp18 KD cells in the presence of cycloheximide (CHX), which is a potent protein synthesis inhibitor and inhibits the replication process (15, 36). The level of NP mRNA in Prp18 KD cells was reduced to ∼30% of that in control cells in the presence of CHX (Fig. 3A, lanes 2 and 4). Next, we carried out complementation experiments for siRNA-transfected cells with a plasmid encoding siRNA-resistant Prp18 (rPrp18)-Myc containing a silent mutation within the siRNA target sequence. The NP mRNA synthesis level was rescued by the expression of rPrp18-Myc (Fig. 3A, compare lane 5 with lane 4). We also confirmed that the expression level of rPrp18-Myc in Prp18 KD cells was similar to that of the endogenous Prp18 protein in control siRNA-treated cells (Fig. 3B, lanes 2 and 5). From these results, we concluded that Prp18 is a positive factor for viral mRNA synthesis in infected cells.
FIG 3.

Effect of Prp18 on primary transcription. (A) HeLa cells were transfected with control siRNA or siPrp18. At 48 h posttransfection, cells were transfected with the pCAGGS-rPrp18-Myc (containing a silent mutation within the siRNA target sequence) and pCAGGS-empty plasmids. After 24 h posttransfection, cells were superinfected with influenza virus at an MOI of 3 and incubated for 4 h in the presence of cycloheximide (CHX). Total RNAs were subjected to quantitative RT-PCR with a primer set specific for segment 5 mRNA. The results are normalized to the level of 18S rRNA. The averages and standard deviations determined from three independent experiments are shown. The level of significance was determined by Student's t test (*, P < 0.01). (B) Western blotting was conducted by using infected cells prepared as described above for panel A. Cell lysates were loaded onto a 10% SDS-PAGE gel and subjected to Western blotting with anti-Prp18 antibody.
The interaction between NP and Prp18 is responsible for stimulation of viral RNA synthesis by Prp18.
To examine which viral factor(s) interacts with Prp18, we carried out immunoprecipitation assays using cell lysates prepared from infected cells expressing Prp18-Myc. As shown in Fig. 4A, Prp18-Myc interacted with both NP and RdR Pol. However, it was not clear whether Prp18 binds to them directly or through some component of vRNP. We then investigated a target of Prp18 using micrococcal nuclease-treated vRNP (mnRNP) (Fig. 4B). Micrococcal nuclease digests viral RNA and produces viral proteins free of RNA. GST-Prp18 interacted with NP and RdR Pol in the absence of viral RNA. It was assumed that Prp18 binds to NP and RdR Pol independently or through NP or RdR Pol, both of which interact with each other (9). Therefore, to examine the interaction between NP and Prp18, we carried out GST pulldown assays with recombinant His-tagged NP (His-NP) and GST-Prp18. Figure 4C shows that NP binds GST-Prp18 in a dose-dependent manner.
FIG 4.

Prp18 interacts with NP and viral polymerase in vivo and in vitro. (A) HEK293T cells were transfected with either pCAGGS or pCAGGS-Prp18-Myc. At 48 h posttransfection, these cells were infected with influenza virus at an MOI of 3 and incubated for 6 h. Cell lysates were prepared and incubated with either control IgG-conjugated (lanes 3 and 5) or anti-Myc antibody-conjugated agarose (lanes 4 and 6). Pulldown products were visualized by immunoblotting using anti-PB1, anti-PB2, anti-PA, anti-NP, and anti-Myc antibodies. (B) GST pulldown assays were carried out by using purified GST-Prp18 (lanes 5 to 7) and either vRNP (lanes 1, 3, and 6) or mnRNP (lanes 2, 4, and 7). Western blot analyses were carried out with anti-GST, anti-PB1, anti-PB2, anti-PA, and anti-NP antibodies. (C) GST pulldown assays were conducted by using purified GST (lane 1, 0.6 pmol), GST-Prp18 (lane 2, 0.6 pmol; lane 3, 2 pmol; lane 4, 6 pmol), and His-NP (lanes 1 to 4, 1.25 pmol). Western blotting was carried out with anti-His and anti-GST antibodies.
To identify the viral protein responsible for the stimulatory activity of viral RNA synthesis by Prp18, we carried out GST pulldown assays using lysates prepared from infected cells expressing GST-fused deletion mutants of Prp18. We constructed C-terminally truncated mutants of Prp18 (Prp18C1, Prp18C2, and Prp18Δ71) (Fig. 5A). Similarly to the wild type, these mutant proteins were localized mainly in the nucleus (Fig. 5B). We found that Prp18C2 hardly interacts with RdR Pol and NP in infected cells (Fig. 5C, lane 4). Prp18C1 and Prp18Δ71 interacted with NP but not RdR Pol, although the interaction level of these mutants was lower than that of wild-type Prp18 (Prp18WT) (Fig. 5C, lanes 3 and 5). To confirm the interaction between NP and Prp18 mutants, we performed GST pulldown assays with lysates prepared from cells expressing NP and each Prp18 mutant. It was revealed that Prp18C1 and Prp18Δ71 interacted with NP in the absence of RdR Pol (Fig. 5D). Thus, we carried out cell-free viral RNA synthesis assays using GST-Prp18C2 and GST-Prp18Δ71 (Fig. 5E and F). These assays showed that GST-Prp18WT and GST-Prp18Δ71, but not Prp18C2, stimulated viral RNA synthesis from exogenous v53 as well as endogenous viral RNAs in a Prp18 dose-dependent manner, suggesting that Prp18 activates viral RNA synthesis through the interaction between Prp18 and NP.
FIG 5.

Prp18 stimulates viral RNA synthesis through interaction with NP. (A) Schematic diagram of Prp18 deletion mutants. a.a, amino acids. (B) Intracellular localization of GST-tagged Prp18 deletion mutants. HeLa cells were transfected with plasmids expressing GST-Prp18, GST-Prp18C1, GST-Prp18C2, and GST-Prp18Δ71. At 24 h posttransfection, cells were subjected to indirect-immunofluorescence assays with anti-GST antibody. DAPI, 4′,6-diamidino-2-phenylindole. (C) GST pulldown assays of GST-Prp18 deletion mutants. HEK293T cells were transfected with plasmids expressing GST-Prp18, GST-Prp18C1, GST-Prp18C2, and GST-Prp18Δ71. At 24 h posttransfection, cells were infected with influenza virus at an MOI of 3. At 6 hpi, cell lysates were prepared and subjected to GST pulldown assays. Coprecipitated proteins were analyzed by Western blotting with anti-PB1, anti-PB2, anti-PA, anti-NP, and anti-GST antibodies. nls, nuclear localization signal. (D) HEK293T cells were transfected with plasmids expressing NP and either GST-Prp18, GST-Prp18C1, GST-Prp18C2, or GST-Prp18Δ71. At 24 h posttransfection, cell lysates were prepared and subjected to a GST pulldown assay. Coprecipitated proteins were analyzed by Western blotting with anti-NP and anti-GST antibodies. (E) Purified recombinant GST, GST-Prp18, GST-Prp18C2, and GST-Prp18Δ71 were separated by 10% SDS-PAGE and visualized by staining with Coomassie brilliant blue. (F) Effect of Prp18 on cell-free RNA synthesis. Cell-free RNA synthesis was carried out with 0.3 pmol (lanes 1, 4, 7, and 10), 1 pmol (lanes 2, 5, 8, and 11), and 3 pmol (lanes 3, 6, 9, and 12) of GST (lanes 1 to 3), GST-Prp18C2 (lanes 4 to 6), GST-Prp18WT (lanes 7 to 9), and GST-Prp18Δ71 (lanes 10 to 12), as described in the legend of Fig. 1.
Prp18 dissociates newly synthesized RNA from the template and stimulates elongation after the early elongation step.
To clarify the role of Prp18 in viral RNA synthesis, we carried out cell-free viral RNA synthesis assays by incubating cells for 0, 1, 2, and 4 min to examine the effect of Prp18 on initiation and elongation reactions. Approximately 60-, 200-, and >300-nt-long viral RNAs were mainly synthesized by 1-, 2-, and 4-min incubations, respectively (Fig. 6A). We found that the amount of shorter newly synthesized RNAs (<200 nt) was not changed by the addition of Prp18 (Fig. 6A, lanes 3, 4, 7, and 8). In contrast, the amount of longer RNAs (>300 nt) was increased in the presence of Prp18 (Fig. 6A, compare lane 9 with lane 5), suggesting that Prp18 stimulates the elongation step but not the initiation step. To confirm the effect of Prp18 on the initiation reaction, we performed a limited elongation reaction in which UTP is omitted from the reaction mixture and, thus, RdR Pol pauses at the first adenine residue on the template. The expected lengths of the limited elongation products are 12 nt for segments 1, 3, and 7; 13 nt for segments 5 and 8; 14 nt for segment 6; 18 nt for segment 4; and 19 nt for segment 2. As expected, we detected comparable amounts of each RNA product in the absence or presence of Prp18 (Fig. 6B). Thus, it is likely that Prp18 is required for a step(s) after the early elongation process, in which short RNAs are synthesized.
FIG 6.

Prp18 stimulates viral RNA synthesis after the early elongation process. (A) Time course of cell-free viral RNA synthesis. Cell-free viral RNA synthesis was carried out in the presence of either GST (lanes 2 to 5) or GST-Prp18 (lanes 6 to 9), as described in the legend of Fig. 1. After 0-, 1-, 2-, and 4-min incubations, samples were collected and analyzed by using 8 M urea–10% PAGE gels. (B) Limited elongation assays were carried out in the presence of either GST (lanes 1 to 3) or GST-Prp18 (lanes 4 to 6) in the absence of UTP. Samples were analyzed by using 8 M urea–15% PAGE gels. (C) RNase T2 digestion assay. Cell-free RNA synthesis was carried out in the presence of either GST (lanes 1 to 3) or GST-Prp18 (lanes 4 to 6). The newly synthesized RNAs were digested with 6.6 U (lanes 2 and 5) and 20 U (lanes 3 and 6) of RNase T2. The digested RNAs were analyzed by using 8 M urea–4% PAGE gels. (D) The signal intensities of each lane were measured by using ImageJ software. Averages and standard deviations determined from three independent experiments are shown.
It has been speculated that without newly synthesized NP and a molecular chaperone such as RAF-2p48/UAP56, the nascent RNA chains hybridize with template strands (11), and this hybridization may result in the premature termination of RNA synthesis. Next, in order to examine the amount of newly synthesized RNA hybridizing to the template in the absence or presence of Prp18, we digested the nascent RNA chains with RNase T2, which is a single-stranded RNA-specific nuclease (Fig. 6C). At 4 min postincubation, the reaction of cell-free viral RNA synthesis was stopped by the addition of 50 mM EDTA, and the reaction mixtures were subjected to RNase T2 digestion. In the absence of GST-Prp18, newly synthesized viral RNAs were resistant to RNase T2 digestion, suggesting that the nascent chains hybridize with template strands (Fig. 6C and D, lanes 1 to 3). In contrast, the Prp18-stimulated longer RNA products were highly sensitive to RNase T2 digestion (Fig. 6C and D, lanes 4 to 6). From these results, it is possible that Prp18 dissociates the newly synthesized RNA from the template after the early elongation step and then stimulates the elongation reaction of viral RNA synthesis.
Prp18 facilitates the formation of NP-RNA complexes.
Based on the structural and functional features of Prp18 described above, we focused our study on the Prp18 function related to NP. We hypothesized that Prp18 may function as an NP chaperone in viral RNA synthesis. First, we performed gel shift assays to examine whether Prp18 plays a role in the formation of NP-RNA complexes (Fig. 7). His-NP and GST-Prp18 expressed in E. coli were purified (Fig. 7A). Gel shift assays with 5′-32P-labeled v53 showed that NP-RNA complexes are formed in an NP dose-dependent manner (Fig. 7B). The complex retained at the gel top, a quite large complex, includes the aggregation of NP and 5′-32P-labeled v53. To examine whether Prp18 facilitates the formation of NP-RNA complexes, His-NP and GST-Prp18 were mixed and incubated at 30°C for 30 min. Each sample was then mixed with v53 and incubated at 30°C for 30 min. In the presence of a limited amount of NP, NP-RNA complex formation was increased by Prp18 (Fig. 7C and D). It is worth noting that Prp18 does not bind to v53 (Fig. 7C, lane 8). Taken together, these results suggest that Prp18 has the NP chaperone activity that facilitates the formation of NP-RNA complexes.
FIG 7.

Prp18 facilitates NP-RNA complex formation in vitro. (A) Purification of the GST-Prp18 and His-NP proteins. Details are described in Materials and Methods. Purified proteins were loaded onto a 10% SDS-PAGE gel and visualized by Coomassie brilliant blue staining. (B) Gel shift assays were carried out with 5′-32P-labeled v53 and His-NP. NP-RNA complexes were separated on a 0.6% agarose gel and detected by autoradiography. (C and D) His-NP (lanes 1 to 7, 12.5 fmol) and increasing amounts of GST-Prp18 (lane 2, 0.013 pmol; lane 3, 0.04 pmol; lane 4, 0.13 pmol; lane 5, 0.4 pmol; lane 6, 1.3 pmol; lanes 7 and 8, 4 pmol) were incubated at 30°C for 30 min. After further incubation with 5′-32P-labeled v53, samples were separated on a nondenaturing gel and subjected to autoradiography. In panel D, the band intensities were quantitatively measured by using ImageJ software, and the averages and standard deviations determined from three independent experiments are shown. The level of significance was determined by Student's t test (*, P < 0.01; **, P < 0.05). AU, arbitrary units.
DISCUSSION
We have identified Prp18 as a stimulatory factor for viral RNA synthesis activity in vitro and in vivo. Prp18 dissociates the newly synthesized RNA from the template after the early elongation step and thus stimulates the elongation reaction of viral RNA synthesis (Fig. 6). We also demonstrated the possibility that Prp18 is a molecular chaperone for NP-RNA complex formation, as are RAF-2p48/UAP56 (26) and Tat-SF1 (27) (Fig. 7).
Prp18 and the previously identified NP chaperones RAF-2p48/UAP56 and Tat-SF1 are related to the splicing reaction (26, 27). It has been reported that spliceosomes are partially destroyed by the influenza virus NS1 protein (26, 37). The release of these splicing factors from spliceosomes is useful for recruiting these factors for viral RNA transcription and replication in the nucleus. The influenza virus RdR Pol recognizes host pre-mRNAs containing the cap structure for the preparation of primers for viral transcription, and viral transcription was found to be dependent on transcriptionally functional host RNA polymerase II complexes (38–40), which are also associated with splicing factors. Splicing occurs cotranscriptionally (41), so vRNP could be associated with these complexes.
It has been reported that RAF-2p48/UAP56 stimulates the encapsidation of nascent RNA chains in concert with viral RNA synthesis (11). The coreplicational encapsidation of nascent chains by NP prevents the premature termination of viral RNA synthesis. It is worth noting that RAF-2p48/UAP56 stimulates viral RNA synthesis from a naked RNA template by recruiting NP to RNA but not from vRNP (26). Here, we showed that Prp18 dissociates newly synthesized RNA from the template after the early elongation step and stimulates the elongation reaction. In contrast to RAF-2p48/UAP56, Prp18 enhances viral RNA synthesis from vRNP, suggesting that Prp18 functions as a chaperone of NP on the virus genome. The binding of NP to the virus genome is required to form the vRNP template for an efficient elongation reaction by RdR Pol (8–12). A recent structural model of RdR Pol of a segmented negative-strand RNA virus led to the proposal that the disruption of the vRNP structure during template reading is restricted around the active center due to the proximity of the entry and exit channels of template (42). Based on these findings, it is possible that Prp18 functions as a molecular chaperone for NP around the active center of RdR Pol to maintain the structural integrity of vRNP for processive template reading. It has been reported that PB2 has NP and PB1 binding sites, and the binding of NP to PB2 could be outcompeted by PB1 (43). It has also been hypothesized that the interaction between NP and RdR Pol might play a role in switching the activity of RdR Pol from transcription to replication (8, 10, 15, 44, 45). To further understand the effect of NP on viral RNA synthesis, biochemical and structural analyses focusing on RdR Pol during template reading are required.
The interaction between NP and RdR Pol is thought to be involved in the targeting of exogenous NP to newly synthesized virus genomes (13). We found that Prp18 depletion reduces the viral polymerase activity (Fig. 2E), but the viral titer was decreased more significantly by Prp18 KD (Fig. 2C). Prp18 was also shown to interact with NP and RdR Pol (Fig. 4). Thus, it is possible that Prp18 is required for postreplicational processes through vRNP encapsidation in addition to the stimulation of viral polymerase activity.
MATERIALS AND METHODS
Cells, virus infection, and transfection.
Monolayer cultures of HeLa cells and MDCK cells were maintained at 37°C in minimal essential medium (MEM) containing 10% fatal bovine serum (FBS) (Gibco). HeLa cells were infected with influenza virus A/Puerto Rico/8/34 at a multiplicity of infection (MOI) of 3. After virus adsorption at 37°C for 1 h, cells were washed twice with serum-free MEM and incubated at 37°C for 3 to 7 h with MEM containing 10% FBS. Transfection was carried out by using Genejuice transfection reagent (Merck Millipore) according to the manufacturer's instructions.
Vector construction.
Full-length human Prp18 cDNA was amplified by PCR using cDNA prepared from total RNA of HeLa cells as the template, using primers 5′-CCGAATTCGCCGCCACCATGGACTACAAGGATGACGACGACAAGGGAATGGACATTCTGAAATCAGAGATCC-3′ and 5′-GGGAATTCTCACAGTGCATTGTACTCCACAC-3′. The PCR fragment was phosphorylated by using T4 polynucleotide kinase (Toyobo) and cloned into SmaI-digested pGEX-6p-1 (pGEX-Prp18). For the construction of the siRNA-resistant Prp18 expression vector, synonymous mutations were introduced into the siRNA-targeting sequence by PCR using a primer with site-directed mutations, 5′-TCCTCCTCTTTGGGTTGGATTTTATACCCGCATCTTTCAAAATA-3′, and primers described above.
Antibodies.
Rabbit anti-PB1, anti-PB2, anti-PA, and anti-NP antibodies were prepared as previously described (28, 46). Mouse polyclonal antibodies against human Prp18 (Sigma-Aldrich), the His tag (Sigma-Aldrich), the GST tag (Nacalai), and the Myc tag (Nacalai) were purchased.
Preparation of recombinant proteins.
GST-Prp18 was purified by using glutathione-Sepharose 4B beads (GE health care) according to the manufacturer's protocols. His-tagged NP (His-NP) was prepared as previously described (11). To reduce contamination by bacterial RNA, recombinant NP was washed with 1 M NaCl. Purified NP with a ratio of the absorbance at 260 nm to that at 280 nm of between 0.57 and 0.59 was used for this study.
Cell-free influenza virus RNA synthesis.
Procedures for the purification of virions and the isolation of vRNP complexes were described previously (19, 23, 26). Cell-free influenza virus RNA synthesis was carried out at 30°C for 1 h in a final volume of 20 μl containing 50 mM HEPES-NaOH (pH 8.0); 3 mM MgCl2; 1.5 mM dithiothreitol; 50 mM KCl; 500 μM each ATP, CTP, and UTP; 25 μM GTP; 5 μCi of [α-32P]GTP (3,000 Ci/mmol); 4 U of an RNase inhibitor; 100 μM ApG dinucleotide; 30 nM 53-nt-long model vRNA template (5′-AGUAGAAACAAGGGUGUUUUUUCAUAUCAUUUAAACUUCACCCUGCUUUUGCU-3′), and vRNP as an enzyme source. RNA products were purified, separated on 10% PAGE gels containing 8 M urea, and detected by autoradiography.
Immunoprecipitation assay.
Cells were suspended in lysis buffer containing 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 30 mM KCl, 1 mM EDTA, and 0.1% NP-40. After sonication with a BioRuptor (Cosmo Bio), the cell lysates were centrifuged at 13,000 × g at 4°C for 5 min. The supernatant fractions were incubated with anti-Myc antibody-conjugated agarose at 4°C for 1 h. After washing with lysis buffer, immunoprecipitated proteins were analyzed by immunoblotting using anti-PB1, anti-PB2, anti-PA, anti-NP, and anti-Myc antibodies.
Gel shift assay.
His-NP and Prp18 were incubated at 30°C for 30 min in a buffer containing 50 mM HEPES-NaOH (pH 8.0), 20 mM KCl, 1.2 mM MgCl2, 0.6 mM dithiothreitol, 0.1% NP-40, 5′-32P-labeled 53-nt-long model vRNA, and 4 U of the RNase inhibitor. Each sample was separated on a 0.6% agarose gel containing 0.5× Tris-borate-EDTA (TBE) buffer.
GST pulldown assay.
GST-Prp18 and GST were incubated with either NP, vRNP, or micrococcal nuclease-treated vRNP (mnRNP) at 25°C for 1 h in a buffer containing 50 mM HEPES-NaOH (pH 8.0), 20 mM KCl, 1.2 mM MgCl2, 0.6 mM dithiothreitol, 0.1% Triton X-100, 4 U of the RNase inhibitor, and glutathione-Sepharose 4B beads. After washing with a buffer containing 20 mM Tris-HCl (pH 8.0), 200 mM NaCl, 1 mM EDTA, and 0.1% Triton X-100, proteins bound to the beads were eluted and analyzed by immunoblotting with anti-His and anti-GST antibodies.
RNase digestion assay.
A cell-free viral RNA synthesis reaction was performed as described above. After incubation for 4 min at 30°C, the reaction was stopped by the addition of 50 mM EDTA, and the mixture was then further incubated with RNase T2 at 30°C for 5 min. RNA was purified by phenol-chloroform extraction and ethanol precipitation and analyzed on 8 M urea–4% PAGE gels.
Gene knockdown experiments.
HeLa cells (2 × 105 cells) were transfected with 50 pmol of Stealth RNAi (catalog number PRPF-HSS112527; Life Technologies) using the Lipofectamine RNAiMAX transfection reagent (Life Technologies) according to the manufacturer's protocols.
Quantitative RT-PCR.
Total RNA was prepared by the acid guanidinium phenol chloroform (AGPC) method and then reverse transcribed with the following primers: 5′-GAGAGAGGAGAAGAGACA-3′ for segment 3 vRNA, 5′-AGTAGAAACAAGGTACTTTTTTGGAC-3′ for segment 3 cRNA, 5′-GACGATGCAACGGCTGGTCTG-3′ for segment 5 vRNA, 5′-AGTAGAAACAAGGGTATTTTTCTTTA-3′ for segment 5 cRNA, 5′-GTCGAAACGTACGTTCTCTCTATC-3′ for segment 7 vRNA, 5′-AGTAGAAACAAGGTAGTTTTTTACTC-3′ for segment 7 cRNA, oligo(dT)20 for viral mRNA, and 5′-GGTGTGTTACAAAGGGCAGGG-3′ for 18S rRNA. Single-stranded cDNAs were then subjected to quantitative real-time PCR with following specific primer sets: 5′-GAGAGAGGAGAAGAGACA-3′ and 5′-TTAATTTTAAGGCATCCATCAGCAGG-3′ for segment 3, 5′-AGCATTGTTCCAACTCCTTT-3′ and 5′-GACGATGCAACGGCTGGTCTG-3′ for segment 5, 5′-GTCGAAACGTACGTTCTCTCTATC-3′ and 5′-TCCCCTTAGTCAGAGGTGAC-3′ for segment 7 and M1 mRNA, 5′-GAGGTCGAAACGCCTAT-3′ and 5′-CTCCAGCTCTATGTTGACAAA-3′ for M2 mRNA, and 5′-AACGGCTACCACATCCAAGG-3 and 5′-GGGAGTGGGTAATTTGCGC-3′ for 18S rRNA, respectively.
ACKNOWLEDGMENTS
We thank K. Takeuchi (Laboratory of Environmental Microbiology, Faculty of Medicine, University of Tsukuba) for critical comments and helpful discussion.
This work was supported in part by a grant-in-aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (24115002 to K.N.). The funder had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Yamanaka K, Ishihama A, Nagata K. 1990. Reconstitution of influenza virus RNA-nucleoprotein complexes structurally resembling native viral ribonucleoprotein cores. J Biol Chem 265:11151–11155. [PubMed] [Google Scholar]
- 2.Honda A, Ueda K, Nagata K, Ishihama A. 1988. RNA polymerase of influenza virus: role of NP in RNA chain elongation. J Biochem 104:1021–1026. [DOI] [PubMed] [Google Scholar]
- 3.Compans RW, Content J, Duesberg PH. 1972. Structure of the ribonucleoprotein of influenza virus. J Virol 10:795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arranz R, Coloma R, Chichon FJ, Conesa JJ, Carrascosa JL, Valpuesta JM, Ortin J, Martin-Benito J. 2012. The structure of native influenza virion ribonucleoproteins. Science 338:1634–1637. doi: 10.1126/science.1228172. [DOI] [PubMed] [Google Scholar]
- 5.Moeller A, Kirchdoerfer RN, Potter CS, Carragher B, Wilson IA. 2012. Organization of the influenza virus replication machinery. Science 338:1631–1634. doi: 10.1126/science.1227270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fodor E, Seong BL, Brownlee GG. 1993. Photochemical cross-linking of influenza A polymerase to its virion RNA promoter defines a polymerase binding site at residues 9 to 12 of the promoter. J Gen Virol 74(Part 7):1327–1333. [DOI] [PubMed] [Google Scholar]
- 7.Nagata K, Kawaguchi A, Naito T. 2008. Host factors for replication and transcription of the influenza virus genome. Rev Med Virol 18:247–260. doi: 10.1002/rmv.575. [DOI] [PubMed] [Google Scholar]
- 8.Portela A, Digard P. 2002. The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J Gen Virol 83:723–734. doi: 10.1099/0022-1317-83-4-723. [DOI] [PubMed] [Google Scholar]
- 9.Biswas SK, Boutz PL, Nayak DP. 1998. Influenza virus nucleoprotein interacts with influenza virus polymerase proteins. J Virol 72:5493–5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newcomb LL, Kuo RL, Ye Q, Jiang Y, Tao YJ, Krug RM. 2009. Interaction of the influenza A virus nucleocapsid protein with the viral RNA polymerase potentiates unprimed viral RNA replication. J Virol 83:29–36. doi: 10.1128/JVI.02293-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawaguchi A, Momose F, Nagata K. 2011. Replication-coupled and host factor-mediated encapsidation of the influenza virus genome by viral nucleoprotein. J Virol 85:6197–6204. doi: 10.1128/JVI.00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shapiro GI, Krug RM. 1988. Influenza virus RNA replication in vitro: synthesis of viral template RNAs and virion RNAs in the absence of an added primer. J Virol 62:2285–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jorba N, Coloma R, Ortin J. 2009. Genetic trans-complementation establishes a new model for influenza virus RNA transcription and replication. PLoS Pathog 5:e1000462. doi: 10.1371/journal.ppat.1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan WH, Ng AK, Robb NC, Lam MK, Chan PK, Au SW, Wang JH, Fodor E, Shaw PC. 2010. Functional analysis of the influenza virus H5N1 nucleoprotein tail loop reveals amino acids that are crucial for oligomerization and ribonucleoprotein activities. J Virol 84:7337–7345. doi: 10.1128/JVI.02474-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vreede FT, Jung TE, Brownlee GG. 2004. Model suggesting that replication of influenza virus is regulated by stabilization of replicative intermediates. J Virol 78:9568–9572. doi: 10.1128/JVI.78.17.9568-9572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takeuchi K, Nagata K, Ishihama A. 1987. In vitro synthesis of influenza viral RNA: characterization of an isolated nuclear system that supports transcription of influenza viral RNA. J Biochem 101:837–845. [DOI] [PubMed] [Google Scholar]
- 17.Beaton AR, Krug RM. 1984. Synthesis of the templates for influenza virion RNA replication in vitro. Proc Natl Acad Sci U S A 81:4682–4686. doi: 10.1073/pnas.81.15.4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.del Rio L, Martinez C, Domingo E, Ortin J. 1985. In vitro synthesis of full-length influenza virus complementary RNA. EMBO J 4:243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Momose F, Handa H, Nagata K. 1996. Identification of host factors that regulate the influenza virus RNA polymerase activity. Biochimie 78:1103–1108. doi: 10.1016/S0300-9084(97)86736-3. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe T, Kawakami E, Shomaker JE, Lopes TJ, Matsuoka Y, Tomita Y, Kozuka-Hata H, Gorai T, Kuwahara T, Takeda E, Nagata A, Takano R, Kiso M, Yamashita M, Sakai-Tagawa Y, Katsura H, Nonaka N, Fujii H, Fujii K, Sugita Y, Noda T, Goto H, Fukuyama S, Waanabe S, Neumann G, Oyama M, Kitano H, Kawaoka Y. 2014. Influenza virus-host interactome screen as a platform for antiviral drug development. Cell Host Microbe 16:795–805. doi: 10.1016/j.chom.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.York A, Hutchison EC, Fodor E. 2014. Interactome analysis of the influenza A virus transcription/replication machinery identifies protein phosphatase 6 as a cellular factor required for efficient virus replication. J Virol 88:13284–13299. doi: 10.1128/JVI.01813-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bradel-Tretheway BG, Mattiacio JL, Krasnoselsky A, Stevenson C, Purdy D, Dewhurst S, Katze MG. 2011. Comprehensive proteomic analysis of influenza virus polymerase complex reveals a novel association with mitochondrial proteins and RNA polymerase accessory factors. J Virol 85:8569–8581. doi: 10.1128/JVI.00496-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimizu K, Handa H, Nakada S, Nagata K. 1994. Regulation of influenza virus RNA polymerase activity by cellular and viral factors. Nucleic Acids Res 22:5047–5053. doi: 10.1093/nar/22.23.5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawaguchi A, Nagata K. 2007. De novo replication of the influenza virus RNA genome is regulated by DNA replicative helicase, MCM. EMBO J 26:4566–4575. doi: 10.1038/sj.emboj.7601881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Momose F, Naito T, Yano K, Sugimoto S, Morikawa Y, Nagata K. 2002. Identification of Hsp90 as a stimulatory host factor involved in influenza virus RNA synthesis. J Biol Chem 277:45306–45314. doi: 10.1074/jbc.M206822200. [DOI] [PubMed] [Google Scholar]
- 26.Momose F, Basler CF, O'Neill RE, Iwamatsu A, Palese P, Nagata K. 2001. Cellular splicing factor RAF-2p48/NPI-5/BAT1/UAP56 interacts with the influenza virus nucleoprotein and enhances viral RNA synthesis. J Virol 75:1899–1908. doi: 10.1128/JVI.75.4.1899-1908.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naito T, Kiyasu Y, Sugiyama K, Kimura A, Nakano R, Matsukage A, Nagata K. 2007. An influenza virus replicon system in yeast identified Tat-SF1 as a stimulatory host factor for viral RNA synthesis. Proc Natl Acad Sci U S A 104:18235–18240. doi: 10.1073/pnas.0705856104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naito T, Momose F, Kawaguchi A, Nagata K. 2007. Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits. J Virol 81:1339–1349. doi: 10.1128/JVI.01917-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horowitz DS, Abelson J. 1993. A U5 small nuclear ribonucleoprotein particle protein involved only in the second step of pre-mRNA splicing in Saccharomyces cerevisiae. Mol Cell Biol 13:2959–2970. doi: 10.1128/MCB.13.5.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horowitz DS, Abelson J. 1993. Stages in the second reaction of pre-mRNA splicing: the final step is ATP independent. Genes Dev 7:320–329. doi: 10.1101/gad.7.2.320. [DOI] [PubMed] [Google Scholar]
- 31.Vijayraghavan U, Abelson J. 1990. PRP18, a protein required for the second reaction in pre-mRNA splicing. Mol Cell Biol 10:324–332. doi: 10.1128/MCB.10.1.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bacikova D, Horowitz DS. 2005. Genetic and functional interaction of evolutionarily conserved regions of the Prp18 protein and the U5 snRNA. Mol Cell Biol 25:2107–2116. doi: 10.1128/MCB.25.6.2107-2116.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He F, Inoue M, Kigawa T, Takahashi M, Kuwasako K, Tsuda K, Kobayashi N, Terada T, Shirouzu M, Guntert P, Yokoyama S, Muto Y. 2012. Solution structure of the splicing factor motif of the human Prp18 protein. Proteins 80:968–974. doi: 10.1002/prot.24003. [DOI] [PubMed] [Google Scholar]
- 34.Bacikova D, Horowitz DS. 2002. Mutational analysis identifies two separable roles of the Saccharomyces cerevisiae splicing factor Prp18. RNA 8:1280–1293. doi: 10.1017/S1355838202023099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horowitz DS, Lee EJ, Mabon SA, Misteli T. 2002. A cyclophilin functions in pre-mRNA splicing. EMBO J 21:470–480. doi: 10.1093/emboj/21.3.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pons MW. 1973. The inhibition of influenza virus RNA synthesis by actinomycin D and cycloheximide. Virology 51:120–128. doi: 10.1016/0042-6822(73)90372-3. [DOI] [PubMed] [Google Scholar]
- 37.Fortes P, Lamond AI, Ortin J. 1995. Influenza virus NS1 protein alters the subnuclear localization of cellular splicing components. J Gen Virol 76(Part 4):1001–1007. [DOI] [PubMed] [Google Scholar]
- 38.Chan AY, Vreede FT, Smith M, Engelhardt OG, Fodor E. 2006. Influenza virus inhibits RNA polymerase II elongation. Virology 351:210–217. doi: 10.1016/j.virol.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 39.Engelhardt OG, Fodor E. 2006. Functional association between viral and cellular transcription during influenza virus infection. Rev Med Virol 16:329–345. doi: 10.1002/rmv.512. [DOI] [PubMed] [Google Scholar]
- 40.Engelhardt OG, Smith M, Fodor E. 2005. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J Virol 79:5812–5818. doi: 10.1128/JVI.79.9.5812-5818.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. 2011. Epigenetics in alternative pre-mRNA splicing. Cell 144:16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerlach P, Malet H, Cusack S, Reguera J. 2015. Structural insights into bunyavirus replication and its regulation by the vRNA promoter. Cell 161:1267–1279. doi: 10.1016/j.cell.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poole E, Elton D, Medcalf L, Digard P. 2004. Functional domains of the influenza A virus PB2 protein: identification of NP- and PB1-binding sites. Virology 321:120–133. doi: 10.1016/j.virol.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 44.Mena I, Jambrina E, Albo C, Perales B, Ortin J, Arrese M, Vallejo D, Portela A. 1999. Mutational analysis of influenza A virus nucleoprotein: identification of mutations that affect RNA replication. J Virol 73:1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beaton AR, Krug RM. 1986. Transcription antitermination during influenza viral template RNA synthesis requires the nucleocapsid protein and the absence of a 5′ capped end. Proc Natl Acad Sci U S A 83:6282–6286. doi: 10.1073/pnas.83.17.6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawaguchi A, Naito T, Nagata K. 2005. Involvement of influenza virus PA subunit in assembly of functional RNA polymerase complexes. J Virol 79:732–744. doi: 10.1128/JVI.79.2.732-744.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]