

ABSTRACT
By using cryo-electron microscopy, expanded 80S-like poliovirus virions (poliovirions) were visualized in complexes with four 80S-specific camelid VHHs (Nanobodies). In all four complexes, the VHHs bind to a site on the top surface of the capsid protein VP3, which is hidden in the native virus. Interestingly, although the four VHHs bind to the same site, the structures of the expanded virus differ in detail in each complex, suggesting that each of the Nanobodies has sampled a range of low-energy structures available to the expanded virion. By stabilizing unique structures of expanded virions, VHH binding permitted a more detailed view of the virus structure than was previously possible, leading to a better understanding of the expansion process that is a critical step in infection. It is now clear which polypeptide chains become disordered and which become rearranged. The higher resolution of these structures also revealed well-ordered conformations for the EF loop of VP2, the GH loop of VP3, and the N-terminal extensions of VP1 and VP2, which, in retrospect, were present in lower-resolution structures but not recognized. These structural observations help to explain preexisting mutational data and provide insights into several other stages of the poliovirus life cycle, including the mechanism of receptor-triggered virus expansion.
IMPORTANCE When poliovirus infects a cell, it undergoes a change in its structure in order to pass RNA through its protein coat, but this altered state is short-lived and thus poorly understood. The structures of poliovirus bound to single-domain antibodies presented here capture the altered virus in what appear to be intermediate states. A careful analysis of these structures lets us better understand the molecular mechanism of infection and how these changes in the virus lead to productive-infection events.
KEYWORDS: 80S, VHH, cryo-electron microscopy, expanded virus, poliovirus, single-domain antibodies, three-dimensional structure
INTRODUCTION
Poliovirus (PV) is a small, nonenveloped, positive-sense, single-stranded RNA (ssRNA) virus that is a member of the enterovirus genus of the picornavirus family (1, 2). The virus consists of an RNA genome that is surrounded by a protein shell. The shell is formed by an icosahedrally symmetric arrangement of 60 copies each of 3 large proteins (VP1, VP2, and VP3), each of which has a wedge-shaped beta barrel core (3), and the small myristoylated protein VP4 (4), which is located on the inner surface of the capsid. To infect its natural host (human epithelial cells in the gut), poliovirus must solve the problem of getting its genome through the host's protective cellular membrane. Details of how this is accomplished are now gradually emerging (5, 6).
Poliovirus infection is initiated when the virus attaches to its receptor, Pvr (7) (a member of the nectin family [8, 9]). When incubated at 37°C, the receptor catalyzes an icosahedrally symmetric expansion in the capsid (10, 11) to form a particle called the 135S particle or A particle (12, 13). Subsequent to the internalization of this particle into the cell, an unknown trigger induces the release of its viral RNA, which results in the formation of an empty particle called the 80S particle.
How poliovirus architecture changes during expansion.
The major capsid proteins of poliovirus (VP1, VP2, and VP3) are simplest to understand as fairly rigid core structures (primarily wedge-shaped antiparallel beta barrels) that are decorated by long movable loops and terminal extensions (3). In native virions, the beta barrels are closely packed and lack holes between subunits, which serves to protect the viral genome (3). The native structure is stabilized by specific interactions between the subunits, including extensive contacts with the N- and C-terminal extensions and flexible loops of neighboring beta barrels.
Upon virus expansion, the beta barrels move, and several of these loops and terminal extensions either are rearranged or become disordered. In this process, many of the intersubunit interactions of native virus become broken, and a smaller set of new stabilizing interactions is formed. These rearrangements open large holes at the 2-fold and quasi-3-fold symmetry axes of the particle (14, 15). During the transition from the native to the 135S conformation, the resulting holes allow the externalization of two normally internal 70-amino-acid-long polypeptides, including VP4, which is myristoylated (4), and the N-terminal extension of VP1. Once externalized, these polypeptides are inserted into membranes (16–18), thereby forming a structure at one of the quasi-3-fold axes that facilitates the release of RNA and its translocation across the host cell membrane (5), resulting in the formation of the 80S particle. Although high-resolution crystal structures have been determined for the expanded states of related viruses (19–21), cryo-electron microscopy (cryo-EM) studies have shown that the expanded state of poliovirus is structurally heterogeneous (14, 15).
VHHs against poliovirus.
VHHs (or Nanobodies) are the variable domains of the single-chain heavy chain antibodies that are found in camelids and sharks (22). Because these domains are stable, expressible in Escherichia coli, nonglycosylated, and approximately one-quarter the size (∼15 kDa) of conventional Fab fragments, they have proven to be useful immunological reagents for a number of laboratory applications.
Tight-binding VHHs directed against poliovirus epitopes were originally developed in the hope that the VHHs would prove to be useful either as therapeutic reagents or for confirming the antigenic integrity of poliovirus vaccine stocks (23). The VHHs were prepared by immunizing a dromedary with poliovirus, preparing cDNA libraries from lymphocytes, and then selecting for tight poliovirus binders by phage display. A selection of these Nanobodies was then expressed in E. coli and screened for their ability to bind and neutralize native 160S poliovirus particles (23). Biochemical and structural studies showed that these VHHs neutralize infectivity by a combination of blocking receptor binding and stabilizing virions in the native conformation (24). Further structural studies showed that all five neutralizing VHHs bind at a site overlapping the receptor binding site and showed that stabilization of the virus was achieved by binding to a set of polypeptide segments that are known to undergo conformational rearrangements during expansion (25).
A second group of five VHHs (having four unique sequences), constituting the focus of the present cryo-EM studies, were selected for their ability to bind tightly and specifically to expanded poliovirus virions (poliovirions) but not to the native virus (26). Expanded poliovirus particles are said to exhibit C or H (for “heated”) antigenicity, as opposed to the D or N antigenicity of “native” virus. In the present study, complexes of these four unique anti-C/anti-H VHHs (PVSP17B, PVSS12B, PVSS10E, and PVSS7A) with heated poliovirus were prepared, plunge-frozen in liquid ethane, and visualized by cryo-EM. Sequencing studies suggested that three of these four Nanobodies are sister clones, with relatively few sequence differences between any pair. In this work, we describe the cryo-EM structures of the complexes of each of the four VHHs with poliovirus in the expanded state. The structures showed that the four antibodies share a binding site on the top surface of VP3 that differs markedly from the binding sites previously found in the anti-N complexes. Additionally, for the three sister clones, none of the sequence differences in the Nanobodies is involved in interactions with the virus particle. Each of the four anti-C VHHs stabilize expanded poliovirions in a fairly homogeneous conformation. Interestingly, the detailed structure of poliovirus in the expanded state is unique for each VHH complex, suggesting that each complex has selected a unique structure from a collection of low-energy structures in the expanded state.
In the VHH-bound expanded structures, several key polypeptide segments (which were disordered in the more heterogeneous structures of uncomplexed expanded virions [14, 15]) now adopt new well-ordered conformations. This is an exciting development, as several of these new conformations provide satisfying structural explanations for mutational data in the poliovirus field that date back many years, which could not be resolved previously, based on structures of either the native virus or the expanded virus at a lower resolution than that in the present study. Much of the present discussion focuses on the implications of new structural features for better understanding the poliovirus life cycle.
RESULTS
Density slices, solid-contour representations, and Fourier shell correlation (FSC) curves for the reconstructions of each of the four VHH-virus complexes are shown in Fig. 1. The FSC curves show that the resolutions of the four structures range from 4.3 to 5.3 Å (at a correlation level of 0.143).
FIG 1.

Amino acid sequences and cryo-EM reconstructions of four anti-C/anti-H-VHH complexes with expanded poliovirus. (Top) Sequence alignment of the four VHHs shows that they are very similar to one another. Differences from PVSS10E are indicated in red lowercase type. For convenience, the locations of beta strands A to G in the PVSP17B structure are indicated. (Second panel) From left to right, the complexes of poliovirus (blue) with PVSS7A (purple), PVSS10E (orange), PVSS12B (cyan), and PVSP17B (yellow) are shown as iso-contour surfaces that are shaded by radius and depth. (Third panel) Representative electron density slices for each of the complexes are shown, each viewed along a 2-fold axis. Note that each VHH binds to the capsid in a similar spot. Also note that many individual polypeptide chains are well resolved. (Bottom) FSC curves show that the resolutions of the four structures range from 4.3 to 5.3 Å (at a correlation level of 0.143 [green dashed line]).
For each VHH-poliovirus complex, atomic models for VHH and viral capsid proteins were repeatedly fitted to the cryo-EM maps (using COOT [27, 28]). The relatively high resolution of the structures resulted in density in both the VHH and virus regions of sufficient quality to orient beta sheets unambiguously and, in favorable regions, to resolve individual polypeptide chains, greatly facilitating model building (Fig. 2). Some side-chain rotamer choices were indicated by chemical considerations or by the shapes of the density contours. The models were then optimized by iterative cycles of manual fitting in COOT and refinement by using Refmac5 (29), until no further improvement could be achieved. This refinement protocol maximizes the agreement between the Fourier amplitudes and phases (which were based on the atomic model with icosahedrally related neighbors present) and the Fourier amplitudes and phases calculated from the corresponding portion of the reference cryo-EM reconstruction. It is designed to obtain a satisfactory fit to the map, using a model that is stereochemically reasonable, chemically plausible, and icosahedrally restrained.
FIG 2.

Quality of the electron density in the reconstructions of anti-C-VHH complexes with poliovirus. (Left) Slice through the VP2 beta barrel illustrating an area where rigid-body fitting of the native atomic model accounts satisfactorily for the PVSP17B experimental electron density map. At a 4.5-Å resolution, rigid-body positioning of beta strands and alpha helices is unambiguous. (Right) A homolog-based atomic model for the VHH, shown from the front and back, can be placed within its density envelope unambiguously. In addition, individual polypeptide chains can often be resolved over short stretches by changing the contour level.
In each atomic model, a substantial subset of the atoms belong to beta barrel “core” regions that are plausibly treated as rigid bodies, based on known crystal structures, in order to limit the number of parameters used. Regions of the map that deviated from the known crystal structures were instead modeled by using individually variable atomic coordinates. The poliovirus capsid proteins (VP1, VP2, and VP3) were each derived from those reported under PDB accession number 1HXS, and single-domain VHHs (with appropriate amino acid substitutions) were each derived from those reported under PDB accession number 2HFF or 4NBZ, which provided the closest available shape matches to the experimental density (see Materials and Methods). Residue ranges that are included in each atomic model and ranges belonging to their rigid core regions are shown in Table 1. Statistics for the final cryo-EM reconstructions and for the fit of each refined atomic model to its density map are summarized in Table 2.
TABLE 1.
Amino acids that form structurally consistent core regionsa
| Complex | Positions |
|||
|---|---|---|---|---|
| PVSP17B | PVSS12B | PVSS10E | PVSS7A | |
| VHH, all ordered | 1–124 | 1–123 | 1–124 | 1–125 |
| VHH coreb | 1–12 | 1–12 | 6–21 | 2–96 |
| 15–31 | 17–31 | 25–27 | ||
| 34–97 | 34–97 | 39–52 | ||
| 57–70 | ||||
| 72–97 | ||||
| 112–120 | 112–120 | 113–122 | ||
| VP1, all ordered | 57–213 | 57–213 | 71–212 | 71–213 |
| 232–279 | 232–279 | 232–279 | 232–279 | |
| VP1 core | 72–94 | 72–94 | 72–94 | 73–93 |
| 107–155 | 107–155 | 107–155 | 107–157 | |
| 172–174 | 172–174 | 172–174 | ||
| 177–180 | 177–180 | 177–180 | ||
| 183–209 | 183–209 | 183–209 | 183–213 | |
| 240–267 | 240–267 | 240–267 | 240–267 | |
| 272–279 | 272–279 | 272–279 | ||
| VP2, all ordered | 1–43 | 1–43 | 1–42 | 10–43 |
| 53–159 | 53–159 | 55–159 | 53–159 | |
| 174–269 | 174–269 | 175–268 | 175–268 | |
| VP2 core | 30–43 | 30–43 | 30–42 | 30–43 |
| 53–100 | 53–100 | 55–100 | 53–132 | |
| 102–133 | 102–133 | 102–132 | ||
| 143–159 | 143–159 | 144–159 | 143–159 | |
| 175–238 | 175–238 | 175–238 | 195–239 | |
| 250–259 | 250–259 | 250–259 | 247–260 | |
| VP3, all ordered | 1–231 | 1–231 | 1–230 | 1–175 |
| 185–230 | ||||
| VP3 core | 1–12c | 1–12c | 1–12c | 1–12c |
| 13–43c | 13–43c | 13–43c | 13–43c | |
| 44–70 | 44–70 | 44–70 | 44–71 | |
| 83–87 | 83–87 | 83–87 | ||
| 95–121 | 95–121 | 95–121 | ||
| 126–133 | 126–133 | 130–133 | 82–135 | |
| 142–177 | 142–177 | 142–172 | 143–147 | |
| 190–201 | 190–201 | 192–193 | 155–156 | |
| 206–225 | 206–225 | 206–225 | 188–226 | |
Residues that were restrained to agree with main-chain coordinates from the structural homolog.
The amino acid residues correspond to the sequence of the antipoliovirus VHH.
N-terminal residues of VP3 are piecewise rigid bodies.
TABLE 2.
Statistics for reconstructions and for refinement of the atomic modelsd
| Parameter | Value for VHH |
|||
|---|---|---|---|---|
| PVSP17B | PVSS12B | PVSS10E | PVSS7A | |
| Resolution range (Å)a | 99–4.2 | 98.6–4.3 | 98.6–4.5 | 98.9–4.5 |
| No. of pseudoreflections | 249,149 | 235,686 | 228,497 | 177,930 |
| wFOMe | 0.503 | 0.410 | 0.435 | 0.403 |
| Rfactor (%) | 44.2 | 47.7 | 48.1 | 48.0 |
| Outer shell | 53.8 | 54.1 | 52.5 | 54.5 |
| FOMe (Refmac5) | 0.745 | 0.741 | 0.742 | 0.752 |
| Bond error (Å) | 0.011 | 0.009 | 0.011 | 0.012 |
| Angle error (°) | 2.09 | 2.02 | 2.16 | 2.16 |
| Resolution (FSC, 0.143)b | 4.5 | 4.3 | 4.8 | 5.3 |
| Grid spacing (Å) | 0.8245 | 0.986 | 0.986 | 0.8245 |
| Sampling grid | 512 × 512 × 512 | 512 × 512 × 512 | 512 × 512 × 512 | 512 × 512 × 512 |
| Submap sizec | 256 × 256 × 240 | 216 × 216 × 200 | 216 × 240 × 200 | 240 × 240 × 240 |
| Submap origin | −98, −93, 0 | −84, −79, 0 | −83, −94, 0 | −92, −76, 0 |
| No. of particles | 14,644 | 17,627 | 13,938 | 17,654 |
| VHH amino acids | 1–124 | 1–123 | 1–124 | 1–125 |
| PDB accession no. of structural homolog | 2HFF | 2HFF | 2HFF | 4NBZ |
| PDB accession no. | 5KU0 | 5KTZ | 5KWL | 5KU2 |
| EMDB accession no. | EMD-8285 | EMD-8284 | EMD-8277 | EMD-8286 |
The resolution range used in the refinement. The high-resolution limit is based on where the amplitudes and phases calculated based on the model correlate with those based on the Fourier transform of the map.
The resolution where the Fourier shell correlation falls below 0.143. For high-resolution structures, this is generally a conservative estimate of the resolution and may have a lower resolution than the apparent resolution based on comparisons of the transforms of the model and map.
The submap size was larger in order to accommodate symmetry copies of newly ordered polypeptides or smaller when those polypeptides were not visible in the map.
The microscope used was an FEI Polara G2 microscope at 300 kV. The emitter used was a field emission gun. The detector used was a Gatan K2 Summit detector.
wFOM is the Fourier-amplitude-weighted average of the cosine of the phase discrepancy, which is analogous to a crystallographic figure of merit (with 1.0 indicating perfect phase agreement and 0.0 indicating a complete lack of correlation). This statistic (and FOM, its unweighted version) was used to compare atomic-model-based Fourier coefficients with the Fourier transform of the reference reconstruction.
How large is expanded poliovirus, and how do the beta barrels move?
The original estimates of the sizes of 135S and 80S particles, 4% larger than native 160S particles, date back to cryo-EM studies by Belnap et al. (11), at a resolution of 18 to 23 Å. At that time, limited resolution as well as limited knowledge of the structure of the expanded virus made it impossible to distinguish rigid expansion from changes in the shape of a protein due to either localized movement or changes in the local degree of order.
Two more recent experiments in our laboratory made it possible to improve older estimates of both the size-scale and rigid-body parameters that describe shifts and rotations in the core regions of the capsid proteins. First, the anti-C-VHH complexes with poliovirus reported here are our first pictures of expanded poliovirus in which individual polypeptide chains can be accounted for (rather than guessed at). As a result, we now know which amino acids in poliovirus properly belong to the rigid cores (as listed in Table 1) and which amino acids should be excluded from the rigid-body analysis. Second, we possess individual micrographs that include both ligand-free “empty” 80S particles and native virus particles (which are “full” of RNA), where the images of the native virus provide us with an absolute calibration of the pixel size. Boxed particle images of these empty capsids were originally collected during a previously reported cryo-EM study of the poliovirus-Pvr complex (30) but were discarded following an automatic classification step using Relion (31), which showed them to be empty, expanded, and undecorated by Pvr. Using Frealign (32), we have now refined their orientation parameters and obtained a class-average reconstruction to a 6.9-Å resolution (which is available under EMDB accession number EMD-8292). In turn, the size-calibrated empty reconstruction was subsequently used to size scale the four anti-C-VHH complexes with expanded poliovirus.
The anti-C-VHH complexes with expanded poliovirus allow us to understand how the beta barrel cores of VP1, VP2, and VP3 differ in their positions and orientations when native and expanded poliovirions are compared. For the core regions of the capsid proteins VP1, VP2, and VP3, expansion to the 80S state causes the capsid protein center of mass to shift outward by 5.5, 2.7, and 3.3%, respectively (compared with the previous overall estimate of 4%). In addition to the outward shift, VP1, VP2, and VP3 each rotate by 4.4° to 5.6°, with VP1 rotating around a radial axis that lies approximately perpendicular to the virus surface and VP2 and VP3 rotating around horizontal axes that lie parallel to the virus surface. Together, these rigid-body motions of the capsid proteins largely account for the opening of holes at the 2-fold and quasi-3-fold axes. These structural rearrangements are very similar to those described previously in crystal structures of the expanded states of enterovirus 71 (EV71) (19), coxsackievirus A16 (CAV16) (20), and human rhinovirus 2 (HRV2) (21). Nevertheless, there are important mechanical differences between poliovirus and the expanded forms of these other viruses that relate to detailed structural changes in the EF loop of VP2. These differences are considered more fully below.
It is interesting to note that the expanded forms of poliovirus, EV71, CAV16, and HRV2 have all expanded by ∼4%. The similarity in size might reflect an evolutionary pressure to limit the size of the holes that are opened in the shell. Limiting the size of these holes prevents double-stranded RNA loops from becoming externalized (at icosahedrally related sites), which would result in the entanglement of the RNA and prevent its successful release.
What kind of expanded poliovirus particle occurs in the anti-C-VHH-poliovirus complexes?
The ability of a VHH (PVSP17B) to bind to poliovirus 160S, 135S, and 80S particles was assessed (see Materials and Methods). Briefly, pure poliovirions, in various states of expansion, were incubated at 25°C, first with the VHH in solution and then with Ni-nitrilotriacetic acid (NTA)-agarose beads that bind to the His6 tail of the VHH. After washing, poliovirus and VHH proteins were eluted with imidazole and visualized as characteristic bands on SDS-PAGE gels (Fig. 3). Consistent with the original procedure for selecting this VHH (26), the VHH was clearly capable of binding to 80S particles but not to 160S native virions. The VHH-mediated binding of 135S particles was similar to 80S binding and well above the level of nonspecific binding. Electron micrographs of the VHH-135S complex stained with uranyl acetate (data not shown) clearly demonstrated that the binding of VHHs to 135S particles did not trigger RNA release.
FIG 3.

Anti-C VHH PVSP17B can bind to purified 80S and 135S poliovirions but not to 160S native virions. Poliovirions, in various states of expansion, were incubated at 25°C, first with the VHH in solution and then with Ni-NTA-agarose beads that bind to the His6 tail of the VHH. After washing, poliovirus and VHH proteins were eluted with imidazole and visualized as characteristic bands on SDS-PAGE gels. Lane 1, EZ ladder size standards (with masses listed in kilodaltons at the left); lane 2, 160S PV particles plus VHH; lane 3, 135S PV particles plus VHH; lane 4, 80S PV particles plus VHH; lane 5, VHH (without PV); lane 6, 135S PV particles (without VHH); lane 7, purified 160S PV particles (control); lane 8, purified VHH (control). Native poliovirus (lane 7) shows characteristic bands for VP1, VP2, VP3, and VP4 (top to bottom).
Given that both 135S and 80S poliovirions can bind to an anti-C VHH, it is important to establish which form of the virus is present in the VHH-virus complexes visualized here by cryo-EM. Like 135S and 80S particles, the virus particles that we see are expanded, having large holes at the 2-fold and quasi-3-fold axes, and lack VP4 (Fig. 4 and 5). Nevertheless, it is absolutely clear that the present VHH complexes have not turned out to be 135S-like because they lack the following two defining attributes of this particle: a full complement of viral RNA and N-terminal extensions of VP1 that are exposed and locked into place at the quasi-3-fold holes (see references 15 and 20). The lack of viral RNA and of VP4 also rules out the reversibly expanded “breathing” virus particle (33) that can be trapped by Fab binding (34). Instead, the lack of a full complement of RNA, together with an open quasi-3-fold hole, indicates that the expanded viruses in all four anti-C-VHH complexes are more 80S-like. The main respect in which the virus differs from known 80S-like particles is in the conformation of the GH loop of VP3 in three of the four complexes, which was apparently the result of intermolecular contacts with the VHH. These atypical GH loop conformations are discussed below in greater detail due to their potential biological implications.
FIG 4.
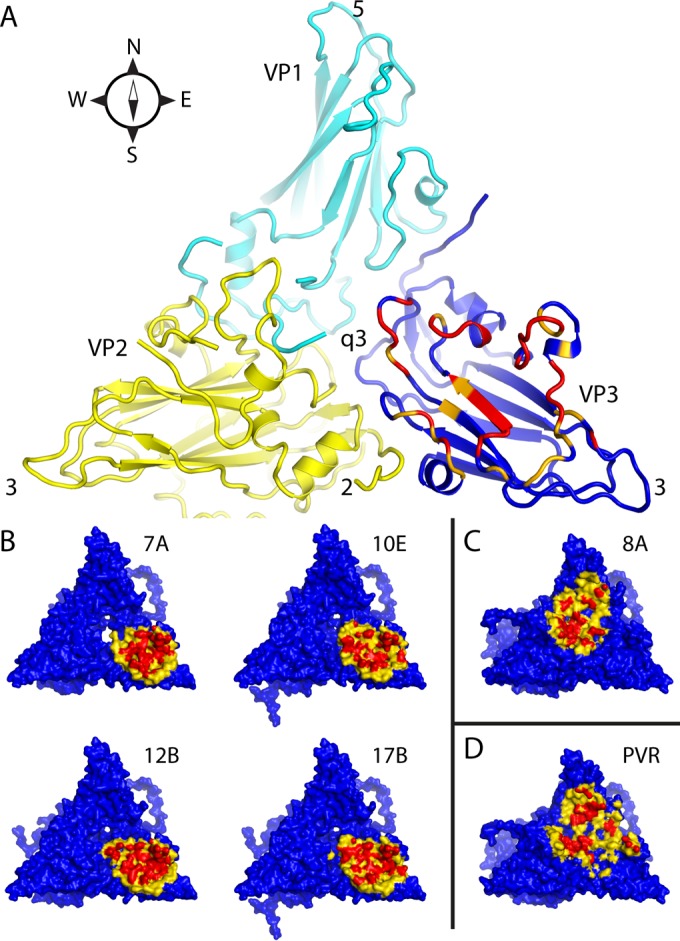
Intermolecular contact areas on the poliovirus surface (“footprints”) that are involved in binding to various proteins. (A) One copy each of VP1 (cyan), VP2 (yellow), and VP3 (mostly dark blue) occupy the “5-3-3 triangle,” which has one 5-fold and two 3-fold symmetry axes at its corners (labeled). Expansion of poliovirus opens holes at the 2-fold axis (labeled) and at the quasi-3-fold axis (labeled). The quasi-3-fold axis lies at the center of the 5-3-3 triangle and is the point at which these three capsid proteins join together in the native virus. By convention, compass directions (north, south, east, and west) indicate the relative positions along the virus surface. This ribbon representation shows the “footprint” of one copy of anti-C-VHH PVSP17B on expanded poliovirus. In VP3, which is the only capsid protein to contact the anti-C VHHs, residues that are responsible for binding are indicated in red (distances of <4.2 Å), and residues with contacts closer than 6.0 Å are orange or yellow. (B to D) Surface representations, color-coded by distance (red, 5 Å or closer; yellow, 5 to 9 Å), show that the binding footprints of the four anti-C VHHs on expanded poliovirus (B) are similar to one another but differ from the footprints (on native poliovirus) of poliovirus receptor (“PVR”) (D) and from the anti-N VHHs (C), of which PVSS8A (“8A”) is typical. Thus, Pvr and all five anti-N VHHs consistently bind to parts of VP1 and VP2 that are close to the quasi-3-fold axis. The surface representations make it apparent that the quasi-3-fold hole is visibly larger in the PVSS7A complex than it is in the other three anti-C VHH complexes.
FIG 5.
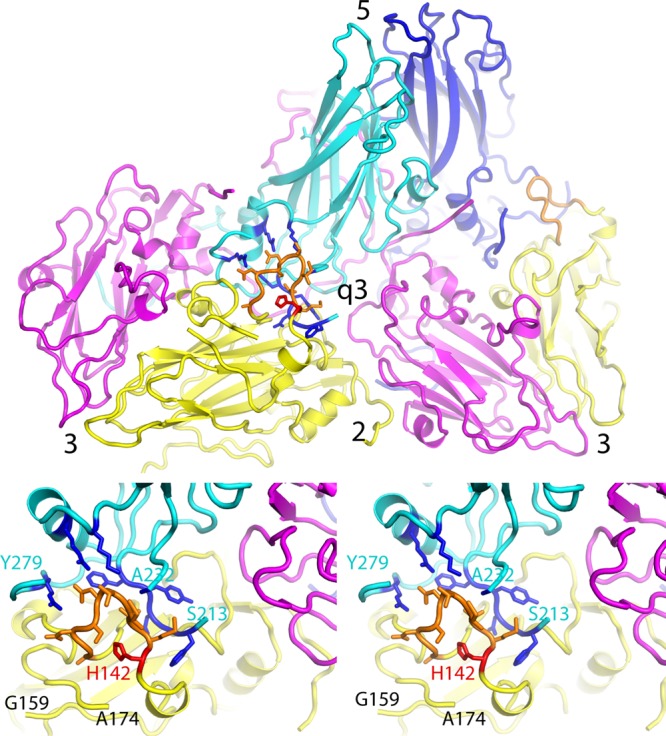
Virus expansion causes dramatic changes in the large EF double loop of VP2. (Top) Overview of the outer surface of the expanded virion, in a ribbon representation, showing how two biological protomers fit together on either side of the quasi-3-fold hole. Each protomer includes one copy of VP3 (magenta), VP2 (yellow), and VP1 (cyan or dark blue, to distinguish them). Axes of symmetry are labeled. The rearranged portion of the EF loop of VP2 is highlighted in orange. (Bottom) Upon virus expansion, a part of the VP2 EF loop (residues 160 to 174) becomes disordered (as indicated by “cut ends” in the model), while another portion (residues 133 to 142) (orange) rearranges to encircle His142 (red) and falls into the gap between VP2 and VP1. This stereo pair focuses on the rearranged portion of the EF loop (orange) and on residues of VP1 (highlighted in dark blue) that stabilize its new conformation. Above the loop, new hydrogen bonds are formed with side chains from the open end of the VP1 beta barrel, including (from left to right) Arg275, Arg119, and Lys113, and with the main chain of Ala232. Additional stabilizing contacts are also made with residues 208 to 212 from the amino end of the VP1 GH loop, which lie behind the orange loop. This rearrangement permits VP1 and VP2 to maintain contacts with one another when their respective beta barrels move apart during expansion.
Binding footprints and intermolecular contacts.
All four VHHs bind to the top of VP3, oriented with their N termini and hypervariable loops facing the virus. The intermolecular contacts of PVSP17B with poliovirus are typical of the anti-C VHHs. Strikingly, the VHH makes no contacts with any capsid protein outside VP3 and avoids the interfaces between capsid proteins (Fig. 4). Only two loops on the VHH are involved in contacts with the virus. These loops are the FG loop (including contacts with Phe100, Leu102, Ser103, Ser105, Tyr107, and Thr108) and the BC loop (at Arg31 and Tyr32), for a total of only 8 amino acids. The binding footprint at the top of VP3 includes several VP3 secondary structures, including the knob-like insertion in the B-beta strand (Asp56), the B-beta strand (Val70), the BC loop (Asp80), the C-beta strand (Leu83, Cys84, and Ser86), the CD helix (Pro93 and Arg84), the EF loop (Pro141 and Lys143), and the GH loop (Asp181, Ser183, Phe184, and Glu186), for a total of 14 amino acids. Thus, each VHH side chain tends to contact multiple residues in VP3. For the FG loop of VHH, such extensive contacts were often achieved by insertion of the VHH side chain into the crevices between VP3 secondary structural elements (Fig. 4). Notably, for Arg31 and Tyr32 of the VHH BC loop, most of the intermolecular contacts involve the GH loop of VP3, which has rearranged to insert under the edge of VHH. (Neither the coiled GH loop conformations seen previously in the native virus nor the extended hairpin-like conformation seen previously in ligand-free 80S and 135S particles would have been able to make contacts with VHH that were this extensive.)
The binding footprint of the anti-C VHHs on the virus surface (at the top of VP3 and avoiding the interfaces between capsid proteins) (Fig. 4B) is markedly different from the footprints of the anti-N VHHs and of Pvr (which include much of the VP1 and VP2 top surfaces and which involve interprotomer interfaces that change dramatically during capsid expansion) (Fig. 4C and D). The binding footprint of the anti-C VHHs is also notable for including certain structures (such as the knob-like insertion and B-beta strand of VP3) that are not solvent exposed in the native virus but instead lie beneath the C-terminal extension of VP1, thus explaining the specificity for expanded particles and the inability to bind native virions.
How do the four anti-C-VHH-poliovirus models compare with one another?
The original selection process for anti-C VHHs (23) was based on randomly choosing five good 80S binders that exhibited minimal cross-reactivity with the native virus. All five of the selected sequences happened to have identical lengths, including 122 amino acids from the dromedary-derived sequence plus a His6 tag at the carboxyl tail. This selection process produced a surprisingly narrow range of VHH sequences (Fig. 1), in which four out of the five sequences differed from one another in the identities of only 0, 1, or 2 amino acids and affected only residues located very far from the virus binding site (so that these few sequence differences are unlikely to be responsible for any structural differences between complexes). The remaining VHH, PVSS7A, was the most dissimilar, differing from each of the other VHHs at 13 or 14 amino acid positions.
After the convergence of model building and refinement, all four atomic models for poliovirus had become closely similar to one another in their core regions (as defined in Table 1). Thus, the PVSP17B, PVSS12B, and PVSS10E complexes (which include sequence-similar VHHs) tended to be the most structurally similar, with average alpha carbon differences of ∼0.3 to 0.4 Å for VP1, VP2, and VP3. Their differences from PVSS7A were only slightly larger, at ∼0.4 to 0.5 Å, provided that the GH loop of VP3 (which was significantly different in PVSS7A) was omitted from the calculation. Although this similarity is due partly to the application of restraints (which encourage similarity to portions of the native capsid proteins), it should be emphasized that restraints were applied only where native-like atomic models were consistent with the experimental maps.
As expected, the VHH models exhibited somewhat greater variability. Thus, the core regions of the three sequence-similar VHHs differed from one another by an average of 0.5 to 0.7 Å and differed from PVSS7A by 0.8 to 0.95 Å. This great variability (relative to poliovirus) likely reflects both the lower quality of the VHH electron density and the need to base structural restraints upon homolog models rather than upon authentic crystal structures.
Structural variability between VHH complexes.
In all four of the VHH complexes, there are several polypeptide segments in the major capsid proteins (VP1, VP2, and VP3) with conformations that differ from those of the native virus (as summarized in Fig. 6). Some of these differences from the native virus are common to all four VHH complexes. These differences include the EF loop of VP2 (residues 129 to 194), wherein all four complexes form essentially the same new structure (see below), and the VP1 GH loop (residues 207 to 238) and C-terminal extension (residues 275 to 302), both of which become disordered in the VHH complexes.
FIG 6.

Overview of polypeptide segments that are disordered or change conformation upon poliovirus expansion. (A) 160S particles of native virus. (B) 80S particles of expanded virus. (Top) Outer surface. (Bottom) Inner surface. In this cartoon, the surface representation of VP3 is in magenta, two copies of VP2 are in yellow, and two copies of VP1 are in cyan and azure (RGB 0,127,255). Symmetry copies of these surfaces are shown in faded colors. In general, a polypeptide segment that is disordered or changes conformation is shown as a colored tube (in at least one symmetry copy) and is labeled. However, note that the N-terminal extension of VP3 (red) does not change significantly upon expansion and is shown only in a tube representation for clarity. Also note that the capsid protein VP4 (which covers much of the inner surface of the native virus) has been omitted for clarity. In VP1, the GH loop, the C-terminal extension, and the N-terminal extension are blue tubes. In VP2, the EF loops are in black, the C-terminal extension is in black, and the N-terminal extension is in forest green. In VP3, the loops that change (GH loop, bottom loop, end spheres, and putative helix) are in orange.
However, a number of areas in the viral capsid differ from one complex to another. As discussed below, the GH loop of VP3 has one nonnative conformation in PVSS7A (which has a unique VHH sequence) and a different nonnative conformation in the other three (sequence-similar) VHH complexes. More surprisingly, several polypeptide segments of the virus adopt new conformations (that are unlike those of the native virus and are well resolved albeit partially occupied) in the PVSP17B and PVSS12B complexes but exhibit disorder in the PVSS10E and PVSS7A complexes. These ordered regions include parts of the N-terminal extension of VP1 (residues 58 to 71), the N-terminal extension of VP2 (residues 1 to 10), and the C-terminal extension of VP2 (residues 264 to 272). In addition, large mounds of ordered density (of unknown composition) are present in the PVSP17B and PVSS12B complexes and located on the inner surface of the VP2 beta barrel on either side of each 2-fold hole (see below). Counterintuitively, the map of the PVSS10E complex does not show a similar ordering of these features despite the near identity of its VHH sequence with those of PVSP17B and PVSS12B.
A similar structural variability was seen previously among populations of ligand-free poliovirus 80S empty capsids in a 9-Å-resolution cryo-EM study by Levy et al. (14). In that study, the classification of each preparation of virus particles into two structural groups (based only on capsid density) improved the resolution markedly. The classes were designated “early” and “late,” with the proportion of late particles increasing with longer incubation times. The most obvious difference between the early and late reconstructions was the appearance in the late empty capsids of large mounds of inward-projecting density features on the inner surface of VP2, similar to the ones that we see in the present study. Clearly, the PVSP17B and PVSS12B complexes correspond to the better-ordered class, and the PVSS10E and PVSS7A complexes correspond to the less-well-ordered class. Both experiments show that physical factors other than the amino acid sequence must be (at least in part) responsible for the structural differences between complexes. Hypothetically, this structural variability might relate to subtle differences in sample handling, incubation time, temperature, or the speed of flash-freezing. Regardless, the conformational repertoire of the virus has been sampled in several distinguishable states, thus providing a valuable window into picornavirus dynamics.
DISCUSSION
The anti-C-VHH complexes with expanded poliovirus have given us a much-higher-resolution view, and a deeper understanding, of the conformational repertoire of the expanded-form poliovirus than ever before. This is a welcome development, as it helps shed light on expansion, which is a key step in poliovirus infection, and explains some structural and mutational questions that were raised in previous studies. For convenience, we first focus on the outer surface of the capsid and describe the concerted rearrangement of the VP2 EF loop and its three supports (the GH loop of VP1, the C-terminal extension of VP1, and the C-terminal extension of VP2) and the VHH-dependent rearrangement of the GH loop of VP3. Following this, we focus on the inner surface of the capsid, where the absence of the capsid protein VP4 permits disordering and partial rearrangement of the N terminus of VP1, the N terminus of VP2, and the “bottom” loop of VP2, along with the development of high-density features that the bottom loop of VP2 may be partly responsible for creating. All of these rearrangements have important implications that help to explain previously known poliovirus biology and suggest some topics for future investigation. In this discussion, we refer the location of features within the triangular icosahedral asymmetric unit that is bounded by a particle 5-fold axis and two 3-fold axes (Fig. 5, top). According to this reference frame, the 5-fold axis is north, the 2-fold axis (bisecting the line between the two 3-fold axes) is south, and features to the left and right of the line between the 2-fold and 5-fold axes are west and east, respectively.
The EF loop of VP2 rearranges, and its structural supports become disordered.
In native poliovirus, the EF loop of VP2 (residues 129 to 194) is a large outward-projecting double-loop structure on top of the VP2 beta barrel that forms the blades of what we have referred to as a three-bladed propeller-like feature surrounding the 3-fold axes of the particle (Fig. 6A). As all four anti-C-VHH complexes show, the rearrangement of the VP2 EF loop is correlated with the disordering of three of its main supports that stabilize the loop in the native virus (see below).
Disorder in the VP1 C-terminal extension is required for binding of anti-C VHHs.
In the native virus, the western support for the VP2 EF loop is formed by the C-terminal extension of VP1 (residues 275 to 302) (Fig. 6A), together with the knot-like insertion (residues 53 to 69) in beta strand B of VP3, which does not become disordered. All four of the anti-C VHHs bind to poliovirus on the top surface of the capsid protein VP3, displacing the C-terminal extension of VP1 that normally occupies this surface in the 160S virions, which then becomes disordered. Similar displacement and disordering were seen in ligand-free 135S and 80S poliovirus structures (14, 15). Curiously, this mobility of the carboxyl terminus is not universal among all picornaviruses, as the carboxyl terminus remains unshifted and well ordered in the expanded forms of EV71 (19), CAV16 (20), and HRV2 (21). In poliovirus, the requirement for the carboxyl terminus to detach before these anti-C VHHs can bind helps to explain why these VHHs are unable to bind to N-antigenic virus, where the C-terminal extension is well ordered and covering the VHH binding site.
The C-terminal extension of VP2 becomes disordered.
The C-terminal extension of VP2 (residues 264 to 272) is well ordered in the native virus (Fig. 6A) and forms the support along the southern edge of the VP2 EF loop. However, in conjunction with the EF loop rearrangement (see below), the C-terminal extension detaches from the top surface of VP2, all the way back to its junction with the VP2 beta barrel (at residue 263), and instead falls into the 2-fold hole, forming a weakly ordered structure in some complexes. A similar ordering of the C terminus within the 2-fold hole was seen in 135S particles (15) and was conjectured to interact with ssRNA during its exit from the capsid.
The GH loop of VP1 becomes disordered.
In the native virus, the GH loop of VP1 (residues 207 to 238) forms a long, twisting, two-stranded hairpin that lies along the top surface of VP2, forming the eastern support of the VP2 EF loop (Fig. 6A). The GH loop of VP1, in its native conformation, is known to interact extensively with both Pvr (30) and neutralizing anti-N VHHs (25). Furthermore, mutational studies have shown that this loop is intimately involved with both Pvr interactions and the control of virus expansion (35). In the present VHH complexes, the GH loop of VP1 has become completely disordered. It is likely that the release of the VP1 GH loop is coupled to its loss of contacts with the EF loop of VP2 when the latter loop rearranges extensively (as described below).
Note that in 135S particles (15), the stem of the GH loop remains ordered on top of the VP2 barrel, while the tip of the GH loop was reported to adopt a new conformation, reaching across the quasi-3-fold hole to contact the VP3 GH loop and the exposed N terminus of VP1. In the anti-C-VHH complexes, the failure of the GH loop of VP1 to form a similar structure near the quasi-3-fold hole is unsurprising given that both the VP3 GH loop and the N terminus of VP1 behave differently. Indeed, in at least three of the VHH complexes, the VP3 GH loop makes VHH-specific contacts and adopts a conformation (as described below) that is unsuitable for binding to the VP1 N terminus.
How the EF loop of VP2 becomes rearranged during expansion.
Previously, in lower-resolution studies of expanded poliovirions (14, 15), atomic models of the capsid protein VP2 were difficult to dock into the reconstructions. Unlike the crystallographic VP1 and VP3 models, VP2 did not fit well, as the upward-projecting tip of the VP2 double EF loop (as seen in native models) (Fig. 6A) consistently projected through the upper density contour. This was surprising, as the double loop in the native virus looks very stable and has its own hydrophobic core. As the anti-C-VHH complexes now show us, one of the two EF loops (residues 160 to 174) becomes disordered, which permits the other loop (residues 133 to 142) to fall over into the gap between VP2 and VP1, near the quasi-3-fold hole, that is created during capsid expansion (Fig. 6B). Seen retrospectively, the broad, featureless density envelopes (in previous low-resolution reconstructions of 135S and 80S particles) were consistent with the current higher-resolution model but were not sufficient to specify this model uniquely.
In its new position, the fallen loop has a well-defined conformation that is stabilized by multiple hydrogen bonds, both within the loop and with neighboring proteins (Fig. 5, bottom). Thus, the main chain of the loop folds around the imidazole side chain of His142, which serves as a nucleus for the new conformation. A number of peptide oxygens from the loop are pointed at the imidazole group and contribute hydrogen bonds and other stabilizing interactions. Additionally, the new EF loop conformation is held in place (in the gap between VP2 and VP1) by charged hydrogen bonds to side chains from VP1 (Fig. 5, bottom). In particular, Asn137 of VP2 (and residues close to it) binds to Lys113 and Arg119 (of the VP1 CD helix), to Arg275 (from the highly conserved CPRPP sequence near the VP1 C-terminal extension), and to Ala232 of the VP1 GH loop (distal to which the GH loop becomes disordered). Importantly, this new arrangement of the VP2 EF loop allows VP2 to maintain contacts with the VP1 CD helix region (at the base of the canyon), even as the VP1 and VP2 beta barrels move apart during expansion (see below).
How the VP2 EF loop rearrangement explains mutational data and old structural puzzles.
The details of the conformational switch, involving the EF loop of VP2, sheds light on two additional long-standing puzzles that were raised by previous structural studies.
First, mutations of VP2 His142 are known to affect Pvr binding and the Pvr-triggered 160S-to-135S transition. These mutations increase the ability of the wild-type receptor to induce the 160S-to-135S transition and also allow the mutated viruses to bind to receptors having mutations that abrogate the ability to bind wild-type virus. However, the crystal structure of virions containing an H2142Y mutation (36) was structurally identical to that of the wild-type virus. The VHH complex structures reveal that His142 plays a key role in the structure of the expanded virus and that no other amino acid would fit in the same way. The existence of the mutant phenotype demonstrates that the newly discovered structural role of His142 must be important functionally. This leads us to propose that the mutation functions to promote receptor-mediated structural changes (30) by stabilizing the structure of expanded virions rather than directly destabilizing the structure of the mature 160S virus.
Second, despite sequence differences, for all three serotypes of poliovirus, but not for most other picornaviruses, the peptide bond between Thr140 and Met141 of VP2 is “strained,” having a highly unfavorable phi-psi combination, in the native virus. A rationale for the existence of this unstable-looking conserved strained bond was provided by the structure of the virus-receptor complex (30), where nearby residues participated in direct contacts with the receptor. This observation led to the proposal that the VP2 EF loop might be primed to rearrange once the 160S-to-135S transition is triggered by Pvr at 37°C. The anti-C-VHH complexes now confirm that the portion of the VP2 EF loop that includes the strained peptide bond indeed rearranges. In the new arrangement, it seems plausible that main-chain torsions along the polypeptide may have relaxed, although the current resolution is not sufficient to directly determine these torsions.
How the VP2 EF loop rearrangement causes poliovirus expansion to differ from the expansion of other enteroviruses.
The icosahedrally symmetric arrangements of beta barrels in native and expanded polioviruses are fairly similar to the arrangements seen in crystal structures of native and expanded forms of other enteroviruses, including EV71 (19), CAV16 (20), and HRV2 (21). However, these other enteroviruses are markedly different from poliovirus in the detailed rearrangements that are made to maintain contacts between VP1 and VP2 in the expanded state. In their original comparison of the expanded and native forms of EV71 (19), Wang et al. described a hinge-like motion (of VP1) that flattens the top surface of VP1 and shifts the VP1 GH loop, C-terminal extension, and CD helices relative to the remainder of VP1. This motion allows these three structural elements (which all remain well ordered in EV71) to maintain their contacts with the VP2 EF loop (which also remains unchanged and well ordered) as the VP1 and VP2 beta barrels move apart during virus expansion. Atomic models of the native and expanded states of CAV16 (20) and of HRV2 (21) show similar hinge-like motions during expansion.
However, in poliovirus 135S (15) and 80S (14) particles, and in the VHH complexes with the expanded virus reported here, no similar hinge motion is needed, so rigid-body superposition works well without deforming the VP1 beta barrel. Instead, in poliovirus, both the GH loop and C terminus of VP1 become disordered upon expansion, and (as described above) the VP2 EF loop becomes rearranged in a way that moves part of it closer to the VP1 CD helix, thus providing an alternative mechanism for maintaining the contacts between VP1 and VP2 in the expanded state. It is not yet clear whether this is a mechanism that is unique to poliovirus or whether it occurs in other members of this genus.
The GH loop of VP3 and its relevance to receptor-triggered poliovirus expansion.
The GH loop of VP3 (residues 169 to 188) (Fig. 6) lies in the center of the icosahedral 5-3-3 triangle and directly to the east of the quasi-3-fold hole in the expanded 135S and 80S structures (14, 15). The GH loop of VP3 is particularly interesting for poliovirus, as it is the only viral loop that normally rearranges upon capsid expansion rather than simply becoming disordered or remaining unchanged. Thus, it goes from a coiled conformation (in 160S particles) that keeps the quasi-3-fold hole plugged to an extended conformation (in 135S and 80S particles) that opens the hole. This hole is the exit site for the N terminus of VP1 in 135S particles of poliovirus and CAV16 (15, 20). The significant rearrangement of the VP3 GH loop upon virus expansion provides an explanation for previously reported mutational data, which showed that Gln178 (in this loop) played a key role in the thermostability of the virion and in the ability of the receptor to facilitate expansion (35). In the PVSS7A VHH-poliovirus complex, it is clear that the stems of the GH loop (residues 170 to 175 and 185 to 188) extend radially outward, with residues 170 to 175 forming an extra beta strand (Fig. 7, top). However, in the PVSP17B, PVSS12B, and PVSS10E complexes (which have VHH sequences that are similar to one another) (Fig. 1), the VP3 GH loop is seen in a wholly new conformation (Fig. 7, top and middle). Here, the innermost portion of the GH loop remains in a native-like arrangement (which continues to obstruct the quasi-3-fold hole), while the outermost tip of the loop (residues 182 to 184) shifts radially outward, forming a gentle arc that contacts the BC and FG loops of the VHH (as detailed in “Binding footprints and intermolecular contacts,” above). Because the opening of the quasi-3-fold hole is key to poliovirus dynamics, we use the anti-C-VHH complexes to model receptor-triggered expansion below.
FIG 7.
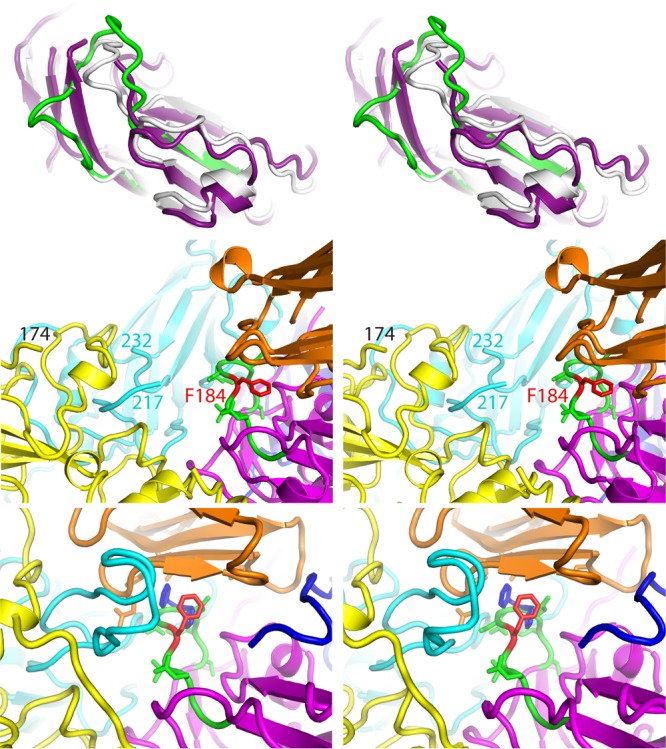
The GH loop of VP3 changes conformation due to both expansion and VHH binding. (Top) A side view of VP3, in a ribbon representation, is shown in a stereo view, strongly depth cued to emphasize changes that occur adjacent to the quasi-3-fold hole. The outer surface of the virus is up, the inner surface is down, and the quasi-3-fold axis is roughly vertical, to the left. The native-like conformation of VP3 (white), as seen in the Pvr-poliovirus complex at low temperature (PDB accession number 3J8F), is shown superimposed on VP3 from the PVSS7A complex (deep purple) and on residues 162 to 194 from the PVSP17B complex (green). In PVSS7A, observe the three beta strands on the left (deep purple), where the third beta strand (residues 170 to 175) results from the uncoiling of the GH loop. This shifts the main chain rightward by as much as 8 Å, which opens the quasi-3-fold hole completely, which is typical of most expanded picornavirus structures (see the text). In contrast, in PVSP17B, the corresponding residues at the bottom of the GH loop (green) remain in the native conformation (and continue to block the quasi-3-fold hole), but the residues at the top of the loop shift outward to contact the VHH. (Middle) Stereo view showing how the quasi-3-fold area rearranges in the PVSP17B complex with expanded poliovirus, as viewed from the 2-fold hole looking toward the 5-fold peak. Observe that the GH loop of VP3 (green [except for Phe184, which is red]) is tucked under the edge of the VHH (orange). The remainder of VP3 is in magenta, VP2 is in yellow, and the two copies of VP1 are in cyan and dark blue. “Cut ends” in the GH loop of VP1 and the EF loop of VP2 (labeled) indicate where these loops become disordered. (Bottom) In a hypothetical docking experiment, shown in a stereo view, VP3 (magenta) from the PVSP17B VHH complex replaces VP3 from the low-temperature complex of poliovirus with Pvr (PDB accession number 3J8F). This positions the upper part of the VP3 GH loop (green) under the lower beta sheet of Pvr (orange). Selected side chains are included. The Phe184 side chain (red) has been rotated by 120° to improve its fit with Pvr. Just behind Phe184, Asp181 and Asp182 (green) occupy the spot that Phe128 and Pro129 (dark blue) would normally bind to in a Pvr-Pvr homodimer (see the text).
The new conformation of the VP3 GH loop is particularly interesting because of its possible relevance to the triggering of viral expansion by its receptor, Pvr. Until recently, it was unclear how Pvr binding at 37°C catalyzes and triggers the irreversible expansion of 160S poliovirions to form 135S particles. However, the Pvr-poliovirus complex at 4°C (30), which is trapped in a native-like state just prior to expansion, sheds some light on the structural mechanism when viewed in light of data from structural studies of Pvr and other nectin family members. Thus, naturally occurring nectin homodimers and heterodimers always include two symmetric or pseudosymmetric binding interactions in which an aromatic side chain from each binding partner becomes bound within a hydrophobic pocket formed on the surface of the other partner (37).
In the Pvr-poliovirus complex, we were struck by the fact that one of these two hydrophobic pockets was empty and positioned directly above the GH loop of VP3 (in its coiled native conformation), which was known to change conformation upon virus expansion (14, 15, 19, 20). We therefore hypothesized that some (as-yet-unknown) conformational rearrangement of the VP3 GH loop might permit either Phe184 or Tyr176 (from the VP3 GH loop) to fill the hydrophobic pocket of Pvr. Hypothetically, the resulting trapping of the GH loop would open the quasi-3-fold hole in the capsid (which is normally plugged by the native coiled conformation of the GH loop) and eliminate some of the intermolecular contacts that normally stabilize the unexpanded virus.
Remarkably, the GH loop conformations that we see in the PVSP17B, PVSS12B, and PVSS10E complexes place Phe184 (along with Asp182 and Ser183) in positions that would be suitable for binding to the underside of Pvr (Fig. 7). Consistent with the above-described hypothesis, the main-chain conformation appears to be roughly shape complementary to the nectin family pocket. This complementarity was established by docking VP3 from the PVSP17B complex, treated as a rigid body, onto VP3 from the Pvr-poliovirus complex (PDB accession number 3J8F) and then looking to see where the superposition would place the rearranged GH loop of VP3 relative to the aromatic binding pocket of the nectin (Fig. 7, bottom). In the Pvr-poliovirus complex, domain 1 of Pvr stands on two wide legs, forming a long arch (a long solvent-filled tunnel) that runs in a south-to-north (2-fold-to-5-fold) direction above the virus surface (30). In the atomic-modeling docking experiment, the top of the VP3 GH loop (residues 182 to 184) runs along the ceiling of the tunnel. Although the Phe184 side chain projects sideward, as we see it, in order to form favorable contacts with the VHH (Fig. 7, middle), it is easy to imagine how the Phe184 side chain could rotate to project outward instead, if Pvr were present to provide a favorable hydrophobic environment for it. Our anti-C-VHH-poliovirus structures thus support our hypothetical triggering mechanism by showing that the required GH loop conformation is a naturally occurring, low-energy conformation that the virus “knows” how to make.
N terminus of VP1.
In the poliovirus complexes with the anti-C-VHHs PVSP17B and PVSS12B, well-resolved density is present for part of the N-terminal extension of VP1 (residues 58 to 71) (Fig. 8, left), which is partly occupied. The observation that these N-terminal residues are ordered is unusual, as this region of VP1 has never been seen in any expanded enterovirus particle when VP4 was absent; that is, normally, N-terminal VP1 ordering is coupled to VP4 ordering. The path of the density differs from that in mature poliovirions (Fig. 6A) and leads from the VP1 core (at the end of the “A helix,” at Arg72), runs along the inner surface of the capsid, and arrives at the edge of the 2-fold hole. Exit of the VP1 N terminus from the capsid, via the 2-fold hole, is known to occur in reversibly breathing virions at 37°C (34). Retraction of the VP1 N terminus back into the capsid interior, via the 2-fold hole, is believed to occur in 80S particles after RNA exit (1, 19). It seems reasonable, therefore, that several copies of the VP1 N terminus (residues 1 to 57) may still exit via the 2-fold hole and remain exposed on the capsid exterior but without forming a consistent external structure that we can see. This would account for the relative abundance of the connecting path on the capsid interior that we see in the PVSP17B and PVSS12B complexes and helps to confirm our developing picture of poliovirus dynamics.
FIG 8.

Newly ordered structures on the inner surface of the capsid. (Left) Across the undersurface of VP3 (magenta), the PVSP17B (and PVSS12B) map includes a continuous, mostly resolved, density feature (black mesh) that has been model built as residues 57 to 71 of VP1 (dark blue), partly because it attaches to the A-alpha helix of VP1 (azure [RGB 0,127,255]). In most structures of expanded picornaviruses, the N-terminal extension is disordered beyond the A-helix. (Middle) On the undersurface of the VP2 beta barrel (yellow), the electron density map for the PVSP17B (and PVSS12B) complex includes a large, roughly cylindrical density feature (black mesh) that the native atomic model does not account for. The orange ribbon is a partly alpha-helical placeholder. The density for the bottom loop of VP2 is weak along its sides and completely disordered between residues 43 and 53 (orange spheres). We postulate that the density mound may result from a rearrangement of the bottom loop (see the text). (Right) VP2 residues Trp38 (red) and Cys61, Cys257, and Cys258 (green) appear to contact the putative helix (orange ribbon). Note that Trp38 was previously implicated in RNA binding (see the text). Additionally, the triangular arrangement of Cys residues and the high level of electron density (gray mesh) suggest that the Cys cluster might be part of a metal binding site.
N terminus of VP2.
In native poliovirus, the N terminus of VP2 (residues 1 to 10) descends inward along the 3-fold axis, extending from a 2-stranded beta hairpin (residues 12 to 28) that is structurally important for holding pentamers together (38). The N terminus itself is not likely to be structurally important in the native virus, because the extent of disorder increases rapidly, further away from the hairpin, and because of a lack of structural conservation among picornaviruses. Although the N terminus of VP2 is similarly disordered in the PVSS7A complex, the main chain can be traced, albeit weakly, in the PVSP17B, PVSS12B, and PVSS10E complexes (Fig. 6B). In these structures, beyond the beta hairpin, the N terminus extends along the inner surface of the capsid, occupying an elongated cavity (one arm of a trefoil-shaped depression) that remains when VP4 and the N-terminal extension of VP1 are no longer present. A similar elongated depression was previously observed in the crystal structure of poliovirus 73S particles (39) (an assembly intermediate in which the immature protein VP0 has not yet been cleaved into VP4 and VP2). Interestingly, at the blind end of the elongated depression, the density corresponding to N-terminal Ser1 of VP2 lies very close (perhaps within hydrogen-bonding distance) to the imidazole side chain of VP2 His195.
Previously, both His195 and the elongated depression were proposed to play roles in the autocatalytic maturation cleavage of VP0 during virus assembly (which is a final step that makes assembly irreversible). Thus, the essential role of His195 was first suggested by its proximity to the scissile bond in uncleaved VP0 (39) and was later confirmed by mutational studies (40). The importance of the elongated depression was suggested by its shape similarity to an analogous cavity in bean pod mottle virus (41) that holds well-ordered RNA. Hypothetically, poliovirus RNA in an analogous position would lie close enough to the scissile bond to participate in a water-bridged general-base-like mechanism (see references 39 and 40 for details).
The anti-C-VHH complexes now suggest the presence of a weak binding site for the flexible N terminus of VP2, which is a cleavage product, on the inner surface of the capsid. Note, however, that the location and orientation of the VP2 N terminus (along the depression) are unlike those of the scissile peptide (across the depression). This suggests that the position of Ser1 (that we now observe) might recapitulate some step in the rearrangement of newly cleaved termini rather than pertaining to the mechanism of autocatalysis per se.
VP2 bottom loop.
In native poliovirus, the VP2 “bottom loop” (residues 26 to 55) (Fig. 6A) is well ordered and wraps all the way around the bottom edges of the VP2 beta barrel, forming a long and elaborate connection between the N-terminal hairpin (residues 12 to 28) and the VP2 core (which starts its “A-helix” at residue 56). However, in all four anti-C-VHH-poliovirus complexes, the tip section of the bottom loop (residues 42 to 53) has become disordered (Fig. 6B). This loop tip disordering is similar to what was observed in the high-resolution crystal structure of (nonexpanded) 73S native-antigenic empty capsids, which similarly lack RNA and natively ordered VP4 (39). Additionally, in the PVSP17B and PVSS12B complexes, the “sides” of the bottom loop (especially residues 54 to 58) appear at a distinctly lower contour level than the remainder of the VP2 beta barrel does, although the main chain remains traceable. We therefore suspect that the bottom loop is dynamic, having a tendency to unravel, and have assigned partial occupancies to these residues.
Mound of inward-projecting density.
The only major density feature that we cannot account for confidently is the inward-projecting mound of density that develops on the bottom surface of VP2. This feature appears as a pair of moderately strong 2-fold-symmetry-related peaks on either side of the 2-fold hole, each roughly large enough (in cross section) to enclose an alpha helix (Fig. 8, middle). These peaks are much larger and stronger in the PVSP17B and PVSS12B complexes, which are also the complexes with the greatest number of newly ordered residues on the inner surface (Table 1). Correspondingly, the complexes of PVSS7A and PVSS10E lack both the mounds and the extra ordered residues (despite the near identity of the sequence of PVSS10E with those of PVSP17B and PVSS12B) (Fig. 1). Recall that similar mounds (and a lack of mounds) were previously seen in the two main structural classes of the poliovirus 80S particle (14). It is not yet clear what role this newly formed density structure might play or at what stage of the poliovirus life cycle. Nevertheless, we find it very intriguing that the new density mound covers both Trp38 and, very close to it, a cluster of three Cys side chains (residues 61, 257, and 258), all of which are on the bottom surface of native VP2 (Fig. 8, right). Trp38 has long been assumed to be a binding site for stacked RNA bases, in the native virus, based on the appearance of planar electron density features that lie parallel to the aromatic side chain in crystal structures of both poliovirus (42) and other picornaviruses (43). In the PVSP17B and PVSS12B complexes, the three Cys side chains now form a small equilateral triangle, with an elevated density level at its center, which is suggestive of a metal binding site. As the cysteines are arranged somewhat differently in the native virus, we presume that the putative metal site could develop only after capsid expansion.
Which residues form the density mound?
Although it is not yet definitive, two lines of evidence strongly suggest that the density mound might include some rearranged residues from the nearby VP2 bottom loop (tip and side, residues 43 to 57). First, we observe that the ordered residues of the bottom loop appear to have stronger density in PVSS7A and PVSS10E, where the mound is much weaker, than in PVSP17B and PVSS12B, where the mound is stronger. Second, we observe that the bottom-loop density level along the side (i.e., for residues 54 to 58) is notably weaker than the mound density is in one subclass (out of five) of the PVSP17B population (called class 2) that was determined automatically by Relion (31) in the initial stages of structure solution. Although that class average is based on only 20% of the PVSP17B population, and is worse in overall resolution (5.0 Å by FSC) than the full PVSP17B reconstruction (at a 4.3-Å resolution), its density for the mound is stronger and better defined, which suggests greater occupancy and homogeneity. Thus, the apparent shift of density from the bottom loop to the mound is exactly what we would expect if rearrangement of the loop polypeptide were required for mound formation and provided at least part of the material.
We report the existence of this “mystery density” feature in the hope that future research will explain why poliovirus 80S particles know how to form it. It remains to be determined whether an analogous rearrangement occurs at some other stage of the poliovirus life cycle, where its role might be biologically important. Its location on the inner surface of the capsid, right next to the 2-fold hole, and its coverage of the Trp and Cys residues invite speculations that, conceivably, the new structural feature might be involved in either catalysis (for example, VP0 cleavage), RNA binding, or RNA unwinding. Thus, during RNA release, the unwinding of secondary structures in the genome (44) is known to be slow (45) and might require catalysis.
Opening of the quasi-3-fold hole and its role in expansion.
In the native-virus-to-135S-particle transition, the N-terminal extension of VP1 must move from an opening at the 2-fold axis (where it has been shown to be located in virus incubated at 37°C in the absence of a receptor [34]) to a newly opened hole at the quasi-3-fold axis. In the native virus, the quasi-3-fold hole is obstructed by a three-layered stack of polypeptide contacts, arranged from bottom to top. The anti-C-VHH complexes with poliovirus shed light on the changes in these three layers that open the quasi-3-fold hole. In forming 135S particles, this opening allows the N-terminal extension of VP1 to first thread between the VP1, VP2, and VP3 cores during its passage from the hole at the 2-fold axis to the new hole at the quasi-3-fold axis and subsequently lock the N-terminal extension of VP1 into place at the quasi-3-fold axis.
Outermost layer.
In the native virus, the outermost layer of the three-layer plug is formed around the aromatic side chain of VP3 Phe184, which belongs to the outer portion of the VP3 GH loop. Thus, in the native virus, Phe184 maintains hydrophobic contacts with the carboxyl end of the GH loop of VP1 at Leu234 and Ala232 and with the amino end of the loop at VP1 Tyr209. Nearby, the VP3 GH loop at Ile180 contacts the EF loop of VP1, around Gly163. In our previous structural study of the low-temperature poliovirus complex with Pvr (30), we hypothesized that the rearrangement of the VP3 GH loop (at 37°C), and subsequent virus expansion, might be triggered by the binding of Phe184 to the underside of Pvr (in a hydrophobic pocket that the nectin family normally uses to bind aromatic side chains of its binding partners). Just as in the anti-C-VHH complexes, VP3 Phe184 would be pulled outward, away from the quasi-3-fold plug, which would eliminate much of the outermost layer of contacts. Indeed, in the VHH complexes, we can see that the shift of Phe184 and capsid expansion are accompanied by a disordering of much of the GH loop of VP1 (Fig. 7, middle) and by a 2.5- to 3-Å shift of the EF loop of VP1 (around residue 163) away from the quasi-3-fold axis. The role of Phe184 in regulating the structure of the native virus is also suggested by the direct contacts that are formed between the top of the VP3 GH loop (around Ile180) and Ser91 of VP3. Notably, the Ser-to-Phe mutation at VP3 residue 91 is known to be critical for the temperature-sensitive phenotype and for attenuation of neurovirulence in the Sabin strain of serotype 3 (46).
Middle layer.
In the middle layer, in the native virus, the lower portion of the VP3 GH loop (around residues 171 to 177) follows a C-shaped path that obstructs the quasi-3-fold hole (Fig. 7, top). This conformation allows it to maintain its contacts (across the hole) with the carboxyl end of the GH loop of VP1 (around residue 202) and with the GH loop of VP2 (around residue 216). In the PVSP17B, PVSS12B, and PVSS10E complexes, this lower portion of the VP3 GH loop behaves very similarly. As a result, even though VP1 (at residue 202) shifts away from the quasi-3-fold axis, it shifts by only 2 Å, which leaves insufficient space for the N terminus of VP1 to be inserted. In contrast, in the PVSS7A complex, where the quasi-3-fold hole is open, both stems of the GH loop of VP3 (residues 170 to 175 and 185 to 188) have rearranged to extend outward, with residues 170 to 175 forming a new beta strand (Fig. 7, top). This rearrangement shifts the VP3 main chain (around residue 173) away from the quasi-3-fold axis by as much as 8 Å. This shift, together with the 2-Å shift in VP1, creates sufficient space for the VP1 N terminus to insert (where it is then locked into place in 135S particles through interactions with the GH loops of VP3 and VP1 [15]).
Innermost layer.
In the native virus, the innermost layer of the quasi-3-fold plug involves contacts of the VP3 N-terminal extension (at residues 32 to 34) with the tip of the VP2 bottom loop (at residue 46) and with the N-terminal extension of VP1 (around residue 67). In anti-C-VHH-poliovirus complexes and in 73S native empty capsids, both of which lack VP4 and RNA, we see that the N-terminal extension of VP1 has become disordered (or rearranged) and that the VP2 bottom-loop tip is disordered (39). Thus, we see evidence for a concerted conformational switch that leaves the innermost layer of the quasi-3-fold plug open prior to VP0 cleavage, closed in the mature virus, and open once again during receptor-triggered expansion of the capsid.
Conclusion.
Research on antipoliovirus VHHs was initiated in the hope of producing therapeutically useful compounds and/or quality control reagents for the production of conventional vaccines. Our previous structural studies of poliovirus complexes with neutralizing anti-N VHHs proved to be informative on the issue of how VHH-based neutralization was accomplished. We showed that those anti-N VHHs interacted directly with the virus's own structural mechanisms for controlling stability and shed some light on the stabilization process itself. In contrast, our current study of poliovirus complexes with anti-C-VHHs has been useful primarily for producing a dramatic improvement in our visualization of poliovirus expansion. This study has sampled the conformational repertoire of the 80S particle, provided information on various stages of the poliovirus life cycle, and suggested directions for future virology research.
MATERIALS AND METHODS
Virus.
The poliovirus type 1 Mahoney strain was grown and purified as described in detail previously (25). The Mahoney strain used differs from the reference Mahoney strain (NCBI GenBank accession number V01149.1) in that it contains the following two substitutions in the capsid proteins: F123S in VP3 and L228I in VP1.
VHHs.
The VHHs used in this study were originally obtained from a dromedary that was immunized with poliovirus type 1. Through the collection of lymphocytes, production of a cDNA library, and phage display panning, several poliovirus binding VHHs were selected. One panel of VHHs was found to bind and neutralize N-antigen, and a second panel selectively bound H-antigen and had no neutralizing activity (23). Four VHHs with the lowest capturing titer (highest affinity) for H-antigen were selected from this panel. Detailed methods for the selection, production, and purification of the VHHs were reported in previous papers (6, 23, 26). The amino acid sequences for the four VHHs are shown in Fig. 1.
Assessment of the ability of VHH to bind to 160S, 135S, and 80S poliovirions.
Native 160S poliovirions at 0.5 mg/ml in low-salt buffer (20 mM Tris-HCl [pH 7.5], 2 mM CaCl2) were either heated at 50°C for 3 min to prepare 135S virions, heated at 56°C for 10 min to prepare 80S virions, or left unheated. Conversion was verified visually by electron microscopy using a uranyl acetate stain. Ten microliters of each poliovirus solution was mixed with 10 μl of VHH (1 mg/ml), and all samples were incubated for 45 min at 25°C. Next, 30 μl of Ni-nitrilotriacetic acid (NTA)-agarose beads (Qiagen) in Tris buffer (50 mM Tris buffer, 300 mM NaCl [pH 8]) containing 20 mM imidazole was added to each sample, and the mixture was incubated for an additional 45 min and washed four times with 60 mM imidazole in Tris buffer. Poliovirus and VHH proteins that remained attached to the beads were then eluted with 250 mM imidazole in Tris buffer and visualized as characteristic bands by SDS-PAGE.
Negative staining of poliovirions.
Three-microliter droplets of each poliovirus (160S, 135S, and 80S) solution were placed onto glow-discharged, 100-mesh, Formvar-carbon-coated grids (EMS), and the excess liquid was removed by blotting with filter paper (Whatman #1) before staining with 1% uranyl acetate. The grids were imaged at 80 kV with a JEOL 1200EX electron microscope equipped with an AMT 2k charge-coupled-device (CCD) camera.
Preparation of VHH-expanded virus complexes.
Ten microliters of VHH (1 mg/ml in 25 mM Tris buffer–137 mM NaCl [pH 7.2]) was added to 10 μl of poliovirus (0.9 mg/ml) in phosphate-buffered saline (PBS) (pH 7.0) at 4°C. This mixture was then heated to 50°C for 5 min prior to freezing in liquid ethane for cryo-EM studies.
Cryo-electron microscopy.
For freezing, a 3-μl droplet of the heated mixture was placed onto a glow-discharged perforated carbon support (C-flat 1.2/1.3; Protochips), and the excess liquid was removed by blotting with filter paper (Whatman #1) before being plunged into liquid ethane slush and stored under liquid nitrogen until microscopic examination. The grids were imaged at 300 kV with a Polara G2 electron microscope (FEI Co.) using a K2 Summit direct detector (Gatan Inc.) in the superresolution mode, such that the final superresolution pixel size was either 0.8245 or 0.986 Å. Twenty-four frames over a span of 10 to 12 s were recorded for each acquisition, using a dose rate of 8 electrons/pixel/s. The frames were aligned and summed by using dosef (47). We discarded the first two frames, as they exhibited the most drift. The particles were picked from the aligned average image by using e2boxer.py (48), and the contrast transfer function (CTF) estimates were obtained with ctffind3 (49). Several rounds of two-dimensional (2D) and 3D classification were carried out with Relion1.3 (31), removing bad particles or smaller inconsistent classes of particles. One of the class averages in the resulting 3D classification was used as a reference for the final iterative alignment routine, which was done by using GeFrealign (50) and Frealign (v9.09) (32). Here, the Fourier terms used for refinement were restricted to include only the information that showed a high correlation between two half-set reconstructions, to avoid the introduction of model bias (51). A correlation value of 0.7 was used as a typical threshold. B-factor sharpening was carried out on the final reconstruction by using the default settings of Frealign.
Construction of the models.
Atomic models have been constructed and refined to fit each of the maps (using COOT [27], SPDBV [52], and Refmac5 [29]), using the Fourier transform of a portion of the reconstruction (both amplitudes and phases) as refinement standards, with symmetry-related neighboring proteins being present, icosahedral symmetry being strongly enforced, and idealized stereochemical standards being applied as restraints. Portions of each model where the density exhibited significant differences from structural homologs (as identified by using VAST [http://structure.ncbi.nlm.nih.gov/Structure/VAST/vastsearch.html]) were subjected to an all-atom refinement, while the core regions that were in good agreement with the homologous model were restrained to resemble rigid-body docking. The residues included in the core regions of the VHHs and VP1, VP2, and VP3 from the virus are listed in Table 1. The resolution limits and statistics for the atomic model refinement of each of the complexes are shown in Table 2.
Accession number(s).
Cryo-EM maps of the complexes of poliovirus type 1 with each of the four anti-C VHHs and their refined atomic coordinates can be found in the Protein Data Bank and the Electron Microscopy Data Bank under the following accession numbers, respectively: 5KU0 and EMD-8285 for PVSP17B, 5KTZ and EMD-8284 for PVSS12B, 5KWL and EMD-8277 for PVSS10E, 5KU2 and EMD-8286 for PVSS7A, and EMD-8292 for expanded poliovirus.
ACKNOWLEDGMENTS
This work was supported by NIH grant AI020566 (to J.M.H.), by Fonds Wetenschappelijk Onderzoek (to L.S.), and by the Alexander von Humboldt-Stiftung (to M.S.).
REFERENCES
- 1.Levy H, Bostina M, Filman DJ, Hogle JM. 2010. Cell entry: a biochemical and structural perspective, p 87–107. In Ehrenfeld E, Domingo E, Roos R (ed), The picornaviruses. ASM Press, Washington, DC. [Google Scholar]
- 2.Racaniello VR. 2013. Picornaviridae: the viruses and their replication, p 453–489. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Hogle JM, Chow M, Filman DJ. 1985. Three-dimensional structure of poliovirus at 2.9 A resolution. Science 229:1358–1365. doi: 10.1126/science.2994218. [DOI] [PubMed] [Google Scholar]
- 4.Chow M, Newman JFE, Filman D, Hogle JM, Rowlands DJ, Brown F. 1987. Myristylation of picornavirus capsid protein VP4 and its structural significance. Nature 327:482–486. doi: 10.1038/327482a0. [DOI] [PubMed] [Google Scholar]
- 5.Strauss M, Levy HC, Bostina M, Filman DJ, Hogle JM. 2013. RNA transfer from poliovirus 135S particles across membranes is mediated by long umbilical connectors. J Virol 87:3903–3914. doi: 10.1128/JVI.03209-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schotte L, Thys B, Strauss M, Filman DJ, Rombaut B, Hogle JM. 2015. Characterization of poliovirus neutralization escape mutants of single-domain antibody fragments (VHHs). Antimicrob Agents Chemother 59:4695–4706. doi: 10.1128/AAC.00878-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mendelsohn CL, Wimmer E, Racaniello VR. 1989. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 56:855–865. doi: 10.1016/0092-8674(89)90690-9. [DOI] [PubMed] [Google Scholar]
- 8.Biederer T. 2006. Bioinformatic characterization of the SynCAM family of immunoglobulin-like domain-containing adhesion molecules. Genomics 87:139–150. doi: 10.1016/j.ygeno.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 9.Ikeda W, Kakunaga S, Takekuni K, Shingai T, Satoh K, Morimoto K, Takeuchi M, Imai T, Takai Y. 2004. Nectin-like molecule-5/Tage4 enhances cell migration in an integrin-dependent, Nectin-3-independent manner. J Biol Chem 279:18015–18025. doi: 10.1074/jbc.M312969200. [DOI] [PubMed] [Google Scholar]
- 10.Tsang SK, McDermott BM, Racaniello VR, Hogle JM. 2001. Kinetic analysis of the effect of poliovirus receptor on viral uncoating: the receptor as a catalyst. J Virol 75:4984–4989. doi: 10.1128/JVI.75.11.4984-4989.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belnap DM, Filman DJ, Trus BL, Cheng N, Booy FP, Conway JF, Curry S, Hiremath CN, Tsang SK, Steven AC, Hogle JM. 2000. Molecular tectonic model of virus structural transitions: the putative cell entry states of poliovirus. J Virol 74:1342–1354. doi: 10.1128/JVI.74.3.1342-1354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fenwick ML, Cooper PD. 1962. Early interactions between poliovirus and ERK cells. Some observations on the nature and significance of the rejected particles. Virology 18:212–223. [DOI] [PubMed] [Google Scholar]
- 13.De Sena J, Mandel B. 1977. Studies on the in vitro uncoating of poliovirus. II. Characteristics of the membrane-modified particle. Virology 78:554–566. [DOI] [PubMed] [Google Scholar]
- 14.Levy HC, Bostina M, Filman DJ, Hogle JM. 2010. Catching a virus in the act of RNA release: a novel poliovirus uncoating intermediate characterized by cryo-electron microscopy. J Virol 84:4426–4441. doi: 10.1128/JVI.02393-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butan C, Filman DJ, Hogle JM. 2014. Cryo-electron microscopy reconstruction shows poliovirus 135S particles poised for membrane interaction and RNA release. J Virol 88:1758–1770. doi: 10.1128/JVI.01949-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fricks CE, Hogle JM. 1990. Cell-induced conformational change of poliovirus: externalization of the amino terminus of VP1 is responsible for liposome binding. J Virol 64:1934–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tuthill TJ, Bubeck D, Rowlands DJ, Hogle JM. 2006. Characterization of early steps in the poliovirus infection process: receptor-decorated liposomes induce conversion of the virus to membrane-anchored entry-intermediate particles. J Virol 80:172–180. doi: 10.1128/JVI.80.1.172-180.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Danthi P, Tosteson M, Li QH, Chow M. 2003. Genome delivery and ion channel properties are altered in VP4 mutants of poliovirus. J Virol 77:5266–5274. doi: 10.1128/JVI.77.9.5266-5274.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Peng W, Ren J, Hu Z, Xu J, Lou Z, Li X, Yin W, Shen X, Porta C, Walter TS, Evans G, Axford D, Owen R, Rowlands DJ, Wang J, Stuart DI, Fry EE, Rao Z. 2012. A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71. Nat Struct Mol Biol 19:424–429. doi: 10.1038/nsmb.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren J, Wang X, Hu Z, Gao Q, Sun Y, Li X, Porta C, Walter TS, Gilbert RJ, Zhao Y, Axford D, Williams M, McAuley K, Rowlands DJ, Yin W, Wang J, Stuart DI, Rao Z, Fry EE. 2013. Picornavirus uncoating intermediate captured in atomic detail. Nat Commun 4:1929. doi: 10.1038/ncomms2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garriga D, Pickl-Herk A, Luque D, Wruss J, Caston JR, Blaas D, Verdaguer N. 2012. Insights into minor group rhinovirus uncoating: the X-ray structure of the HRV2 empty capsid. PLoS Pathog 8:e1002473. doi: 10.1371/journal.ppat.1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N, Hamers R. 1993. Naturally occurring antibodies devoid of light chains. Nature 363:446–448. doi: 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- 23.Thys B, Schotte L, Muyldermans S, Wernery U, Hassanzadeh-Ghassabeh G, Rombaut B. 2010. In vitro antiviral activity of single domain antibody fragments against poliovirus. Antiviral Res 87:257–264. doi: 10.1016/j.antiviral.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 24.Schotte L, Strauss M, Thys B, Halewyck H, Filman DJ, Bostina M, Hogle JM, Rombaut B. 2014. Mechanism of action and capsid-stabilizing properties of VHHs with an in vitro antipolioviral activity. J Virol 88:4403–4413. doi: 10.1128/JVI.03402-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strauss M, Schotte L, Thys B, Filman DJ, Hogle JM. 2016. Five of five VHHs neutralizing poliovirus bind the receptor-binding site. J Virol 90:3496–3505. doi: 10.1128/JVI.03017-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thys B, Saerens D, Schotte L, De Bleeser G, Muyldermans S, Hassanzadeh-Ghassabeh G, Rombaut B. 2011. A simple quantitative affinity capturing assay of poliovirus antigens and subviral particles by single-domain antibodies using magnetic beads. J Virol Methods 173:300–305. doi: 10.1016/j.jviromet.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. 2011. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strauss M, Filman DJ, Belnap DM, Cheng N, Noel RT, Hogle JM. 2015. Nectin-like interactions between poliovirus and its receptor trigger conformational changes associated with cell entry. J Virol 89:4143–4157. doi: 10.1128/JVI.03101-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scheres SH. 2012. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grigorieff N. 2007. FREALIGN: high-resolution refinement of single particle structures. J Struct Biol 157:117–125. doi: 10.1016/j.jsb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 33.Li Q, Yafal AG, Lee YM-H, Hogle J, Chow M. 1994. Poliovirus neutralization by antibodies to internal epitopes of VP4 and VP1 results from reversible exposure of these sequences at physiological temperature. J. Virol 68:3965–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin J, Lee LY, Roivainen M, Filman DJ, Hogle JM, Belnap DM. 2012. Structure of the Fab-labeled “breathing” state of native poliovirus. J Virol 86:5959–5962. doi: 10.1128/JVI.05990-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chow M, Basavappa R, Hogle JM. 1997. The role of conformational transitions in poliovirus pathogenesis, p 157–186. In Chiu W, Burnette R, Garcea R (ed), Structural biology of viruses. Oxford University Press, Oxford, United Kingdom. [Google Scholar]
- 36.Wien MW, Curry S, Filman DJ, Hogle JM. 1997. Structural studies of poliovirus mutants that overcome receptor defects. Nat Struct Biol 4:666–674. doi: 10.1038/nsb0897-666. [DOI] [PubMed] [Google Scholar]
- 37.Harrison OJ, Vendome J, Brasch J, Jin X, Hong S, Katsamba PS, Ahlsen G, Troyanovsky RB, Troyanovsky SM, Honig B, Shapiro L. 2012. Nectin ectodomain structures reveal a canonical adhesive interface. Nat Struct Mol Biol 19:906–915. doi: 10.1038/nsmb.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Filman DJ, Syed R, Chow M, Macadam AJ, Minor PD, Hogle JM. 1989. Structural factors that control conformational transitions and serotype specificity in type 3 poliovirus. EMBO J 8:1567–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Basavappa R, Syed R, Flore O, Icenogle JP, Filman DJ, Hogle JM. 1994. Role and mechanism of the maturation cleavage of VP0 in poliovirus assembly: structure of the empty capsid assembly intermediate at 2.9Å resolution. Protein Sci 3:1651–1669. doi: 10.1002/pro.5560031005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hindiyeh M, Li QH, Basavappa R, Hogle JM, Chow M. 1999. Poliovirus mutants at histidine 195 of VP2 do not cleave VP0 into VP2 and VP4. J Virol 73:9072–9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Z, Stauffacher C, Li Y, Schmidt T, Bomu W, Kamer G, Shanks M, Lomonossoff G, Johnson JE. 1989. Protein-RNA interactions in an icosahedral virus at 3.0 Å resolution. Science 245:154–159. doi: 10.1126/science.2749253. [DOI] [PubMed] [Google Scholar]
- 42.Flore O, Fricks CE, Filman DJ, Hogle JM. 1990. Conformational changes in poliovirus assembly and cell entry, p 429–438. In Hogle JM. (ed), Seminars in virology, 1st ed WB Saunders Co, Philadelphia, PA. [Google Scholar]
- 43.Hadfield AT, Lee W, Zhao R, Oliveira MA, Minor I, Rueckert RR, Rossmann MG. 1997. The refined structure of human rhinovirus 16 at 2.15 A resolution: implications for the viral life cycle. Structure 5:427–441. doi: 10.1016/S0969-2126(97)00199-8. [DOI] [PubMed] [Google Scholar]
- 44.Brandenburg B, Lee LY, Lakadamyali M, Rust MJ, Zhuang X, Hogle JM. 2007. Imaging poliovirus entry in live cells. PLoS Biol 5:e183. doi: 10.1371/journal.pbio.0050183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang Y, Hogle JM, Chow M. 2000. Is the 135S poliovirus particle an intermediate during cell entry? J Virol 74:8757–8761. doi: 10.1128/JVI.74.18.8757-8761.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Westrop GD, Evans DMA, Minor PD, Magrath D, Schild GC, Almond JW. 1986. Investigation of the molecular basis of attenuation in the Sabin type 3 vaccine using novel recombinant polioviruses constructed from infectious cDNA, p 53–60. In Rowlands DJ, Mahy BWJ, Mayo M (ed), The molecular biology of the positive strand RNA viruses. Academic Press, London, United Kingdom. [Google Scholar]
- 47.Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. 2013. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods 10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. 2007. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 49.Mindell JA, Grigorieff N. 2003. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 142:334–347. doi: 10.1016/S1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 50.Li X, Grigorieff N, Cheng Y. 2010. GPU-enabled FREALIGN: accelerating single particle 3D reconstruction and refinement in Fourier space on graphics processors. J Struct Biol 172:407–412. doi: 10.1016/j.jsb.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scheres SH, Chen S. 2012. Prevention of overfitting in cryo-EM structure determination. Nat Methods 9:853–854. doi: 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guex N, Peitsch MC. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]