

ABSTRACT
The dependence of adenovirus on the host pre-RNA splicing machinery for expression of its complete genome potentially makes it vulnerable to modulators of RNA splicing, such as digoxin and digitoxin. Both drugs reduced the yields of four human adenoviruses (HAdV-A31, -B35, and -C5 and a species D conjunctivitis isolate) by at least 2 to 3 logs by affecting one or more steps needed for genome replication. Immediate early E1A protein levels are unaffected by the drugs, but synthesis of the delayed protein E4orf6 and the major late capsid protein hexon is compromised. Quantitative reverse transcription-PCR (qRT-PCR) analyses revealed that both drugs altered E1A RNA splicing (favoring the production of 13S over 12S RNA) early in infection and partially blocked the transition from 12S and 13S to 9S RNA at late stages of virus replication. Expression of multiple late viral protein mRNAs was lost in the presence of either drug, consistent with the observed block in viral DNA replication. The antiviral effect was dependent on the continued presence of the drug and was rapidly reversible. RIDK34, a derivative of convallotoxin, although having more potent antiviral activity, did not show an improved selectivity index. All three drugs reduced metabolic activity to some degree without evidence of cell death. By blocking adenovirus replication at one or more steps beyond the onset of E1A expression and prior to genome replication, digoxin and digitoxin show potential as antiviral agents for treatment of serious adenovirus infections. Furthermore, understanding the mechanism(s) by which digoxin and digitoxin inhibit adenovirus replication will guide the development of novel antiviral therapies.
IMPORTANCE Despite human adenoviruses being a common and, in some instances, life-threating pathogen in humans, there are few well-tolerated therapies. In this report, we demonstrate that two cardiotonic steroids already in use in humans, digoxin and digitoxin, are potent inhibitors of multiple adenovirus species. A synthetic derivative of the cardiotonic steroid convallotoxin was even more potent than digoxin and digitoxin when tested with HAdV-C5. These drugs alter the cascade of adenovirus gene expression, acting after initiation of early gene expression to block viral DNA replication and synthesis of viral structural proteins. These findings validate a novel approach to treating adenovirus infections through the modulation of host cell processes.
KEYWORDS: RNA processing, adenovirus, cardiotonic steroids, therapy
INTRODUCTION
Adenovirus infections are common in the human population, as indicated by the high seroprevalence of antiadenovirus antibodies (ranging from 80 to 90% in sub-Saharan Africa to 30 to 70% in Europe and North America) (1). At least 70 types of human adenoviruses (HAdVs) have been recognized to date and classified into seven species (A to G) based on their genetic relatedness (1, 2). Although most adenovirus infections are likely to be subclinical, infection can be manifested by relatively mild upper respiratory tract disease as well as more serious bronchiolitis and pneumonia and by diarrhea and conjunctivitis. Some adenovirus types infect specific tissues, whereas other types can infect multiple sites (1). For example, HAdV-D8 and HAdV-D19 predominantly infect the eye, causing serious epidemic keratoconjunctivitis (EKC) that can lead to visual impairment (3). HAdV-C5 typically causes mild upper respiratory tract infections but can spread to cause disseminated disease in immunocompromised hosts. Certain species B adenoviruses, including types 3, 7, and 21, tend to cause more severe respiratory disease, even in immunocompetent hosts. Case fatality rates for disseminated disease and for severe pneumonia can be more than 50% (4, 5). At present, there is no approved treatment for adenoviral infection. Cidofovir (a deoxycytosine monophosphate analog) and brincidofovir (CMX001) (a lipid ester form of cidofovir), which target the viral DNA polymerase, have shown success in the clinic but are associated with significant toxicity to the kidney and gastrointestinal tract, respectively (6–10). Consequently, new strategies are required to control these infections.
The cardiotonic steroids digoxin and digitoxin have been used to treat heart failure for more than 200 years, initially as powdered leaves of digitalis (foxglove) plants and later in purified form (11–14). Over the past few decades, their potential for treatment of cancer has been recognized, with studies showing that patients taking these drugs have a significant reduction in relapse after the initial cancer diagnosis (15–18). Their potential as antiviral agents was recognized by Hartley et al. (19), who reasoned that ionic changes within the cell would prevent replication of selected viruses, including herpesvirus and adenovirus. Here, we expand on the initial observation (19) that showed inhibition of adenovirus replication in a plaque reduction assay with digoxin. Both digoxin and digitoxin block the Na+/K+ ATPase (NKA) on the cell surface (20–24). Treatment with either drug increases intracellular levels of Na+, which subsequently increases intracellular Ca2+ levels due to effects on the Na+/Ca2+ exchanger (NCX) used to efflux Ca2+ (21, 23, 25). Although effects of cardiotonic steroids previously were attributed to alterations in intracellular K+ and Ca2+ levels, it is now recognized that the NKA is an important signaling molecule for regulation of cell function (26, 27). Binding of cardiotonic steroids to the NKA induces multiple signaling cascades through modulation of neighboring tyrosine kinases, including src, ultimately affecting gene expression (15, 24, 27).
Signaling pathways can affect RNA splicing by modulating the activity of splicing factors (28). Evidence linking cardiotonic steroids and splicing came from screens for small-molecule modulators of RNA splicing (29). The cardiotonic steroids induce alterations of a subset of SR proteins (SRSF3 and Tra2β) that are known to regulate RNA processing and thereby affect a limited subset of host splicing events (30). In the context of HIV-1, cardiotonic steroids are potent inhibitors of virus replication due to the modulation of viral RNA processing. Specifically, they enhance splicing of the primary viral transcript, resulting in loss of RNAs encoding the HIV-1 structural proteins (31). Repression of HIV-1 replication by cardiotonic steroids depends on NKA inhibition but does not require the associated increase in levels of intracellular Ca2+, consistent with inhibition mediated by cellular signaling pathways that modulate host factors involved in splicing (32).
Adenoviruses rely heavily on host RNA splicing machinery to express the full complement of viral proteins. Indeed, pre-mRNA splicing was first discovered in adenovirus transcripts (33, 34). The adenovirus genome is 30 to 36 kb in length (35) but generates more than 50 proteins in a regulated fashion through a combination of transcriptional and posttranscriptional control (36, 37). Five early transcription units (E1A, E1B, E2, E3, and E4) and two intermediate transcription units (protein IX and IVa2), each with its own promoter, are used for expression of 34 mRNAs generated largely by alternative splicing of primary transcripts (38). Late gene expression begins with the onset of genome replication (39). From that time onwards, 17 viral proteins are all produced from a single major late promoter through alternative polyadenylation and splicing of a common transcript (36, 38, 40). Given the dependence of adenoviruses on host RNA splicing machinery, it seemed reasonable that small-molecule modulators of RNA splicing might be effective antiviral agents in this context. Accordingly, this study was initiated to examine the effects of digoxin and digitoxin on adenovirus replication, beyond the plaque reduction data of Hartley et al. (19).
RESULTS
Digoxin and digitoxin inhibit adenovirus replication.
We examined the extent to which digoxin and digitoxin affect adenovirus replication in A549 cells, given that adenovirus replication depends on regulation of RNA processing and that cardiotonic steroids alter RNA splicing (29) and potently inhibit HIV-1 replication (31) by altering viral RNA processing. Initial experiments screened digoxin and digitoxin, at a range of concentrations, for their ability to inhibit replication of HAdV-C5 in A549 cells. Each drug, when added at the end of the adsorption period, showed a concentration-dependent reduction in the number of infected cells, as determined by immunodetection of hexon (major capsid protein) at 24 h postinfection (p.i.), with no apparent damage to the cells (Fig. 1). In subsequent experiments to measure the yield of infectious progeny virus, each drug reduced virus yield by at least 2 logs (Fig. 2). Furthermore, antiviral activity was observed for adenoviruses from four different species (HAdV-A31, HAdV-B35, HAdV-C5, and a species D isolate from a case of conjunctivitis). The two drugs showed similar dose responses, with maximum inhibition at a concentration of 100 to 150 nM. The cellular toxicity of digoxin and digitoxin was assessed using alamarBlue to reflect metabolic activity and using viable counts with trypan blue (Fig. 3). Metabolic activity after 24 h of exposure to 100 to 150 nM digoxin and digitoxin was reduced to 75 to 80% and 55 to 60%, respectively, of that of untreated cells. Despite reduced metabolic activity, there was no apparent effect on cell viability by trypan blue exclusion, and cell counts were comparable to those for untreated cells.
FIG 1.

Initial screening identifies digoxin and digitoxin as potential adenovirus inhibitors. A549 cells in 96-well plates were infected at 1 day postseeding with HAdV-C5 and treated with different concentrations of digoxin or digitoxin at the end of the adsorption period, in parallel with uninfected cells. Cells were fixed at 24 h p.i. for immunodetection of hexon (green) as described in Materials and Methods. Nuclei were stained with DAPI (blue). The image shown is representative of cells treated with digitoxin.
FIG 2.
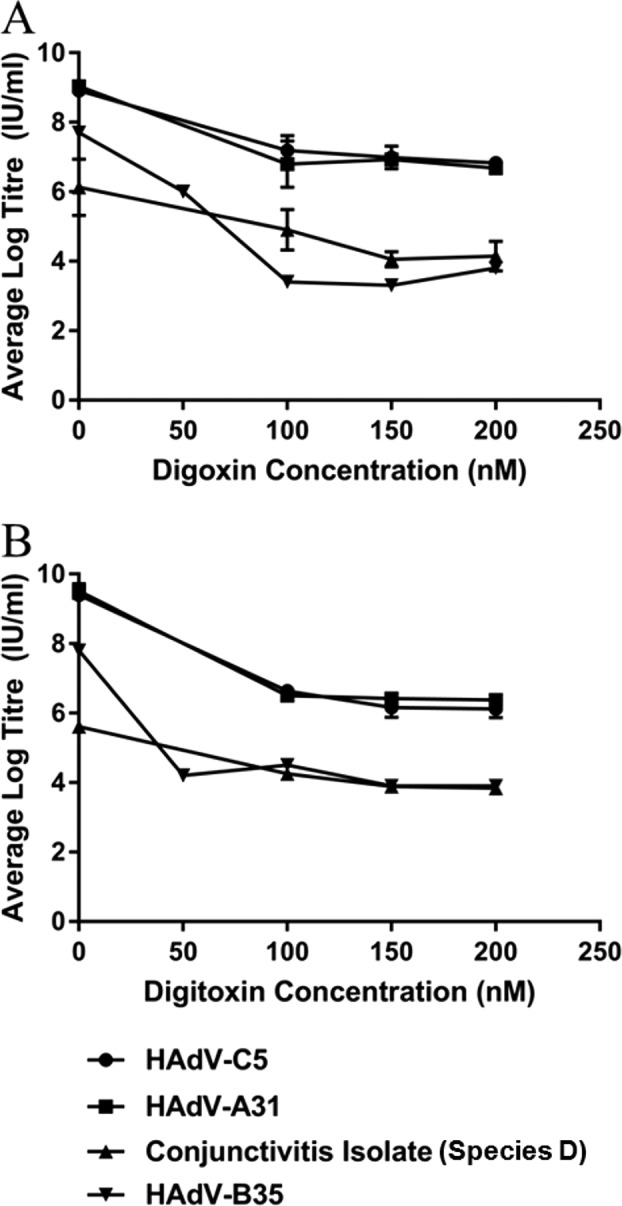
Digoxin and digitoxin suppress replication of multiple adenovirus species. A549 cells in 6-well plates were infected at 1 day postseeding. After 60 min of adsorption at 37°C, the inoculum was removed and replaced with culture medium containing DMSO, digoxin, or digitoxin. Cells and media were collected together at 24 h p.i. for titration of total virus by endpoint dilution in 293 cells. Data points represent average titers of duplicate samples. Error bars represent standard error of the mean from three experiments for HAdV-C5 with each drug and HAdV-A31 with digoxin and for two experiments for HAdV-A31 with digitoxin and HAdV-D (conjunctivitis isolate) with digoxin. Only one experiment was done for HAdV-D with digitoxin and HAdV-B35 with each drug.
FIG 3.

Digoxin and digitoxin have minimal cytotoxic effects on A549 cells. Cells treated with digoxin (A) or digitoxin (B) were compared to DMSO-treated cells in an alamarBlue metabolic assay and by viable counts with trypan blue exclusion. Cells in a 96-well plate were treated at 1 day postseeding with digoxin or digitoxin at different concentrations (with duplicate wells at each concentration), and alamarBlue was added 24 h later. For viable counts, cells seeded in a 24-well plate were treated at 1 day postseeding with DMSO or with digoxin or digitoxin at different concentrations, in duplicate. After 24 h of drug exposure, cells were trypsinized and the suspension diluted with culture medium and then added to an equal volume of 0.3% trypan blue. Cells were counted with disposable hemocytometers containing 10 grids. More than 99% of the cells excluded trypan blue. The viable cell count in each well containing drug was normalized to the count in the DMSO-treated wells in the same experiment. Error bars in both panels represent the standard error of the mean from three experiments.
Digoxin and digitoxin block replication prior to viral DNA synthesis.
Initial experiments to define the block in replication analyzed expression of the immediate early protein E1A and the major late capsid protein hexon by Western blot analysis and immunofluorescence (IF) staining of infected cells. Consistent with initial screening experiments (Fig. 1), hexon expression at 24 h p.i. was detectable in fewer than 10% of the treated cells in the presence of either digoxin or digitoxin (Fig. 4A). E1A was detectable by IF in untreated cells as early as 2 h p.i. as a dull signal in a minority of the population (less than 1%) (data not shown). The signal intensity and the proportion of positive cells increased with time up to 8 h p.i., when most cells showed a strong E1A signal (Fig. 4B). There was only a limited effect of digoxin or digitoxin on E1A expression or on the proportion of cells expressing detectable E1A, as determined by Western blot analysis and IF staining of infected cultures, respectively, at 8 h p.i. (Fig. 4B), indicating that the drugs did not compromise genome delivery or immediate early gene expression but blocked one or more steps prior to late gene expression. The delayed early protein E4orf6 was affected to some degree (Fig. 5). About 30% of untreated cells were positive for E4orf6 expression by IF at 8 h p.i., with 12% and 6% cells staining positive in digoxin- and digitoxin-treated cultures, respectively (Fig. 5A). By 24 h p.i., the majority of cells (>85%) stained positive for E4orf6, with or without drug, though the signal appeared to be somewhat weaker in treated cells (Fig. 5B).
FIG 4.

Differential effect of drug treatment on expression of hexon and E1A proteins. A549 cells infected with HAdV-C5 were harvested for immunodetection of hexon at 24 h p.i. (A) or of E1A at 8 h p.i. (B) by fluorescence microscopy and Western blot analysis. Blots were reprobed for α-tubulin to verify protein loading. Numbers below the E1A blot indicate the relative expression of the viral protein normalized to the level in DMSO-treated cells. The graphs show the proportion of cells expressing the protein of interest as determined by counting the number of antibody-stained cells and the total number of nuclei (DAPI) in four or five random fields in each of two experiments for E1A and four to eight random fields in each of three experiments for hexon. Data are based on more than 200 cells per condition for E1A and more than 1,000 cells per condition for hexon. Error bars show the standard error of the mean from two (E1A) or three (hexon) experiments. ns, not significant in an unpaired, 2-tailed Student t test.
FIG 5.

Effect of digoxin and digitoxin on adenoviral E4orf6 protein expression. A549 cells were infected with HAdV-C5, treated with digoxin or digitoxin immediately after the adsorption period, and then fixed at 8 h p.i. (A) and at 24 h p.i. (B) for immunodetection of E4orf6. Nuclei are stained with DAPI. The proportion of cells positive for E4orf6 protein expression is based on total counts of ∼500 cells in 12 to 14 random fields for each condition in panel A and 300 to 400 cells in 8 or 9 random fields for each condition in panel B. Error bars show the standard error of the mean from two (B) or three (A) experiments. An unpaired, 2-tailed Student t test was used to determine P values.
In light of the effects of digoxin and digitoxin on viral protein expression, we examined what was happening at the level of viral RNA accumulation/processing. Splicing of the E1A transcript results in three major species, 13S, 12S, and 9S, generated by the use of different 5′ splice sites and a common 3′ splice site (41, 42). The 13S and 12S species are predominant at early times postinfection whereas the 9S species becomes predominant late in infection (37), as seen with the untreated samples in our study (Fig. 6A). Specifically, at 8 h p.i., 13S and 12S E1A RNAs were present at comparable levels and were the predominant forms detected, with only ∼10% being 9S (Fig. 6A). By 24 h p.i., the 9S form represented >95% of the E1A RNAs detected. Treatment with either drug altered the relative proportion of E1A RNA isoforms. At 8 h p.i., the proportion of E1A 13S RNA was increased in the presence of drug at the expense of both 12S and 9S RNAs. At 24 h p.i., 9S RNA was predominant with and without drug, but in the drug-treated cells, 12S and 13S RNAs together accounted for 20 to 30% of the total E1A transcripts, compared to barely detectable amounts in untreated cells. Consistent with the reduced expression of hexon (Fig. 1 and 4), RNAs coding for late proteins 100K, fiber, hexon, and penton base (Fig. 6B) were barely detectable in drug-treated cells.
FIG 6.
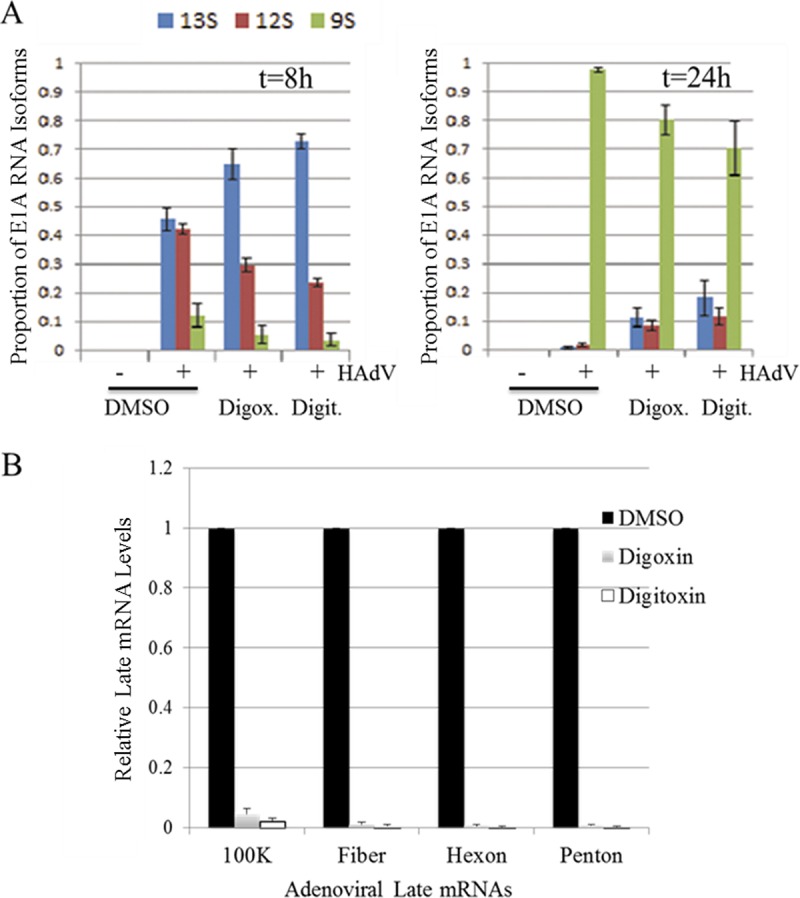
Digoxin and digitoxin alter the accumulation of adenoviral mRNAs. A549 cells were infected with HAdV-C5 and treated with digoxin or digitoxin (100 nM) immediately after the adsorption period. Total RNA was extracted from cells collected at 8 h and 24 h p.i. (A) Amplicons from RT-PCR were fractionated on polyacrylamide gels, and the relative abundance of E1A RNA isoforms was determined by integration of signal for amplicons corresponding to spliced isoforms. (B) Relative abundance of mRNAs encoding 100K, fiber, hexon, or penton base protein, as determined by qRT-PCR. Values shown are normalized to levels in samples treated with DMSO alone. Error bars show the standard error of the mean from three experiments.
Since the adenovirus genome must undergo replication before it can serve as a template for late gene expression (37, 39), it was of interest to determine whether reduced expression of late viral proteins was due to reduced genome replication. As determined by quantitative PCR (qPCR), treatment with either digoxin or digitoxin drastically reduced genome levels at 20 and 22 h p.i., respectively (Fig. 7). Given that adenovirus infection also results in alterations in the subcellular distribution of the splicing regulatory SR proteins due to E4orf4 expression, we examined whether digoxin/digitoxin could block this response. As shown in Fig. 8, adenovirus infection did induce a marked alteration in the subnuclear distribution of the SR-related protein Tra2β, a response that was blocked upon addition of either digoxin or digitoxin.
FIG 7.
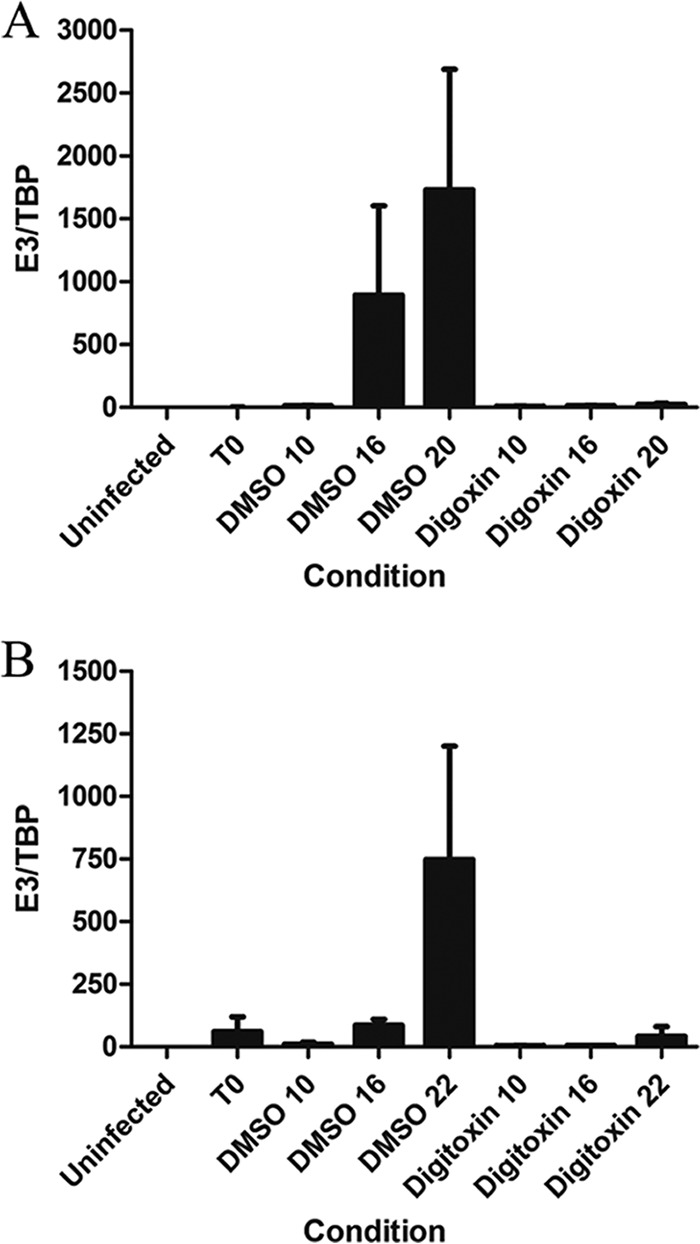
Digoxin and digitoxin block adenovirus genome replication. A549 cells were infected with HAdV-C5 and treated with 100 nM digoxin (A) or 100 nM digitoxin (B) immediately after the adsorption period. Cells were collected at 0, 10, 16, and 20 or 22 h p.i., total DNA was extracted, and the level of adenovirus DNA was determined by qPCR. The relative amount of viral DNA at each time point is plotted as the ratio of viral DNA to cellular TBP DNA (E3/TBP). The results represent three independent experiments for digoxin (A) and two independent experiments for digitoxin (B), with each sample analyzed in duplicate. Error bars show the standard error of the mean.
FIG 8.
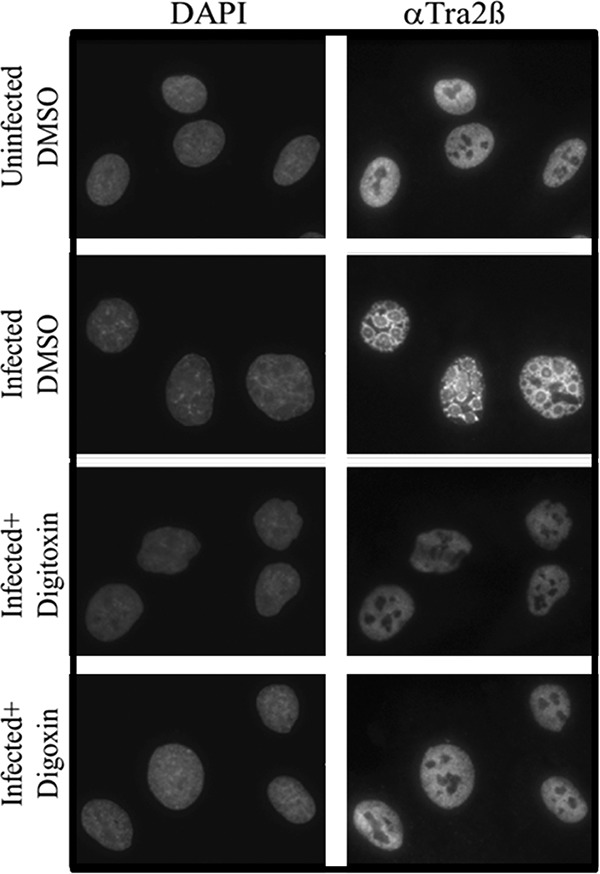
Addition of digoxin or digitoxin inhibits redistribution of host SR proteins in infected cells. A549 cells were either uninfected or infected with HAdVC5 and treated with DMSO, digitoxin (100 nM), or digoxin (100 nM) immediately after removal of the virus inoculum. Cells were fixed at 24 h p.i. and stained for DNA (DAPI) or for Tra2β. Images were taken using a 63× oil objective. Shown are representative images from three independent experiments.
Effect of time of addition of digoxin/digitoxin on modulation of adenovirus gene expression.
Given that E1A expression was only moderately affected by addition of drugs at the end of the adsorption period, it was of interest to determine whether E1A expression could be affected to a greater degree by pretreatment of cells prior to infection. Pretreatment for as little as 4 h not only reduced hexon expression (data not shown), as expected, but also reduced levels of E1A protein provided that drug was still present after infection (Fig. 9A). The effect of pretreatment for as long as 24 h prior to infection was lost when drug was removed at the time of infection, indicating that the drugs were not toxic and that any effect on the host cell was reversible.
FIG 9.

Effect of pretreatment and time of addition of digoxin and digitoxin on adenovirus E1A and hexon expression. (A) A549 cells were incubated with 100 nM drug for 24 h (24PT), 12 h (12PT), or 4 h (4PT) prior to infection with HAdV-C5. After 60 min of adsorption, the inoculum was replaced with medium containing DMSO alone (no posttreatment) or containing 100 nM digoxin or digitoxin (+ posttreatment). Cells were harvested at 8 h p.i., and levels of E1A expression were determined by Western blot analysis. The relative expression levels of E1A, normalized to the level in untreated cells, are shown below the blot. (B) Cells were infected with HAdV-C5, and drugs (100 nM) were added either immediately after the adsorption period (t = 0 h p.i.) or at 2 or 8 h p.i. (t = 2 h p.i. or t = 8 h p.i.). Cells were fixed at 24 h p.i. for immunodetection of hexon by fluorescence microscopy. (C) The proportion of cells staining positive for hexon is based on counts from four to eight random fields in each of three independent experiments, with the exception of digitoxin added at 2 h p.i. (six random fields in one experiment). The total cell count for each condition exceeded 1,000. Error bars represent the standard error of the mean from three experiments.
Time-of-addition experiments also indicated that digoxin and digitoxin have maximal effect on virus replication between 2 and 8 h p.i. Either compound could be added at 2 h p.i. and still compromise viral protein expression to the same degree as drug added immediately at the end of the adsorption period (T0). When added at 8 h p.i., the drugs still blocked hexon expression in some cells, though in fewer cells than when drug was added immediately after virus adsorption or at 2 h p.i. (Fig. 9B and C).
The cardiac glycoside derivative RIDK34 has enhanced antiviral activity.
In the search for drugs with greater potency and potentially reduced toxicity in vivo, we tested RIDK34 (Fig. 10A), a synthetic derivative of convallotoxin (43). RIDK34 at only 12.5 nM achieved the same 3.5-log reduction in virus yield that was achieved with 150 nM digoxin (Fig. 10B). Metabolic activity at concentrations above 12.5 nM was 60% of that in untreated cells, but viable counts with trypan blue showed no evidence of cell death (Fig. 10D). Similar to the case for digoxin and digitoxin, RIDK34 had only a limited effect on E1A expression when added at the end of the adsorption period but dramatically reduced hexon synthesis (Fig. 10C), attributable to reduced viral genome replication (Fig. 10E).
FIG 10.

The analog RIDK34 has increased antiviral activity relative to that of digoxin. (A) Structure of RIDK34. (B) Yield reduction assay. Cells were infected with HAdV-C5, treated with RIDK34 or digoxin, and harvested at 24 h p.i. for assay of total virus by endpoint dilution. Data represent the results of three experiments. Error bars show the standard error of the mean. (C) E1A and hexon expression in HAdV-C5 infected cells incubated with DMSO or 12.5 nM RIDK34 and harvested at 8 h p.i. (E1A) or 24 h p.i. (hexon). Data are representative of results from three experiments. (D) Effect of RIDK34 on cell metabolism and viability. Following infection with HAdV-C5, cells were treated with the indicated concentrations of RIDK34. After 24 h, metabolic activity was assessed using alamarBlue and cell viability by counts with trypan blue. More than 99% of the cells excluded trypan blue. The results shown are the averages from three independent assays. (E) qPCR analysis of adenovirus genome amplification in cells incubated with DMSO or 12.5 nM RIDK34 and harvested at 0, 10, 16, and 22 h p.i. The relative amount of viral DNA at each time point is plotted as the ratio of viral DNA to cellular TBP DNA (E3/TBP). Results represent two independent experiments, with each sample analyzed in duplicate. Error bars represent the standard error of the mean.
DISCUSSION
The potential of cardiotonic steroids as antiviral agents was recognized by Hartley et al. (19), who reported that replication of adenovirus and three herpesviruses (herpes simplex virus [HSV], varicella-zoster virus [VZV], and cytomegalovirus [CMV]) was inhibited by digoxin. The overall effect was attributed to an inhibition of viral DNA synthesis due to lowered levels of intracellular K+, though genome replication was not assessed directly. Using a different approach, our study confirmed the antiviral effect of digoxin on adenovirus replication and identified the time frame during which digoxin, digitoxin, and RIDK34 exert their antiviral effect. Expression of immediate early E1A protein was not markedly affected unless cells were pretreated with drug, suggesting that the onset of E1A expression normally began before the drug took effect. Expression of the delayed early E4orf6 protein was compromised at 8 h p.i., but the difference in drug-treated and untreated cells was minimal at 24 h p.i. The extent to which reduced E4orf6 levels contributed to subsequent events is not known. It was clear, however, that genome synthesis was blocked. Without replication, the genome cannot serve as a template for late transcription (39, 44). Consequently, the block in late gene expression and lack of relocalization of the host factor Tra2β were likely secondary to the block in genome replication.
The time-of-addition experiments give some insight into the timing of inhibition. A 4-h pretreatment, with continued presence of drug after infection, was sufficient to reduce expression of immediate early E1A expression, showing that antiviral activity was established within 4 h. Addition of drug at 2 h p.i., with no pretreatment, had the same inhibitory effect on hexon expression as addition of drug at the end of the adsorption period. Together, this evidence suggests that the antiviral state is established within 2 to 4 h after the drug is added. Given that the drug can still have some effect when added as late as 8 h p.i., it appears that the drug exerts its antiviral effects by compromising processes taking place in the interval from 2 to about 10 h p.i., following the onset of E1A expression and prior to genome synthesis. Knowing that drug added to cells 24 h prior to infection can inhibit E1A protein expression means that the drug was still active after 24 h. The inhibitory effect of pretreatment was lost unless the drug was also present after infection, indicating that drug-induced changes can be rapidly reversed.
The mechanism by which digoxin and digitoxin inhibit adenovirus replication has yet to be fully characterized. If the drugs are modulating signal transduction, it might be expected that virus replication would be compromised, given the importance of signal transduction for virus entry and trafficking as well as subsequent events in the virus replication cycle (45–49). In the current study, digoxin and digitoxin usually were added at the end of a 60-min adsorption period, by which time HAdV-C5 genome delivery is complete (50). Digoxin has been shown to increase the formation of promyelocytic leukemia (PML) bodies (51), which are known to have an antiviral function (52, 53), but E1A expression in the presence of digoxin argues against inhibition by PML bodies (51–53). However, reduced uptake, genome delivery, and/or increased formation of PML bodies may have contributed to the reduction in E1A expression when cells were pretreated with drug.
It is possible that the inhibitory effect of the drugs reflects altered splicing, as seen with HIV (31). The change in the relative proportions of 13S, 12S, and 9S E1A mRNAs at 8 h p.i. in treated compared to untreated cells is consistent with altered RNA processing, but the extent to which these changes influence subsequent steps is not known. Continued low-level expression of 13S and 12S RNAs at 24 h p.i., in the presence of drugs, may reflect altered RNA processing but cannot be responsible for the reduced yield of virus harvested 24 h p.i. Analysis of the effect of digoxin/digitoxin on expression of E2 and E4 proteins is warranted. Whereas E2 proteins, including DNA polymerase, single-stranded DNA binding protein, and preterminal protein, are essential for genome replication, certain E4 proteins are known to alter the activity of host splicing factors associated with changes in viral RNA processing during the course of the replication cycle (54, 55). Regulation of splice site usage by SR proteins is modulated by the extent of their phosphorylation (28, 56–58). E4orf4 recruits the PP2a phosphatase to dephosphorylate SR proteins, affecting their ability to regulate RNA processing (54). Of particular note, responses to cardiac glycoside addition in our study mimicked effects seen upon overexpression of the SR protein ASF/SF2 (SRSF1). SRSF1 overexpression impaired the late accumulation pattern of E1 transcripts and blocked viral DNA replication, attributed to a block in splicing of E2B transcripts, with a two-log reduction in infectious virus yield (59).
Regardless of the precise mechanism of action, digoxin and digitoxin reduce the yields of four different species (A to D) of adenoviruses by at least 2 logs. Given the absence of well-tolerated therapies for adenovirus infection, repurposing digoxin as an antiviral agent is of interest, particularly given the wealth of clinical data that have accumulated over the years during which digoxin has been used to treat heart disease. Although digoxin has been associated with toxicity (20, 60), the use of cardiac glycosides as antivirals would be short term, in contrast to the chronic use in patients with heart disease. In this study, digoxin and digitoxin reduced virus yields by 2 to 4 logs when used at concentrations of 100 to 150 nM, which are below the tissue concentrations associated with toxicity in vivo (61, 62). Although metabolic activity was compromised to some degree (Fig. 3), there was no change in appearance of the cells and no evidence of cell death in cultures exposed to these drugs for 24 h or more. Accordingly, the reduced yield of progeny virus cannot be attributed to cell death.
Due to possible resistance to using digoxin in the clinic, it was of interest to identify a drug with the potential of having a better selectivity index (SI) in cell culture, as a predictor of a better therapeutic index in vivo. RIDK34 (43) was more potent than digoxin in terms of antiviral activity, achieving a 3.5-log reduction in virus yield at a concentration of only 12.5 nM, but did not show reduced cytotoxicity. RIDK34 compromised metabolic activity to a greater degree than did digoxin at the effective antiviral concentration (Fig. 11). As with digoxin, however, there was no evidence of cell death (Fig. 10D). Neither drug reduced metabolic activity by as much as 50%, even at concentrations up to 1,000 nM (Fig. 11). It is difficult to predict how reduced metabolic activity might affect toxicity in vivo, though it might be predicted that cells would recover normal metabolism after removal of the drug.
FIG 11.
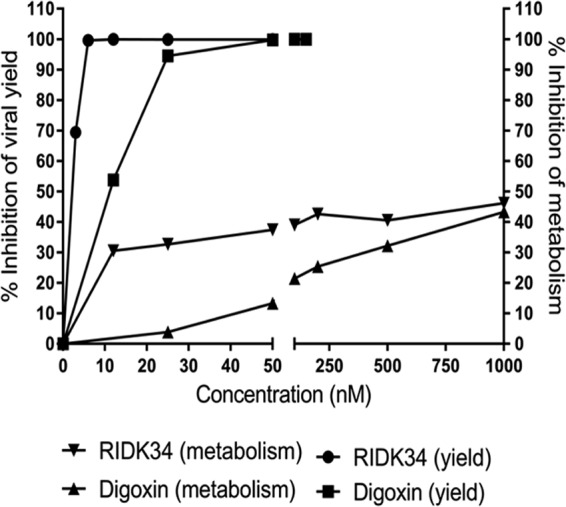
Comparative effects of RIDK34 and digoxin on virus yield and metabolic activity in A549 cells. The digoxin data from Fig. 2 and 3, along with the RIDK34 data from Fig. 10, are shown as percent inhibition of virus yield (left axis) and percent inhibition of metabolic activity as determined with alamarBlue (right axis).
Apart from use to treat severe respiratory and disseminated disease, these drugs seem attractive as potential agents for topical treatment of epidemic keratoconjunctivitis and, even prophylactically, to treat close contacts of patients with EKC. It is not clear why a phase 2 trial for use of digoxin in adenovirus keratoconjunctivitis was discontinued (http://adisinsight.springer.com/drugs/800018650). A more recent phase 2 trial testing digoxin, alone and in combination with furosemide (19), for treatment of cutaneous warts due to human papillomavirus has been completed (https://clinicaltrials.gov/show/NCT02333643), but results have not been reported. The documented antiviral activity of cardiotonic steroids against HIV-1 (31), multiple DNA viruses (adenovirus, human papillomavirus [HPV], and herpesvirus [19, 63]), and several unrelated RNA viruses (alphaviruses, reoviruses, and vesicular stomatitis virus) (64) suggests that they mediate their antiviral activity through a common host cell function, such as RNA processing, required for viral gene expression. Greater understanding of the mechanism(s) by which digoxin and digitoxin act to effect such a dramatic reduction in virus replication will guide the development of novel agents, with little to no toxicity, for the treatment of multiple viral infections.
MATERIALS AND METHODS
Compounds.
Digoxin and digitoxin were purchased from Sigma-Aldrich. RIDK34 was derived synthetically from convallotoxin, as described by De Gouveia (43). Drugs were dissolved in dimethyl sulfoxide (DMSO) at a stock concentration of 10 mM and stored at −20 C.
Viruses and cells.
A549 cells (human lung carcinoma) were obtained from the American Type Culture Collection (ATCC) at passage level 76 and used between passages 89 and 110. HEK 293 (human embryonic kidney) cells (65, 66) were obtained from F. Graham, McMaster University, Hamilton, Ontario, Canada, at passage 24 and were used between passages 58 and 90. All cells were maintained in minimal essential medium (MEM) supplemented with 10% fetal calf serum (FCS) plus penicillin (100 U/ml) and streptomycin (100 μg/ml). alamarBlue metabolic assays were performed according to the manufacturer's recommendations (ThermoFisher). Hybridoma cells (2Hx-2) were obtained from the ATCC and cultured in MEM as described for A549 cells but supplemented with additional glucose (final concentration, 0.5%). HAdV-C5 was initially obtained from the ATCC. Other viruses were isolated from clinical specimens, specifically, stool of a pediatric patient with diarrhea (HAdV-A31), lung tissue from a fatal infection in a neonate (HAdV-B35), and an eye swab from an adult patient with uncomplicated conjunctivitis (HAdV-D).
Virus propagation and assay.
All viruses were propagated in 293 cells and used in experiments at passage level 3 following primary isolation from clinical samples or receipt from the ATCC (HAdV-C5). 293 cells in tissue culture flasks were infected at input multiplicity of infection (MOI) of <0.1 and harvested when cytopathic effect (CPE) was complete. Cells were collected by centrifugation for 10 min at 2,000 × g, resuspended in a small volume of culture medium, and then disrupted with five cycles of freezing and thawing to produce cell lysates. The lysate was clarified by centrifugation for 10 min at 2,000 × g. Clarified lysate and culture fluid were assayed for infectious virus by endpoint dilution in 293 cells, using 60-well Terasaki plates (Sarstedt), as described previously (67). Titers were calculated by the statistical method described by Reed and Muench (68) and expressed as infectious units (IU)/ml (67).
Effect of drugs on adenovirus yield.
A549 cells were seeded in 6-well plates at a density of 500,000 cells per well. Cells were infected at 1 day postseeding at input MOIs of 100 to 400 for HAdV-C5 and -A31, 5 for HAdV-B35, and 0.1 for the conjunctival isolate HAdV-D. After 1 h of adsorption at 37°C, the inoculum was removed and replaced with fresh culture medium containing DMSO (solvent control) or drug dissolved in DMSO (duplicate wells per condition). Progeny virus was harvested at 24 h postinfection (p.i.) by scraping the cells into the culture fluid and then freeze-thawing the suspension five times with vortexing. The lysate was clarified by centrifugation at 500 × g for 5 min and titrated by endpoint dilution in 293 cells (67).
Collection of protein and RNA samples.
A549 cells were seeded on glass coverslips in 6-cm plates at a density of 1 × 106 cells per plate and infected at 1 day postseeding. At 8 or 24 h p.i., the coverslip was removed and cells were fixed in 3.7% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 15 min for subsequent staining. The culture medium was removed from the well, the remaining cells were detached in 1× PBS–2 mM EDTA, and then the suspension was divided into two tubes and cells were pelleted by centrifugation at 800 × g for 3 min. For protein analysis, cell pellets were resuspended with radioimmunoprecipitation assay (RIPA buffer) (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS), and then the lysate was clarified by centrifugation at 9,000 × g for 5 min and stored at −20°C. For RNA analysis, the pellet was resuspended in lysis buffer provided in the Aurum total RNA extraction kit (Aurum). Total RNA was extracted as per the manufacturer's instruction (Bio-Rad) and analyzed by reverse transcription-PCR (RT-PCR) or quantitative RT-PCR (qRT-PCR) as described below.
IF staining.
For immunofluorescence (IF) staining, cells were fixed at the indicated times postinfection with 3.7% PFA for 15 min, washed with 1× PBS, permeabilized for 15 min with 0.1% Triton X-100 in PBS (PBT), and then blocked for 45 min with 5% bovine serum albumin (BSA) in PBT (BSA-PBT). Cells were incubated with primary antibody for 45 min, washed three times with 1× PBS, and then incubated with secondary antibody for 45 min. Cells were washed twice with PBT and then twice with 1× PBS, and coverslips were mounted in PBS containing DAPI (4′,6′-diamidino-2-phenylindole) at 0.25 μg/ml. Primary antibodies were as follows: monoclonal antibody M73 (ThermoFisher) for E1A, undiluted culture fluid from 2Hx-2 hybridoma cells for hexon, rabbit polyclonal antibody for E4orf6 (a gift from G. Ketner [69]), and rabbit polyclonal antibody for endogenous Tra2β (Abcam 31353). The secondary antibody was either goat anti-mouse or anti-rabbit antibody labeled with Alexa Fluor 488, fluorescein isothiocyanate (FITC), or Texas Red and used at 1:200 dilution. Cells were viewed with a 40× objective or 63× oil immersion objective using a Leica DMR microscope, and images were captured with Openlab imaging software version 2.0.7.
Western blot analysis.
To assess hexon and E1A expression, cell lysates (prepared in RIPA buffer as described above) were separated on 7 or 10% SDS-polyacrylamide gels and transferred onto polyvinylidene difluoride (PVDF) membranes using the Bio-Rad TurboBlot system according to the manufacturer's protocol. For hexon analysis, blots were blocked with 5% skim milk diluted in 0.05% Tween20 in 1× PBS (PBS-T) for 1 h and then incubated overnight with undiluted hybridoma (2Hx-2) supernatant containing antihexon antibody. Following three washes with PBS-T, secondary anti-mouse antibody conjugated with horseradish peroxidase (HRP) (1:5,000) was added for 1 h. For analysis of E1A protein, blots were blocked with 5% skim milk diluted in TBS-T (0.05% Tween 20 in 1× Tris-buffered saline [TBS]) for 1 h and then incubated overnight with polyclonal E1A antibody (Santa Cruz, sc-430) diluted 1:2,000 in TBS-T. Following at least 5 washes, secondary anti-rabbit antibody conjugated with HRP (diluted 1:5,000 in TBS-T) was added and left for 1 h. Both E1A and hexon bands were visualized using ECL+ chemiluminescence solution or Bio-Rad Clarity ECL Western blotting substrate and imaged with Bio-Rad ChemiDoc. Blots were subsequently probed for tubulin or GAPDH (glyceraldehyde-3-phosphate dehydrogenase) as loading controls.
Adenoviral RNA and DNA analysis.
To examine E1A RNA splicing, analysis was performed as previously outlined by Yomoda et al. (70). To analyze late gene expression, qRT-PCR was performed using a forward primer common to all adenovirus late mRNAs (AdC-L1-5-qF, 5′-CGAGAAAGGCGTCTAACCAG-3′) and reverse primers specific to the mRNA of interest: Hexon-qR (5′-CCCGAGATGTGCATGTAAGA-3′), Fiber-qR (5′-GAAAAGGCACAGTTGGAGGA-3′), Penton-qR (5′-GCTCACCACGCTCTCGTAG-3′), or 100K-qR (5′-GACGGGGAAGGTGGTAGG-3′). Results were normalized to those for GAPDH generated using GAPDH-qF (5′-CAATGACCCCTTCATTGACC-3′) and GAPDH-qR (5′-GACAAGCTTCCCGTTCTCAG-3′).
For DNA analysis, cells were seeded on 6-cm plates at a density of 1 × 106 cells per plate. The next day, cells were infected with HAdV-C5 for 1 h, and the inoculum was removed and replaced with medium containing DMSO or 100 nM digoxin/digitoxin or 12 nM RIDK34. At the indicated times postinfection, cells were collected and washed in 1× PBS. Cells were lysed using 200 μl lysis buffer (10 mM Tris-HCl [pH 8.0], 75 mM NaCl, 0.1% SDS, 0.5% NP-40, 0.5% Tween 20, 0.5 mg/ml proteinase K) at 56°C for 4 to 5 h, and the mixture was boiled for 15 min. Samples were centrifuged at 13,000 × g, and the supernatant was collected. All samples from a given experiment were processed at the same time. Adenovirus DNA was amplified with primers specific for the adenovirus E3 region (71). The reference gene encoding TATA box binding protein (TBP) was amplified with forward primer 5′-GATGCCTTATGGCACTGGAC-3′ and reverse primer 5′-GCCTTTGTTGCTCTTCCAAA-3′ (gifts from Lucy Osborne). PCR mixtures contained 0.4 μl of Taq DNA polymerase (5 U/μl; NEB, catalog number M0267L), 2.5 μl of ThermoPol buffer, 1.5 μl of 10× SYBR green I (Sigma-Aldrich, catalog number S9430), 2.5 μl of 2.5 mM deoxynucleoside triphosphates (dNTPs), 1.0 μl of 5′ primer (0.1 μg/μl), 1.0 μl of 3′ primer (0.1 μg/μl), 11.1 μl H2O, and 5 μl of DNA. Standard curves were made using serial 10-fold dilutions of DNA collected at 20 h p.i. from infected cells treated with DMSO. Extracted DNA was diluted 1/10 and 1/100 for amplification with TBP and E3 primers, respectively. Parameters for E3 primers were 50.0°C for 2.00 min, 95.0°C for 10.00 min, and 35 cycles of 95.0°C for 15 s, 65.0°C for 1.00 min, and 72.0°C for 1.00 min. Parameters for TBP primers were 50.0°C for 2.00 min, 95.0°C for 10.00 min, and 40 cycles of 95.0°C for 15 s, 60.0°C for 1.00 min, and 72.0°C for 30 s. qPCR was done using a Bio-Rad MyiQ single-color real-time PCR detection system, standard edition, and data were collected with iQ5 Optical System software, version 2.1.
ACKNOWLEDGMENTS
We acknowledge the contributions of Jingwei Chen, an undergraduate student who was involved in the initial screening experiments (Fig. 1). We appreciate the gift of antibody against E4orf6 from Gary Ketner (Johns Hopkins Bloomberg School of Public Health, Baltimore, MD).
The RNA analysis in Fig. 6 was done by P.S. Tra2β staining (Fig. 8) was done by A.C. RIDK34 was synthesized by C.L. F.G. did all of the other experiments, contributing to the design of the experiments and analysis of results. A.C. and M.B. contributed equally to the design of the experiments, analysis of results, and writing of the manuscript.
F.G. was supported in part by a University of Toronto Open Scholarship. The work was supported by an operating grant (HOP-134065) to A.C. from the Canadian Institutes of Health Research (CIHR).
REFERENCES
- 1.Wold WSM, Ison MG. 2013. Adenoviruses, p 1732–1767. In Knipe DM, Howley PM (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA, USA. [Google Scholar]
- 2.Hage E, Gerd Liebert U, Bergs S, Ganzenmueller T, Heim A. 2015. Human mastadenovirus type 70: a novel, multiple recombinant species D mastadenovirus isolated from diarrhoeal faeces of a haematopoietic stem cell transplantation recipient. J Gen Virol 96:2734–2742. doi: 10.1099/vir.0.000196. [DOI] [PubMed] [Google Scholar]
- 3.Dawson CR, O'Day D, Vastine D. 1975. Adenovirus 19, a cause of epidemic keratoconjunctivitis, not acute hemorrhagic conjunctivitis. N Engl J Med 293:45–46. [DOI] [PubMed] [Google Scholar]
- 4.Lindemans CA, Leen AM, Boelens JJ. 2010. How I treat adenovirus in hematopoietic stem cell transplant recipients. Blood 116:5476–5485. doi: 10.1182/blood-2010-04-259291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lion T. 2014. Adenovirus infections in immunocompetent and immunocompromised patients. Clin Microbiol Rev 27:441–462. doi: 10.1128/CMR.00116-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Clercq E. 2011. The clinical potential of the acyclic (and cyclic) nucleoside phosphonates: the magic of the phosphonate bond. Biochem Pharmacol 82:99–109. doi: 10.1016/j.bcp.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 7.De Clercq E, Holy A. 2005. Acyclic nucleoside phosphonates: a key class of antiviral drugs. Nat Rev Drug Discov 4:928–940. doi: 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]
- 8.Hostetler KY. 2009. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: current state of the art. Antiviral Res 82:A84–98. doi: 10.1016/j.antiviral.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kinchington PR, Romanowski EG, Jerold Gordon Y. 2005. Prospects for adenovirus antivirals. J Antimicrob Chemother 55:424–429. doi: 10.1093/jac/dki057. [DOI] [PubMed] [Google Scholar]
- 10.Naesens L, Lenaerts L, Andrei G, Snoeck R, Van Beers D, Holy A, Balzarini J, De Clercq E. 2005. Antiadenovirus activities of several classes of nucleoside and nucleotide analogues. Antimicrob Agents Chemother 49:1010–1016. doi: 10.1128/AAC.49.3.1010-1016.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rietbrock N, Woodcock B. 1985. Two hundred years of foxglove therapy. Trends Pharmacol Sci 6:267–269. doi: 10.1016/0165-6147(85)90123-3. [DOI] [Google Scholar]
- 12.Wade OL. 1986. Digoxin 1785-1985. I. Two hundred years of digitalis. J Clin Hosp Pharm 11:3–9. [DOI] [PubMed] [Google Scholar]
- 13.Withering W. 1785. An account of the foxglove and some of its medical uses with practical remarks on dropsy and other diseases. Birmingham, London, United Kingdom. [Google Scholar]
- 14.Pliny the Elder. 77. The squill: twenty-three remedies. In Bostock J, Riley HT (ed), The natural history of Pliny, 1856 ed. G. Bell & Sons, London, United Kingdom. [Google Scholar]
- 15.Prassas I, Diamandis EP. 2008. Novel therapeutic applications of cardiac glycosides. Nat Rev Drug Discov 7:926–935. doi: 10.1038/nrd2682. [DOI] [PubMed] [Google Scholar]
- 16.Prassas I, Karagiannis GS, Batruch I, Dimitromanolakis A, Datti A, Diamandis EP. 2011. Digitoxin-induced cytotoxicity in cancer cells is mediated through distinct kinase and interferon signaling networks. Mol Cancer Ther 10:2083–2093. doi: 10.1158/1535-7163.MCT-11-0421. [DOI] [PubMed] [Google Scholar]
- 17.Newman RA, Yang P, Pawlus AD, Block KI. 2008. Cardiac glycosides as novel cancer therapeutic agents. Mol Interv 8:36–49. doi: 10.1124/mi.8.1.8. [DOI] [PubMed] [Google Scholar]
- 18.Wang H-Y, Doherty G. 2012. Modulators of Na/K-ATPase: a patent review. Expert Opin Ther Patents 22:587–605. doi: 10.1517/13543776.2012.690033. [DOI] [PubMed] [Google Scholar]
- 19.Hartley C, Hartley M, Pardoe I, Knight A. 2006. Ionic contra-viral therapy (ICVT); a new approach to the treatment of DNA virus infections. Arch Virol 151:2495–2501. doi: 10.1007/s00705-006-0824-x. [DOI] [PubMed] [Google Scholar]
- 20.Hauptman PJ, Kelly RA. 1999. Digitalis Circulation. 99:1265–1270. [DOI] [PubMed] [Google Scholar]
- 21.Schoner W, Scheiner-Bobis G. 2007. Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism, and cell growth. Am J Physiol Cell Physiol 293:C509–C536. doi: 10.1152/ajpcell.00098.2007. [DOI] [PubMed] [Google Scholar]
- 22.Wasserstrom JA, Aistrup GL. 2005. Digitalis: new actions for an old drug. Am J Physiol Heart Circ Physiol 289:H1781–1793. doi: 10.1152/ajpheart.00707.2004. [DOI] [PubMed] [Google Scholar]
- 23.Satoh H, Ginsburg KS, Qing K, Terada H, Hayashi H, Bers DM. 2000. KB-R7943 block of Ca(2+) influx via Na(+)/Ca(2+) exchange does not alter twitches or glycoside inotropy but prevents Ca(2+) overload in rat ventricular myocytes. Circulation 101:1441–1446. doi: 10.1161/01.CIR.101.12.1441. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Tian J, Haas M, Shapiro JI, Askari A, Xie Z. 2000. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J Biol Chem 275:27838–27844. [DOI] [PubMed] [Google Scholar]
- 25.Iwamoto T, Watanabe Y, Kita S, Blaustein MP. 2007. Na+/Ca2+ exchange inhibitors: a new class of calcium regulators. Cardiovasc Hematol Disorders Drug Targets 7:188–198. doi: 10.2174/187152907781745288. [DOI] [PubMed] [Google Scholar]
- 26.Matchkov V, Krivoi I. 2016. Specialized functional diversity and interactions of the Na,K-ATPase. Front Physiol 7:179. doi: 10.3389/fphys.2016.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie Z, Cai T. 2003. Na+-K+–ATPase-mediated signal transduction: from protein interaction to cellular function. Mol Interv 3:157–168. doi: 10.1124/mi.3.3.157. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Z, Fu XD. 2013. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 122:191–207. doi: 10.1007/s00412-013-0407-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stoilov P, Lin CH, Damoiseaux R, Nikolic J, Black DL. 2008. A high-throughput screening strategy identifies cardiotonic steroids as alternative splicing modulators. Proc Natl Acad Sci U S A 105:11218–11223. doi: 10.1073/pnas.0801661105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anderson ES, Lin CH, Xiao X, Stoilov P, Burge CB, Black DL. 2012. The cardiotonic steroid digitoxin regulates alternative splicing through depletion of the splicing factors SRSF3 and TRA2B. RNA 18:1041–1049. doi: 10.1261/rna.032912.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong RW, Balachandran A, Ostrowski MA, Cochrane A. 2013. Digoxin suppresses HIV-1 replication by altering viral RNA processing. PLoS Pathog 9:e1003241. doi: 10.1371/journal.ppat.1003241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laird G, Eisele E, Rabi S, Nikolaeva D, Silicano R. 2014. A novel cell-based high-throughput screen for inhibitors of HIV-1 gene expression and budding identifies the cardiac glycosides. J Antimicrob Chemother 69:988–994. doi: 10.1093/jac/dkt471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berget SM, Moore C, Sharp PA. 1977. Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proc Natl Acad Sci U S A 74:3171–3175. doi: 10.1073/pnas.74.8.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chow LT, Gelinas RE, Broker TR, Roberts RJ. 1977. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 12:1–8. doi: 10.1016/0092-8674(77)90180-5. [DOI] [PubMed] [Google Scholar]
- 35.Davison AJ, Benko M, Harrach B. 2003. Genetic content and evolution of adenoviruses. J Gen Virol 84:2895–2908. doi: 10.1099/vir.0.19497-0. [DOI] [PubMed] [Google Scholar]
- 36.Biasiotto R, Akusjarvi G. 2015. Regulation of human adenovirus alternative RNA splicing by the adenoviral L4-33K and L4-22K proteins. Int J Mol Sci 16:2893–2912. doi: 10.3390/ijms16022893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berk AJ. 2013. Adenoviridae, p 1704–1731. In Knipe DM, Howley PM (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA, USA. [Google Scholar]
- 38.Zhao H, Chen M, Pettersson U. 2014. A new look at adenovirus splicing. Virology 456-457:329–341. doi: 10.1016/j.virol.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 39.Thomas GP, Mathews MB. 1980. DNA replication and the early to late transition in adenovirus infection. Cell 22:523–533. doi: 10.1016/0092-8674(80)90362-1. [DOI] [PubMed] [Google Scholar]
- 40.Akusjarvi G. 2008. Temporal regulation of adenovirus major late alternative RNA splicing. Front Biosci 13:5006–5015. [DOI] [PubMed] [Google Scholar]
- 41.Himmelspach M, Cavaloc Y, Chebli K, Stevenin J, Gattoni R. 1995. Titration of serine/arginine (SR) splicing factors during adenoviral infection modulates E1A pre-mRNA alternative splicing. RNA 1:794–806. [PMC free article] [PubMed] [Google Scholar]
- 42.Chow LT, Broker TR, Lewis JB. 1979. Complex splicing patterns of RNAs from the early regions of adenovirus-2. J Mol Biol 134:265–303. doi: 10.1016/0022-2836(79)90036-6. [DOI] [PubMed] [Google Scholar]
- 43.De Gouveia P. 2010. Cardiac glycosides, a novel treatment for neuroblastoma: efficacy and mechanism. M.Sc. thesis University of Toronto, Toronto, Ontario, Canada. [Google Scholar]
- 44.Larsson S, Kreivi JP, Akusjarvi G. 1991. Control of adenovirus alternative RNA splicing: effect of viral DNA replication on RNA splice site choice. Gene 107:219–227. doi: 10.1016/0378-1119(91)90322-3. [DOI] [PubMed] [Google Scholar]
- 45.Diehl N, Schaal H. 2013. Make yourself at home: viral hijacking of the PI3K/Akt signaling pathway. Viruses 5:3192–3212. doi: 10.3390/v5123192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dunn EF, Connor JH. 2012. HijAkt: The PI3K/Akt pathway in virus replication and pathogenesis. Prog Mol Biol Transl Sci 106:223–250. doi: 10.1016/B978-0-12-396456-4.00002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kong K, Kumar M, Taruishi M, Javier RT. 2015. Adenovirus E4-ORF1 dysregulates epidermal growth factor and insulin/insulin-like growth factor receptors to mediate constitutive Myc expression. J Virol 89:10774–10785. doi: 10.1128/JVI.01463-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolfrum N, Greber UF. 2013. Adenovirus signalling in entry. Cell Microbiol 15:53–62. doi: 10.1111/cmi.12053. [DOI] [PubMed] [Google Scholar]
- 49.Yamauchi Y, Greber UE. 2016. Principles of virus uncoating: cues and the snooker ball. Traffic 17:569–592. doi: 10.1111/tra.12387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greber UF, Willetts M, Webster P, Helenius A. 1993. Stepwise dismantling of adenovirus 2 during entry into cells. Cell 75:477–486. doi: 10.1016/0092-8674(93)90382-Z. [DOI] [PubMed] [Google Scholar]
- 51.Milutinovic S, Heynen-Genel S, Chao E, Dewing A, Solano R, Milan L, Barron N, He M, Diaz PW, Matsuzawa S, Reed JC, Hassig CA. 2016. Cardiac glycosides activate the tumor suppressor and viral restriction factor promyelocytic leukemia protein (PML). PLoS One 11:e0152692. doi: 10.1371/journal.pone.0152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schreiner S, Wimmer P, Sirma H, Everett RD, Blanchette P, Groitl P, Dobner T. 2010. Proteasome-dependent degradation of Daxx by the viral E1B-55K protein in human adenovirus-infected cells. J Virol 84:7029–7038. doi: 10.1128/JVI.00074-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schreiner S, Martinez R, Groitl P, Rayne F, Vaillant R, Wimmer P, Bossis G, Sternsdorf T, Marcinowski L, Ruzsics Z, Dobner T, Wodrich H. 2012. Transcriptional activation of the adenoviral genome is mediated by capsid protein VI. PLoS Pathog 8:e1002549. doi: 10.1371/journal.ppat.1002549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Estmer Nilsson C, Petersen-Mahrt S, Durot C, Shtrichman R, Krainer AR, Kleinberger T, Akusjarvi G. 2001. The adenovirus E4-ORF4 splicing enhancer protein interacts with a subset of phosphorylated SR proteins. EMBO J 20:864–871. doi: 10.1093/emboj/20.4.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kanopka A, Muhlemann O, Petersen-Mahrt S, Estmer C, Ohrmalm C, Akusjarvi G. 1998. Regulation of adenovirus alternative RNA splicing by dephosphorylation of SR proteins. Nature 393:185–187. doi: 10.1038/30277. [DOI] [PubMed] [Google Scholar]
- 56.Long JC, Caceres JF. 2009. The SR protein family of splicing factors: master regulators of gene expression. Biochem J 417:15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 57.Sanford JR, Ellis J, Caceres JF. 2005. Multiple roles of arginine/serine-rich splicing factors in RNA processing. Biochem Soc Trans 33:443–446. doi: 10.1042/BST0330443. [DOI] [PubMed] [Google Scholar]
- 58.Zahler AM, Lane WS, Stolk JA, Roth MB. 1992. SR proteins: a conserved family of pre-mRNA splicing factors. Genes Dev 6:837–847. doi: 10.1101/gad.6.5.837. [DOI] [PubMed] [Google Scholar]
- 59.Molin M, Akusjarvi G. 2000. Overexpression of essential splicing factor ASF/SF2 blocks the temporal shift in adenovirus pre-mRNA splicing and reduces virus progeny formation. J Virol 74:9002–9009. doi: 10.1128/JVI.74.19.9002-9009.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Campbell TJ, MacDonald PS. 2003. Digoxin in heart failure and cardiac arrhythmias. Med J Aust 179:98–102. [DOI] [PubMed] [Google Scholar]
- 61.Schentag J, Bang A, Kozinski-Tober J. 2006. Principles of therapeutic drug monitoring, p 410 In Burton M, Shaw L, Schentag J, Evans W (ed), Applied pharmokinetics and pharmacodynamics. Lippincott Williams & Wilkins, Philadelphia, PA, USA. [Google Scholar]
- 62.Wettrell G, Andersson KE. 1977. Clinical pharmacokinetics of digoxin in infants. Clin Pharmacokinet 2:17–31. doi: 10.2165/00003088-197702010-00002. [DOI] [PubMed] [Google Scholar]
- 63.Dodson AW, Taylor TJ, Knipe DM, Coen DM. 2007. Inhibitors of the sodium potassium ATPase that impair herpes simplex virus replication identified via a chemical screening approach. Virology 366:340–348. doi: 10.1016/j.virol.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ashbrook A, AJ L, Zamora P, Silva L, May N, Bauer J, Morrison T, Dermody T. 2016. Antagonism of the sodium-potassium ATPase impairs Chikungunya virus infection. mBio 7:e00693-16. doi: 10.1128/mBio.00693-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Graham FL, Smiley J, Russell WC, Nairn R. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol 36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 66.Shaw G, Morse S, Ararat M, Graham FL. 2002. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J 16:869–871. [DOI] [PubMed] [Google Scholar]
- 67.Brown M. 1985. Selection of nonfastidious adenovirus species in 293 cells inoculated with stool specimens containing adenovirus 40. J Clin Microbiol 22:205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent end-points. Am J Hyg (Lond) 27:493–497. [Google Scholar]
- 69.Mohammadi ES, Ketner EA, Johns DC, Ketner G. 2004. Expression of the adenovirus E4 34k oncoprotein inhibits repair of double strand breaks in the cellular genome of a 293-based inducible cell line. Nucleic Acids Res 32:2652–2659. doi: 10.1093/nar/gkh593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yomoda J, Muraki M, Kataoka N, Hosoya T, Suzuki M, Hagiwara M, Kimura H. 2008. Combination of Clk family kinase and SRp75 modulates alternative splicing of adenovirus E1A. Genes Cells 13:233–244. doi: 10.1111/j.1365-2443.2008.01163.x. [DOI] [PubMed] [Google Scholar]
- 71.Ying B, Toth K, Spencer JF, Meyer J, Tollefson AE, Patra D, Dhar D, Shashkova EV, Kuppuswamy M, Doronin K, Thomas MA, Zumstein LA, Wold WS, Lichtenstein DL. 2009. INGN 007, an oncolytic adenovirus vector, replicates in Syrian hamsters but not mice: comparison of biodistribution studies. Cancer Gene Ther 16:625–637. doi: 10.1038/cgt.2009.6. [DOI] [PMC free article] [PubMed] [Google Scholar]