

ABSTRACT
While the entry of infectious bursal disease virus (IBDV) is initiated by the binding of the virus to the two major receptors integrin and HSP90, the signaling events after receptor binding and how they contribute to virus entry remain elusive. We show here that IBDV activates c-Src by inducing the phosphorylation of the Y416 residue in c-Src both in DF-1 chicken fibroblasts and in vivo in the bursa of Fabricius from specific-pathogen-free (SPF) chickens. Importantly, inactivated IBDV fails to stimulate c-Src Y416 phosphorylation, and a very virulent IBDV strain induces a much higher level of c-Src Y416 phosphorylation than does an attenuated strain. Inhibition of c-Src activation by an Src kinase inhibitor or expression of a c-Src dominant negative mutant results in a significant decrease in the internalization of IBDV but has little effect on virus adhesion. Furthermore, short hairpin RNA (shRNA) downregulation of integrin, either the α4 or β1 subunit, but not HSP90 remarkably attenuates IBDV-induced c-Src Y416 phosphorylation, resulting in a decrease in IBDV internalization but not virus adhesion. Moreover, interestingly, inhibition of either c-Src downstream of the phosphatidylinositol 3-kinase (PI3K)/Akt-RhoA signaling cascade or actin rearrangement leads to a significant decrease in IBDV internalization irrespective of the IBDV-induced high levels of c-Src phosphorylation. Cumulatively, our results suggest a novel feed-forward model whereby IBDV activates c-Src for benefiting its cell entry via an integrin-mediated pathway by the activation of downstream PI3K/Akt-RhoA signaling and cytoskeleton actin rearrangement.
IMPORTANCE While IBDV-caused immunosuppression is highly related to viral invasion, the molecular basis of the cellular entry of IBDV remains elusive. In this study, we demonstrate that IBDV activates c-Src by inducing the phosphorylation of the Y416 residue in c-Src to promote virus internalization but not virus adhesion. The ability to induce the level of c-Src Y416 phosphorylation correlates with the pathogenicity of an IBDV strain. IBDV-induced c-Src Y416 activation is α4β1 integrin but not HSP90 dependent and involves the activation of the downstream PI3K/Akt-RhoA GTPase-actin rearrangement cascade. Thus, our findings provide new insights into the IBDV infection process and the potential for c-Src as a candidate target for the development of IBDV therapeutic drugs.
KEYWORDS: IBDV, c-Src, internalization, integrin
INTRODUCTION
Infectious bursal disease virus (IBDV), a member of the genus Avibirnavirus of the family Birnaviridae, is the causative agent of infectious bursal disease, which is an acute and highly contagious disease of young chickens manifested as inflammation and subsequent atrophy of the bursa of Fabricius (BF) and immunosuppression (1). IBDV targets mainly the BF, the major chicken immune organ, resulting in immunosuppression and an increased susceptibility to secondary infections in young chickens (2).
IBDV is a nonenveloped, icosahedral virus with a double-stranded RNA (dsRNA) genome consisting of two segments, segment A (3.2 kb) and segment B (2.8 kb). While segment B has only one open reading frame (ORF) that encodes VP1, the putative RNA-dependent RNA polymerase of IBDV (3), segment A is composed of two overlapping ORFs, ORF1 and ORF2, that encode the viral nonstructural protein VP5, which may be involved in virus egress (4, 5), and a precursor polyprotein (pVP2-VP4-VP3), respectively (6, 7). While VP4 is a serine protease that cleaves the polyprotein (VP2-VP4-VP3) to form VP2, VP3, and VP4, the major viral structural protein VP2 assembles into 260 trimers to form a T=13 icosahedral IBDV capsid (8). In addition, VP4 inhibits the expression of type I interferon (IFN) by binding to the glucocorticoid-induced leucine zipper (9). Our previous study showed that VP3 competes with MDA5 to bind intracellular viral genomic dsRNA to block IFN-β induction (10).
IBDV infection begins with virus attachment to the putative cellular receptors on the surface of host cells. So far, three putative IBDV receptors on host cells have been identified, including IgM in chicken B lymphocytes (11), integrin in mouse NIH 3T3 cells (12), and HSP90 (heat shock protein 90) in chicken fibroblasts (13). While integrins are cell surface-expressed membrane proteins, HSP90, a highly conserved molecular chaperone, can be present inside the cell, expressed on the cell surface (13, 14), and secreted into the extracellular space (15). Beside its role as a cell attachment receptor for IBDV, which confers susceptibility to nonpermissive cells in IBDV infection (13), surface HSP90 was recently shown to initiate the autophagy cascade by interacting with VP2 of IBDV (14). The capsid protein VP2 is the primary immunogen of IBDV and is displayed as trimeric clusters of outer-protruding structures on IBDV capsids (16). This protein contains three distinct domains, namely, the base (B), shell (S), and projection (P) domains (17, 18). The P domain contains a conserved putative α4β1 integrin binding motif of Ile-Asp-Ala (IDA), which mediates the interaction of IBDV with integrin receptors on the surface of permissive cells (19). After receptor binding, IBDV actively modulates and engages cytoskeletons and is then internalized to enter target cells via a mechanism of adsorptive or receptor-mediated endocytosis that is independent of clathrin (20). Endosomal penetration by IBDV is mediated by a pep46-dependent pore-forming mechanism. pep46, a membrane permeabilization protein present in the virus capsid, is generated from the self-cleavage of precursor VP2 during VP2 maturation. This protein interacts with the lipid bilayer to induce pores with a diameter of <10 nm in endosomal membranes, resulting in the release of viral particles from the endosome into the cytoplasm (21). However, the signaling events that occur after IBDV particles attach to cellular receptors and the cellular factors that regulate these virus entry events have not yet been clear.
c-Src kinase is a multifunctional nonreceptor tyrosine kinase involved in regulating a diverse spectrum of biological activities such as proliferation, survival, metastasis, and angiogenesis (22). c-Src has been implicated in the regulation of the life cycles of various viruses. A number of viruses, including herpes simplex virus 8 (human herpesvirus 8 [HHV8]) (23), Japanese encephalitis virus (JEV) (a member of the genus Flavivirus of the family Flaviviridae) (24), and bovine ephemeral fever virus (BEFV) (a member of the genus Ephemerovirus of the family Rhabdoviridae) (25), stimulate c-Src phosphorylation to facilitate virus entry. One of the possible mechanisms is that c-Src bridges between the upstream surface α4β1 integrin receptors and downstream phosphatidylinositol-3-kinase (PI3K)/Akt signaling cascades. Through the activation of downstream RhoA, a small G protein, activated PI3K/Akt regulates the actin cytoskeleton, which is essential for virus entry and the virus life cycle (26).
The regulation of c-Src activity involves the phosphorylation of two key tyrosine sites on c-Src, Y416 and Y527 (27). Under basal conditions, c-Src is phosphorylated at Y527, which enables c-Src to be maintained in an inactive configuration by multiple intramolecular interactions. Tyrosine 416 is present in the activation loop, and intermolecular autophosphorylation of this site promotes kinase activity (28, 29).
IBDV actively modulates and engages cytoskeletons during its entry into cells, and inhibitions or depletions of c-Src kinase lead to a significant reduction of IBDV cell entry (20), suggesting that c-Src plays a role in IBDV infection, especially in virus entry. Despite these indications, however, it is not clear whether the activation of c-Src signaling could be moderated during IBDV infection and how it contributes to IBDV infection. In the present study, we have thus determined the effect of IBDV on host c-Src phosphorylation/activation and its role in IBDV invasion at an early stage. Our results demonstrate that IBDV infection rapidly induces the tyrosine phosphorylation of c-Src kinase at Y416, which is a prerequisite for efficient IBDV internalization but not for virus-receptor binding. IBDV-induced c-Src tyrosine phosphorylation and activation are α4β1 integrin dependent and involve the downstream activation of the PI3K-RhoA GTPase-actin rearrangement cascade. This is the first report demonstrating a novel feed-forward model whereby IBDV activates c-Src kinase via an integrin pathway but not HSP90 to facilitate its entry into cells.
RESULTS
IBDV infection elicits host c-Src Y416 phosphorylation/activation in a virulence-dependent manner both in vitro and in vivo.
c-Src, a tyrosine protein kinase, has been reported to play an important role in virus entry into host target cells (30). The activation of c-Src is characterized by the intermolecular autophosphorylation of tyrosine 416 in the activation loop (31). To investigate whether c-Src kinase could also play a role during IBDV infection, we examined c-Src expression and its tyrosine phosphorylation at the activation-related Y416 site by Western blotting of the IBDV-infected or mock-infected chicken fibroblast cell line DF-1 over a time course of 4 h. The kinetics of c-Src Y416 phosphorylation was plotted by using the quantified band intensity of c-Src phosphorylation against whole c-Src expression in IBDV-infected DF-1 cells. Upon IBDV infection of DF-1 cells, c-Src phosphorylation at Y416 showed a rapid increase, peaked at 1 h postinfection (hpi), and decreased thereafter (Fig. 1A). As a control, total c-Src protein levels were not changed (Fig. 1A, middle). We next examined whether the induction of c-Src phosphorylation at Y416 requires the infectivity of IBDV by infecting DF-1 cells for 1 h with either live IBDV or heat-inactivated virus that was rendered noninfectious. Whereas live-IBDV infection induced a >3-fold increase in c-Src kinase phosphorylation at Y416, infection by heat-inactivated IBDV induced only a slight increase in c-Src kinase phosphorylation at Y416 (Fig. 1B). To investigate whether IBDV infection induces c-Src Y416 phosphorylation in vivo, specific-pathogen-free (SPF) chickens were challenged with two IBDV strains, a very virulent strain (vvIBDV) and an attenuated IBDV strain (aIBDV). We chose four postinoculation intervals, i.e., 1, 2, 3, and 4 days postinfection (dpi), to include the time points ranging from the early appearance of infection to maximum infection by both vvIBDV and aIBDV before death. Infected BFs were harvested and subjected to hematoxylin and eosin (H&E) staining and Western blot analysis of c-Src tyrosine phosphorylation at Y416. As expected, H&E staining of histological sections of BFs showed that vvIBDV caused moderate hemorrhage, severe necrosis, and depletion of lymphocytes, whereas aIBDV caused slight hemorrhage and atrophy (Fig. 1D, right versus middle). As observed for the DF-1 cell line, both the vvIBDV and aIBDV strains induced significant and comparable increases in c-Src phosphorylation at Y416 in BFs from virus-infected SPF chickens at 1 dpi (Fig. 1C). However, the level of c-Src Y416 phosphorylation induced by vvIBDV continued to increase and peaked at 2 dpi, whereas in aIBDV-infected BFs, c-Src Y416 phosphorylation started to decline at 2 dpi (Fig. 1C, lane 3 versus lane 7). To further confirm the correlation between the pathogenicity of IBDV strains and elevated levels of c-Src Y416 phosphorylation in vivo, histological sections of BFs were subjected to immune staining using specific antibodies against VP3 and chicken c-Src-phosphorylated Y416, followed by confocal image analysis. As expected, there were significantly more viral components, i.e., VP3 (Fig. 1E, green) detected in vvIBDV-infected BFs than in aIBDV-infected BFs. Staining of c-Src Y416 phosphorylation (Fig. 1E, red) was also much more abundant in vvIBDV-infected BFs than in aIBDV-infected BFs (Fig. 1E). Taken together, c-Src kinase phosphorylation is induced during IBDV infection both in a cell line and in vivo, and the intensity of c-Src kinase phosphorylation appears to correlate with the extent of virus dissemination and pathogenicity of IBDV strains.
FIG 1.
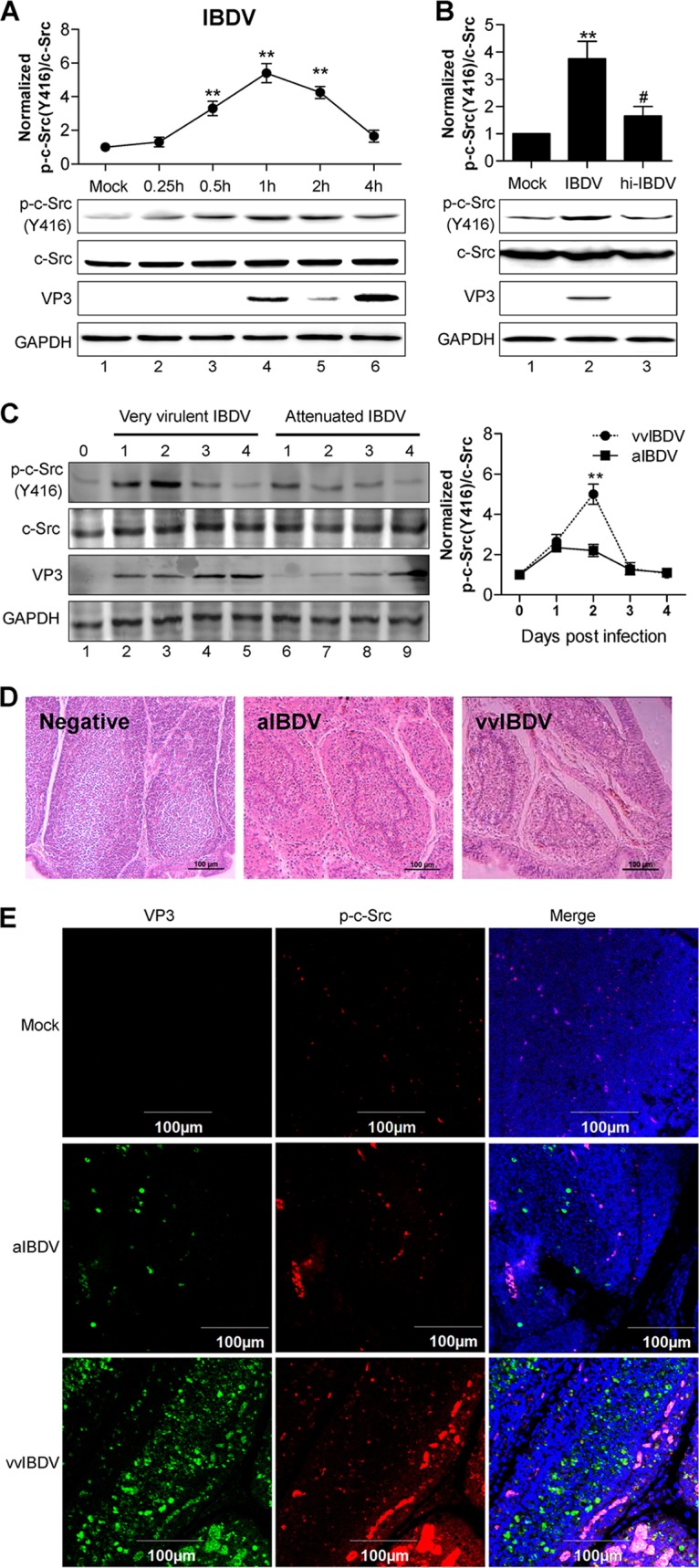
IBDV infection elicits host c-Src tyrosine phosphorylation and activation in a cell line and in vivo. (A) Time course of c-Src Y416 phosphorylation upon IBDV infection. DF-1 cells were either mock infected or infected with IBDV (MOI = 50) for different times (0.25 to 4 h), and Western blot analysis was then performed by using antibodies against phospho-c-Src (Y416), VP3, and total c-Src. GAPDH was used as a loading control. The line graph shows data from densitometry analysis of the ratio of phospho-c-Src/total c-Src. (B) DF-1 cells were mock infected or incubated with live IBDV or with heat-inactivated IBDV for 1 h, and Western blot analysis was then performed by using antibodies against phospho-c-Src, VP3, and total c-Src. The histogram shows data from densitometry analysis of the ratio of phospho-c-Src/total c-Src. (C) SPF chickens inoculated with vvIBDV or aIBDV were sacrificed at the indicated times (days postinfection), and the BFs were collected for Western blotting to analyze the phosphorylation level of c-Src at Y416. (D) H&E staining of BF sections for assessment of pathological lesions. (E) Representative confocal microscopy images of BF sections from SPF chickens inoculated with aIBDV or vvIBDV coimmunostained with anti-VP3 plus anti-mouse Ig Alexa Fluor 488 or anti-phospho-c-Src plus anti-rabbit Ig Alexa Fluor 568, respectively. Nuclei (blue) were stained with DAPI (4′,6-diamidino-2-phenylindole). All the data are presented as means ± SDs from three independent experiments. **, P < 0.01; #, P > 0.05.
Inhibition of c-Src kinase phosphorylation leads to decreased internalization, but not adhesion, of IBDV.
Given the observation that IBDV rapidly induced c-Src tyrosine phosphorylation and activation in the DF-1 cell line as well as in vivo in the bursa of Fabricius, we next examined the effect of c-Src tyrosine phosphorylation and activation on the early stages of IBDV infection, including viral particle adhesion and entry into target cells. DF-1 cells were either mock treated or pretreated with the c-Src inhibitor AZD0530 for 1 h, prior to IBDV infection for 1 h, and the effect of c-Src kinase inhibition by AZD0530 treatment on IBDV adhesion and internalization was assessed by various methods, including Western blotting, plaque assays, reverse transcription-quantitative PCR (qRT-PCR) analysis of the 5′ untranslated region (UTR) of segment A, and immunofluorescence assays (IFAs). In contrast to the marked increase of c-Src kinase phosphorylation at Y416 in mock-treated DF-1 cells, AZD0530 pretreatment led to a reduction of c-Src phosphorylation in a dose-dependent manner (Fig. 2A). To confirm the reduction in virus internalization by c-Src inhibition, we further analyzed the expression of IBDV VP3 proteins at 1 h and 4 h postinfection using Western blot analysis. At all time points, there was significantly less VP3 protein detected in the AZD0530 treatment group (Fig. 2B, lane 4 versus lane 2 and lane 5 versus lane 3). Furthermore, the effect of c-Src inhibition by treatment with AZD0530 on the entry of IBDV was assessed by a plaque assay. The viral titer was monitored in IBDV-infected DF-1 cells treated with dimethyl sulfoxide (DMSO) or with AZD0530 at the time points of 1, 4, 12, and 24 hpi. The results clearly indicate that a significant decrease in IBDV internalization by AZD0530 treatment occurred by as early as 1 h postinfection, which suggested an early role of IBDV-induced c-Src signaling in the early steps of the entry of infectious virus (Fig. 2C). The effect of c-Src inhibition by treatment with AZD0530 on IBDV entry was assessed by immunofluorescence staining of the IBDV structural protein VP3 (Fig. 2D, green, and a to c). Given that in the cells at 1 hpi, VP3 was derived mostly from the internalized parent IBDVs rather than from the newly synthesized progeny viruses, VP3 displayed a small-dotted staining pattern. There were considerably many more dot-like VP3 signals detected specifically in DMSO-treated DF-1 cells than in AZD0530-treated cells (Fig. 2D, b versus c). In addition, comparison of the quantitative score in percentages of VP3-positive cells indicated that there was a significantly lower number of VP3-positive cells in cells treated with AZD0530 than in control cells treated with DMSO (Fig. 2Dd). The effects on both IBDV entry and IBDV binding to target cells with AZD0530 treatment were further assessed in parallel by using both qRT-PCR analysis of the 5′ UTR of segment A (Fig. 2E) and a plaque assay (Fig. 2F). In both assays, while the adhesion of IBDV to target cells showed little difference between the mock and AZD0530 treatment groups (Fig. 2E and F, open bars), IBDV internalization was reduced by about one-half in the AZD0530 treatment group in comparison to that in the mock treatment group (Fig. 2E and F, filled bars). Furthermore, to investigate whether there is a direct link between c-Src Y416 phosphorylation and IBDV internalization, we examined the effect of a c-Src dominant negative (DN) mutant on IBDV internalization. In DF-1 cells, transfection of the c-Src DN mutant led to a 60% reduction in virus internalization (Fig. 2G and H, filled bars) but little change in virus adhesion (Fig. 2G and H, open bars). These results suggest that c-Src phosphorylation at Y416 and activation are required for IBDV entry into cells but not for adhesion to host cells.
FIG 2.

Inhibition of c-Src activation by treatment with AZD0530 or overexpression of a c-Src dominant negative (DN) mutant leads to decreased internalization but not adhesion of IBDV. (A) DF-1 cells pretreated with either DMSO or different concentrations of AZD0530 were mock infected or infected with IBDV for 1 h. The whole-cell lysates were then extracted and subjected to Western blot analysis using anti-phospho-c-Src, followed by anti-c-Src and anti-GAPDH as the loading controls. (B) DF-1 cells pretreated with either DMSO or 20 μM AZD0530 were mock infected or infected with IBDV for either 1 h or 4 h, and Western blot analysis was then performed by using anti-VP3. GAPDH was included as a loading control. (C) DF-1 cells pretreated with either DMSO or 20 μM AZD0530 were infected with IBDV for either 1 h, 4 h, 12 h, or 24 h. The viral titer under each condition was then measured by a viral plaque assay. (D) Representative confocal microscopy images of immunostaining of VP3 (green) in DF-1 cells that were pretreated with either DMSO or 20 μM AZD0530, followed by infection with IBDV or mock infection for 1 h (a to c). Nuclei were stained with DAPI (blue). The bar graph indicates the quantitative score in the percentage of VP3-positive cells determined by counting 150 cells per group (d). (E and F) The effects of AZD0530 treatment on both IBDV entry and IBDV binding to target cells were further assessed in parallel by using both a plaque assay (F) and qRT-PCR analysis of the 5′ UTR of segment A (E), with values normalized against the value for GADPH. (G and H) The effect of the expression of c-Src DN on IBDV binding and internalization was measured in parallel by using qRT-PCR (G) and a plaque assay (H). The expression of the Flag-tagged c-Src DN mutant was verified by Western blotting (G, right). All the data are presented as means ± SDs from at least three independent experiments. **, P < 0.01.
IBDV-induced c-Src phosphorylation and activation are mediated by membrane surface α4β1 integrin but not HSP90.
Upon infection, IBDV first needs to make an attachment to target cells and subsequently progresses to cell internalization (20). The cell surface molecule α4β1 integrin links to its downstream c-Src via focal adhesion kinase (FAK) (22) in the integrin signaling pathway. Integrin and HSP90 are two major IBDV binding receptors (12, 13). To address the role of integrin and HSP90 in IBDV-induced c-Src Y416 phosphorylation and activation, we screened for effective short hairpin RNAs (shRNAs) by Western blotting (Fig. 3Aa to c) and confirmed the effect of shRNA knockdown of the integrin α4 or β1 subunit or HSP90 by qRT-PCR (Fig. 3Ad). Knockdown of either the integrin α4 or β1 subunit but not HSP90 significantly reduced IBDV-enhanced c-Src Y416 phosphorylation (Fig. 3B and D). Furthermore, the effect of α4β1 integrin knockdown on IBDV adhesion and internalization was examined. While the level of IBDV adhesion to target cells was hardly affected (Fig. 3C), knockdown of either the α4 or β1 subunit of integrin led to a significant 50% decrease in IBDV internalization, as determined by qRT-PCR analysis of the 5′ UTR of segment A in DF-1 cells. In contrast, despite having little effect on c-Src Y416 phosphorylation, knockdown of HSP90 led to significant reductions in not only IBDV internalization but also virus attachment (Fig. 3E), supporting that HSP90 serves as the major binding receptor for IBDV attachment. These data indicate that IBDV-induced c-Src Y416 phosphorylation is mediated through the host cell surface molecule integrin but not HSP90, which is required specifically for c-Src-mediated IBDV internalization but not for virus adhesion to target cells.
FIG 3.
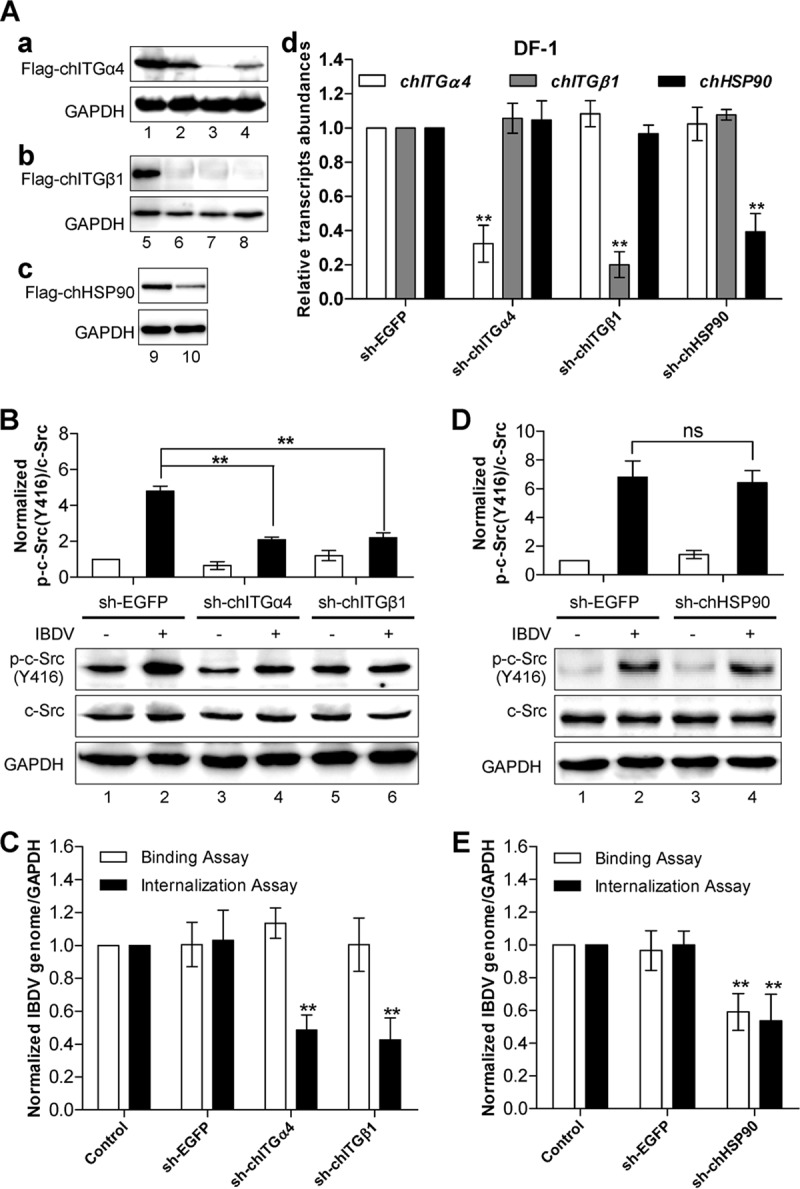
IBDV-induced c-Src phosphorylation and activation are dependent on host cell surface integrin α4β1, which is required for IBDV internalization but not for IBDV adhesion. (A) Generation of stable DF-1 cell pools with lentivirus-mediated expression of integrin ITGα4 shRNAs, integrin ITGβ1 shRNAs, HSP90 shRNA, and nontargeting EGFP shRNA as a control. shRNA-ITGα4#2 (a, lane 3), shRNA-ITGβ1#3 (b, lane 8), and shRNA-HSP90 (c, lane 10) effectively suppressed the expression of Flag-tagged ITGα4, shRNA-ITGβ1, and HSP90, respectively. GAPDH was detected as a loading control. The knockdown of the integrin subunits chITGA4 and chITGB1 or HSP90 in the stable line was confirmed by qRT-PCR, and values were normalized to the value for GAPDH (d). (B) DF-1 cells that stably expressed shRNAs against integrin α4, integrin β1, or EGFP (control) were infected with IBDV for 1 h, and the activation of c-Src was examined by Western blotting using anti-phospho-c-Src, followed by anti-c-Src and anti-GAPDH as the loading controls. The histogram shows data from densitometry analysis of the ratio of phospho-c-Src/total c-Src. (C) IBDV binding and internalization assays were performed in DF-1 cells stably expressing shRNAs against integrin α4, integrin β1, or EGFP. (D) Knockdown of HSP90 has little effect on IBDV-induced phosphorylation of c-Src. DF-1 cells with stable expression of HSP90 shRNA or control EGFP shRNA were infected with IBDV for 1 h, and the phosphorylation of c-Src was examined by Western blotting using anti-phospho-c-Src, followed by anti-c-Src and anti-GAPDH as the loading controls. The histogram shows data from densitometry analysis of the ratio of phospho-c-Src/total c-Src. (E) IBDV particle adhesion and internalization assays were performed by using DF-1 cells with stable expression of shRNA against HSP90 or EGFP. All the data are presented as means ± SDs from three independent experiments. **, P < 0.01.
Inhibition of c-Src downstream PI3K/Akt-RhoA signaling and cytoskeleton actin rearrangement prevents c-Src activation-mediated IBDV internalization.
Given that IBDV-induced c-Src activation promotes integrin-mediated IBDV internalization, the next question is which c-Src downstream targets are involved in IBDV internalization. It was reported previously that IBDV could activate the PI3K/Akt signaling pathway (32), one of the downstream targets of c-Src. To examine whether c-Src Y416 phosphorylation and PI3K/Akt activation upon IBDV infection are two separate or coupled events, we thus investigated the effect of c-Src inhibition on IBDV-induced PI3K/Akt activation and the effect of PI3K/Akt inhibition on IBDV-induced c-Src Y416 phosphorylation. PI3K activity upon IBDV infection was measured by analysis of Akt phosphorylation at S473 in IBDV-infected DF-1 cells (33). In line with data from a previous report (32), Akt phosphorylation at S473 was greatly increased upon IBDV infection (Fig. 4A, lane 2). Treatment with the c-Src inhibitor AZD0530 significantly reduced not only IBDV-induced Y416 phosphorylation of c-Src (Fig. 4A, lane 4 versus lane 2) but also IBDV-induced S473 phosphorylation of Akt (Fig. 4A, lane 4 versus lane 2). In contrast, treatment with the Akt inhibitor LY294002 inhibited only IBDV-induced Akt phosphorylation at S473 (Fig. 4A, lane 6 versus lane 2) but had little effect on IBDV-induced phosphorylation of c-Src at Y416 (Fig. 4A, lane 6 versus lane 2). These results suggest that IBDV-induced c-Src Y416 phosphorylation is an event that occurs earlier than IBDV-induced Akt activation, and PI3K/Akt signaling is a downstream target of activated c-Src upon IBDV infection.
FIG 4.

c-Src and the downstream molecule PI3K/Akt in the integrin signaling pathway are required for IBDV internalization. (A) DF-1 cells were pretreated with the indicated inhibitors and then infected with IBDV at an MOI of 50 for 1 h. Western blot analysis of cell lysates was performed by using the indicated antibodies. The ratio of the signal intensity of phosphorylated c-Src to that of total c-Src and the ratio of the signal intensity of phosphorylated Akt to that of total Akt were quantitated from three independent experiments and are plotted in a histogram (right). (B) Time course of RhoA GTPase activity in DF-1 cells that were infected with IBDV at an MOI of 50 for various time periods (0.5 to 1.5 h). GTP-RhoA was analyzed by a pulldown assay, and the ratio between GTP-RhoA and RhoA was quantified from three independent experiments and plotted in a histogram. (C) DF-1 cells were pretreated with the indicated inhibitors and then infected with IBDV at an MOI of 50 for 1 h, and RhoA GTPase activity was then measured by using a G-LISA assay. (D and F) DF-1 cells pretreated with the c-Src inhibitor AZD0530, the PI3K inhibitor LY294002, RhoA inhibitor I, as well as the cytoskeleton actin inhibitors cytochalasin D and jasplakinolide were subsequently used for measuring IBDV binding and internalization as described in the legend of Fig. 2. The values plotted in the bar chart were normalized to 1 for the mock treatment control. (E) DF-1 cells pretreated with the c-Src inhibitor AZD0530, the PI3K inhibitor LY294002, or DMSO were infected with IBDV at an MOI of 50 for 1 h, and coimmunofluorescence staining with TRITC-labeled phalloidin and anti-VP3 was performed to detect F-actin (red) and the viral protein VP3 (green). Nuclei were stained with DAPI. Images were captured by using a confocal microscope. All the data are presented as means ± SDs from three independent experiments. **, P < 0.01.
RhoA is one of the small GTPases downstream of PI3K/Akt signaling and is the master regulator of cytoskeleton rearrangement for virus entry into cells (26). To examine whether enhanced c-Src activation upon IBDV infection has an effect on the activation of these signaling molecules involved in cytoskeleton rearrangement, we measured the activity of RhoA GTPase by Western blotting and a G-LISA assay. Upon IBDV infection, the RhoA GTPase activity was significantly increased, peaked at 1 h postinfection, and then decreased (Fig. 4B). This result was further confirmed by a G-LISA showing that RhoA activity was upregulated by nearly 3-fold in DF-1 cells upon IBDV infection compared to that in mock-treated cells (Fig. 4C). Interestingly, IBDV-induced RhoA activation was inhibited not only by pretreatment of target cells with RhoA inhibitor I but also by pretreatment with either the c-Src inhibitor AZD0530 or the PI3K/Akt inhibitor LY294002 (Fig. 4C). As expected, all these inhibitors had an inhibitory effect on IBDV internalization but not on virus adhesion (Fig. 4D).
To further investigate whether actin rearrangement also plays a role in IBDV infection, we stained cytoskeleton actin with tetramethyl rhodamine isocyanate (TRITC)-labeled phalloidin in DF-1 cells that were mock infected or infected with IBDV. As shown in Fig. 4E, while the stress fibers crossing the cytoplasm were prominent in control DF-1 cells (left), the stress fibers nearly disappeared and the actin filaments accumulated in the periphery of cells infected with IBDV (middle left). Interestingly, IBDV failed to induce actin rearrangement in cells that were pretreated with either the c-Src inhibitor AZD0530 or the PI3K/Akt inhibitor LY294002 prior to IBDV infection (Fig. 4E, middle right and right). In line with the above-described result, the amount of internalized IBDV (Fig. 4E, green) was reduced significantly in DF-1 cells pretreated with AZD0530 or LY294002 compared to that in cells that were mock treated or treated with the vehicle DMSO (Fig. 4E, top). Furthermore, pretreatment of host DF-1 cells with a cellular actin dynamic inhibitor, either cytochalasin D (34) or jasplakinolide (35), inhibited IBDV internalization in a dose-dependent manner (Fig. 4F). Thus, IBDV induces cellular cytoskeleton actin rearrangement for its internalization, which is dependent on the earlier event of c-Src activation, and inhibition of cytoskeleton actin rearrangement also prevents c-Src activation-mediated IBDV internalization.
Taken together, these results suggest that the elevated c-Src activity induced by IBDV promotes IBDV internalization by targeting the PI3K/Akt-RhoA signaling cascade and cytoskeleton actin rearrangement.
DISCUSSION
IBDV-caused immunosuppression is highly related to virus invasion. However, the molecular processes and signaling events involved in IBDV cellular entry are poorly characterized. In this study, we show that IBDV infection stimulates α4β1 integrin-dependent c-Src tyrosine phosphorylation at Y416 in both the chicken DF-1 cell line and bursa of Fabricius from SPF chickens, which is required for IBDV internalization but not for adhesion. Our data support a model where IBDV infection rapidly activates c-Src via an integrin pathway as an essential feed-forward mechanism to facilitate the entry of IBDV into cells. Thus, modulation of the phosphorylation dynamics of c-Src downstream of integrin by IBDV constitutes an essential mechanism for the rapid and efficient entry of the virus into host cells.
Modulation of c-Src tyrosine activation following virus infection represents an important mechanism that is involved in the virus life cycle (36). Our analysis of the effect of IBDV infection on c-Src tyrosine phosphorylation shows that IBDV could rapidly trigger c-Src tyrosine phosphorylation at an early stage of infection in both the chicken DF-1 cell line and bursa of Fabricius from SPF chickens. Notably, heat-inactivated IBDV failed to induce an increase in c-Src Y416 phosphorylation, suggesting that the infectious ability of IBDV is required for its induction of c-Src tyrosine phosphorylation. This was supported by the finding that the highly pathogenic IBDV strain induced a higher level of c-Src Y416 phosphorylation than did the attenuated lowly pathogenic strain (Fig. 1C and E), suggesting that the ability to induce the level of c-Src Y416 phosphorylation correlates with the pathogenicity of a particular IBDV strain. Thus, IBDV-induced c-Src Y416 phosphorylation can potentially serve as a hallmark of viral infection.
What are the major benefits of IBDV triggering a rapid surge in the tyrosine phosphorylation of c-Src kinase at an early stage when interacting with target cells? The finding that virus internalization but not adhesion was significantly reduced in cells pretreated with the c-Src inhibitor compared with that in mock-treated cells points to the prerequisite of an increase in c-Src Y416 phosphorylation for efficient internalization of IBDV, which was not needed for attachment. This was further supported by the finding that the expression of the c-Src dominant negative mutant specifically disrupted IBDV internalization but not virus adhesion. These data suggest that the attachment and internalization of IBDV are separate phases that are dependent on different membrane surface molecules. So far, there have been three reported putative IBDV receptors on host cells, which are HSP90 in chicken fibroblast (13), integrin in mouse NIH 3T3 cells (12), and IgM in chicken B lymphocytes (11). shRNA downregulation of either the α4 or β1 subunit of surface integrin but not HSP90 disrupted IBDV-induced c-Src tyrosine phosphorylation (Fig. 3B), suggesting that the stimulatory effects of IBDV infection on c-Src phosphorylation are mediated by integrin but not by HSP90. Because of the ablation of IBDV-induced c-Src tyrosine phosphorylation, as expected, knockdown of integrin resulted in a significant decrease in the level of IBDV internalization. However, surprisingly, the level of IBDV attachment was not affected upon the knockdown of α4 or β1 integrin (Fig. 3C), one of the putative IBDV receptors in mouse NIH 3T3 cells (12). Given that IBDV has multiple binding receptors, one potential scenario that could account for the unchanged virus adhesion to integrin knockdown cells is that when the integrin molecules are knocked down, the other major IBDV attachment receptors, e.g., HSP90, could compensate to mediate virus attachment. In support of this interpretation, in contrast with the knockdown of integrin, HSP90 shRNA downregulation caused a significant reduction in IBDV attachment (Fig. 3E). IBDV internalization in HSP90 knockdown cells was also disrupted, but this was most likely a consequence of the loss of virus binding, supporting that HSP90 serves as the major binding receptor for IBDV attachment, although the possibility that HSP90 could also play a direct role in IBDV internalization is not excluded. Thus, these data argue that there is a discrepancy between integrin and HSP90 in mediating IBDV binding. While HSP90 serves as a major IBDV binding receptor that supports virus adhesion to host cells, integrin-mediated IBDV attachment initiates the downstream signaling cascades that mainly regulate IBDV internalization.
The decrease in IBDV internalization by AZD0530 treatment occurred by as early as 1 h postinfection. However, AZD0530 treatment also yielded an even more significant decrease in virus internalization at 24 h postinfection than at 12 h postinfection (Fig. 2C). One possible explanation for this finding is that besides its early role in promoting virus internalization, IBDV-induced c-Src signaling has potential late functions in other steps of the virus replication cycle (e.g., postinternalization). For example, it is likely that the actin arrangement driven by IBDV-induced c-Src signaling will have an impact on IBDV endosomal penetration to promote the release of IBDV particles from endosomes to the cytosol (21) and the postendocytic trafficking of virus-containing endosomal vesicles (Fig. 5).
FIG 5.
Suggested model scheme of the c-Src signaling pathway being induced and hijacked by IBDV to facilitate its infectious entry. Subsequent to binding to cell receptors, IBDV takes advantage of clathrin-independent endocytosis to be internalized (20) and is subsequently unlocked from endosomes through a viral structural peptide pep46-mediated endosomal membrane penetration mechanism (21). Our data suggest that IBDV activates and hijacks the integrin-dependent c-Src–PI3K/Akt–RhoA–actin signaling axis, which plays an essential role in promoting these early processes of IBDV entry. First, actin rearrangement can mediate membrane ruffling and blebbing and create protrusions that encompass viral particles for endocytosis. Second, it is also likely that the actin rearrangement driven by IBDV-induced c-Src signaling may have an impact on IBDV endosomal penetration to promote the release of IBDV particles from endosomes to the cytosol (21). Third, IBDV-induced c-Src–PI3K/Akt–RhoA–actin signaling may play a role in other steps of the IBDV life cycle, e.g., promoting postendocytic trafficking of the virus-containing endosomal vesicles and influencing the spread of the viruses via the regulation of intercellular connections (45).
However, the exact viral component of IBDV that is responsible for inducing c-Src tyrosine phosphorylation remains unknown. Given that the ability of IBDV to induce c-Src Y416 phosphorylation is associated with viral infectivity, the component should serve as a viral virulence factor to confer to IBDV the ability to induce rapid integrin-dependent c-Src tyrosine phosphorylation for viral cellular entry. Furthermore, the component could either indirectly trigger the activation of the integrin signaling pathway or serve as a direct ligand for integrin to initiate the activation of the integrin signaling pathway. It is more likely that VP2, which possesses a surface integrin molecule binding motif, mediates virus infection (34). However, our results suggest that the induced c-Src phosphorylation cannot be attributed entirely to the sole component of a viral protein such as VP2, as heat-inactivated IBDV lost the ability to induce c-Src kinase phosphorylation (Fig. 1B). Given this finding and as IBDV is a nonenveloped, icosahedral virus, it is most likely that the induction of c-Src Y416 phosphorylation may involve multiple components of IBDV, which also need to be maintained in an optimal conformation. Thus, the exact component(s) of IBDV that is responsible for the induction of c-Src tyrosine phosphorylation and the activation pathway needs to be further determined in the future.
The major effects on the entry of IBDV into cells were attributed previously to the process of Akt phosphorylation and downstream cytoskeleton rearrangement. It is possible that the role of c-Src activation in promoting IBDV internalization is simply to enable cytoskeleton rearrangement via the downstream PI3K-RhoA signaling cascade. Supporting this interpretation is the reduction of IBDV-induced c-Src Y416 phosphorylation and IBDV internalization but not adhesion in cells expressing shRNA against integrin or by treatment with the c-Src-specific inhibitor (Fig. 3C and 4D). While pep46-mediated pore formation for endosomal penetration by IBDV occurs after the virus is internalized into endosomes, our work has focused mainly on the signaling aspect associated with IBDV entry at the step of endocytic internalization of the virus. We have shown that IBDV induces and hijacks integrin-dependent c-Src phosphorylation/activation for facilitating its cellular entry but not viral cell binding. This action is via c-Src activation of its downstream signaling axis of c-Src-PI3K/Akt-RhoA GTPase, resulting in actin skeleton rearrangement. Thus, this work extends and nicely complements the existing IBDV endosomal membrane penetration model. In addition to the effect on the endocytotic internalization of IBDV, actin rearrangement driven by c-Src signaling may also have an impact on IBDV endosomal penetration, which could contribute to IBDV replication by an enhancement of the release of IBDV from endosomes.
In summary, our results demonstrate that the activation of the phosphorylation of integrin-mediated c-Src kinase is indispensable for the IBDV entry process, and the phosphorylation level is related to virus virulence. Our model favors the possibility that IBDV activates the integrin–c-Src–PI3K signaling axis by specifically inducing Y416 phosphorylation of c-Src, which serves as a component of the switch apparatus that controls IBDV cell entry (Fig. 5). Thus, our findings provide new insights into the IBDV infection process and c-Src as a potential candidate target for the development of IBDV therapeutic drugs.
MATERIALS AND METHODS
Cell lines, virus strains, and reagents.
HEK293T (ATCC CRL-11268) cells and the chicken fibroblast cell line DF-1 (ATCC CRL-12203) were routinely maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Gibco). IBDV very virulent strain ZJ2000 (vvIBDV) and attenuated strain HZ2 (aIBDV) (adapted for growth in DF-1 cells) were gifts from Yaowei Huang (37). The very virulent strain and the attenuated strain of IBDV were propagated in SPF chicken embryos and DF-1 cells. All propagated virus was concentrated by ultracentrifugation. Mock- or IBDV-infected BFs from SPF chickens were prepared as described previously (38). Mouse anti-Flag monoclonal antibody was purchased from Sigma-Aldrich (St. Louis, MO). Rabbit anti-Src (Y416), mouse anti-Src, rabbit anti-Akt (S493), and mouse anti-Akt antibodies were purchased from Cell Signaling Technology (Danvers, MA). Mouse anti-VP3 polyclonal antibody was generated by immunization with prokaryotic purified VP3. Rabbit polyclonal antibody against GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was purchased from Hangzhou GoodHere Biotechnology Co. Ltd. (Hangzhou, China). The G-LISA biochemical kits for RhoA, Rac1, and Cdc42 activation assays were purchased from Cytoskeleton Inc. (Denver, CO, USA). The pulldown kits for the RhoA activation assay were purchased from Merck Millipore Corporation (Billerica, MA, USA). TRITC-labeled phalloidin used for actin filament staining was purchased from Sigma-Aldrich.
Virus infection and Western blotting.
Monolayers of DF-1 cells were incubated with DMEM without serum for 24 h at 37°C prior to virus infection. The cells were mock infected or infected with concentrated IBDV at a multiplicity of infection (MOI) of 50 at 37°C for 1 h after mock or chemical inhibitor treatment at 37°C for 1 h. The total cell lysates were extracted by using radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) at 4°C for 30 min, followed by centrifugation at 12,000 × g at 4°C for 30 min. Equivalent amounts of the cell lysate were subjected to 12% SDS-PAGE and transferred onto nitrocellulose membranes. After blocking with 5% bovine serum albumin in phosphate-buffered saline (PBS) containing 0.1% Tween 20 (PBST) at room temperature for 1 h, the membranes were incubated with primary antibodies at 4°C overnight, followed by incubation with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG or goat anti-rabbit IgG (Sigma-Aldrich) at 37°C for 1 h. The blots were developed with ECL Prime Western blotting detection reagent (Amersham). Finally, the blots were scanned, and densitometric analysis was performed by using Bio-Rad Quantity One software.
Measurements of GTPase activity.
DF-1 cells were incubated with DMEM (without FBS) for 24 h at 37°C, and the cells were then mock infected or infected with purified IBDV at an MOI of 50 at 37°C for different times. The activity of the small GTPase RhoA was determined by a pulldown assay according to the manufacturer's instructions (Merck Millipore, Billerica, MA, USA). A G-LISA was carried out to determine RhoA activity in DF-1 cells under conditions of IBDV or mock infections with or without a combination of different chemical inhibitors according to the instructions provided with a kit purchased from Cytoskeleton Inc. (Denver, CO, USA).
Plasmids and transfection.
The full-length cDNA of c-Src was amplified from DF-1 cells by two-step RT-PCR according to standard RT-PCR protocols and generated Flag-tagged c-Src expression plasmid Flag-c-Src. The c-Src dominant mutant (39) was generated by QuikChange site-directed mutagenesis using Flag–c-Src as the template, as described previously (10). All c-Src mutants were confirmed by Sanger sequencing. Transfections of DF-1 cells and HEK293T cells were performed by using the Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
qRT-PCR.
Total RNA of the samples was prepared with the TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Reverse transcription of 1 μg total RNA was carried out by using a SuperScript first-strand synthesis system (Fermentas, Pittsburgh, PA) according to the manufacturer's protocol. The amplification of the target gene was used to determine the transcript abundance, and the transcripts of gapdh were used as the internal control. The qRT-PCR assay was performed with an ABI 7500 sequence detector system (Applied Biosystems, Carlsbad, CA) by using the SYBR Premix Ex Taq reagent (TaKaRa, Dalian, China) (40). The primer sequences for qRT-PCR are available upon request.
RNAi and generation of the stable knockdown DF-1 cells.
shRNA target sequences for integrin α4 (4.1 [5′-GCATCTGAATGTTCTGTTTGA-3′], 4.2 [5′-GGAAAGTACCATCAGAGAAGA-3′], and 4.3 [5′-GCACAGCAAGTCAGTACTTCT-3′]) and integrin β1 (1.1 [5′-GCTGTCCTGAGCAAGATATAG-3′], 1.2 [5′-GCATACAATTCCCTTTCTTCA-3′], and 1.3 [5′-GCGATCGATCAAACGGTTTGA-3′]), selected by using the RNA interference (RNAi) target sequence selector available on the GenScript website, and a validated shRNA against chHSP90 (5′-GGACCACTTGGCTGTCAAACA-3′) (41) were generated as described previously (10). An shRNA targeting enhanced green fluorescent protein (EGFP) mRNA, which was not found in the chicken database, was used as a negative control. An shRNA-mediated in vitro screening assay was used to generate and validate the stable specific knockdown cell lines as reported previously (42, 43). In detail, we cotransfected shRNAs expressed in plasmids against the chicken integrin subunit chITGA4 or chITGB1 or chHSP90 together with the corresponding plasmids expressing the Flag-tagged full-length chicken integrin subunit chITGA4 or chITGB1 or chHSP90 into HEK293T cells. The interference efficiency of each shRNA clone in silencing the expressed relevant protein was assessed by Western blotting with anti-Flag antibody. The highest-efficiency shRNA was then packaged into a lentivirus that was used to infect DF-1 cells. After puromycin selection, the stable DF-1 cell line with the downregulated integrin subunit chITGA4 or chITGB1 or chHSP90 was established after continuous selection for 3 weeks, when the control mock-infected DF-1 cells died out completely. The reduction in the mRNA level of chITGA4, chITGB1, or chHSP90 in knockdown DF-1 cells was further validated by qRT-PCR.
Plaque assay.
Series of 10-fold dilutions of virus were inoculated onto DF-1 cell monolayers in 6-well plates. Generally, 1 h later, the monolayers were covered with 1% low-melting-point agarose after 3 washes with PBS. The plates were stained with 1% crystal violet after another 72 h of incubation. The wells containing between 10 and 100 plaques were counted to calculate the titer.
Virus adhesion and internalization assays.
The protocol used for virus adhesion and internalization assays was described previously, with minor modifications (20, 44). The monolayers of DF-1 cells in 6-well plates were mock treated or treated with chemical inhibitors for 1 h at 37°C. The cells were washed 3 times with ice-cold PBS, and 2 ml serum-free DMEM was then added to each well. After the plate was placed at 4°C for 30 min for precooling, cells were infected with IBDV at an MOI of 50, the plate was then placed at 4°C for virus binding, and 1 h later, the supernatant was collected by freeze-thawing three times. Alternatively, the cells placed at 4°C for virus binding were washed with ice-cold PBS 3 times and detached by incubation with Tris-EDTA (TE) at 37°C for 20 min, and the collected cells were then seeded into wells with 2 ml complete DMEM for virus internalization assays. The supernatant from the cells for the adhesion assay or the internalization assay with 3 freeze-thaw cycles was subjected to a plaque assay to measure the virus titer. Alternatively, total RNA was extracted to perform qRT-PCR to assess the binding and internalization of IBDV by amplification of the 5′ UTR of segment A.
Cell staining and confocal laser scanning microscopy.
DF-1 cells in chamber slides were incubated for 24 h with DMEM without serum at 37°C and were then incubated at 37°C for another 1 h with DMSO or the chemical inhibitors AZD0530 and LY294002. The cells were washed 3 times with DMEM without serum and were then mock treated or infected with purified IBDV at an MOI of 50 for 1 h. The cells were fixed in a 4% formaldehyde solution in PBS for 30 min, permeabilized with 0.1% Triton X-100 in PBS after extensive washing, and stained with 50 μg/ml TRITC-labeled phalloidin at room temperature for 1 h. The confocal images were acquired on a Zeiss LSM 510 confocal microscope with excitation wavelengths of 488 nm for Alexa Fluor 488 (green) and 543 nm for Alexa Fluor 568 (red).
Animal experiment.
Thirty 4-week-old SPF chickens (purchased from Hangzhou Jianliang Biotechnology Ltd.) were divided into 3 groups randomly: one group was mock treated as a negative control, and the other two groups were subjected to infection with 104 50% egg infectious doses (EID50) of very virulent IBDV strain ZJ2000 and 104 PFU attenuated IBDV strain HZ2, respectively, via nasal drops. Two pieces of BF were collected from different animals sacrificed at 24, 48, 72, and 96 h postinfection. The negative BFs from mock-treated chickens were collected at the same time. Two pieces of one-third of the BFs were mixed to extract the whole lysate to perform Western blotting to analyze the phosphorylation level of c-Src at Y416 in different groups, and 4 pieces of negative BFs at each time point were mixed to extract the whole lysate for use as the negative control. The remaining BFs were fixed in a 4% formaldehyde solution, and histological sections were taken to perform a double-immune-staining assay to analyze viral protein expression and c-Src phosphorylation by confocal microscopy. The animal experiments were carried out with the approval of the ethics committee of Zhejiang A&F University (ZAFU).
Statistical analysis.
All data are presented as means ± standard deviations (SDs) for each group and were analyzed by using SPSS13.0 (IBM, Armonk, NY, USA). Student's t test was used for comparisons between two groups. A P value of <0.05 was considered statistically significant.
ACKNOWLEDGMENTS
We thank Yongping Wu and Jing Sun for the assistance in the animal experiment and plaque assay.
This work is supported by National Natural Science Foundation of China grant 31502061, Natural Scientific Foundation of Zhejiang Province grant LQ15C180003, and ZAFU Research and Development Fund grants 2014FR019 and 2015FR008.
REFERENCES
- 1.Sharma JM, Kim I-J, Rautenschlein S, Yeh H-Y. 2000. Infectious bursal disease virus of chickens: pathogenesis and immunosuppression. Dev Comp Immunol 24:223–235. doi: 10.1016/S0145-305X(99)00074-9. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberger J, Gelb J Jr. 1978. Response to several avian respiratory viruses as affected by infectious bursal disease virus. Avian Dis 22:95–105. doi: 10.2307/1589512. [DOI] [PubMed] [Google Scholar]
- 3.Ursula I, Gorbalenya AE, Schirrmeier H, Behrens S-E, Letzel T, Mundt E. 2004. VP1 of infectious bursal disease virus is an RNA-dependent RNA polymerase. J Gen Virol 85:2221–2229. doi: 10.1099/vir.0.19772-0. [DOI] [PubMed] [Google Scholar]
- 4.Lombardo E, Maraver A, Espinosa I, Fernández-Arias A, Rodriguez JF. 2000. VP5, the nonstructural polypeptide of infectious bursal disease virus, accumulates within the host plasma membrane and induces cell lysis. Virology 277:345–357. doi: 10.1006/viro.2000.0595. [DOI] [PubMed] [Google Scholar]
- 5.Wu Y, Hong L, Ye J, Huang Z, Zhou J. 2009. The VP5 protein of infectious bursal disease virus promotes virion release from infected cells and is not involved in cell death. Arch Virol 154:1873–1882. doi: 10.1007/s00705-009-0524-4. [DOI] [PubMed] [Google Scholar]
- 6.Kibenge FS, Jackwood DJ, Mercado CC. 1990. Nucleotide sequence analysis of genome segment A of infectious bursal disease virus. J Gen Virol 71:569–577. doi: 10.1099/0022-1317-71-3-569. [DOI] [PubMed] [Google Scholar]
- 7.Spies U, Müller H, Becht H. 1989. Nucleotide sequence of infectious bursal disease virus genome segment A delineates two major open reading frames. Nucleic Acids Res 17:7982–7982. doi: 10.1093/nar/17.19.7982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee CC, Ko TP, Chou CC, Yoshimura M, Doong SR, Wang MY, Wang AH. 2006. Crystal structure of infectious bursal disease virus VP2 subviral particle at 2.6A resolution: implications in virion assembly and immunogenicity. J Struct Biol 155:74–86. doi: 10.1016/j.jsb.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 9.Li Z, Wang Y, Li X, Li X, Cao H, Zheng SJ. 2013. Critical roles of glucocorticoid-induced leucine zipper in infectious bursal disease virus (IBDV)-induced suppression of type I interferon expression and enhancement of IBDV growth in host cells via interaction with VP4. J Virol 87:1221–1231. doi: 10.1128/JVI.02421-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ye C, Jia L, Sun Y, Hu B, Wang L, Lu X, Zhou J. 2014. Inhibition of antiviral innate immunity by birnavirus VP3 protein via blockage of viral double-stranded RNA binding to the host cytoplasmic RNA detector MDA5. J Virol 88:11154–11165. doi: 10.1128/JVI.01115-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo J, Zhang H, Teng M, Fan J-M, You L-M, Xiao Z-J, Yi M-L, Zhi Y-B, Li X-W, Zhang G-P. 2010. Surface IgM on DT40 cells may be a component of the putative receptor complex responsible for the binding of infectious bursal disease virus. Avian Pathol 39:359–365. doi: 10.1080/03079457.2010.506211. [DOI] [PubMed] [Google Scholar]
- 12.Delgui L, Ona A, Gutierrez S, Luque D, Navarro A, Caston JR, Rodriguez JF. 2009. The capsid protein of infectious bursal disease virus contains a functional alpha 4 beta 1 integrin ligand motif. Virology 386:360–372. doi: 10.1016/j.virol.2008.12.036. [DOI] [PubMed] [Google Scholar]
- 13.Lin TW, Lo CW, Lai SY, Fan RJ, Lo CJ, Chou YM, Thiruvengadam R, Wang AHJ, Wang MY. 2007. Chicken heat shock protein 90 is a component of the putative cellular receptor complex of infectious bursal disease virus. J Virol 81:8730–8741. doi: 10.1128/JVI.00332-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu B, Zhang Y, Jia L, Wu H, Fan C, Sun Y, Ye C, Liao M, Zhou J. 2015. Binding of the pathogen receptor HSP90AA1 to avibirnavirus VP2 induces autophagy by inactivating the AKT-MTOR pathway. Autophagy 11:503–515. doi: 10.1080/15548627.2015.1017184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eustace BK, Sakurai T, Stewart JK, Yimlamai D, Unger C, Zehetmeier C, Lain B, Torella C, Henning SW, Beste G, Scroggins BT, Neckers L, Ilag LL, Jay DG. 2004. Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nat Cell Biol 6:507–514. doi: 10.1038/ncb1131. [DOI] [PubMed] [Google Scholar]
- 16.Bottcher B, Kiselev NA, Stel'Mashchuk VY, Perevozchikova NA, Borisov AV, Crowther RA. 1997. Three-dimensional structure of infectious bursal disease virus determined by electron cryomicroscopy. J Virol 71:325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coulibaly F, Chevalier C, Gutsche I, Pous J, Navaza J, Bressanelli S, Delmas B, Rey FA. 2005. The birnavirus crystal structure reveals structural relationships among icosahedral viruses. Cell 120:761–772. doi: 10.1016/j.cell.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 18.Garriga D, Querol-Audi J, Abaitua F, Saugar I, Pous J, Verdaguer N, Caston JR, Rodriguez JF. 2006. The 2.6-angstrom structure of infectious bursal disease virus-derived T=1 particles reveals new stabilizing elements of the virus capsid. J Virol 80:6895–6905. doi: 10.1128/JVI.00368-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mould AP, Humphries MJ. 1991. Identification of a novel recognition sequence for the integrin alpha 4 beta 1 in the COOH-terminal heparin-binding domain of fibronectin. EMBO J 10:4089–4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yip CW, Hon CC, Zeng F, Leung FC. 2012. Cell culture-adapted IBDV uses endocytosis for entry in DF-1 chicken embryonic fibroblasts. Virus Res 165:9–16. doi: 10.1016/j.virusres.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 21.Galloux M, Libersou S, Morellet N, Bouaziz S, Da Costa B, Ouldali M, Lepault J, Delmas B. 2007. Infectious bursal disease virus, a non-enveloped virus, possesses a capsid-associated peptide that deforms and perforates biological membranes. J Biol Chem 282:20774–20784. doi: 10.1074/jbc.M701048200. [DOI] [PubMed] [Google Scholar]
- 22.Mitra SK, Schlaepfer DD. 2006. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol 18:516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 23.Veettil MV, Sharma-Walia N, Sadagopan S, Raghu H, Sivakumar R, Naranatt PP, Chandran B. 2006. RhoA-GTPase facilitates entry of Kaposi's sarcoma-associated herpesvirus into adherent target cells in a Src-dependent manner. J Virol 80:11432–11446. doi: 10.1128/JVI.01342-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang C-M, Lin C-C, Lee I-T, Lin Y-H, Yang CM, Chen W-J, Jou M-J, Hsiao L-D. 2012. Japanese encephalitis virus induces matrix metalloproteinase-9 expression via a ROS/c-Src/PDGFR/PI3K/Akt/MAPKs-dependent AP-1 pathway in rat brain astrocytes. J Neuroinflammation 9:12. doi: 10.1186/1742-2094-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng CY, Huang WR, Chi PI, Chiu HC, Liu HJ. 2015. Cell entry of bovine ephemeral fever virus requires activation of Src-JNK-AP1 and PI3K-Akt-NF-κB pathways as well as Cox-2-mediated PGE2/EP receptor signalling to enhance clathrin-mediated virus endocytosis. Cell Microbiol 17:967–987. doi: 10.1111/cmi.12414. [DOI] [PubMed] [Google Scholar]
- 26.Taylor MP, Koyuncu OO, Enquist LW. 2011. Subversion of the actin cytoskeleton during viral infection. Nat Rev Microbiol 9:427–439. doi: 10.1038/nrmicro2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bjorge JD, Jakymiw A, Fujita DJ. 2000. Selected glimpses into the activation and function of Src kinase. Oncogene 19:5620–5635. doi: 10.1038/sj.onc.1203923. [DOI] [PubMed] [Google Scholar]
- 28.Boggon TJ, Eck MJ. 2004. Structure and regulation of Src family kinases. Oncogene 23:7918–7927. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- 29.Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. 1999. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell 3:629–638. doi: 10.1016/S1097-2765(00)80356-1. [DOI] [PubMed] [Google Scholar]
- 30.Mercer J, Helenius A. 2009. Virus entry by macropinocytosis. Nat Cell Biol 11:510–520. doi: 10.1038/ncb0509-510. [DOI] [PubMed] [Google Scholar]
- 31.Wenqing X, Harrison S, Eck M. 1997. Three-dimensional structure of the tyrosine kinase c-Src. Nature 385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- 32.Wei L, Hou L, Zhu S, Wang J, Zhou J, Liu J. 2011. Infectious bursal disease virus activates the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway by interaction of VP5 protein with the p85alpha subunit of PI3K. Virology 417:211–220. doi: 10.1016/j.virol.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 33.Reddy MA, Prasadarao NV, Wass CA, Kim KS. 2000. Phosphatidylinositol 3-kinase activation and interaction with focal adhesion kinase in Escherichia coli K1 invasion of human brain microvascular endothelial cells. J Biol Chem 275:36769–36774. doi: 10.1074/jbc.M007382200. [DOI] [PubMed] [Google Scholar]
- 34.Casella JF, Flanagan MD, Lin S. 1981. Cytochalasin D inhibits actin polymerization and induces depolymerization of actin filaments formed during platelet shape change. Nature 293:302–305. doi: 10.1038/293302a0. [DOI] [PubMed] [Google Scholar]
- 35.Senderowicz AM, Kaur G, Sainz E, Laing C, Inman WD, Rodriguez J, Crews P, Malspeis L, Grever MR, Sausville EA. 1995. Jasplakinolide's inhibition of the growth of prostate carcinoma cells in vitro with disruption of the actin cytoskeleton. J Natl Cancer Inst 87:46–51. doi: 10.1093/jnci/87.1.46. [DOI] [PubMed] [Google Scholar]
- 36.Pagano MA, Tibaldi E, Palù G, Brunati AM. 2013. Viral proteins and Src family kinases: mechanisms of pathogenicity from a “liaison dangereuse.” World J Virol 2:71–78. doi: 10.5501/wjv.v2.i2.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu L, Li JR, Huang YW, Dikki J, Deng R. 2001. Molecular characteristics of full-length genomic segment A of three infectious bursal disease viruses in China: two attenuated strains and one virulent field strain. Avian Dis 45:862–874. doi: 10.2307/1592866. [DOI] [PubMed] [Google Scholar]
- 38.Wu Y, Peng C, Xu L, Zheng X, Liao M, Yan Y, Jin Y, Zhou J. 2012. Proteome dynamics in primary target organ of infectious bursal disease virus. Proteomics 12:1844–1859. doi: 10.1002/pmic.201100479. [DOI] [PubMed] [Google Scholar]
- 39.Gonzalez L, Agullo-Ortuno MT, Garcia-Martinez JM, Calcabrini A, Gamallo C, Palacios J, Aranda A, Martin-Perez J. 2006. Role of c-Src in human MCF7 breast cancer cell tumorigenesis. J Biol Chem 281:20851–20864. doi: 10.1074/jbc.M601570200. [DOI] [PubMed] [Google Scholar]
- 40.Bustin SA, Mueller R. 2005. Real-time reverse transcription PCR (qRT-PCR) and its potential use in clinical diagnosis. Clin Sci 109:365–379. doi: 10.1042/CS20050086. [DOI] [PubMed] [Google Scholar]
- 41.Yuan W, Zhang X, Xia X, Sun H. 2012. Inhibition of infectious bursal disease virus infection by artificial microRNAs targeting chicken heat-shock protein 90. J Gen Virol 93:876–879. doi: 10.1099/vir.0.039172-0. [DOI] [PubMed] [Google Scholar]
- 42.Chih B, Engelman H, Scheiffele P. 2005. Control of excitatory and inhibitory synapse formation by neuroligins. Science 307:1324–1328. doi: 10.1126/science.1107470. [DOI] [PubMed] [Google Scholar]
- 43.Karpala AJ, Lowenthal JW, Bean AG. 2008. Activation of the TLR3 pathway regulates IFNbeta production in chickens. Dev Comp Immunol 32:435–444. doi: 10.1016/j.dci.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 44.Yamada Y, Liu DX. 2009. Proteolytic activation of the spike protein at a novel RRRR/S motif is implicated in furin-dependent entry, syncytium formation, and infectivity of coronavirus infectious bronchitis virus in cultured cells. J Virol 83:8744–8758. doi: 10.1128/JVI.00613-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roberts KL, Manicassamy B, Lamb RA. 2015. Influenza A virus uses intercellular connections to spread to neighboring cells. J Virol 89:1537–1549. doi: 10.1128/JVI.03306-14. [DOI] [PMC free article] [PubMed] [Google Scholar]