

ABSTRACT
Adeno-associated virus (AAV) vectors have made great progress in their use for gene therapy; however, fundamental aspects of AAV's capsid assembly remain poorly characterized. In this regard, the discovery of assembly-activating protein (AAP) sheds new light on this crucial part of AAV biology and vector production. Previous studies have shown that AAP is essential for assembly; however, how its mechanistic roles in assembly might differ among AAV serotypes remains uncharacterized. Here, we show that biological properties of AAPs and capsid assembly processes are surprisingly distinct among AAV serotypes 1 to 12. In the study, we investigated subcellular localizations and assembly-promoting functions of AAP1 to -12 (i.e., AAPs derived from AAV1 to -12, respectively) and examined the AAP dependence of capsid assembly processes of these 12 serotypes using combinatorial approaches that involved immunofluorescence and transmission electron microscopy, barcode-Seq (i. e., a high-throughput quantitative method using DNA barcodes and a next-generation sequencing technology), and quantitative dot blot assays. This study revealed that AAP1 to -12 are all localized in the nucleus with serotype-specific differential patterns of nucleolar association; AAPs and assembled capsids do not necessarily colocalize; AAPs are promiscuous in promoting capsid assembly of other serotypes, with the exception of AAP4, -5, -11, and -12; assembled AAV5, -8, and -9 capsids are excluded from the nucleolus, in contrast to the nucleolar enrichment of assembled AAV2 capsids; and, surprisingly, AAV4, -5, and -11 capsids are not dependent on AAP for assembly. These observations highlight the serotype-dependent heterogeneity of the capsid assembly process and challenge current notions about the role of AAP and the nucleolus in capsid assembly.
IMPORTANCE Assembly-activating protein (AAP) is a recently discovered adeno-associated virus (AAV) protein that promotes capsid assembly and provides new opportunities for research in assembly. Previous studies on AAV serotype 2 (AAV2) showed that assembly takes place in the nucleolus and is dependent on AAP and that capsids colocalize with AAP in the nucleolus during the assembly process. However, through the investigation of 12 different AAV serotypes (AAV1 to -12), we find that AAP is not an essential requirement for capsid assembly of AAV4, -5, and -11, and AAP, assembled capsids, and the nucleolus do not colocalize for all the serotypes. In addition, we find that there are both serotype-restricted and serotype-promiscuous AAPs in their assembly roles. These findings challenge widely held beliefs about the importance of the nucleolus and AAP in AAV assembly and show the heterogeneous nature of the assembly process within the AAV family.
KEYWORDS: assembly-activating protein, adeno-associated virus, capsid assembly, gene therapy, nucleolus, parvovirus
INTRODUCTION
Capsid assembly of icosahedral viruses has been an important area of research with an impact on multiple fields. Foremost is the basic biology behind how pathogenic viral proteins hijack the host cell to aid in their assembly and how viral capsid proteins fit together with the ultimate end of devising antiviral therapies (1). While there are a multitude of morphologies that viruses can have, icosahedral symmetry is found broadly in perhaps half of the known viral families (2). A model icosahedral virus with T=1 symmetry (3) is adeno-associated virus (AAV), a parvovirus that belongs to the genus Dependoparvovirus of the family Parvoviridae. AAV has recently become a well-regarded vector for in vivo gene therapy with successful clinical trials for hemophilia B (4), lipoprotein lipase deficiency (5), and Leber congenital amaurosis (6, 7), among others (reviewed by Mingozzi and High [8]), thus making the study of its capsid assembly an attractive pursuit both for gene therapy applications and for furthering our knowledge of parvovirus biology.
AAV is a small, nonenveloped virus with a single-stranded DNA genome of 4.7 kb containing two genes, rep and cap, between two inverted terminal repeats. The rep gene produces nonstructural Rep proteins essential for viral genome replication and packaging. The cap gene produces the three structural proteins VP1, VP2, and VP3, translated from different start codons in a single open reading frame (ORF). Alternative mRNA splicing and the combined use of an ATG codon and an alternative start codon for the initiation of VP protein translation lead to appropriate capsid stoichiometry at a VP1/VP2/VP3 ratio of approximately 1:1:10 (9, 10). It was long believed that the AAV genome encodes only the Rep and VP proteins, until 2010, when a second +1-frameshifted open reading frame (ORF) that encodes a 204-amino-acid-long nonstructural protein was identified within the cap gene of AAV serotype 2 (AAV2) (11). This new AAV protein has been named assembly-activating protein (AAP), after the role that it plays in capsid assembly (11).
The AAP ORFs have been found in all parvoviruses that belong to the genus Dependoparvovirus (11, 12), and among them, AAP derived from AAV2 (i.e., AAP2) has been the main focus of the studies to date. AAP2 is a nucleolus-localizing protein essential for AAV2 capsid assembly (11, 13). When the AAV2 VP proteins are expressed in cultured cells in the absence of AAP, the VP proteins can be found in both the cytoplasm and the nucleus but are excluded from the nucleolus, and there is no detectable capsid assembly (11, 13). When the AAV2 VP proteins and AAP2 are coexpressed in cells, the VP proteins translocate to and accumulate in the nucleolus together with AAP2 and assemble into capsids (11, 13). AAP2 can form high-molecular-weight oligomers and change the conformation of a wide range of VP protein oligomer intermediates, leading to the formation of capsid-specific antibody-positive oligomers before the capsids are fully assembled (14). This, together with the demonstration of AAP2-AAV2 VP3 interactions through hydrophobic regions, may suggest that AAP functions as a scaffolding protein in the capsid assembly reaction as well as a transporter, targeting VP proteins to the nucleolus for assembly (14, 15). As for the role of AAPs derived from other AAV serotypes, Sonntag et al. demonstrated that AAP is essential for AAV1, -8, and -9 assembly by showing that the expression of VP3 alone does not yield capsids but that assembly can be restored by the coexpression of heterologous AAP2 (12). They also showed that such a cross-complementation of capsid assembly with heterologous AAP2 does not easily extend to AAV5, one of the most divergent serotypes (12). In addition, those researchers found that AAP1, -2, and -5 protein expression levels could be significantly affected by the nature of the coexpressed homologous and heterologous VP proteins (12). While these findings are intriguing, a number of questions remain about the roles of AAP in capsid assembly and elsewhere in the life cycle.
In this study, to further understand the roles of AAPs in AAV capsid assembly, we comprehensively characterized AAPs derived from AAV1 to -12 (AAP1 to -12, respectively). To this end, we investigated the subcellular localizations of AAP1 to -12 and their assembly-promoting abilities for homologous and heterologous AAV VP3 proteins derived from all 12 serotypes by immunofluorescence microscopy, a comprehensive AAP-VP3 cross-complementation assay, and transmission electron microscopy (TEM). These analyses revealed AAP's various capabilities for nucleolar enrichment and heterologous capsid assembly among the serotypes and serotype-dependent differences in the sites of capsid assembly inside the nucleus. The most striking finding was that VP proteins derived from AAV4, -5, and -11 could assemble without requiring AAP, in contrast to the VP proteins from the other nine serotypes, which required AAP for assembly. A recombinant AAV5 vector, produced in the absence of AAP, was found to be infectious and capable of transducing cells, indicating that AAP is not a necessary component for the production of infectious virions for some AAV serotypes.
RESULTS
Successful expression of AAP1 to -12 in cultured cells.
Initial work by Sonntag et al. found that the expression level of AAP in transfected cells depended on the serotype and was especially low for AAP5. In an effort to try and increase AAP production in our transfections, we used the 750-bp cytomegalovirus immediate early (CMV-IE) enhancer-promoter fused with the 132-bp intervening sequence (IVS) that is known to enhance the stability of mRNA (16) to drive the expression of FLAG-tagged codon-optimized AAP2. We also changed the native non-ATG start codon to the strong ATG start codon. Previously, we found that this expression plasmid construction led to strong steady-state levels of AAP2 (13). Prompted by this observation, we constructed a panel of FLAG-tagged AAP expression plasmids for the AAPs from AAV1 to -12, pCMV3-FLAG-cmAAPx (where x is 1 to 12), using the same plasmid backbone. All the AAPs were readily detected by Western blot analysis of plasmid-transfected human embryonic kidney 293 (HEK 293) cells in the absence of coexpressed VP proteins, with each AAP showing primarily a discrete single band, except for AAP8, -9, and -10, which had faint secondary bands (Fig. 1). Interestingly, the molecular masses of FLAG-tagged AAPs that were experimentally determined by Western blot analysis were higher than the theoretical molecular masses by 14% to 63%, with AAP4 showing the largest discrepancy (Table 1). An increase of the molecular masses by the addition of a FLAG tag, which is only 1 kDa, does not account for this discrepancy. Such a discrepancy was previously observed for AAP5 (12). The slower-than-expected migration might be due simply to differences in amino acid sequences, although the possibility of posttranslational modification has not been ruled out.
FIG 1.
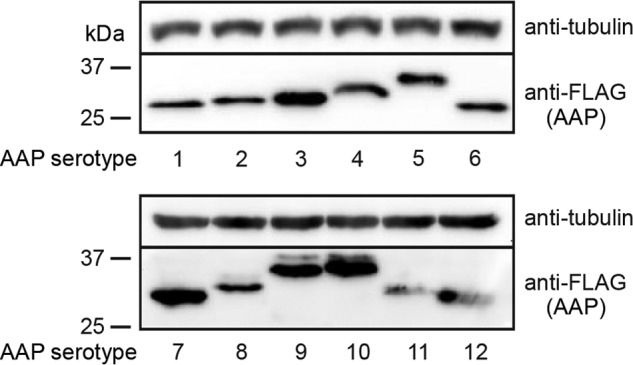
Expression of AAP1 to -12 in HEK 293 cells. HEK 293 cells were transiently transfected with a plasmid expressing the respective FLAG-tagged AAPs indicated below each panel, and AAP expression in transfected cells was analyzed by Western blotting using anti-FLAG antibody. α-Tubulin was used as a loading control. Molecular mass markers (kilodaltons) are shown at the left.
TABLE 1.
Theoretically and experimentally determined molecular masses of FLAG-tagged AAPs
| AAP | Theoretical molecular mass (kDa) | Mean experimentally determined molecular mass (kDa) ± SDa | % difference between theoretical and experimentally determined molecular massesb |
|---|---|---|---|
| 1 | 22 | 27.3 ± 1.7 | 24 |
| 2 | 24 | 27.3 ± 1.6 | 14 |
| 3B | 23 | 28.5 ± 1.2 | 24 |
| 4 | 19 | 31.0 ± 1.1 | 63 |
| 5 | 22 | 33.2 ± 0.9 | 51 |
| 6 | 22 | 27.3 ± 1.3 | 24 |
| 7 | 22 | 27.8 ± 1.7 | 26 |
| 8 | 22 | 30.4 ± 1.5 | 38 |
| 9 | 22 | 33.7 ± 0.7 | 53 |
| 10 | 22 | 34.4 ± 0.7 | 56 |
| 11 | 20 | 29.7 ± 0.6 | 48 |
| 12 | 20 | 28.6 ± 0.7 | 43 |
The molecular mass of each FLAG-tagged AAP was determined by Western blot analysis in four biological replicate experiments.
Percent increase of the molecular mass as determined by Western blot analysis compared to the theoretical molecular mass.
AAPs show various subnuclear localizations.
Our previous work on identifying amino acids involved in AAP2 nuclear and nucleolar localization identified five clusters of basic amino acids (AAP2 basic region 1 [BR1] to AAP2 BR5) near its C terminus that contained overlapping nuclear and nucleolar localization signals (NLS-NoLS) (13). While these regions are generally well conserved among the AAPs of different AAV serotypes (AAP BR1 to AAP BR5) (Fig. 2), certain AAPs lack some or nearly all of the lysine and arginine residues found in their corresponding protein sequences (Fig. 2). For instance, AAP5 has more proline residues and fewer basic residues than does AAP2 in these regions, leading us to hypothesize that AAP5 might be excluded from the nucleolus, as discussed in our previous publication (13). To test this hypothesis and determine the subcellular localization of AAP1 to -12, HeLa cells were transiently transfected with each FLAG-tagged AAP-expressing pCMV3-FLAG-cmAAPx plasmid (where x is 1 to 12) individually, and localization was determined 48 h later by using an anti-FLAG antibody. To assess statistically the degree of nucleolar association of AAP1 to -12, we counted 50 nuclei with the anti-FLAG antibody signal and categorized the nuclear staining patterns into the following two groups: “No+,” showing nucleolar association, and “No−,” showing decreased or no nucleolar association. All of the AAPs were found in the nucleus, but their nucleolar localization varied by serotype (Fig. 3). AAP5 and AAP9, which have the least basic charge between BR1 and BR5, exhibited a substantially decreased nucleolar association compared to those of the other 10 serotypes (adjusted P value of <0.001 for all possible pairwise comparisons by using Fisher's exact test) (Tables 2 to 4). The other 10 AAPs showed unambiguous nucleolar localization to various degrees (Fig. 3). In a random model in which only two AAPs show a distinctively decreased nucleolar association among a total of 12 AAPs, the probability that the 2 AAPs showing the lowest pI values are also the 2 AAPs that exhibit a distinctively decreased nucleolar association is 0.015. This indicates a strong correlation between low pI values and decreased nucleolar association. There is another basic amino-acid-rich region further down toward the C terminus (AAP BR6); however, such a strong relationship between low pI and decreased nucleolar association was not observed in the larger segment containing AAP BR6 (Table 4). Therefore, the amino acid stretch containing only AAP BR1 to AAP BR5 is the most likely determinant of nucleolar localization. These results demonstrate that, while all 12 AAPs are localized to the nucleus, the strong nucleolar association observed for AAP2 is not a general characteristic of AAPs and that AAPs with lower pI values in the well-conserved basic amino-acid-rich region near the C terminus show decreased nucleolar association.
FIG 2.

Sequence alignment of the C termini of AAP1 to -12. Sequences of the basic amino-acid-rich C termini of AAP1 to -12 are aligned. The basic amino-acid-rich regions BR1 to BR6 are indicated with overlines. Positively charged lysine (K) and arginine (R) residues are indicated with red letters. These 6 BRs are defined arbitrarily based on sequence alignment data. BR1 to BR5 were previously identified as the NLS-NoLS in the context of AAP2.
FIG 3.

Intracellular localization of AAP1 to -12. HeLa cells were transiently transfected with a plasmid expressing the respective FLAG-tagged AAPs indicated in each panel. The cells were fixed at 48 h posttransfection, immunostained with anti-FLAG antibody (green) and antinucleostemin antibody (red), and counterstained with DAPI (blue) before being imaged on a Zeiss LSM 710 confocal microscope with a 100×/1.46-NA objective. Two representative fields of view are shown for each AAP. (A) AAP1; (B) AAP2; (C) AAP3B; (D) AAP4; (E) AAP5; (F) AAP6; (G) AAP7; (H) AAP8; (I) AAP9; (J) AAP10; (K) AAP11; (L) AAP12. All AAPs are enriched in the nucleus. AAP5 and -9 show a pattern of decreased nucleolar association, while nucleolar signals are observed with other AAPs to various degrees. Note that the ring-shaped anti-FLAG antibody staining pattern with a central hollow (A to D, F, G, and J to L) is most likely a staining artifact caused by the overexpression of a nucleolus-localizing protein and does not necessarily indicate that nucleolar expression is enriched in the nucleolar periphery (13).
TABLE 2.
Nucleolar association of AAPs
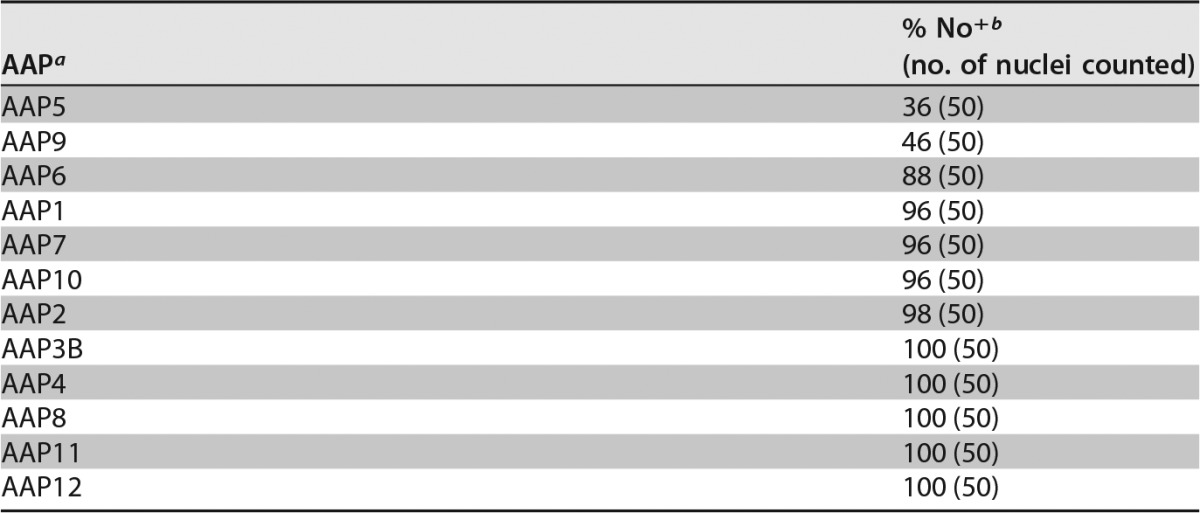
a The order of AAPs is sorted by the No+ percentage.
b % No+ is the percentage of the nuclei showing a nucleolar association of AAP among the total nuclei counted that were stained with the anti-FLAG antibody (for detection of FLAG-tagged AAP).
TABLE 3.
Biochemical properties of AAP BR1 to BR5
| AAPa | No. of residuesb |
pI | |
|---|---|---|---|
| H, K, or R | D or E | ||
| AAP5 | 8 | 0 | 11.45 |
| AAP9 | 11 | 1 | 12.37 |
| AAP3B | 15 | 0 | 12.43 |
| AAP8 | 12 | 0 | 12.43 |
| AAP7 | 13 | 0 | 12.52 |
| AAP10 | 14 | 0 | 12.52 |
| AAP6 | 13 | 0 | 12.65 |
| AAP2 | 15 | 0 | 12.70 |
| AAP11 | 13 | 0 | 12.70 |
| AAP4 | 14 | 0 | 12.78 |
| AAP1 | 12 | 0 | 12.96 |
| AAP12 | 13 | 0 | 13.04 |
The order of AAPs is sorted by pI (lowest to highest). pI values were determined by using BioPerl pICalculator.
D, aspartic acid; E, glutamic acid; H, histidine; K, lysine; R, arginine.
TABLE 4.
Biochemical properties of AAP BR1 to BR6 toward the C-terminus
| AAPa | No. of residuesb |
pI | |
|---|---|---|---|
| H, K, or R | D or E | ||
| AAP5 | 15 | 1 | 11.86 |
| AAP2 | 18 | 2 | 12.26 |
| AAP8 | 15 | 0 | 12.26 |
| AAP3B | 18 | 1 | 12.40 |
| AAP7 | 16 | 0 | 12.48 |
| AAP10 | 17 | 0 | 12.48 |
| AAP9 | 14 | 1 | 12.52 |
| AAP6 | 16 | 0 | 12.57 |
| AAP11 | 13 | 0 | 12.70 |
| AAP1 | 15 | 0 | 12.74 |
| AAP4 | 14 | 0 | 12.78 |
| AAP12 | 13 | 0 | 13.04 |
The order of AAPs is sorted by pI (lowest to highest). pI values were determined by using BioPerl pICalculator.
D, aspartic acid; E, glutamic acid; H, histidine; K, lysine; R, arginine.
Investigation of assembly of capsids of various serotypes without AAP.
It is well established that AAV2 capsid assembly requires AAP (11, 13). As for other serotypes, Sonntag et al. previously reported that AAV1, -5, -8, and -9 capsid assembly also requires AAP (12). To reproduce the observations by Sonntag et al. and investigate AAP-independent assembly for other serotypes, we expressed AAV1 to -12 VP3 proteins from pCMV1-AAVxVP3 plasmids (where x is 1 to 12) in HEK 293 cells in the presence or absence of cognate AAP expressed from a separate plasmid. In this transfection system, we also provided the components necessary for the packaging of recombinant AAV genomes encoding enhanced green fluorescent protein (eGFP) into assembled capsids. We then quantified packaged viral genomes as surrogates of assembly by quantitative dot blotting in an experiment performed in biological duplicates. In the presence of AAP, all serotypes showed assembled capsids at readily detectable levels, as expected (Fig. 4). Surprisingly, in the absence of AAP, the signals from AAV4, -5, and -11 were clearly beyond the background level, in contrast to the other serotypes (Fig. 4). This result strongly indicates that AAV4, -5, and -11 could assemble without AAP. As for the other serotypes, signals were below the lowest standard signal and near or below the background signal. This indicates that capsid assembly of the other serotypes did not occur or was significantly impaired.
FIG 4.
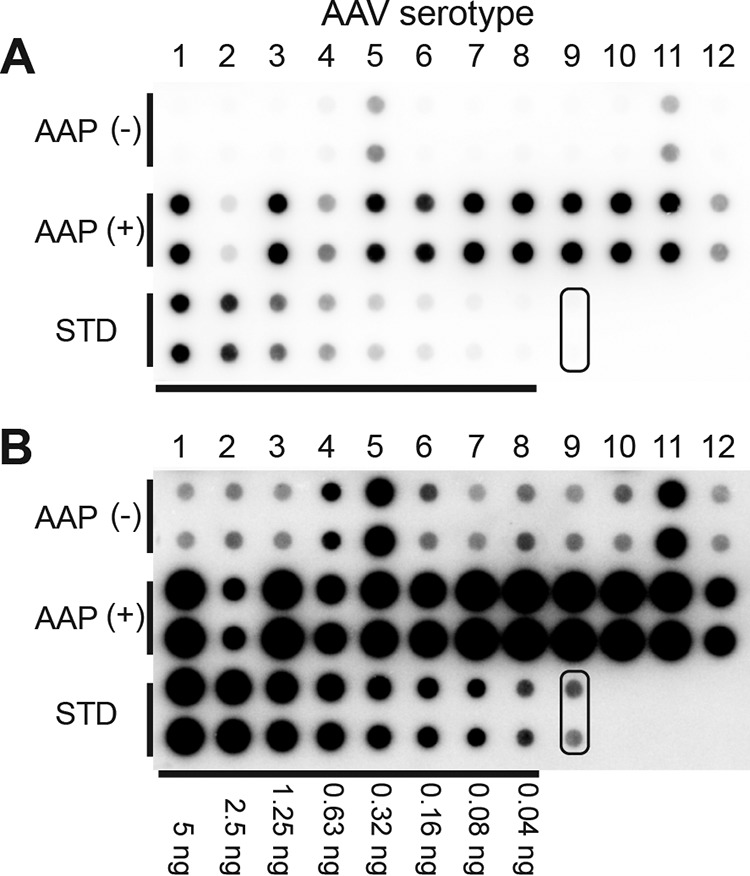
Quantitative dot blot assay to determine titers of VP3 capsids produced in the presence or absence of AAP. (A) AAP-independent assembly of VP3 proteins of AAV1 to -12 assessed by a quantitative dot blot assay in an experiment performed in biological duplicates. VP3-only particles derived from AAV1 to -12 that contained a double-stranded AAV-CMV-GFP genome were produced in 6-well plates in the presence or absence of their cognate AAPs. Benzonase-resistant DNA was recovered from 7% of the samples obtained from each well, blotted onto a nylon membrane together with standards (STD) (i.e., linearized pEMBL-CMV-GFP plasmid), and probed with a 32P-labeled GFP probe. Pairs of dots in each combination represent the results obtained from two separate transfections. The pair of dots indicated with a rounded rectangle are negative controls. (B) Same blot as the one shown in panel A. The signals were intensified equally across the entire image by using ImageJ.
AAV5 VP3 can assemble capsids without AAP.
To examine potential AAP-independent assembly further, we chose to study AAV5 in depth because there are commercially available reagents for this serotype that allow verification of capsid assembly on multiple levels and because, contrary to our experience, AAV5 assembly was previously reported to require cognate AAP5 (12). In all the experiments described above, the VP3 plasmids used still retained the C-terminal 80% of the AAP ORFs. Without its start codon, AAP should not be expressed, but it was important to rule out any possibility that an unidentified cryptic start codon could have provided a functional phenotype through the expression of N-terminally truncated AAP5. To exclude this possibility, we created plasmids carrying codon-modified AAV2 (cmAAV2) and cmAAV5 VP3 ORFs (pCMV1-AAV2cmVP3 and pCMV1-AAV5cmVP3, respectively) and used them in subsequent experiments. These mutated AAP ORFs within the cmVP3 ORFs should not express any peptides that exhibit the capsid assembly-promoting function at detectable levels (see Discussion). To verify that the AAV5 cmVP3 proteins could also assemble capsids without AAP, we transiently transfected HeLa cells with pCMV1-AAV2cmVP3 or pCMV1-AAV5cmVP3 together with either the cognate AAP-expressing plasmid (pCMV3-FLAG-cmAAP2 or pCMV3-FLAG-cmAAP5) or pCMV3-GFP, a GFP-expressing control plasmid devoid of AAP expression. Forty-eight hours after transfection, the cells were stained with anti-FLAG and anti-AAV capsid antibodies (A20 to detect assembled AAV2 VP3 capsids and ADK5a to detect assembled AAV5 VP3 capsids). As previously shown (11), AAV2 VP3 was able to assemble capsids only when AAP2 was supplied in trans (Fig. 5A). In contrast, AAV5 VP3 was able to produce capsid antibody-positive staining regardless of the presence or absence of AAP5 (Fig. 5A). The ADK5a antibody is an anti-AAV5 antibody that should be specific for only intact AAV5 capsids and not monomers or oligomers of AAV5 VP proteins (17). Thus, positive staining with ADK5a strongly indicates that AAV5 VP3 can assemble into capsids without a need for AAP. The same approach was used to investigate capsid assembly for AAV8 VP3 and AAV9 VP3 in the presence or absence of the cognate AAP. For this analysis, we used pCMV1-AAV8VP3 or pCMV1-AAV9VP3 together with the cognate AAP-expressing plasmids. Assembled capsid-specific signals were readily detected in cells expressing AAP; however, none of the cells had assembled AAV8 or AAV9 VP3 capsid signals in the absence of AAP expression (Fig. 5A). These observations complement the dot blot results and strongly support that capsid assembly requires AAP for the AAV2, AAV8, and AAV9 VP3 proteins.
FIG 5.

Capsid assembly of AAV2, -4, -5, -8, and -9 in HeLa cells. HeLa cells were transiently transfected with plasmids expressing VP3 proteins derived from AAV2,-4, -5, -8, or -9 in the presence (+) or absence (−) of a cotransfected plasmid expressing their cognate AAP. The groups that received no AAP-expressing plasmid were transfected with pCMV3-GFP instead to ensure successful transfection and maintain the total quantity of transfected DNA constant across the groups. The cells were fixed at 48 h posttransfection, immunostained with the antibodies indicated in each panel, and counterstained with DAPI. The images were obtained by using a Zeiss LSM 710 confocal microscope with a 63×/1.4-NA objective. Three representative fields of view are shown for each condition. (A) Cells were stained with anti-AAV capsid antibody (A20 for AAV2, ADK4 for AAV4, ADK5a for AAV5, ADK8 for AAV8, and ADK8/9 for AAV9) (red) and anti-FLAG antibody (white). (B) Cells were stained with antinucleostemin antibody (white), anti-AAV capsid antibodies (green), and anti-FLAG antibody (red). Bar, 10 μm.
Finally, TEM was used to visualize directly the assembly (or not) of the AAV5 “VP3-only” capsid in the presence or absence of AAP5. HEK 293 cells were transfected by using either plasmid pCMV1-AAV2cmVP3, with or without pCMV3-FLAG-cmAAP2, or pCMV1-AAV5cmVP3, with or without pCMV3-FLAG-cmAAP5. At 5 days posttransfection, the medium and cells were harvested and subjected to three rounds of purification by cesium chloride (CsCl) density gradient ultracentrifugation to prepare samples for TEM. In the final round, a discrete band was found at refractive indices (RIs) of 1.3640 to 1.3645, except for AAV2 VP3 without AAP2 (Fig. 6A and D). After fractions were collected, an enzyme-linked immunosorbent assay (ELISA) specific for intact capsids was performed on each fraction to confirm the presence of assembled AAV2 or AAV5. Assembled AAV2 capsids were not detected in the absence of AAP2 but were present when AAP2 was coexpressed (Fig. 6B), consistent with the observed CsCl banding and results of immunofluorescence microscopy. AAV5 capsids were detected in samples of both AAV5 VP3 without AAP5 and AAV5 VP3 with AAP5 by an ELISA (Fig. 6E). An analysis of TEM images of the fractions showing high ELISA optical density (OD) values confirmed the presence of assembled capsids, while the sample of AAV2 VP3 without AAP2 with the corresponding RI (1.3640) did not contain any capsids (Fig. 6C and F). AAV5 VP3 capsids produced in the absence of coexpressed AAP5 were morphologically indistinguishable from those produced in the presence of AAP5 (Fig. 6F). Thus, we concluded that the AAV5 VP3 proteins are capable of forming capsids in the absence of AAP.
FIG 6.

Transmission electron microscopy of AAV VP3-only capsids produced with or without AAP. HEK 293 cells were transfected with plasmids expressing VP3 proteins and AAP proteins as indicated. At 5 days posttransfection, capsids were purified by three rounds of CsCl density gradient ultracentrifugation. (A, D, G, and I) Negative-image photos and accompanying cartoons showing bands formed after the third CsCl ultracentrifugation. AAV2 VP3 with AAP2 [AAP2(+)] contained two bands at RIs of 1.3630 and 1.3645. The following ELISA indicated that the heavier of the two bands contained more capsids, and this was the band used for TEM imaging. (B and E) Assembled AAV capsids positive for anti-AAV capsid antibody in each CsCl fraction were assessed by an AAV capsid-specific ELISA. Relative optical density (OD) values at 450 nm are plotted against RIs of CsCl fractions. The OD at 450 nm obtained in the peak fraction was set to 1.0. (C, F, H, and J) Representative TEM images of samples prepared with or without AAP. The CsCl fractions showing RIs of 1.3645, 1.3640, 1.3645, 1.3640, 1.3635, 1.3640, 1.3635, and 1.3640 were used for TEM imaging of samples of AAV2 VP3 with and without AAP2 [AAP2(+) and AAP2(−), respectively], AAV5 VP3 with and without AAP5 [AAP5(+) and AAP5(−), respectively], AAV4 VP3 with and without AAP4 [AAP4(+) and AAP4(−), respectively], and AAV11 VP3 with and without AAP11 [AAP11(+) and AAP11(−), respectively]. Bar, 100 nm. (A to C) AAV2; (D to F) AAV5; (G and H) AAV4; (I and J) AAV11.
Infectious AAV5 virions can be produced without AAP.
While AAV capsids can be composed entirely of VP3, VP1 is required for viral infectivity and cell transduction (18–20). Having established that AAV5 VP3 could assemble capsids in the absence of coexpressed AAP, we sought to determine if infectious AAV5 virions could be produced by expressing all of the AAV5 VP proteins, VP1, VP2, and VP3, without supplying the AAP5 protein. To this end, we utilized two types of AAV5 helper plasmids, pHLP-AAV5(AAP5+) and pHLP-AAV5(AAP5−). The pHLP-AAV5(AAP5+) plasmid is our standard AAV5 helper plasmid for vector production that expresses AAV2 Rep and the AAV5 VP1, VP2, VP3, and AAP5 proteins. pHLP-AAV5(AAP5−) expresses all the AAV proteins except AAP5 due to extensive codon modification of the AAP-VP-overlapping ORFs (Fig. 7A). Western blot analysis of the cell lysates obtained from HEK 293 cells transfected with pHLP-AAV5(AAP5−) or pHLP-AAV5(AAP5+) with or without pCMV3-FLAG-cmAAP5 showed that the AAV5 VP3 protein was detectable in small amounts when AAP5 was not expressed and that the steady-state level of expression of AAV5 VP proteins could be increased substantially when AAP5 was coexpressed (Fig. 7B). Using the adenovirus-free plasmid transfection method (21) with pHLP-AAV5(AAP5−), we successfully produced AAV5 vectors containing a recombinant AAV vector genome in the absence of AAP5 expression, although the vector yield was an order of magnitude lower than the yields that could be obtained when AAP5 was expressed (Fig. 7C).
FIG 7.

Production and characterization of AAV5 VP1/VP2/VP3 particles produced with and without AAP5. (A) Schematic representation of plasmids expressing AAV5 VP proteins. These plasmids were used for the experiments shown in panels B to D. The VP1, VP2, and VP3 translation start sites are indicated with black vertical lines and arrows from left to right. The AAP translation start site is indicated with red vertical lines and a red arrow. The AAP ORF is shown with red boxes. The codon-modified region is indicated with yellow boxes. pA, polyadenylation signal; p5(G7), AAV2 p5 promoter with the TATA box sequence TATTTAA replaced with GGGGGGG. (B) HEK 293 cells were transiently transfected with an AAV5 helper plasmid, pHLP-AAV5(AAP5+) or pHLP-AAV5(AAP5−), with or without a plasmid expressing AAP5. All the groups were also transfected with an adenovirus helper plasmid, pHelper, to induce protein expression from the AAV5 helper plasmids. At 48 h posttransfection, the AAV5 VP1, VP2, and VP3 proteins were probed with anti-AAV VP antibody (B1) by Western blotting. Cyclophilin A (CPA) was used as a loading control. (C) HEK 293 cells were transfected with the plasmids indicated to produce AAV5 VP1/VP2/VP3 particles or VP3-only particles containing a double-stranded AAV-CMV-GFP vector genome in the presence or absence of AAP5 expressed from a separate plasmid, pCMV3-FLAG-cmAAP5. At 5 days posttransfection, the medium and cells were harvested, and Benzonase-resistant viral genomes in each sample were quantified by a dot blot assay in an experiment performed in biological triplicates. The y axis shows the AAV vector titers (vector genomes) per well in a 6-well-plate format. (D) CHO-K1 cells were infected with either the AAV5(AAP5+)-CMV-luc or AAV5(AAP5−)-CMV-luc vector, which was produced with or without AAP5, respectively, at an MOI of 106. Luciferase activity was measured at 46 h postinfection in an experiment performed in biological triplicates. The negative-control group received the luciferase-containing samples prepared in the same manner except for the absence of AAV5 VP protein expression, which provides a measure of pseudotransduction. For the pseudotransduction control, the same sample volume as that for the AAV5(AAP5−)-CMV-luc vector preparation was used. The y axis shows relative light units (RLUs). Error bars represent standard deviations. An asterisk indicates statistical significance with a P value of <0.05 (two-tailed Welch's t test). ns, not significant.
We then produced two types of recombinant AAV5 vectors expressing firefly luciferase, AAV5(AAP5−)-CMV-luc and AAV5(AAP5+)-CMV-luc, using pHLP-AAV5(AAP5−) and pHLP-AAV5(AAP5+), respectively, without supplying AAP5 in trans from the pCMV3-FLAG-cmAAP5 plasmid. The two vector preparations were added to Chinese hamster ovary K1 (CHO-K1) cells at a multiplicity of infection (MOI) of 106, and luciferase activity was measured at 46 h postinfection in an experiment performed in biological triplicates. The results showed that the AAV5(AAP5−)-CMV-luc vector was able to transduce CHO-K1 cells, and there was no statistically significant difference in transduction efficiency between the AAV5(AAP5−)-CMV-luc and AAV5(AAP5+)-CMV-luc vectors (P = 0.18 by two-tailed Welch's t test) (Fig. 7D). Taken together, our data demonstrate that AAV5 does not require AAP5 for the assembly of infectious virions containing a recombinant viral genome.
AAV4 and -11 can assemble capsids without AAP.
Having determined that AAV5 was capable of assembling infectious virions in the absence of AAP, we next sought to determine conclusively if AAV4 and AAV11 could also assemble capsids without AAP. AAV4 has a commercially available mouse monoclonal antibody against the assembled AAV4 capsid, ADK4, and thus, we were able to use immunofluorescence microscopy to determine if capsids could be made from AAV4 VP3 without AAP4. However, this approach was not applicable to AAV11 because an antibody specific for the assembled AAV11 capsid is currently not available.
In the immunofluorescence microscopy approach, we transfected HeLa cells with pCMV1-AAV4VP3 and either pCMV3-FLAG-cmAAP4 or pCMV3-GFP. At 48 h posttransfection, the cells were fixed and stained with an anti-FLAG antibody or anti-AAV4 capsid antibody (ADK4). This analysis confirmed that AAV4 VP3 was also able to assemble antibody-positive capsids regardless of whether AAP4 was coexpressed (Fig. 5A). In the TEM approach, we followed the same procedure as the one that we used for the AAV5 TEM experiment described above. In brief, to completely abolish any possible functional AAP expression, we constructed pCMV1-AAV4cmVP3 and pCMV1-AAV11cmVP3, in which the AAP-VP-overlapping ORFs were extensively codon modified compared to the native nucleotide sequences. These mutated AAP ORFs within the cmVP3 ORFs should not retain the AAP function at detectable levels (see Discussion). These two plasmids were used to produce viral capsids in HEK 293 cells in the presence or absence of coexpressed AAP, and the TEM samples were then prepared by three rounds of CsCl density gradient ultracentrifugation. In the final round of ultracentrifugation, a discrete band could be seen in each tube for both AAV4 and AAV11 regardless of the presence or absence of coexpressed AAP (Fig. 6G and I). TEM analysis revealed that these bands contained capsid structures of the expected size and shape for AAV (Fig. 6H and J). These observations provide direct evidence that AAV4 and AAV11 VP3s are capable of assembling capsids without AAP.
Heterologous AAPs efficiently promote capsid assembly, except for AAP4, -5, -11, and -12.
The promiscuity of AAPs in assembling capsids of heterologous AAV serotypes was previously shown with combinations of AAPs and AAV VP3 proteins derived from AAV1, -2, -5, -8, and -9 (12). In that study, Sonntag et al. reported that AAP2 could stimulate the assembly of heterologous AAV1, -8, and -9 capsids but not the AAV5 capsid; AAP1 could stimulate AAV2 capsid assembly; and AAP5 could weakly support AAV1 capsid assembly. While the observations obtained from these limited combinations have given us a glimpse of the nature of AAP-VP compatibility in capsid assembly, more expansive investigation of numerous different AAP and VP3 combinations for heterologous capsid assembly is imperative for a deeper understanding of the role of AAP. In order to fill this knowledge gap, we performed an AAP-VP3 cross-complementation study in which we investigated all 121 possible combinations of AAP1 to -11 and the VP3 proteins of AAV1 to -11 in an experiment performed in biological triplicates. Including the no-AAP controls used to determine the background levels for the assay, a total of 396 assessments of capsid formation was required for this comprehensive experiment. For this reason, we applied a massively parallel capsid assembly assay using an Illumina sequencing-based AAV barcode-Seq approach reported previously (22). In this approach, packaged viral genomes were quantified as surrogates of assembly (see Materials and Methods). A subset of 7 combinations was validated by quantitative dot blotting in an experiment performed in biological replicates (n = 2 to 4). This validation experiment revealed that the results obtained by these two different methods were primarily concordant (Fig. 8). As for AAP12, which was not included in this analysis, we determined the yields by quantitative dot blot analysis. The results showed that AAPs derived from AAV1, -2, -3B, -6, -7, -8, -9, and -10 could promote heterologous serotype capsid assembly at least 30% as efficiently as the native combinations (Fig. 9). As observed in the previous study (12), AAP5 also weakly supported the assembly of many heterologous VP3 proteins. AAP4, -11, and -12 were found to be least efficient for heterologous capsid assembly (Fig. 9). In particular, AAP12 supported assembly efficiently only for AAV11 besides its cognate capsid. AAV12 VP3, which could not assemble without AAP, formed capsids exclusively with AAP4 or AAP11 besides its cognate AAP12 (Fig. 9). The capsid assembly-promoting role of heterologous AAPs was difficult to assess for the AAV4, -5, and -11 VP3 proteins because of the high background signals due to AAP-independent assembly (Fig. 9). Nonetheless, the decreased ability of AAP4, -5, -11, and -12 to promote the assembly of VP3 capsids of heterologous serotypes is congruent with previously reported data showing that AAP4, -5, -11, and -12 were the most phylogenetically dissimilar AAPs compared to the other AAPs derived from AAV1 to -13 (14). These observations establish that the AAPs promiscuously promote the efficient assembly of capsids among wide groups of closely related serotypes but that promiscuity does not extend to more distantly related serotypes such as AAV4, -5, -11, and -12.
FIG 8.
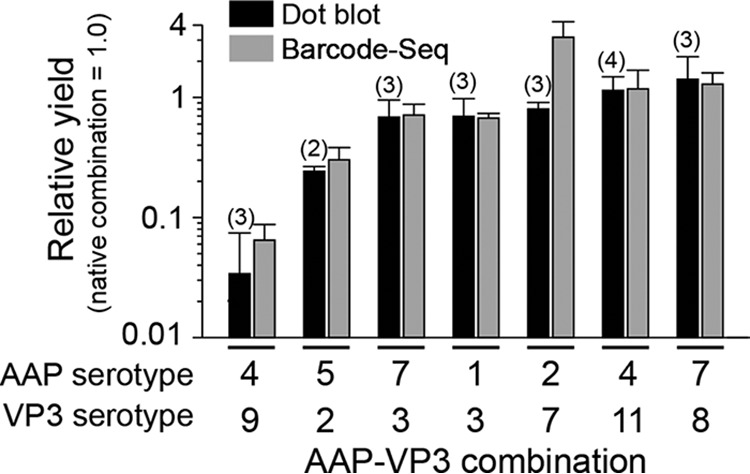
AAP-VP3 cross-complementation analysis by a quantitative dot blot assay. A total of 7 AAP-VP3 combinations were analyzed for cross-complementation of capsid assembly by a quantitative dot blot assay in an experiment performed in biological replicates. The numbers of replicates for each AAP-VP combination are indicated in parentheses. The results were compared to those obtained by AAV barcode-Seq, as shown in Fig. 9. Values represent relative AAV VP3-only particle yields with each AAP-VP combination relative to those obtained with the native AAP-VP3 combination. Error bars represent standard deviations, except for the AAP5-AAV2 VP3 combination. For the AAP5-AAV2 VP3 combination, the error bar represents the difference between each value and the mean value because the data were collected from samples in biological duplicates.
FIG 9.

AAP-VP3 cross-complementation among 12 different AAV serotypes. (A) Data from AAV barcode-Seq analysis showing the ability of each AAP to assemble homologous and heterologous VP3 proteins derived from AAV1 to -11. No-AAP controls are shown as AAP (−) to the left of each panel. The y axis shows AAV VP3-only particle yields in each AAP-VP combination relative to the yields obtained with the native AAP-VP3 combination. The values obtained with the native combination are set as 1.0. The data were collected from an experiment performed in biological triplicates. Error bars represent standard deviations. Asterisks indicate that values are higher than those for the no-AAP controls with a P value of <0.05 (one-tailed Mann-Whitney U test). (B) Matrix heat map showing the ability of each AAP to cross-complement assembly. The data for AAV1 to -11 were obtained from the data shown in panel A. The data for AAV12 were obtained by a quantitative dot blot assay in an experiment performed in biological duplicates. The values obtained by the native combination are set as 1.0.
Concordant and discordant subnuclear localization between AAPs, capsids, and nucleostemin.
During microscopic analyses, we observed both concordant and discordant subcellular localization patterns between AAPs, assembled capsids, and nucleostemin. In the case of AAV2, AAP2, assembled AAV2 VP3 capsids, and nucleostemin were tightly associated with each other and localized to the nucleoli and subnuclear bodies (Fig. 5B). AAP, assembled capsids, and nucleostemin were also colocalized in the case of AAV4 (Fig. 5B). In contrast, in the case of AAV5, AAP, assembled capsids, and nucleostemin were localized in different subnuclear compartments (Fig. 5B). The AAV5 VP3 capsid signals formed discrete spheres and globules, while the AAP5 signals were diffuse and different from the AAV5 VP3 capsid localization, and nucleostemin was not associated with these viral elements. AAV8 and AAV9 VP3 capsids also showed no association with nucleostemin (Fig. 5B). These observations demonstrate that the sites of AAP enrichment do not necessarily correspond to the sites of capsid assembly. In addition, our data presented here challenge a long-believed notion that tight association with the nucleolus is a hallmark of AAV capsid assembly. Because assembled capsids for certain serotypes such as AAV5, -8, and -9 are found outside the nucleolus and are not associated with the nucleolar protein nucleostemin, the involvement of the nucleolus and nucleolar proteins seen with AAV2 capsid assembly is not generalizable to all AAV serotypes.
DISCUSSION
In this study, we comprehensively analyzed AAPs derived from 12 AAV serotypes, AAV1 to -12, for their subcellular localization and their ability to promote the assembly of capsids derived from homologous and heterologous serotypes. For nearly a half-century, AAV2 has been the prototype for studies of the AAV life cycle, including infection, replication, and assembly, and much of what is currently known about AAV comes from experiments with this serotype. Although all the currently known AAV serotypes and variants display significant similarity to each other in their viral protein amino acid sequences and virion structures, studies of AAVs as gene delivery vectors for the last 2 decades have delineated that there are substantial differences in their biological properties, including cell and tissue tropisms (23). This has raised the possibility that certain aspects of fundamental AAV biology are also diverse among different serotypes, and what we had learned from the prototype AAV2 might not be applicable to the basic biology of the entire AAV family, including the process of capsid assembly. In this regard, the study reported here has clearly demonstrated that (i) AAP, which had been believed to be essential for capsid assembly based on observations obtained with AAV2, -8, and -9 (11, 12), is not necessarily essential for capsid assembly for other serotypes; (ii) a tight association between AAP and assembled capsids in the nucleolus during or after assembly, which has been shown for AAV2, is not always the case for other serotypes; and (iii) targeting to the nucleolus, which serves as an indispensable organelle for many viruses, including AAV2 (15, 24–29), may not be required for certain AAV serotypes. To be more concrete, we demonstrated here the dispensability of AAP for the assembly of AAV4, -5, and -11 capsids and for the production of infectious virions, at least for AAV5; serotype-specific concordant (in the cases of AAV2, -4, -8, and -9) and discordant (in the case of AAV5) associations between AAP and assembled capsids; and nucleolar exclusion of assembled AAV5, -8, and -9 capsids as opposed to the tight nucleolar association observed for AAV2 capsids.
The structural organization of AAP has been partially revealed in previous studies (13, 14). AAP has two hydrophobic domains of ∼15 amino acids near the N terminus. The first domain closer to the N terminus consists of four short hydrophobic motifs that are evolutionarily highly conserved among AAV1, -2, -3B, -6, -7, -8, -9, -10, and -13 and partially conserved among AAV4, -5, -11, and -12. The second domain, which is closer to the C terminus, is highly conserved across AAV1 to -13 and termed the “conserved core” of AAP (14). A coimmunoprecipitation analysis using a panel of AAP2 and AAV2 VP3 mutants showed that these two hydrophobic domains in the AAP proteins play a crucial role in interacting with the capsid VP proteins through a highly conserved hydrophobic patch near the VP C terminus that includes I682 (14) (note that this amino acid position is based on the AAV2 capsid protein). This high level of conservation of amino acids that constitute the interface of the AAP-VP interaction might allow an AAP derived from one serotype to assist in the assembly of AAV capsids of other serotypes. Therefore, the promiscuity of AAPs in cross-complementing the assembly of heterologous capsids that we observed in this study conforms well to the inference drawn from the evolutionarily conserved interfaces in the AAP-VP complex. Although the mechanism underlying why AAP4, -5, -11, and -12 show no or limited promiscuity in heterologous capsid assembly has yet to be elucidated, the decreased hydrophobicity in the first hydrophobic region found only in these four AAPs might correspondingly decrease the ability of the AAPs to interact with the hydrophobic C-terminal regions in VP proteins. A note of interest in the context of AAV capsid engineering by DNA shuffling (23) is that while, overall, AAPs are generally able to cross-complement many different serotypes, capsid libraries that include AAV4, -5, -11, and -12 might benefit from providing their cognate AAPs during capsid assembly reactions.
To our surprise, our study convincingly demonstrated that AAV4, -5, and -11 can assemble capsids without a need for AAP. This was not anticipated, because a previous study had shown that AAV5 capsid assembly required AAP (12). Although the reasons for this discrepancy are still unknown, there are a few possible explanations. First, there was a difference in the AAV5 VP3 expression plasmid constructs used in these studies. The plasmid used in the previous study contained an extra 154 bp after the stop codon derived from the AAV5 genome. Second, in our experiment, AAV5 VP3 might have been expressed in cells at a higher level than that in the previous study. As detailed in Materials and Methods, we used a strong CMV-IE enhancer-promoter with an enhancing element to express VP proteins. Taking this into account, a higher concentration of AAV5 VP3 proteins might allow AAP-independent capsid assembly. Interestingly, AAV12 VP3 was found to require AAP for assembly. This was also unexpected because AAV12 VP3 is evolutionarily closely related to AAV4 and AAV11 (12), which exhibit AAP-independent assembly, and because the subcellular localization and specificity in the cross-complementation of capsid assembly of heterologous serotypes are conserved among these serotypes. Further studies will be needed to delineate the AAP-independent assembly properties seen in the AAV4 and AAV11 VP3 proteins, which will lead to a better understanding of AAV assembly mechanisms.
One might argue that the +1-frameshifted mutated AAP ORFs within the cmVP3 ORFs could still express peptides that promote capsid assembly, allowing AAV4, AAV5, and AAV11 to assemble even in the absence of AAP supplied in trans. Naumer et al. (14) showed that (i) a C-terminal deletion of AAP2 including the right half of the basic region (BR) (Fig. 10) can abolish capsid assembly, (ii) the left half of the hydrophobic region of AAP2 (Fig. 10) is essential for capsid assembly, and (iii) amino acid mutations within the conserved region (CC) (Fig. 10) impair capsid assembly. When those previously reported observations are taken into account, one can conclude that the mutations in the AAP ORF should be sufficient to abrogate or impair assembly substantially, because the encoded peptides embody the deleterious alterations described above (Fig. 10). Complete abrogation of the AAP function by the codon modification has been proven experimentally by using a plasmid carrying the AAV2 cmVP3 ORF since no assembled AAV2 VP3 capsids were observed for AAV2 cmVP3 (Fig. 5 and 6). The amino acid sequences of the essential regions identified previously by Naumer et al. (14) are well conserved across AAPs derived from various serotypes (14); therefore, it is extremely unlikely that the mutated AAP ORFs in our cmVP3 plasmid constructs retain the assembly-promoting function.
FIG 10.

Sequence alignment of wild-type AAPs and the mutated AAPs encoded by the AAP ORFs within the cmVP3 ORFs. Amino acid sequences encoded by the mutated AAP ORFs in the pCMV1-AAVxcmVP3 plasmids (where x is 2, 4, 5, and 11) (knockout [KO]) are aligned with the wild-type (WT) AAPs, showing extensive amino acid alterations with new stop codons in the mutated AAP ORFs within the cmVP3 ORFs. Black and red letters indicate amino acids with and without changes, respectively. Underlining indicates stop codons. Annotations and the indicated amino acid positions were described previously by Naumer et al. (14). HR, hydrophobic region; CC, conserved core; PR, proline rich; TSR, T/S rich; BR, basic region.
It is worth noting that despite the dispensable nature of AAP5 for infectious AAV5 virion formation, the capsid assembly-promoting role of AAP5 is still obvious, as the abrogation of AAP5 expression from the capsid gene substantially decreased AAV5 vector production (Fig. 7C). AAP4 and AAP11 also shared this role (Fig. 9), although they were also not essential for capsid formation. These observations led us to speculate that AAP might contribute to the promotion of capsid assembly through mechanisms other than its above-described essential role. In this respect, it is intriguing that when we used the pCMV1-AAV5cmVP3 plasmid to express AAV5 VP3 in HEK 293 cells (Fig. 7A), we were able to produce assembled AAV5 VP3 capsids at equivalent levels irrespective of the presence or absence of coexpressed AAP5 (Fig. 7C), while this was not the case when we used the pCMV1-AAV5VP3 plasmid (Fig. 4). The pCMV1-AAV5cmVP3 plasmid differs from the pCMV1-AAV5VP3 plasmid in that the AAV5 VP3 ORF had been codon optimized for human cell expression. Hence, the concentration of VP proteins in cells may be rate limiting in the AAP-independent assembly process of the AAV5 capsid. At a lower concentration, the AAV5 VP3 proteins might require the presence of AAP5 to become stabilized and accumulate at the sites where capsids assemble through the interaction with AAP5. Therefore, it is plausible to propose a stabilization-accumulation mechanism via AAP-VP interactions, through which capsid assembly could be further enhanced. Such a mechanism might be postulated not only for AAV5 but also for other serotypes, although AAP-dependent serotypes still require AAP for its essential role in the assembly of capsids.
For many years, the nucleolus has been viewed as an organelle important for the AAV2 life cycle. Early work showed that AAV2 capsids are first seen in the nucleolus and subnuclear bodies during replication (15, 27). Several studies have found a close association between nucleolar proteins and AAV2 capsids (27, 28), and it was found that AAV2 capsids are trafficked to the nucleolus following infection (29). These observations led the field to speculate a role for the nucleolus or nucleolar proteins in AAV2 replication. A proposed hypothesis is that AAV capsid assembly occurs in the nucleolus and that capsids are subsequently moved to the nucleoplasm for genome packaging in a Rep-dependent manner (27). The identification of the nucleolus as the site of AAV2 capsid assembly was further supported by the identification of the nucleolus-localizing AAP2 protein (11). However, the role of the nucleolus had not been examined for other AAV serotypes. Our study addressed this question and revealed that AAP5 and AAP9 display a markedly decreased association with the nucleolus and that AAV5, -8, and -9 capsids do not accumulate in the nucleolus or associate with nucleostemin. Thus, the question arises as to whether the nucleolus plays an important role in capsid assembly for only a subset of AAV serotypes. It is possible that certain AAV serotypes require only a transient interaction with the nucleolus for capsid assembly and that the assembled capsids leave the nucleolus for packaging viral genomes in the nucleoplasm. Alternatively, for certain serotypes such as AAV5, assembly of the capsid may not have to rely upon factors in the nucleolus. Thus, our observations challenge the generalized view on the significance of roles of the nucleolus in the AAV life cycle and highlight a potential heterogeneity of the mechanisms of viral capsid assembly and replication among different AAV serotypes.
In summary, we show that capsid assembly in the nucleolus and its strict dependence on AAP are not universal phenomena applicable to all AAV serotypes. A potential caveat of our study is that we used an artificial plasmid transient-transfection system to study AAPs, which might not necessarily mimic the AAV infection and replication that take place in nature. Nonetheless, this study reveals that the processes and mechanisms involved in the AAV life cycle are more heterogeneous among different serotypes than previously thought. Further study into the roles and functions of AAPs in the AAV life cycle will advance our foundational knowledge of icosahedral capsid assembly mechanisms and lead to improved methods for the production of AAV vectors for gene therapy.
MATERIALS AND METHODS
Plasmid construction.
The pCMV3-FLAG-cmAAPx plasmids (where x is 1 to 12) are plasmids expressing codon-modified versions of AAP with an N-terminal FLAG tag under the control of the human CMV-IE gene enhancer-promoter and an ATG start codon. Each AAP ORF was codon modified to optimize expression in human cells and cloned into the pCMV3-FLAG-cmAAP2 parental plasmid used in our previous study (13) by replacing the cmAAP2 ORF with a new cmAAPx ORF. Plasmid pCMV3-FLAG-AAP2 carries the native AAP2 ORF sequence in place of the cmAAP2 ORF. Plasmid pCMV3-GFP is a plasmid expressing eGFP under the control of the same CMV-IE enhancer-promoter. The pCMV1-AAVxVP3 plasmids (where x is 1 to 12) are plasmids expressing each VP3 protein from the native ORF initiating at the ATG start codon. The pCMV1-AAVxcmVP3 plasmids (where x is 2, 4, 5, and 11) are plasmids expressing each VP3 protein from a codon-modified ORF in which the AAP-VP-overlapping ORFs were codon modified to optimize VP3 expression in human cells. The modification resulted in extensive changes in amino acids encoded by each AAP ORF, with identities and numbers of new stop codons being 37% and 6 for AAP2, 48% and 5 for AAP4, 47% and 4 for AAP5, and 44% and 8 for AAP11, respectively. Each VP3 ORF was cloned in the pCMV1-AAV2VP3 parental plasmid used in our previous study (13) by replacing the AAV2VP3 ORF with a new VP3 ORF. The CMV-IE enhancer-promoters that we used in this study contained an intervening sequence (IVS) consisting of a splice donor, an intron, and a splice acceptor from pIRES (Clontech, Mountain View, CA) for the AAP-expressing plasmids or the IVS from pAAV-MCS (Agilent, Santa Clara, CA) for the VP3-expressing plasmids. An adenovirus helper plasmid, pHelper, was purchased from Agilent. pHLP-AAV5(AAP5+) and pHLP-AAV5(AAP5−) are AAV5 helper plasmids carrying the AAV2 rep gene and the AAV5 cap gene. pHLP-AAV5(AAP5+) is the same as pHLP19-5 (30), which has been used for recombinant AAV5 vector production in our laboratory. In the pHLP19-5 helper plasmid, the AAV2 p5 promoter is moved from the native location to the downstream region of the AAV2 polyadenylation signal, and the TATA box sequence in the p5 promoter, TATTTAA, is replaced with the sequence GGGGGGG to reduce the expression of the large Rep proteins. pHLP-AAV5(AAP5−) is a derivative of pHLP-AAV5(AAP5+) that carries the codon-modified AAP-VP ORFs, which abolishes functional AAP5 expression while preserving the expression of the wild-type AAV5 VP1, VP2, and VP3 proteins. The identity of AAP5 amino acids encoded by the native and codon-modified AAP-VP ORFs is 44% with 4 new stop codons being introduced into the AAP5 ORF. pHLP-Rep is a plasmid that expresses all the AAV2 Rep proteins in HEK 293 cells in the presence of cotransfected pHelper. pHLP-Rep was constructed by removing a 1.8-kb XhoI-XcmI fragment from the wild-type AAV2 genome contained in our standard AAV2 helper plasmid pHLP19-2 (30) and expressed only AAV2 Rep proteins. pEMBL-CMV-GFP is an AAV vector plasmid for the production of a double-stranded AAV vector expressing eGFP under the control of the CMV-IE enhancer-promoter and was a gift from X. Xiao. pAAV-CMV-luc is an AAV vector plasmid for the production of a single-stranded AAV vector expressing firefly luciferase under the control of the CMV-IE enhancer-promoter and was created from pAAV-MCS. The pdsAAV-U6-VBCx plasmids (where x is an integer identification number indicating each different DNA barcode contained in each pdsAAV-U6-VBCx plasmid) are all double-stranded AAV vector plasmids carrying a 135-bp DNA fragment (nucleotide positions 4445 to 4579 of pAAV9-SBBANN-VBCLib [GenBank accession number KF032296]) that harbors a pair of 12-nucleotide-long DNA barcodes (virus barcodes [VBCs]). Besides the DNA barcodes and flaking PCR primer binding sites, the pdsAAV-U6-VBCx vector plasmids contain a human U6 snRNA promoter-driven nonfunctional noncoding RNA expression cassette of 0.6 kb and a 1.0-kb stuffer DNA derived from the bacterial lacZ gene between the two AAV2 inverted terminal repeats. The pdsAAV-U6-VBCx vector plasmids were designed and created for a separate study, and their feature of noncoding RNA expression was not utilized in this study. We confirmed that the expression of noncoding RNA from the pdsAAV-U6-VBCx plasmids does not affect AAV vector production in HEK 293 cells. pCMV1 and pCMV3 are control empty plasmids carrying no transgene in the pCMV1 and pCMV3 backbones, respectively. The native capsid ORFs used for plasmid construction were derived from pHLP19-1 to -6 (30); AAV7, -8, and -9 helper plasmids were provided by J. M. Wilson and G. Gao; AAV10 and AAV11 helper plasmids were provided by S. Mori; and a plasmid containing a de novo-synthesized AAV12 cap ORF was provided by Voyager Therapeutics. The codon-modified AAP and VP3 ORFs were synthesized by GenScript.
Cells.
HEK 293 cells (AAV293) were purchased from Stratagene. The HeLa human cervical cancer cell line and the CHO-K1 cell line were obtained from the American Type Culture Collection (ATCC). HEK 293 cells and HeLa cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Lonza, Basel, Switzerland) supplemented with 10% fetal bovine serum (FBS), l-glutamine, and penicillin-streptomycin. CHO-K1 cells were grown in F-12K medium supplemented with 10% FBS.
AAV particle production.
AAV VP1/VP2/VP3 particles and VP3-only particles were produced in HEK 293 cells by using an adenovirus-free plasmid transfection method (21), with modifications. In brief, we changed the complete culture medium to serum-free medium immediately before transfection, transfected cells with a mixture of the required amount of each plasmid DNA with polyethyleneimine (PEI) at a DNA/PEI weight ratio of 1:2, and harvested both medium and cells for viral particle recovery at 5 days posttransfection. The plasmids used for the production of each viral particle preparation are described in each relevant section. The AAV5(AAP5−)-CMV-luc and AAV5(AAP5+)-CMV-luc vectors were produced by using one 225-cm2 flask and concentrated from an initial volume of 25 ml to a final volume of 250 μl by using Amicon Ultra Centrifugal Filter units with a molecular mass cutoff of 100 kDa. For TEM, we produced VP3-only virus-like particles using 15 225-cm2 flasks. The harvested medium and cells underwent one cycle of freezing and thawing, and the cell debris was removed by centrifugation at 10,000 × g for 15 min. The culture medium supernatants were made with 8% polyethylene glycol 8000 (PEG 8000) and 0.5 M NaCl, incubated on ice for 3 h, and spun at 10,000 × g for 30 min to precipitate viral particles. The pellets were resuspended in a buffer containing 50 mM Tris-HCl (pH 8.5) and 2 mM MgCl2, treated with Benzonase (EMD Millipore, Darmstadt, Germany) at a concentration of 200 U per ml for 1 h, and subjected to purification by three rounds of CsCl density gradient ultracentrifugation (30) followed by dialysis with a buffer (25 mM HEPES, 150 mM NaCl [pH 7.4]) for TEM.
Cell infection.
CHO-K1 cells seeded onto a 96-well plate were infected with AAV5(AAP5−)-CMV-luc or AAV5(AAP5+)-CMV-luc at an MOI of 106 in the absence of a helper virus. At 46 h postinfection, luciferase activity was quantified by using the Bright-Glo luciferase assay system (Promega, Madison, WI) and the CentroXS LB960 plate reader (Berthold, Bad Wildbad, Germany). The data were collected from an experiment performed in biological triplicates.
Immunofluorescence microscopy.
HeLa cells were seeded onto coverslips in 12-well plates and transfected with plasmid DNA by using PEI. Forty-eight hours after transfection, the cells were fixed with 4% paraformaldehyde at room temperature, permeabilized with 0.2% Tween 20, and blocked with 8% bovine serum albumin (BSA). For AAP localization, the cells were stained with mouse monoclonal anti-FLAG M2 antibody (catalog number F1804; Sigma-Aldrich, St. Louis, MO) and rabbit polyclonal antinucleostemin antibody (catalog number sc-67012; Santa Cruz Biotechnology, Dallas, TX), followed by DAPI (4′,6-diamidino-2-phenylindole), Alexa Fluor 488-AffiniPure goat anti-mouse IgG antibody (catalog number 115-545-166; Jackson ImmunoResearch, West Grove, PA), and Cy3-AffiniPure goat anti-rabbit IgG antibody (catalog number 111-165-144; Jackson ImmunoResearch). For imaging of AAV capsids, cells were stained with mouse monoclonal anti-AAV2 capsid antibody (A20 clone, catalog number 03-61055; American Research Products Inc., Waltham, MA), mouse monoclonal anti-AAV4 capsid antibody (ADK4 clone, 03-610147; American Research Products Inc.), mouse monoclonal anti-AAV5 capsid antibody (ADK5a clone, catalog number 03-61048; American Research Products Inc.), mouse monoclonal anti-AAV8 capsid antibody (ADK8 clone, catalog number 03-651160; American Research Products Inc.), or mouse monoclonal anti-AAV8/9 capsid antibody (ADK8/9 clone, catalog number 03-651161; American Research Products Inc.) and rat monoclonal anti-DYKDDDDK (FLAG) antibody (catalog number NBP1-06712; Novus Biological, Littleton, CO), followed by DAPI, Alexa Fluor 488-AffiniPure goat anti-mouse IgG antibody (catalog number 115-545-166; Jackson ImmunoResearch), and Cy3-AffiniPure donkey anti-rat IgG antibody (catalog number 712-165-153; Jackson ImmunoResearch). The nucleolus was visualized by using rabbit polyclonal antinucleostemin antibody (catalog number sc-67012; Santa Cruz Biotechnology) and Alexa Fluor 647-AffiniPure goat anti-rabbit IgG antibody (catalog number 111-605-144; Jackson ImmunoResearch). GFP was directly visualized by fluorescence microscopy. The cells were imaged on a Zeiss LMS 710 laser scanning confocal microscope using either a 63×/1.4-numerical-aperture (NA) or a 100×/1.46-NA objective. To perform a quantitative assessment of the degree of nucleolar association of AAP1 to -12, cells were imaged on a Zeiss Axio Imager 2 microscope using a 40×/1.3-NA objective, and a virtual large image was reconstructed from 49 individual image tiles by using an ApoTome.2 device attached to the microscope. We categorized the nuclear staining patterns into the following two groups: No+ and No−. The nuclei of the No+ group have nucleoli that are stained with the anti-FLAG antibody at the same level as that of the nucleoplasm or show a pattern of nucleolar enrichment. The nucleoli that do not belong to the No+ group are categorized as No−. We counted 50 nuclei for each AAP serotype for a statistical comparison.
Quantitative dot blotting.
HEK 293 cells were seeded onto 6-well plates 1 day before transfection. We changed complete culture medium to serum-free medium before transfection and transfected cells with 0.4 μg each of the following 5 plasmids using PEI: pCMV3-FLAG-cmAAPx (where x is 1 to 12) or pCMV3 (an empty plasmid as a no-AAP control), pCMV1-AAVxVP3 (where x is 1 to 12), pEMBL-CMV-GFP, pHLP-Rep, and pHelper. At 5 days posttransfection, we harvested both medium and cells, disrupted cells by one cycle of freezing and thawing, and released viral particles into the culture medium. After cell debris was removed by centrifugation at 21,100 × g for 5 min, 200 μl of the culture medium supernatant was subjected to nuclease treatment with 200 U per ml of Benzonase at 37°C for 4 h, followed by proteinase K treatment at 55°C for 1 h. Viral genome DNA was purified by phenol-chloroform extraction, ethanol precipitated, and dissolved in 1× Tris-HCl–EDTA (TE) buffer (pH 8.0). The viral DNA and linearized standard plasmid DNA were then denatured with 0.4 N NaOH, blotted onto a Zeta Probe nylon membrane (Bio-Rad, Hercules, CA), and hybridized with a 32P-labeled GFP probe. The hybridized signals were imaged and quantified by using a Typhoon FLA7000 scanner (GE Healthcare Bio-Science, Uppsala, Sweden). The negative control contained double-stranded AAV-CMV-GFP genomes that had undergone AAV2 Rep-mediated replication but were not protected by viral capsids. The negative control ensured efficient nuclease digestion in the assay. However, the Benzonase-treated negative control still generates a low level of background signal; therefore, the dot blot assay by itself cannot completely rule out the possibility of the presence of viral particles at very low levels. To create dot blot images for figures, the tiff images were imported to ImageJ, and the dot intensities were adjusted equally across the entire image by using the ImageJ Brightness & Contrast function.
AAP-VP cross-complementation analysis.
For AAP-VP cross-complementation analysis, we used two methods: Illumina sequencing-based AAV barcode-Seq (22) and the conventional quantitative dot blot assay as described above. For AAV barcode-Seq, HEK 293 cells were seeded onto 12-well plates 1 day before transfection, and VP3-only particles containing a DNA-barcoded viral genome were produced essentially in the same manner as described above except that we used 0.24 μg of each plasmid, pdsAAV-U6-VBCx in place of pEMBL-CMV-GFP, and pCMV3-FLAG-AAP2 in place of pCMV3-FLAG-cmAAP2. There were a total of 132 AAP and VP combinations, and each combination received a different DNA-barcoded AAV vector plasmid, pdsAAV-U6-VBCx (where x is 1 to 132). At 5 days posttransfection, we harvested both medium and cells and pooled them in a bottle. We performed this procedure in triplicate and produced three pooled samples. Viral genome DNA was extracted from 200 μl of each pooled sample, purified by phenol-chloroform extraction, ethanol precipitated, and dissolved in 20 μl of 1× TE buffer (pH 8.0). We then PCR amplified both the left VBCs (lt-VBCs) and the right VBCs (rt-VBCs) separately using two different sets of PCR primers and 2 μl each of the resulting DNA solution. The PCR primers are as follows: lt-VBC-For (frameshifting nucleotide [FSN]-sample-specific barcode [SBC]-ACCTACGTACTTCCGCTCAT), lt-VBC-Rev (FSN-SBC-TCCCGACATCGTATTTCCGT), rt-VBC-For (FSN-SBC-ACGGAAATACGATGTCGGGA), and rt-VBC-Rev (FSN-SBC-CTTCTCGTTGGGGTCTTTGC). Each primer contained a 7-nucleotide-long SBC and 1 to 5 FSNs at the 5′ end. The primer combinations of lt-VBC-For plus lt-VBC-Rev and rt-VBC-For plus rt-VBC-Rev were used to amplify the lt-VBCs and rt-VBCs, respectively, in each of the biological triplicates of the experiment. The resulting six PCR products were mixed at an equimolar ratio and subjected to multiplexed Illumina sequencing as previously described (22), together with other PCR products prepared in the same manner in separate AAV barcode-Seq studies. One to five micrograms of PCR products attached to Illumina sequencing adaptors was sent to Elim Biopharmaceuticals Inc. (Hayward, CA) and sequenced with a 50-cycle single-end run on an Illumina HiSeq 2500 instrument. The quality measures of Illumina raw sequence reads determined by FastQC (i.e., per-base sequence quality, per-sequence quality scores, per-base N content, and sequence length distribution) were all met in the data set used in this study. We analyzed the Illumina sequencing data at the Pittsburgh Supercomputing Center using an algorithm that we developed. In this experimental scheme, a pair of the DNA barcodes carried by each AAV vector plasmid could provide a measure of AAV vector yield from each individual transfection by means of AAV barcode-Seq.
Barcode-Seq data analysis.
We determined relative viral particle yields in each of the triplicate sets of the experiment using the same principle as that used for our previous study (22). First, we globally normalized Illumina raw sequence read numbers for all the 132 VBCs to obtain relative read number data for each of the lt-VBCs and rt-VBCs. We then adjusted the relative read number data for each of the lt-VBCs and rt-VBCs by each VBC-specific PCR amplification efficiency factor. The VBC-specific PCR amplification efficiency factor was determined in the following manner. We created two sets of an equimolar mixture of 379 pdsAAV-U6-VBCx plasmids (where x is 1 to 379) independently. The 132 pdsAAV-U6-VBCx plasmids (where x is 1 to 132) used in this study were included in each equimolar plasmid mixture. We then PCR amplified 379 lt-VBCs together using primers lt-VBC-For and lt-VBC-Rev and PCR amplified 379 rt-VBCs together by using primers rt-VBC-For and rt-VBC-Rev, in each of the two equimolar plasmid mixtures. The resulting four PCR products were mixed at an equimolar ratio and subjected to multiplexed Illumina sequencing together with other PCR products prepared in the same manner in separate AAV barcode-Seq studies as described above. This gave us raw sequence read numbers for all 132 lt-VBCs and 132 rt-VBCs in each set. We then globally normalized the sequence read numbers and determined a relative quantity value for each lt-VBC and each rt-VBC in each set. The relative quantity values for each VBC obtained from the duplicate sets of the experiment were averaged and used as the PCR amplification efficiency factor. Since the experiment was done in triplicate and two DNA barcodes, lt-VBC and rt-VBC, were used, we obtained a total of 6 relative quantity values that could quantify the AAV vector yield with each AAP-VP3 combination. Among the 6 values for each AAP-VP3 combination, we excluded outliers showing values more than three times the interquartile range beyond the upper and lower quartiles. The AAV vector yield of each AAP-VP3 combination relative to the native combination was determined for each serotype. The AAV vector yield for the native AAP-VP3 combination was set as 1.0. It should be noted that Benzonase-treated samples containing replicated AAV vector genomes unprotected by capsid coats generate positive barcode PCR signals due to the high sensitivity of this PCR-based assay. This often generates a background signal higher than that of the quantitative dot blot assay described above. We have found that the background signals can be as high as ∼0.2; therefore, positive values of up to ∼0.2 do not necessarily indicate capsid assembly. This represents the limitation of this assay.
Transmission electron microscopy.
Viral particle preparations, purified by three rounds of CsCl density gradient ultracentrifugation followed by dialysis, were spun at 6,100 × g for 10 min to remove any viral precipitate. Carbon-coated copper grids (Cu-300CN; Pacific Grid-Tech, San Francisco, CA) were glow discharged for 25 s at 15 mA by using the Pelco easiGlow glow discharge cleaning system (Ted Pella Inc., Redding, CA) immediately prior to use. Four microliters of each sample was placed onto the grids for 3 min and manually blotted with Whatman filter paper (catalog number 1001-125; GE Healthcare, Pittsburg, PA). The grids were then placed face down onto 45-μl drops of a buffer (50 mM HEPES, 25 mM MgCl2, 50 mM NaCl [pH 7.4]) for 30 s, washed three times similarly with distilled water, and then blotted. The samples on the grids were stained with 5.5 μl of 0.75% uranyl formate (pH 4.5) for 30 s and washed with distilled water, followed by blotting. The grids were allowed to dry and then stored at room temperature in a petri dish sealed with Parafilm until imaging.
Enzyme-linked immunosorbent assay.
After viral particle preparations were purified by three rounds of CsCl density gradient ultracentrifugation, 0.04 μl of each fraction was subjected to a capsid-specific ELISA using the AAV2 Titration ELISA kit or the AAV5 Titration ELISA kit (Progen, Heidelberg, Germany) according to the manufacturer's instructions.
Western blotting.
We seeded HEK 293 cells onto 6-well plates and transfected them with a total of 2 μg of a plasmid or a mixture of plasmids using PEI. The plasmids used in each experiment are described in each relevant section. At 48 h posttransfection, we lysed HEK 293 cells in radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitors (Complete Mini; Roche, Indianapolis, IN), sonicated the cells, and determined protein concentrations in the cell lysates with a DC Protein Assay kit (Bio-Rad, Hercules, CA). The same amount of total cell lysates (40 μg per lane) along with a molecular weight marker were separated on a 10% SDS-PAGE gel, transferred onto a polyvinylidene difluoride (PVDF) membrane, and reacted with mouse monoclonal anti-FLAG M2 antibody and mouse anti-α-tubulin antibody (catalog number sc-32293; Santa Cruz Biotechnology) or mouse monoclonal anti-AAV VP1/VP2/VP3 antibody (B1) (catalog number 03-61058; American Research Products) and rabbit polyclonal anti-cyclophilin A antibody (Cell Signaling Technology, Danvers, MA) followed by goat polyclonal anti-mouse IgG antibody (catalog number sc-2055; Santa Cruz Biotechnology) or goat polyclonal anti-rabbit IgG antibody (catalog number sc-2004; Santa Cruz Biotechnology) conjugated to horseradish peroxidase (HRP). The signals on the blots were visualized with the Immobilon Western chemiluminescent HRP substrate (EMD Millipore) and imaged on X-ray films or by using the FluorChem M system (ProteinSimple, Santa Clara, CA). Molecular weights of FLAG-tagged AAP1 to -12 were determined in quadruplicate Western blots. In brief, tiff images of the blots were imported into ImageJ, and the positions of the peak of each AAP band were identified by densitometry and used for the calculation of the migration distance for each AAP. The molecular weights of AAPs were then determined by interpolation using the migration distances of molecular weight markers and their log-transformed molecular weights. The Western blot images for figures were created in the same manner as described above for the dot blot images by using ImageJ.
Statistics.
In the cross-complementation study using AAV barcode-Seq, the null hypothesis that there was no enhancement of assembly by AAP was examined by a one-tailed Mann-Whitney U test for each serotype. For the quantitative assessment of the nucleolar association of AAPs, we used Fisher's exact test. Because 12 AAPs were compared pairwise, P values were adjusted by Bonferroni correction. In the AAV5 vector production and infection assays, we used two-tailed Welch's t test to assess differences in the mean values between two groups. P values of <0.05 were considered statistically significant.
ACKNOWLEDGMENTS
We thank Xiao Xiao at the University of North Carolina at Chapel Hill for providing us with the pEMBL-CMV-GFP plasmid; Guangping Gao and James M. Wilson for helper plasmids for AAV7, -8, and -9; Seiichiro Mori for the helper plasmid for AAV10 and AAV11; Voyager Therapeutics for a plasmid containing the de novo-synthesized AAV12 cap gene ORF; and Christoph Kahl and Michelle Gomes for technical assistance.
This work was supported by Public Health Service grants (R01 DK078388, R01 NS 088399, R01 GM066875, T32 GM071338, T32 AI007472, and T32 EY232113) and in part by the National Institutes of Health through resources provided by the National Resource for Biomedical Supercomputing (P41 RR06009), which is part of the Pittsburgh Supercomputing Center.
REFERENCES
- 1.Lingappa VR, Hurt CR, Garvey E. 2013. Capsid assembly as a point of intervention for novel anti-viral therapeutics. Curr Pharm Biotechnol 14:513–523. doi: 10.2174/13892010113149990201. [DOI] [PubMed] [Google Scholar]
- 2.Mateu MG. 2013. The structural basis of virus function, p 3–51. In Mateu MG. (ed), Structure and physics of viruses: an integrated textbook. Springer, New York, NY. [Google Scholar]
- 3.Caspar DL, Klug A. 1962. Physical principles in the construction of regular viruses. Cold Spring Harb Symp Quant Biol 27:1–24. doi: 10.1101/SQB.1962.027.001.005. [DOI] [PubMed] [Google Scholar]
- 4.Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, Della Peruta M, Lheriteau E, Patel N, Raj D, Riddell A, Pie J, Rangarajan S, Bevan D, Recht M, Shen YM, Halka KG, Basner-Tschakarjan E, Mingozzi F, High KA, Allay J, Kay MA, Ng CY, Zhou J, Cancio M, Morton CL, Gray JT, Srivastava D, Nienhuis AW, Davidoff AM. 2014. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carpentier AC, Frisch F, Labbe SM, Gagnon R, de Wal J, Greentree S, Petry H, Twisk J, Brisson D, Gaudet D. 2012. Effect of alipogene tiparvovec (AAV1-LPL(S447X)) on postprandial chylomicron metabolism in lipoprotein lipase-deficient patients. J Clin Endocrinol Metab 97:1635–1644. doi: 10.1210/jc.2011-3002. [DOI] [PubMed] [Google Scholar]
- 6.Jacobson SG, Cideciyan AV, Ratnakaram R, Heon E, Schwartz SB, Roman AJ, Peden MC, Aleman TS, Boye SL, Sumaroka A, Conlon TJ, Calcedo R, Pang JJ, Erger KE, Olivares MB, Mullins CL, Swider M, Kaushal S, Feuer WJ, Iannaccone A, Fishman GA, Stone EM, Byrne BJ, Hauswirth WW. 2012. Gene therapy for Leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol 130:9–24. doi: 10.1001/archophthalmol.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pierce EA, Bennett J. 2015. The status of RPE65 gene therapy trials: safety and efficacy. Cold Spring Harb Perspect Med 5:a017285. doi: 10.1101/cshperspect.a017285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mingozzi F, High KA. 2011. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet 12:341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- 9.Becerra SP, Koczot F, Fabisch P, Rose JA. 1988. Synthesis of adeno-associated virus structural proteins requires both alternative mRNA splicing and alternative initiations from a single transcript. J Virol 62:2745–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trempe JP, Carter BJ. 1988. Alternate mRNA splicing is required for synthesis of adeno-associated virus VP1 capsid protein. J Virol 62:3356–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sonntag F, Schmidt K, Kleinschmidt JA. 2010. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc Natl Acad Sci U S A 107:10220–10225. doi: 10.1073/pnas.1001673107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sonntag F, Kother K, Schmidt K, Weghofer M, Raupp C, Nieto K, Kuck A, Gerlach B, Bottcher B, Muller OJ, Lux K, Horer M, Kleinschmidt JA. 2011. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J Virol 85:12686–12697. doi: 10.1128/JVI.05359-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Earley LF, Kawano Y, Adachi K, Sun XX, Dai MS, Nakai H. 2015. Identification and characterization of nuclear and nucleolar localization signals in the adeno-associated virus serotype 2 assembly-activating protein. J Virol 89:3038–3048. doi: 10.1128/JVI.03125-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naumer M, Sonntag F, Schmidt K, Nieto K, Panke C, Davey NE, Popa-Wagner R, Kleinschmidt JA. 2012. Properties of the adeno-associated virus assembly-activating protein. J Virol 86:13038–13048. doi: 10.1128/JVI.01675-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wistuba A, Kern A, Weger S, Grimm D, Kleinschmidt JA. 1997. Subcellular compartmentalization of adeno-associated virus type 2 assembly. J Virol 71:1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang MT, Gorman CM. 1990. Intervening sequences increase efficiency of RNA 3′ processing and accumulation of cytoplasmic RNA. Nucleic Acids Res 18:937–947. doi: 10.1093/nar/18.4.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuck D, Kern A, Kleinschmidt JA. 2007. Development of AAV serotype-specific ELISAs using novel monoclonal antibodies. J Virol Methods 140:17–24. doi: 10.1016/j.jviromet.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Girod A, Wobus CE, Zadori Z, Ried M, Leike K, Tijssen P, Kleinschmidt JA, Hallek M. 2002. The VP1 capsid protein of adeno-associated virus type 2 is carrying a phospholipase A2 domain required for virus infectivity. J Gen Virol 83:973–978. doi: 10.1099/0022-1317-83-5-973. [DOI] [PubMed] [Google Scholar]
- 19.Stahnke S, Lux K, Uhrig S, Kreppel F, Hosel M, Coutelle O, Ogris M, Hallek M, Buning H. 2011. Intrinsic phospholipase A2 activity of adeno-associated virus is involved in endosomal escape of incoming particles. Virology 409:77–83. doi: 10.1016/j.virol.2010.09.025. [DOI] [PubMed] [Google Scholar]
- 20.Nicolson SC, Samulski RJ. 2014. Recombinant adeno-associated virus utilizes host cell nuclear import machinery to enter the nucleus. J Virol 88:4132–4144. doi: 10.1128/JVI.02660-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsushita T, Elliger S, Elliger C, Podsakoff G, Villarreal L, Kurtzman GJ, Iwaki Y, Colosi P. 1998. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther 5:938–945. doi: 10.1038/sj.gt.3300680. [DOI] [PubMed] [Google Scholar]
- 22.Adachi K, Enoki T, Kawano Y, Veraz M, Nakai H. 2014. Drawing a high-resolution functional map of adeno-associated virus capsid by massively parallel sequencing. Nat Commun 5:3075. doi: 10.1038/ncomms4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asokan A, Schaffer DV, Samulski RJ. 2012. The AAV vector toolkit: poised at the clinical crossroads. Mol Ther 20:699–708. doi: 10.1038/mt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salvetti A, Greco A. 2014. Viruses and the nucleolus: the fatal attraction. Biochim Biophys Acta 1842:840–847. doi: 10.1016/j.bbadis.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pederson T. 2011. The nucleolus. Cold Spring Harb Perspect Biol 3:a000638. doi: 10.1101/cshperspect.a000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matthews DEE, Hiscox J. 2011. Viruses and the nucleolus, p 321–345. In Olson MO. (ed), The nucleolus. Springer, New York, NY. [Google Scholar]
- 27.Bevington JM, Needham PG, Verrill KC, Collaco RF, Basrur V, Trempe JP. 2007. Adeno-associated virus interactions with B23/nucleophosmin: identification of sub-nucleolar virion regions. Virology 357:102–113. doi: 10.1016/j.virol.2006.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiu J, Brown KE. 1999. A 110-kDa nuclear shuttle protein, nucleolin, specifically binds to adeno-associated virus type 2 (AAV-2) capsid. Virology 257:373–382. doi: 10.1006/viro.1999.9664. [DOI] [PubMed] [Google Scholar]
- 29.Johnson JS, Samulski RJ. 2009. Enhancement of adeno-associated virus infection by mobilizing capsids into and out of the nucleolus. J Virol 83:2632–2644. doi: 10.1128/JVI.02309-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grimm D, Zhou S, Nakai H, Thomas CE, Storm TA, Fuess S, Matsushita T, Allen J, Surosky R, Lochrie M, Meuse L, McClelland A, Colosi P, Kay MA. 2003. Preclinical in vivo evaluation of pseudotyped adeno-associated virus vectors for liver gene therapy. Blood 102:2412–2419. doi: 10.1182/blood-2003-02-0495. [DOI] [PubMed] [Google Scholar]