

ABSTRACT
Signal transducer and activator of transcription 3 (STAT3) is a pleiotropic signaling mediator of many cytokines, including interleukin-6 (IL-6) and IL-10. STAT3 is known to play critical roles in cell growth, proliferation, differentiation, immunity and inflammatory responses. The objective of this study was to determine the effect of porcine reproductive and respiratory syndrome virus (PRRSV) infection on the STAT3 signaling since PRRSV induces a weak protective immune response in host animals. We report here that PRRSV infection of MARC-145 cells and primary porcine pulmonary alveolar macrophages led to significant reduction of STAT3 protein level. Several strains of both PRRSV type 1 and type 2 led to a similar reduction of STAT3 protein level but had a minimal effect on its transcripts. The PRRSV-mediated STAT3 reduction was in a dose-dependent manner as the STAT3 level decreased, along with incremental amounts of PRRSV inocula. Further study showed that nonstructural protein 5 (nsp5) of PRRSV induced the STAT3 degradation by increasing its polyubiquitination level and shortening its half-life from 24 h to ∼3.5 h. The C-terminal domain of nsp5 was shown to be required for the STAT3 degradation. Moreover, the STAT3 signaling in the cells transfected with nsp5 plasmid was significantly inhibited. These results indicate that PRRSV antagonizes the STAT3 signaling by accelerating STAT3 degradation via the ubiquitin-proteasomal pathway. This study provides insight into the PRRSV interference with the JAK/STAT3 signaling, leading to perturbation of the host innate and adaptive immune responses.
IMPORTANCE The typical features of immune responses in PRRSV-infected pigs are delayed onset and low levels of virus neutralizing antibodies, as well as weak cell-mediated immunity. Lymphocyte development and differentiation rely on cytokines, many of which signal through the JAK/STAT signaling pathway to exert their biological effects. Here, we discovered that PRRSV antagonizes the JAK/STAT3 signaling by inducing degradation of STAT3, a master transcription activator involved in multiple cellular processes and the host immune responses. The nsp5 protein of PRRSV is responsible for the accelerated STAT3 degradation. The PRRSV-mediated antagonizing STAT3 could lead to suppression of a broad spectrum of cytokines and growth factors to allow virus replication and spread in host animals. This may be one of the reasons for the PRRSV interference with the innate immunity and its poor elicitation of protective immunity. This finding provides insight into PRRSV pathogenesis and its interference with the host immune responses.
KEYWORDS: porcine reproductive and respiratory syndrome virus (PRRSV), signal transducer and activator of transcription 3, STAT3, JAK/STAT signaling, innate immunity, PRRSV nsp5
INTRODUCTION
Porcine reproductive and respiratory syndrome (PRRS) has been an economically important viral disease in the swine industry since it was first reported in 1987, leading to an estimated annual loss of $664 million in the United States alone (1). The causative agent of the devastating disease is PRRS virus (PRRSV), a small enveloped RNA virus belonging to the genus Arterivirus, family Arteriviridae, order Nidovirales (2, 3). There are two PRRSV species in the newly proposed taxonomy, PRRSV-1 and PRRSV-2, corresponding to the currently known genotype type 1 (European) and type 2 (North American) PRRSV, respectively (3). PRRSV virions contain a single-stranded, positive-sense RNA genome in a size of ∼15 kb. The PRRSV genome encodes over 10 open reading frames (ORFs) (2, 4, 5). PRRSV mainly targets pulmonary alveolar macrophages (PAMs) and some lineages of monocytes in pigs (6). PRRSV propagation in vitro is generally conducted on MARC-145 cells, which are derived from epithelial cells of a monkey kidney (7).
Typical features of the immune response to PRRSV infection in pigs are delayed onset and low titers of virus-neutralizing antibodies, as well as a weak cell-mediated immune response (8, 9). PRRSV infection is characterized by prolonged viremia, followed by persistent viral replication in regional lymph nodes for as long as 250 days (10). One of the possible reasons for the weak protective immune response is that PRRSV interferes with the innate immunity, such as inhibition of the synthesis and downstream signaling of type I interferons (IFNs) (11–14). Cytokines, including type I IFNs, that are produced at the site of infection, stimulate and coordinate the innate and adaptive immune responses against the invading pathogen (15–17). Many of the cytokines initiate functions by binding to specific receptors on cells to activate the Janus kinase (JAK) signal transducers and activators of transcription (STAT) signal pathway (18, 19). STATs are a family of transcription factors that regulate cell growth, differentiation, proliferation, apoptosis, immunity, inflammatory responses, and angiogenesis. There are seven STAT proteins (STAT1, -2, -3, -4, -5A, -5B, and -6) in mammalians. Each STAT member responds to a defined set of cytokines, although some of the cytokines can induce signaling via several STAT proteins (19–21). STAT proteins are activated by JAK phosphorylation of specific tyrosine residues, followed by homodimer or heterodimer formation and nuclear translocation to activate transcription of a specific set of genes.
Among all the STAT proteins, STAT3 is known as highly pleiotropic in mediating the expression of a variety of genes in response to both cytokines and growth factors and thus plays a pivotal role in numerous cellular processes, including cell survival, proliferation, embryogenesis, and the immune response (19, 22, 23). Numerous cytokines, including interlukin-5 (IL-5), IL-6, IL-9, IL-10, IL-11, IL-12, IL-21, IL-22, IL-27, oncostatin M (OSM), gamma interferon (IFN-γ), tumor necrosis factor alpha (TNF-α), and leukemia inhibitory factor (LIF), trigger STAT3 activation (20, 24). The IL-6 family cytokines, including IL-6, OSM, and LIF, bind to the gp130 receptor and activate STAT3, known as the gp130/JAK/STAT3 signaling. IL-6, a pleiotropic cytokine, plays important roles in triggering the acute phase response of the body to injury or inflammation. OSM, a multifunctional cytokine produced by activated T lymphocytes, monocytes, and dendritic cells, enhances the antiviral effects of IFN-α and plays a role in the induction of the adaptive immune response to pathogens (25, 26). STAT3 is found to be a central regulator of lymphocyte differentiation and function (27). Mutations in STAT3 cause autosomal-dominant hyper-IgE syndrome, a rare multisystem primary immunodeficiency characterized by recurrent bacterial infections in skin and lung and with abnormally high levels of IgE (28, 29).
PRRSV blocks the nuclear translocation of STAT1 and STAT2 to inhibit the IFN signaling pathway via the nonstructural protein 1β (nsp1β) (13, 14). However, PRRSV effect on the gp130/JAK/STAT3 signaling remains unknown. In the present study, our objective was to determine the PRRSV effect on STAT3 signaling. We discovered that PRRSV inhibits the STAT3 signaling via inducing STAT3 degradation via nsp5. Infection of MARC-145 cells by several PRRSV type 1 and type 2 strains resulted in a reduction of STAT3 protein without affecting its transcript level. The addition of the proteasome inhibitor MG132 to the PRRSV-infected cells restored the STAT3 protein levels. Transient expression of nsp5 led to a reduction of STAT3 protein via degradation through the ubiquitin-proteasome pathway. The C-terminal portion of nsp5 is responsible for the induction of STAT3 degradation. In consequence, nsp5 inhibited the OSM-activated gp130/JAK/STAT3 signaling pathway. This finding provides further insight into PRRSV pathogenesis and its interference with the host immune response.
RESULTS
OSM inhibits PRRSV replication, and PRRSV infection reduces STAT3 without affecting its transcript level.
OSM, a member of the IL-6 family, was found to synergistically inhibit the replication of hepatitis C virus in combination with IFN-α (25, 26). We sought to determine whether OSM alone had any antiviral role against PRRSV. MARC-145 cells were pretreated with OSM for 24 h before inoculation with the PRRSV strain VR-2385. The real-time PCR result showed that the OSM treatment significantly reduced PRRSV RNA replication in comparison to mock-treated control, whereas OSM had a minimal effect on cell viability (Fig. 1A). Based on our early study of the IFN-activated JAK/STAT signaling (14), we reasoned that PRRSV would antagonize the OSM-activated gp130/JAK/STAT3 signaling. We first determined the OSM-activated STAT3 phosphorylation in PRRSV-infected cells. The result showed that the STAT3 phosphorylation level in the PRRSV-infected cells was 15% of the mock-infected cells after OSM stimulation (Fig. 1B). The STAT3 protein level in the infected cells was significantly reduced to 0.26- or 0.36-fold in the absence or presence of OSM, respectively, compared to mock-infected cells.
FIG 1.

PRRSV infection reduces STAT3 in MARC-145 and PAM cells. (A) OSM treatment of MARC-145 cells inhibits PRRSV replication and has a minimal effect on cell viability. The cells were pretreated with OSM for 24 h before inoculation with PRRSV VR-2385 at a multiplicity of infection (MOI) of 1. The cells were harvested at 24 h postinfection (hpi). The relative percentage of PRRSV RNA levels determined by real-time PCR are shown. Significant differences from the untreated cells are denoted by asterisks (**) for P < 0.01. (B) PRRSV reduces STAT3 phosphorylation in MARC-145 cells upon OSM stimulation. The cells were infected with VR-2385 at an MOI of 1, incubated for 48 h, and treated with OSM for 30 min before harvested for Western blotting (WB) with antibody against STAT3, phosphorylated STAT3 (pSTAT3), and tubulin. The relative levels of pSTAT3 and STAT3 are shown below the images after normalization with tubulin in densitometry analysis. (C) PRRSV reduces the STAT3 protein level in MARC-145 cells but has a minimal effect on the STAT1 protein. The relative levels of STAT3 are shown below the images after normalization with GAPDH. (D) PRRSV reduces STAT3 protein level in PAM cells but has a minimal effect on the STAT1 protein. The cells were infected with VR-2385 at an MOI of 1 and harvested for WB at 16 hpi.
To exclude the possibility that PRRSV reduced STAT3 nonspecifically, we examined STAT1 protein level. Compared to mock-infected cells, STAT3 level in the infected cells was reduced to 0.36-fold, whereas the STAT1 level was unchanged (Fig. 1C). Since MARC-145 cells are monkey derived though they are PRRSV permissible, we wondered whether PRRSV had the same effect on STAT3 in primary PAM cells, which are main target cells in pigs during acute PRRSV infection. PAM cells were infected with VR-2385 and harvested 16 hpi for immunoblotting. Similarly, STAT3 level in the PRRSV-infected PAM cells was reduced to 0.37-fold in comparison to mock-infected control, whereas STAT1 remained steady (Fig. 1D). The harvesting of infected PAMs was done at 16 hpi since VR-2385 infection in these cells progresses faster than that in MARC-145 cells.
To exclude the possibility that the STAT3 reduction is the consequence from infection of PRRSV strain VR-2385 only, we included strains from both PRRSV type 1 and type 2 in the test. MARC-145 cells were infected with PRRSV type 2 strains VR-2385, Ingelvac PRRS MLV, and VR-2332 or PRRSV type 1 strain Lelystad. Compared to mock-infected cells, the STAT3 protein levels in cells infected with VR-2385, MLV, VR-2332, and Lelystad were reduced 0.37-, 0.33-, 0.24-, and 0.33-fold, respectively, whereas STAT1 remained steady (Fig. 2A and B). We reasoned that STAT3 reduction could be due to a decrease in transcription and/or translation or to accelerated protein degradation. To determine the mRNA level of STAT3 in the cells with PRRSV infection, we conducted reverse transcription quantitative PCR (RT-qPCR). The results showed that there was no significant difference in the mRNA levels of endogenous STAT3 between the PRRSV-infected and mock-infected cells (Fig. 2C). These results indicated that the PRRSV-induced reduction of STAT3 protein level was not due to perturbation of its transcripts.
FIG 2.
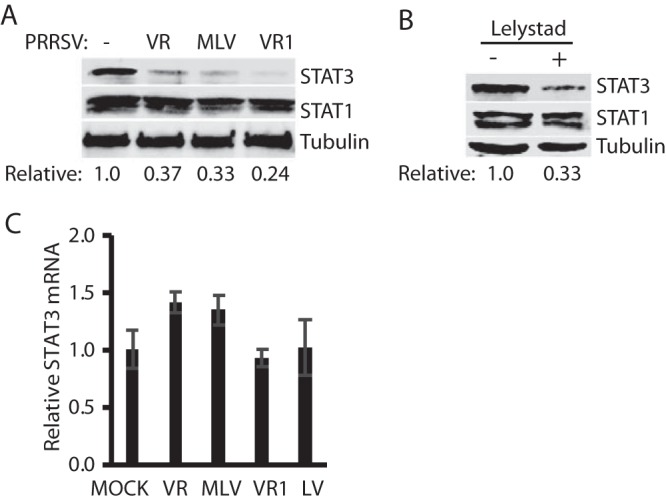
Infection of MARC-145 cells with PRRSV type 1 and type 2 strains reduces the STAT3 protein level but has a minimal effect on STAT3 transcripts. (A) Reduction of STAT3 protein level but no effect on STAT1 protein level in MARC-145 cells infected with PRRSV type 2 strains. The cells were infected with PRRSV type 2 strains VR-2385 (VR), Ingelvac PRRS MLV, and VR-2332 (VR1). At 48 hpi, the cells were harvested for WB with antibody against STAT3, STAT1, and tubulin. The relative levels of STAT3 are shown below the images after normalization with tubulin. (B) Reduction of STAT3 protein level by PRRSV type 1 strain Lelystad. (C) STAT3 mRNA levels in PRRSV-infected cells. The relative STAT3 mRNA levels are shown compared to the mock-infected cells. Error bars represent standard errors of the means of three repeated experiments. LV, Lelystad.
PRRSV reduces STAT3 protein level in a dose- and time-dependent manner.
To further study the PRRSV-mediated reduction of STAT3, we inoculated MARC-145 cells with a different amount of VR-2385. Along with the incremental amount of VR-2385 inoculation, the STAT3 protein level was reduced in a dose-dependent manner, whereas the STAT1 level remained relatively stable (Fig. 3A). Densitometry analysis showed that the STAT3 levels in MARC-145 cells inoculated at multiplicities of infection (MOIs) of 0.01, 0.1, 1, and 10 were reduced 0.43-, 0.38-, 0.27-, and 0.22-fold, respectively, compared to the mock-infected control. VR-2385 RNA levels in the infected cells significantly increased, along with the incremental inocula (Fig. 3B), as expected.
FIG 3.
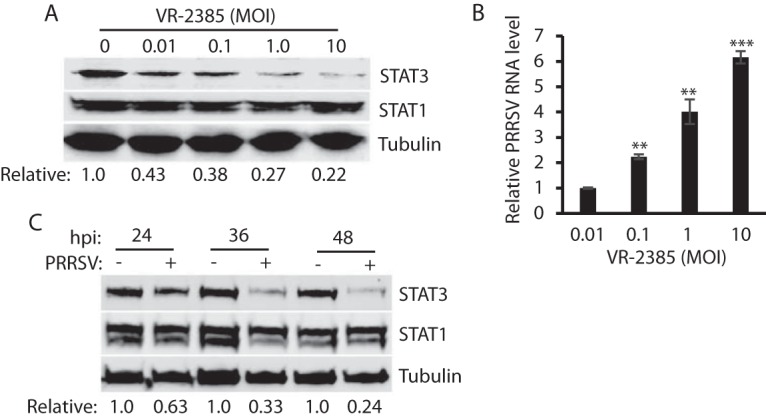
PRRSV reduces STAT3 protein level in a dose- and time-dependent manner. (A) Dose-dependent reduction of STAT3 by PRRSV strain VR-2385. MARC-145 cells were inoculated with incremental MOIs of VR-2385 and harvested for WB with antibodies against STAT3, STAT1, and tubulin at 48 hpi. The relative levels of STAT3 are shown below the images after normalization with tubulin. (B) PRRSV RNA levels in infected cells. The cells were harvested for RNA isolation and RT-qPCR at 24 hpi. Error bars represent the standard errors of the results of repeated experiments. Significant differences in RNA level from an MOI of 0.01 are denoted by asterisks (**, P < 0.01; ***, P < 0.001). (C) Temporal kinetics of STAT3 levels in PRRSV-infected cells. MARC-145 cells were infected with VR-2385 at an MOI of 1. At the different time points, the cells were harvested for WB. The relative levels of STAT3 protein are shown below the images after normalization with tubulin at each time point.
To examine the PRRSV effect on the temporal kinetics of STAT3, we infected MARC-145 cells with VR-2385 and harvested the cells 24, 36, and 48 hpi. Compared to the mock-infected cells at each time point, the STAT3 levels in the virus-infected cells at 24, 36, and 48 hpi were reduced 0.63-, 0.33-, and 0.24-fold, respectively (Fig. 3C). This result demonstrated that PRRSV infection reduces STAT3 protein level in a time-dependent manner.
PRRSV mediates STAT3 reduction via the ubiquitin-proteasomal degradation pathway.
The results presented above showed that PRRSV infection reduced the level of STAT3 protein but had a minimal effect on its transcript level. We reasoned that the STAT3 reduction could possibly be due to accelerated degradation by the ubiquitin-proteasomal pathway. To test this, MG132, a proteasome inhibitor, was added to the cells at 30 h after VR-2385 infection. The cells were harvested 6 h later for STAT3 examination. The MG132 treatment of the infected cells resulted in the restoration of STAT3 to a level similar to the mock-infected cells (Fig. 4A). PRRSV RNA levels in the cells with or without MG132 treatment are similar (Fig. 4B). This result indicates that PRRSV mediates the STAT3 reduction via ubiquitin-proteasomal degradation pathway.
FIG 4.
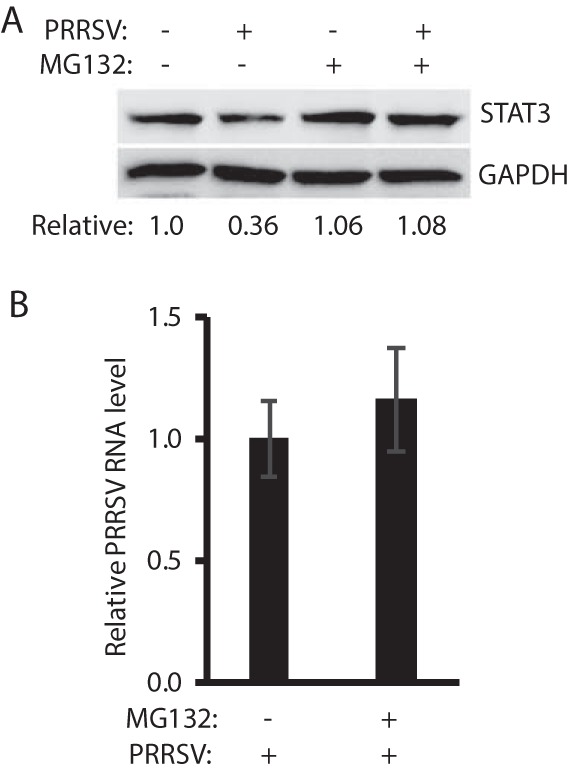
PRRSV mediates STAT3 reduction via the ubiquitin-proteasomal degradation pathway. (A) MG132 treatment restores STAT3 level in PRRSV-infected cells. MARC-145 cells were infected with VR-2385 at an MOI of 1. At 30 hpi, the cells were treated with MG132 for 6 h and then harvested for Western blotting with antibodies against STAT3 and GAPDH. Nontreated and mock-infected cells were included as controls. The relative levels of STAT3 protein are shown as fold values below the images after normalization with GAPDH. (B) PRRSV replication is not affected by the MG132 treatment. The relative PRRSV RNA levels determined by real-time PCR are shown compared to mock-treated cells.
PRRSV nsp5 reduces STAT3 protein level.
Having demonstrated that PRRSV infection reduces the STAT3 protein level, we wondered which protein of PRRSV was responsible for the reduction. HeLa cells were transfected with plasmids encoding PRRSV VR-2385 nsp or structural proteins to assess their effects on STAT3 levels. Western blotting results showed that cells transfected with nsp5 plasmid had a 0.4-fold STAT3 protein level compared to the cells transfected with green fluorescent protein (GFP), whereas the cells transfected with the other PRRSV plasmids had no significant reduction (Fig. 5). nsp2, nsp3, nsp9, and nsp10 are much larger than the other nsp and were analyzed separately by immunoblotting. The structural proteins had a minimal effect on STAT3 and were not examined further in this study (data not shown). Therefore, nsp5 was selected for further analysis.
FIG 5.
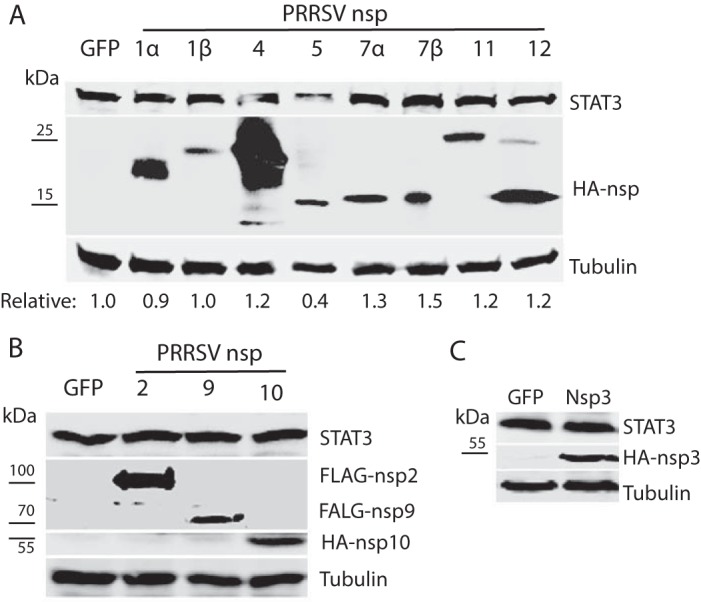
Screening of PRRSV nsp to identify the viral protein that is responsible for reduction of STAT3 protein level. (A) nsp5 reduces STAT3 in HeLa cells, whereas other PRRSV nsp have a minimal effect. The cells were transfected with individual HA-nsp plasmids. A GFP plasmid was included as a control. At 36 h posttransfection (hpt), WB analyses against STAT3, HA, and tubulin were performed to detect the endogenous STAT3 protein levels in cells with transient expression of the nsp. The relative levels of STAT3 protein are shown below the images. Molecular mass markers are added on the left of image of HA blotting. (B) nsp2, nsp9, and nsp10 have a minimal effect on STAT3 level in HeLa cells. The cells were transfected with FLAG-nsp2, FLAG-nsp9, HA-nsp10, or GFP plasmid. (C) nsp3 has a minimal effect on STAT3 level in HeLa cells. The cells were transfected with HA-nsp3 or GFP plasmid.
The nsp5 plasmid was used to transfect HEK293 cells to confirm the effect on STAT3. Compared to the cells transfected with empty vector, the cells transfected with nsp5 plasmid reduced STAT3 protein level in a dose-dependent manner (Fig. 6A). Densitometry analysis showed that the STAT3 levels in HEK293 cells transfected with 0.25, 0.5, 1.0, and 2.0 μg of nsp5 plasmid were reduced 0.64-, 0.40-, 0.33-, and 0.05-fold, respectively, compared to the empty vector control. The nsp5 protein level increased, along with the incremental amount of the plasmid DNA in the transfection.
FIG 6.
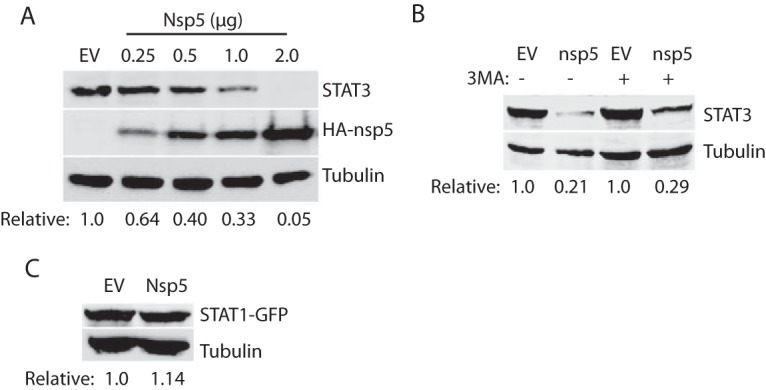
PRRSV nsp5 reduces the STAT3 protein level. (A) nsp5 reduces STAT3 protein level in a dose-dependent manner. HEK293 cells were transfected with nsp5 plasmid in incremental amounts. WB analyses with antibodies against STAT3, HA, and tubulin were conducted at 48 hpt. The relative levels of STAT3 are shown below the images after normalization with tubulin. (B) 3-MA treatment is unable to rescue STAT3 protein level in HEK293 cells cotransfected with 0.5 μg of HA-nsp5 and 0.5 μg of STAT3 plasmid. Mock-treated cells were included as controls. The cells were treated with 3-MA for 24 h before being harvested for WB. The relative levels of STAT3 protein are shown below the images. (C) nsp5 has no effect on STAT1 protein level. HEK293 cells were transfected with 0.5 μg of pCAGEN-HA-nsp5 plasmid and 0.5 μg of pEGFP-C1-STAT1 plasmid. WB against STAT1 antibody was performed at 36 hpt. The relative levels of STAT1-GFP are shown below the images.
nsp5 is known to induce autophagy (30, 31). To exclude the possibility that nsp5 reduces STAT3 via autophagy pathway, we treated the cells with autophagy inhibitor 3-methyladenine (3-MA) (32). Compared to cells transfected with the empty vector, the cells with nsp5 had a similar lower STAT3 level in the presence or absence of 3-MA (Fig. 6B). Like the whole virus infection, nsp5 alone had no effect on the STAT1 level (Fig. 6C).
PRRSV nsp5 leads to an elevation in STAT3 ubiquitination and a shortening of its half-life.
Since MG132 treatment restored STAT3 levels in cells with PRRSV infection, we reasoned that the STAT3 ubiquitination levels in the cells with nsp5 expression would increase. HEK293 cells were cotransfected with STAT3 and nsp5 plasmids. The cells were lysed for immunoprecipitation (IP) with an antibody against STAT3. Western blotting with ubiquitin antibody showed that ubiquitinated STAT3 in the cells with nsp5 expression was 12.6-fold greater than that in the cells transfected with the empty vector (Fig. 7A), which indicates that there was more polyubiquitination of STAT3 in the cells with nsp5 expression. Blotting with HA antibody failed to determine any nsp5 band in the IP complex lane, which suggests that STAT3 did not interact with nsp5 under our testing conditions. In addition, the input of whole-cell lysate was included in the blotting and results showed that the total ubiquitination level in the cells with nsp5 expression was similar to the cells transfected with empty vector (Fig. 7A). These results demonstrated that nsp5 induced the elevation of STAT3 ubiquitination in the cells.
FIG 7.
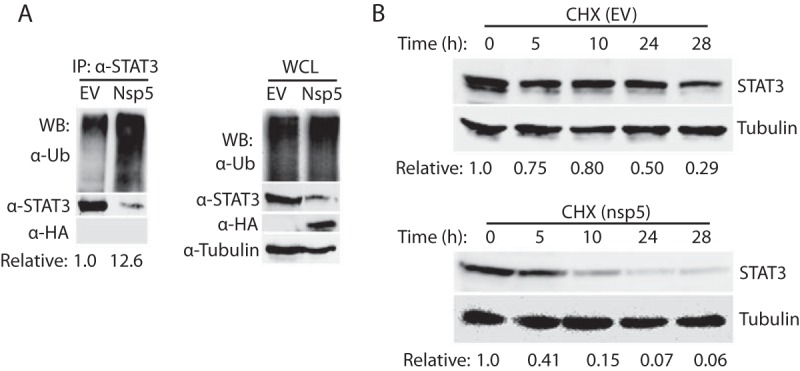
PRRSV nsp5 leads to STAT3 degradation by the ubiquitin-proteasomal pathway. (A) Presence of nsp5 leads to elevation of STAT3 polyubiquitination level. IP analyses with STAT3 antibody and then WB with antibodies against ubiquitin (Ub), STAT3, and HA were performed. Samples of whole-cell lysate (WCL) were included as controls. The relative levels of ubiquitinated STAT3 are shown below the images after normalization with STAT3 level. (B) nsp5 shortens STAT3 half-life from around 24 to 3.5 h. HEK293 cells were transiently cotransfected with 0.5 μg of STAT3 and nsp5 plasmids. The cells were treated with cycloheximide (CHX) at 100 μg/ml at 24 hpt and harvested at indicated times in hours posttreatment, followed by WB with antibodies against STAT3 and tubulin. The relative levels of STAT3 are shown below the images after normalization with tubulin.
We next tested whether the half-life of STAT3 would be shortened. HEK293 cells were cotransfected with STAT3 and nsp5 plasmids and treated with cycloheximide, a translation inhibitor, followed by harvesting and immunoblotting at different time points. In the presence of nsp5, the STAT3 level decreased at a higher rate than that in the cells transfected with the empty vector (Fig. 7B). At 5, 10, 24, and 28 h after the cycloheximide treatment, the STAT3 levels in the cells with nsp5 expression were reduced by 59, 85, 93, and 94%, respectively, whereas STAT3 in the cells transfected with the empty vector were reduced by 25, 20, 50, and 71%, separately (Fig. 7B). The densitometry analysis was normalized with β-tubulin, which has a long half-life of 50 h (33). The expression of nsp5 induced a shortening of the STAT3 half-life from 24 h to approximately 3.5 h. This result is consistent with the elevation of STAT3 polyubiquitination in the cells with nsp5 expression.
The C-terminal domain of nsp5 appears to be responsible for the induced degradation of STAT3.
To map the domain of nsp5 involved in inducing the degradation of STAT3, deletion constructs of VR-2385 nsp5 were prepared. Based on analysis of nsp5 polypeptide sequence using Lasergene, four fragments of nsp5 were cloned into the pCDNA3-VenusC1 vector (Fig. 8A). Overexpression of the nsp5 truncation constructs in HEK293 cells was confirmed by immunoblotting with GFP antibody (Fig. 8B).
FIG 8.

The C-terminal domain of nsp5 is required for inducing STAT3 degradation. (A) Schematic illustration of truncation mutants nsp5D1, nsp5D2, nsp5D3, and nsp5D4. The numbers above the lines indicate amino acid residues of nsp5. (B) nsp5D2, nsp5D3, and nsp5D4 lead to STAT3 degradation, whereas nsp5D1 has a minimal effect. HEK293 cells were transfected with 0.5 μg of YFP-tagged plasmids and harvested at 36 hpt for immunoblotting with antibodies against STAT3, YFP, and tubulin. The relative levels of STAT3 are shown below the images after normalization with tubulin. Molecular mass markers are given to the left of the YFP-nsp5 image. EV, empty vector; FL, full length. (C) Predicted membrane protein topology of nsp5 determined using TMHMM. The numbers on the x axis denote the nsp5 residues. The bars in different colors indicate the topology. Loops inside or outside the membrane are indicated by different colors. (D) Alignment of nsp5 amino acid sequences for PRRSV VR-2385, VR-2332, MN184, and Lelystad. The rectangle highlights the highly conserved residues. The numbers below the consensus sequences and on the left denote the amino acid residue number of nsp5. Residues that are identical to the consensus at the top are not shown.
Immunoblotting results showed that the cells transfected with YFP-nsp5D2 (amino acids [aa] 59 to 170), YFP-nsp5D3 (aa 1 to 122), and YFP-nsp5D4 (aa 86 to 170) had considerably lower STAT3 protein levels than the cells transfected with the empty vector (Fig. 8B). Densitometry analysis showed that the cells with YFP-nsp5D1, YFP-nsp5D2, YFP-nsp5D3, and YFP-nsp5D4 had STAT3 levels reduced by 8, 76, 58, and 67%, respectively, compared to the cells transfected with the empty vector. The results indicate that the nsp5D1 (aa 1 to 85) corresponding to the N-terminal domain has a minimal effect on the STAT3 level, whereas the C-terminal domain (aa 86 to 170) of nsp5 is responsible for the induced degradation of STAT3.
Sequence analysis suggests that nsp5 polypeptides are highly hydrophobic and may be membrane associated. Analysis with TMHMM, a membrane protein topology prediction method (34), showed that nsp5 has five transmembrane domains spanning residues aa 13 to 35, 45 to 67, 79 to 101, 121 to 143, and 150 to 169 (Fig. 8C). There are two outside-oriented loops spanning residues aa 36 to 44 and aa 102 to 120 and three inside-oriented loops spanning residues aa 1 to 12, 68 to 78, and 144 to 149 in nsp5. The outside-oriented loop spanning aa 102 to 120 is longer than the other loops and is located in the C-terminal domain.
Sequence alignment of nsp5 showed that VR-2385 nsp5 has 96.5, 85.3, and 69.4% identical amino acid residues with VR-2332, MN184, and Lelystad, respectively. Among the 52 aa variations between nsp5 of VR-2385 and Lelystad, 13 aa distribute in the N-terminal half of nsp5, and the remaining 39 aa are in the C-terminal half. There is a highly conserved stretch of residues, aa 110 to 118, in the outside-oriented loop spanning aa 102 to 120 (Fig. 8D). Among the nine residues in aa 110 to 118 of nsp5 from the four PRRSV strains, eight are identical, which needs further study to determine whether they correlate with the nsp5 induction of STAT3 reduction.
nsp5 inhibits gp130/JAK/STAT3 signaling.
As a transcription activator, STAT3 mediates multiple important signaling pathways related to immune responses, cell growth, and cell survival. Thus, we speculated that nsp5 would inhibit the STAT3 signaling as it reduced STAT3 protein level. First, we tested whether it inhibits the OSM-mediated STAT3 activation. As expected, the STAT3 phosphorylation in HEK293 cells transfected with nsp5 plasmid was much lower than in cells with empty vector (Fig. 9A). The relative phosphorylated STAT3 levels in the cells with 0.25 and 1 μg of nsp5 were reduced by 60 and 74%, respectively, compared to the control cells with empty vector. The relative STAT3 levels in the cells with 0.25 and 1 μg of nsp5 were reduced by 50 and 72%, respectively, compared to the control cells. The results indicate that the nsp5 inhibition of STAT3 phosphorylation was due to the reduction of STAT3 protein level.
FIG 9.
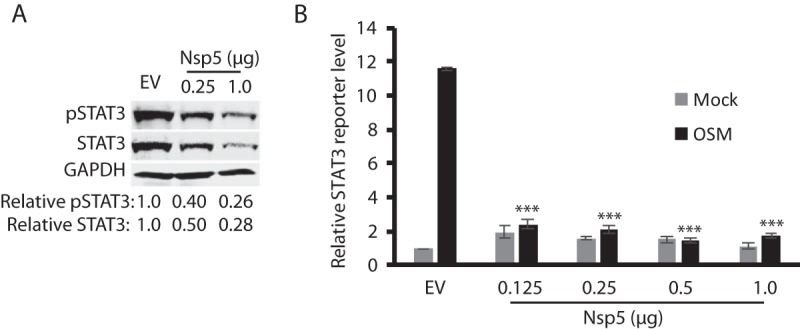
PRRSV nsp5 inhibits STAT3 signaling. (A) HEK293 cells transfected with nsp5 have a much lower OSM-mediated STAT3 phosphorylation level than that of the control cells with empty vector (EV). The cells were treated with OSM for 30 min before harvested for WB. The relative levels of phosphorylated STAT3 (pSTAT3) and total STAT3 are shown below the images. (B) nsp5 inhibits OSM-stimulated STAT3 reporter expression. HEK293 cells were cotransfected with 0.5 μg of nsp5 plasmid, 0.5 μg of STAT3 reporter, and 0.05 μg of Renilla plasmid. At 24 hpt, the cells were treated with OSM and harvested for luciferase activity assay 24 h later. The relative levels of firefly luciferase activity are shown as fold values compared to the EV control after normalization with the Renilla activity. A significant difference in firefly luciferase activity from the EV+OSM control is denoted by asterisks (***, P < 0.001).
We next tested whether nsp5 inhibits STAT3 reporter expression. HEK293 cells were cotransfected with the incremental amount of nsp5 plasmid DNA, along with STAT3 reporter plasmid 4×M67 pTATA TK-Luc and pRL-TK. The cells were then treated with OSM for 24 h before harvested for luciferase assay. OSM induced obvious transactivation of 4×M67 pTATA TK-Luc in the cells transfected with the empty vector (Fig. 9B), whereas the luciferase activities in cells transfected with nsp5 plasmid from 0.125 to 1.0 μg were significantly (P < 0.001) lower. This result demonstrated that nsp5 led to the inhibition of the OSM-mediated gp130/JAK/STAT3 signaling by inducing degradation of the STAT3 protein.
DISCUSSION
Our results in this study demonstrated that PRRSV inhibits the STAT3 signaling pathway by inducing the degradation of STAT3. The JAK/STAT pathway is critical in relaying signals from cytokines and growth factors to regulate gene expression in biological processes including apoptosis, angiogenesis, differentiation, cell proliferation, inflammatory response, and immunity (18, 19). In particular, STAT3 is a central regulator of lymphocyte differentiation and function (27). It can regulate CD4+ T cell differentiation and control Th17 cells differentiation by IL-6 and IL-23. STAT3 deficiency affects the generation of memory CD8+ T cells (35, 36) and memory B cells (37, 38). Therefore, STAT3 is indispensable in the host immune system. PRRSV infection induces a weak cell-mediated immune response, in which PRRSV-specific T cells transiently appear 2 weeks after infection without a change in frequency of CD4+ and CD8+ T cells (9). The STAT3 antagonizing may be one of the reasons for PRRSV interference with the development of protective immune response.
We discovered that PRRSV inhibits the STAT3 signaling by inducing degradation of STAT3. The PRRSV-mediated degradation of STAT3 is specific as PRRSV has no effect on the STAT1 protein level in MARC-145 and PAM cells. The PRRSV-induced reduction of STAT3 also occurs in primary PAMs, the main target cells in infected pigs. This reduction appears to be an intrinsic property of PRRSV since several strains from both PRRSV type 1 and type 2, including a vaccine strain, are able to do so in MARC-145 cells. The STAT3 decrease was found due to degradation via the ubiquitin-mediated proteasome pathway because blocking this degradation pathway with MG132 resulted in the restoration of STAT3 levels in the cells infected with PRRSV.
Macrophages are important immune effector cells and can respond to endogenous stimuli generated by injury or infection (39). PRRSV infection of PAMs leads to the inhibition of IFN induction (11) and the suppression of the JAK/STAT signaling (14), which may result in the poor generation of stimuli to other cells and consequently an ineffectual immune response. PRRSV infection in pigs leads to the elevation of IL-10 (40, 41) and induces lung lesions with inflammatory cell infiltration (42). IL-10 signaling via mediator STAT3 results in the generation of regulatory macrophages, which have an anti-inflammatory activity to dampen immunopathology. PRRSV antagonizing the STAT3 signaling could interfere with the IL-10 regulatory function and leads to inflammation.
PRRSV nsp5 was found to be responsible for the STAT3 reduction. nsp5 induces the elevation of the STAT3 polyubiquitination and shortens its half-life from 24 h to approximately 3.5 h. Our data suggest that nsp5 enhances STAT3 degradation via the ubiquitin-proteasomal pathway. There seems no direct interaction between nsp5 and STAT3 because IP with an antibody against STAT3 did not pull down nsp5. Likewise, immunofluorescence assay and confocal microscopy did not determine colocalization of nsp5 and STAT3 in cells cotransfected with both plasmids (data not shown). These results suggest that STAT3 and nsp5 either have no direct interaction or, if any, only transient interaction. We presume that nsp5 activates an ubiquitin E3 ligase of STAT3 and accelerates proteasome degradation of the transcription activator. The E3 ligase for the STAT3 proteasome degradation remains unknown, although TMF/ARA 160, a Golgi-resident protein, was found to mediate the degradation of STAT3 (43). However, nsp5 was found to colocalize with the endoplasmic reticulum, not the Golgi apparatus (data not shown). nsp5 induces autophagic cell death when it is overexpressed (31). However, treatment of the cells with 3-MA to block autophagy in this study did not rescue the STAT3 level, indicating that autophagy was not the reason for the STAT3 reduction.
nsp5 is a hydrophobic transmembrane protein and can form a membranous structure in the cytoplasm that could be the site for PRRSV replication (5). The C-terminal domain (aa 86 to 170) of nsp5 might be responsible for the STAT3 degradation since the deletion constructs nsp5D2, nsp5D3, and nsp5D4 containing the full or partial C-terminal domain led to the reduction, whereas nsp5D1 (aa 1 to 85) failed to do so. This C-terminal domain contains a mixture of hydrophobic and hydrophilic residues with a potential surface location motif, as shown by TMHMM analysis. In the outside-oriented loop in the C-terminal domain, there is a stretch of residues that are highly conserved across the two PRRSV species. This suggests that these conserved residues might play a role in the nsp5-induced STAT3 reduction. Further work is needed to address the speculation.
The nsp5-mediated degradation of STAT3 led to blocking of the STAT3 signaling. In the OSM-dependent transactivation of the reporter plasmid of the STAT3 binding promoter, the presence of nsp5 resulted in a significant reduction of the reporter expression. This result substantiates that PRRSV nsp5 mediates the inhibition of the gp130-dependent signaling via its inducing STAT3 degradation.
Due to its importance in the host innate and adaptive immune responses, STAT3 has been found to be the target of some viral pathogens. Measles virus V protein inhibits the IL-6-mediated STAT3 signaling (44). The V protein of mumps virus prevents responses to IL-6 and v-Src by inducing STAT3 ubiquitination and degradation (45). The rabies virus interferon antagonist P protein inhibits the gp130 receptor signaling by interacting with activated STAT3 (46). Inhibition of the STAT3 signaling by these viruses and PRRSV can lead to inhibition of a broad spectrum of cytokines and growth factors to thwart host antiviral responses and allow virus replication and spread in vivo. PRRSV was shown to inhibit the IFN-activated JAK/STAT signaling by blocking STAT1 nuclear translocation (14). The blocking was found to be due to PRRSV nsp1β-mediated degradation of karyopherin α1 (KPNA1; importin α5) (13). Here, we found that PRRSV interferes with the gp130/JAK/STAT3 signaling via nsp5. This sheds further light on PRRSV antagonizing the JAK/STAT signaling, which generates a conducive environment for virus replication and spread. Our results demonstrate that PRRSV infection induces STAT3 degradation via the ubiquitin-proteasomal pathway. PRRSV nsp5 was found to be responsible for the STAT3 reduction and for blocking its signaling. This finding provides insights for PRRSV pathogenesis and its interference with the host immune response.
MATERIALS AND METHODS
Cells, viruses, and chemicals.
MARC-145 (7), HEK293 (ATCC CRL-1573) and HeLa (ATCC CCL-2) cells were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum. Primary PAM cells were prepared and cultured in RPMI 1640 medium as previously described (47). The cryopreserved PAM cells were revived and precultured for 24 h before used for virus inoculation.
PRRSV strains VR-2385 (48), VR-2332 (49), Ingelvac PRRS MLV (50), and Lelystad (4) were used to inoculate MARC-145 cells at an MOI of 1. The median tissue culture infectious dose of PRRSV was determined in MARC-145 cells (51).
MG132 (Sigma-Aldrich, St. Louis, MO), a proteasome inhibitor, was used to treat cells at a final concentration of 10 mM for 6 h prior to harvesting for further analysis. In order to determine the half-life of STAT3, cycloheximide (Sigma) was added to cultured cells at a final concentration of 100 μg/ml to block protein translation. OSM (R&D Systems, Inc., Minneapolis, MN) was used to activate the gp130/JAK/STAT3 signaling pathway at a final concentration of 10 ng/ml. The cells were harvested at the time points indicated in Results or in the figure legends for Western blotting. The half-life of STAT3 was determined using nonlinear regression in Prism (GraphPad Software, Inc., La Jolla, CA) in the analysis of the densitometry data of the immunoblotting results. 3-Methyladenine (3-MA; Thermo Fisher Scientific, Waltham, MA), an inhibitor of autophagy by blocking autophagosome formation (32), was used at 5 mM in the treatment of cells for 12 to 24 h.
A cell viability assay was performed with a CellTiter-Glo luminescent cell viability assay according to the manufacturer's instructions (Promega, Mandison, WI). The cells to be tested were placed into 96-well culture plate and treated as described above.
Plasmids.
The nsp5 of PRRSV VR-2385 was cloned into a pCAGEN-HA and pCDNA3-VenusC1 vectors (13) with the primers 32nsp5F1 and 85nsp5R2 (Table 1). The nsp5 deletion mutants were cloned into pCDNA3-VenusC1 vector using the following primers: 32nsp5F1 and 85nsp5R7 for nsp5D1, 85nsp5F3 and 85nsp5R2 for nsp5D2, 32nsp5F1 and 85nsp1R5 for nsp5D3, and 85nsp5F6 and 85nsp5R2 for nsp5D4 (Table 1). The resulting recombinant plasmids were confirmed by restriction enzyme digestion and DNA sequencing. The construction of the pCDNA3-Ubiquitin-Myc plasmid and other PRRSV nsp plasmids was described previously (52, 53).
TABLE 1.
Primers used in this study
| Primera | Sequence (5′–3′)b | Target gene |
|---|---|---|
| 32nsp5F1 | CGGAATTCGGAGGCCTCTCCACCGTCC | nsp5 |
| 85nsp5R2 | CCGCTCGAGTTACTCGGCAAAGTACCGCAGG | nsp5 |
| 85nsp5R7 | GCTCGAGTTAGCTGAAAAAGGCAAGTGAC | nsp5D1 |
| 85nsp5F3 | GGAATTCCCATGGTCTGCGCAAGTTC | nsp5D2 |
| 85nsp5R5 | GCTCGAGTTACACAACCATCATCCGAGGCAG | nsp5D3 |
| 85nsp5F6 | CGGAATTCAGCCTTGGTGCGGTGACCGG | nsp5D4 |
| R-STAT3-F1 | GTGATGCTTCCCTGATTGTG | STAT3 |
| R-STAT3-R1 | GCAAGGAGTGGGTCTCTAGG | STAT3 |
F, forward primer; R, reverse primer. “32” or “85” before a primer name indicates that the primer is based on sequences of PRRSV VR-2332 (GenBank accession no. U87392) and VR-2385 (GenBank accession no. JX044140), respectively. An “R” before a primer name indicates the primer was designed for real-time PCR.
Italicized letters indicate restriction enzyme cleavage sites for cloning.
The pEGFP-C1-STAT1 was a gift from Alan Perantoni (Addgene, plasmid 12301) (54). The pCDNA3-STAT3 was a gift from Jim Darnell (Addgene, plasmid 8706) (55). The pRc/CMV-FLAG-STAT3 was a gift from Jim Darnell (Addgene, plasmid 8707) (56). The 4×M67 pTATA TK-Luc was a gift from Jim Darnell (Addgene, plasmid 8688) (57) and used for the STAT3 reporter assay.
Western blot analysis.
Protein samples were separated by SDS-PAGE and analyzed by Western blotting as described previously (58, 59). The primary antibodies used in this study were against STAT3 (Santa Cruz Biotechnology, Inc., Dallas, TX), phospho-STAT3 (Tyr705, clone EP2147Y; Thermo Fisher Scientific), ubiquitin (Santa Cruz), hemagglutinin (HA; Thermo Fisher Scientific), FLAG (Sigma), GFP (Rockland Immunochemicals, Inc., Gilbertsville, PA), glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Santa Cruz), and β-tubulin (Sigma). The secondary antibodies used in this study were goat anti-rabbit or goat anti-mouse IgG conjugated with horseradish peroxidase (Rockland Immunochemicals). The chemiluminescence signal acquisition and densitometry analysis were conducted using the Quantity One program (v4.6) in a Chemi-Doc XRS imaging system (Bio-Rad Laboratories, Hercules, CA).
IP.
Immunoprecipitation (IP) was conducted as previously described (13). The clarified cell lysate was incubated with the STAT3 antibody (Santa Cruz), followed by incubation with protein G-agarose (KPL, Inc., Gaithersburg, MD). The IP samples were subjected to Western blotting with antibodies against ubiquitin, STAT3, and HA. To determine ubiquitinated STAT3, ubiquitin aldehyde (Boston Biochem, Inc., Cambridge, MA), a specific inhibitor of ubiquitin C-terminal hydrolases, was included in the lysis buffer at a final concentration of 2.53 μM.
RNA isolation and real-time PCR.
Total RNA was isolated from HEK293 and MARC-145 cells with TRIzol reagent (Thermo Fisher Scientific) in accordance with the manufacturer's instructions. RT-qPCR analyses were conducted as described previously (47, 60). The real-time PCR primers used for STAT3 were R-STAT3-F1 and R-STAT3-R1 (Table 1). Transcripts of ribosomal protein L32 (RPL32) were also amplified and used to normalize the total input RNA. The primers used for PRRSV and RPL32 were described previously (13). The relative transcript levels were shown as fold values compared to the mock-treated control after RPL32 normalization.
Reporter assay.
HEK293 cells were transfected with the STAT3 reporter plasmid 4×M67 pTATA TK-Luc (57) and nsp5 plasmids. The Renilla luciferase vector pRL-TK (Promega, Madison, WI), which expresses Renilla luciferase under the control of a constitutively active promoter, was also transfected for normalization. At 24 h after transfection, OSM was added into the cells at a final concentration of 10 ng/ml. After incubation for another 24 h, the cells were lysed for the luciferase activity assay of firefly and Renilla luciferases according to the manufacturer's instructions (Promega). Lysates of the cells without OSM treatment were included as a control for the assessment of the STAT3 activation level. The relative firefly luciferase activity is shown after normalization with the Renilla level, as described previously (53).
Statistical analysis.
Differences in indicators between the treatment group and control were assessed by using the Student t test. A two-tailed P value of <0.05 was considered significant.
ACKNOWLEDGMENT
We are grateful to Joseph F. Urban at Human Nutrition Research Center (USDA, Beltsville, MD) for providing of the lung lavage of piglets.
Z.M., R.W., and S.L. were partially sponsored by Chinese Scholarship Council. Y.X. and Y.W. were partially supported by Shandong Bureau of Education. This study was partially funded by internal fund of the University of Maryland.
REFERENCES
- 1.Holtkamp DJ, Kliebenstein JB, Neumann EJ, Zimmerman J, Rotto H, Yoder TK, Wang C, Yeske P, Mowrer C, Haley C. 2013. Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on U.S. pork producers. J Swine Health Prod 21:72–84. [Google Scholar]
- 2.Faaberg KS, Balasuriya UB, Brinton MA, Gorbalenya AE, Leung FC-C, Nauwynck H, Snijder EJ, Stadejek T, Yang H, Yoo D. 2012. Family Arteriviridae, p 796–805. In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed), Virus taxonomy: classification and nomenclature of viruses: ninth report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, San Diego, CA. [Google Scholar]
- 3.Kuhn JH, Lauck M, Bailey AL, Shchetinin AM, Vishnevskaya TV, Bao Y, Ng TF, LeBreton M, Schneider BS, Gillis A, Tamoufe U, Diffo Jle D, Takuo JM, Kondov NO, Coffey LL, Wolfe ND, Delwart E, Clawson AN, Postnikova E, Bollinger L, Lackemeyer MG, Radoshitzky SR, Palacios G, Wada J, Shevtsova ZV, Jahrling PB, Lapin BA, Deriabin PG, Dunowska M, Alkhovsky SV, Rogers J, Friedrich TC, O'Connor DH, Goldberg TL. 2016. Reorganization and expansion of the nidoviral family Arteriviridae. Arch Virol 161:755–768. doi: 10.1007/s00705-015-2672-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conzelmann KK, Visser N, Van Woensel P, Thiel HJ. 1993. Molecular characterization of porcine reproductive and respiratory syndrome virus, a member of the arterivirus group. Virology 193:329–339. doi: 10.1006/viro.1993.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lunney JK, Fang Y, Ladinig A, Chen N, Li Y, Rowland B, Renukaradhya GJ. 2016. Porcine reproductive and respiratory syndrome virus (PRRSV): pathogenesis and interaction with the immune system. Annu Rev Anim Biosci 4:129–154. doi: 10.1146/annurev-animal-022114-111025. [DOI] [PubMed] [Google Scholar]
- 6.Rossow KD, Collins JE, Goyal SM, Nelson EA, Hennings CJ, Benfield DA. 1995. Pathogenesis of porcine reproductive and respiratory syndrome virus infection in gnotobiotic pigs. Vet Pathol 32:361–373. doi: 10.1177/030098589503200404. [DOI] [PubMed] [Google Scholar]
- 7.Kim HS, Kwang J, Yoon IJ, Joo HS, Frey ML. 1993. Enhanced replication of porcine reproductive and respiratory syndrome (PRRS) virus in a homogeneous subpopulation of MA-104 cell line. Arch Virol 133:477–483. doi: 10.1007/BF01313785. [DOI] [PubMed] [Google Scholar]
- 8.Labarque GG, Nauwynck HJ, Van Reeth K, Pensaert MB. 2000. Effect of cellular changes and onset of humoral immunity on the replication of porcine reproductive and respiratory syndrome virus in the lungs of pigs. J Gen Virol 81:1327–1334. doi: 10.1099/0022-1317-81-5-1327. [DOI] [PubMed] [Google Scholar]
- 9.Xiao Z, Batista L, Dee S, Halbur P, Murtaugh MP. 2004. The level of virus-specific T-cell and macrophage recruitment in porcine reproductive and respiratory syndrome virus infection in pigs is independent of virus load. J Virol 78:5923–5933. doi: 10.1128/JVI.78.11.5923-5933.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wills RW, Doster AR, Galeota JA, Sur JH, Osorio FA. 2003. Duration of infection and proportion of pigs persistently infected with porcine reproductive and respiratory syndrome virus. J Clin Microbiol 41:58–62. doi: 10.1128/JCM.41.1.58-62.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Albina E, Carrat C, Charley B. 1998. Interferon-alpha response to swine arterivirus (PoAV), the porcine reproductive and respiratory syndrome virus. J Interferon Cytokine Res 18:485–490. doi: 10.1089/jir.1998.18.485. [DOI] [PubMed] [Google Scholar]
- 12.Buddaert W, Van Reeth K, Pensaert M. 1998. In vivo and in vitro interferon (IFN) studies with the porcine reproductive and respiratory syndrome virus (PRRSV). Adv Exp Med Biol 440:461–467. doi: 10.1007/978-1-4615-5331-1_59. [DOI] [PubMed] [Google Scholar]
- 13.Wang R, Nan Y, Yu Y, Zhang YJ. 2013. Porcine reproductive and respiratory syndrome virus Nsp1β inhibits interferon-activated JAK/STAT signal transduction by inducing karyopherin-α1 degradation. J Virol 87:5219–5228. doi: 10.1128/JVI.02643-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel D, Nan Y, Shen M, Ritthipichai K, Zhu X, Zhang YJ. 2010. Porcine reproductive and respiratory syndrome virus inhibits type I interferon signaling by blocking STAT1/STAT2 nuclear translocation. J Virol 84:11045–11055. doi: 10.1128/JVI.00655-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez-Navajas JM, Lee J, David M, Raz E. 2012. Immunomodulatory functions of type I interferons. Nat Rev Immunol 12:125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takaoka A, Yanai H. 2006. Interferon signalling network in innate defence. Cell Microbiol 8:907–922. doi: 10.1111/j.1462-5822.2006.00716.x. [DOI] [PubMed] [Google Scholar]
- 17.Iwasaki A, Medzhitov R. 2015. Control of adaptive immunity by the innate immune system. Nat Immunol 16:343–353. doi: 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stark GR, Darnell JE Jr. 2012. The JAK-STAT pathway at twenty. Immunity 36:503–514. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. 2015. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med 66:311–328. doi: 10.1146/annurev-med-051113-024537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuchipudi SV. 2015. The complex role of STAT3 in viral infections. J Immunol Res 2015:272359. doi: 10.1155/2015/272359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Shea JJ, Plenge R. 2012. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 36:542–550. doi: 10.1016/j.immuni.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, Yoshimura A, Hennighausen L, O'Shea JJ. 2006. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A 103:8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Villarino AV, Kanno Y, Ferdinand JR, O'Shea JJ. 2015. Mechanisms of Jak/STAT signaling in immunity and disease. J Immunol 194:21–27. doi: 10.4049/jimmunol.1401867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garbers C, Aparicio-Siegmund S, Rose-John S. 2015. The IL-6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr Opin Immunol 34:75–82. doi: 10.1016/j.coi.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Ikeda M, Mori K, Ariumi Y, Dansako H, Kato N. 2009. Oncostatin M synergistically inhibits HCV RNA replication in combination with interferon-alpha. FEBS Lett 583:1434–1438. doi: 10.1016/j.febslet.2009.03.054. [DOI] [PubMed] [Google Scholar]
- 26.Larrea E, Aldabe R, Gonzalez I, Segura V, Sarobe P, Echeverria I, Prieto J. 2009. Oncostatin M enhances the antiviral effects of type I interferon and activates immunostimulatory functions in liver epithelial cells. J Virol 83:3298–3311. doi: 10.1128/JVI.02167-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kane A, Deenick EK, Ma CS, Cook MC, Uzel G, Tangye SG. 2014. STAT3 is a central regulator of lymphocyte differentiation and function. Curr Opin Immunol 28:49–57. doi: 10.1016/j.coi.2014.01.015. [DOI] [PubMed] [Google Scholar]
- 28.Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, Anderson VL, Darnell DN, Welch PA, Kuhns DB, Frucht DM, Malech HL, Gallin JI, Kobayashi SD, Whitney AR, Voyich JM, Musser JM, Woellner C, Schaffer AA, Puck JM, Grimbacher B. 2007. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 357:1608–1619. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 29.Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, Metin A, Karasuyama H. 2007. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 448:1058–1062. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 30.Cottam EM, Maier HJ, Manifava M, Vaux LC, Chandra-Schoenfelder P, Gerner W, Britton P, Ktistakis NT, Wileman T. 2011. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 7:1335–1347. doi: 10.4161/auto.7.11.16642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang L, Wang R, Ma Z, Wang Y, Zhang Y. 2015. Inducing autophagic cell death by nsp5 of porcine reproductive and respiratory syndrome virus. Austin Virol Retrovirol 2015:1014. [Google Scholar]
- 32.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. 2000. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 275:992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 33.Caron JM, Jones AL, Kirschner MW. 1985. Autoregulation of tubulin synthesis in hepatocytes and fibroblasts. J Cell Biol 101:1763–1772. doi: 10.1083/jcb.101.5.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 35.Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. 2011. An interleukin-21/interleukin-10/STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity 35:792–805. doi: 10.1016/j.immuni.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siegel AM, Heimall J, Freeman AF, Hsu AP, Brittain E, Brenchley JM, Douek DC, Fahle GH, Cohen JI, Holland SM, Milner JD. 2011. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity 35:806–818. doi: 10.1016/j.immuni.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Avery DT, Deenick EK, Ma CS, Suryani S, Simpson N, Chew GY, Chan TD, Palendira U, Bustamante J, Boisson-Dupuis S, Choo S, Bleasel KE, Peake J, King C, French MA, Engelhard D, Al-Hajjar S, Al-Muhsen S, Magdorf K, Roesler J, Arkwright PD, Hissaria P, Riminton DS, Wong M, Brink R, Fulcher DA, Casanova JL, Cook MC, Tangye SG. 2010. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med 207:155–171. doi: 10.1084/jem.20091706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deenick EK, Avery DT, Chan A, Berglund LJ, Ives ML, Moens L, Stoddard JL, Bustamante J, Boisson-Dupuis S, Tsumura M, Kobayashi M, Arkwright PD, Averbuch D, Engelhard D, Roesler J, Peake J, Wong M, Adelstein S, Choo S, Smart JM, French MA, Fulcher DA, Cook MC, Picard C, Durandy A, Klein C, Holland SM, Uzel G, Casanova JL, Ma CS, Tangye SG. 2013. Naive and memory human B cells have distinct requirements for STAT3 activation to differentiate into antibody-secreting plasma cells. J Exp Med 210:2739–2753. doi: 10.1084/jem.20130323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mosser DM, Edwards JP. 2008. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chung HK, Chae C. 2003. Expression of interleukin-10 and interleukin-12 in piglets experimentally infected with porcine reproductive and respiratory syndrome virus (PRRSV). J Comp Pathol 129:205–212. doi: 10.1016/S0021-9975(03)00036-7. [DOI] [PubMed] [Google Scholar]
- 41.Suradhat S, Thanawongnuwech R. 2003. Upregulation of interleukin-10 gene expression in the leukocytes of pigs infected with porcine reproductive and respiratory syndrome virus. J Gen Virol 84:2755–2760. doi: 10.1099/vir.0.19230-0. [DOI] [PubMed] [Google Scholar]
- 42.Halbur PG, Paul PS, Frey ML, Landgraf J, Eernisse K, Meng XJ, Lum MA, Andrews JJ, Rathje JA. 1995. Comparison of the pathogenicity of two US porcine reproductive and respiratory syndrome virus isolates with that of the Lelystad virus. Vet Pathol 32:648–660. doi: 10.1177/030098589503200606. [DOI] [PubMed] [Google Scholar]
- 43.Perry E, Tsruya R, Levitsky P, Pomp O, Taller M, Weisberg S, Parris W, Kulkarni S, Malovani H, Pawson T, Shpungin S, Nir U. 2004. TMF/ARA160 is a BC-box-containing protein that mediates the degradation of Stat3. Oncogene 23:8908–8919. doi: 10.1038/sj.onc.1208149. [DOI] [PubMed] [Google Scholar]
- 44.Ulane CM, Rodriguez JJ, Parisien JP, Horvath CM. 2003. STAT3 ubiquitylation and degradation by mumps virus suppress cytokine and oncogene signaling. J Virol 77:6385–6393. doi: 10.1128/JVI.77.11.6385-6393.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ulane CM, Kentsis A, Cruz CD, Parisien JP, Schneider KL, Horvath CM. 2005. Composition and assembly of STAT-targeting ubiquitin ligase complexes: paramyxovirus V protein carboxyl terminus is an oligomerization domain. J Virol 79:10180–10189. doi: 10.1128/JVI.79.16.10180-10189.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lieu KG, Brice A, Wiltzer L, Hirst B, Jans DA, Blondel D, Moseley GW. 2013. The rabies virus interferon antagonist P protein interacts with activated STAT3 and inhibits Gp130 receptor signaling. J Virol 87:8261–8265. doi: 10.1128/JVI.00989-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patel D, Opriessnig T, Stein DA, Halbur PG, Meng XJ, Iversen PL, Zhang YJ. 2008. Peptide-conjugated morpholino oligomers inhibit porcine reproductive and respiratory syndrome virus replication. Antiviral Res 77:95–107. doi: 10.1016/j.antiviral.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meng XJ, Paul PS, Halbur PG. 1994. Molecular cloning and nucleotide sequencing of the 3′-terminal genomic RNA of the porcine reproductive and respiratory syndrome virus. J Gen Virol 75(Pt 7):1795–1801. doi: 10.1099/0022-1317-75-7-1795. [DOI] [PubMed] [Google Scholar]
- 49.Collins JE, Benfield DA, Christianson WT, Harris L, Hennings JC, Shaw DP, Goyal SM, McCullough S, Morrison RB, Joo HS, Gorcyca D, Chladek D. 1992. Isolation of swine infertility and respiratory syndrome virus (isolate ATCC VR-2332) in North America and experimental reproduction of the disease in gnotobiotic pigs. J Vet Diagn Invest 4:117–126. doi: 10.1177/104063879200400201. [DOI] [PubMed] [Google Scholar]
- 50.Yuan S, Nelsen CJ, Murtaugh MP, Schmitt BJ, Faaberg KS. 1999. Recombination between North American strains of porcine reproductive and respiratory syndrome virus. Virus Res 61:87–98. doi: 10.1016/S0168-1702(99)00029-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang YJ, Stein DA, Fan SM, Wang KY, Kroeker AD, Meng XJ, Iversen PL, Matson DO. 2006. Suppression of porcine reproductive and respiratory syndrome virus replication by morpholino antisense oligomers. Vet Microbiol 117:117–129. doi: 10.1016/j.vetmic.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nan Y, Ma Z, Wang R, Yu Y, Kannan H, Fredericksen B, Zhang YJ. 2014. Enhancement of interferon induction by ORF3 product of hepatitis E virus. J Virol 88:8696–8705. doi: 10.1128/JVI.01228-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang R, Nan Y, Yu Y, Yang Z, Zhang YJ. 2013. Variable interference with interferon signal transduction by different strains of porcine reproductive and respiratory syndrome virus. Vet Microbiol 166:493–503. doi: 10.1016/j.vetmic.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 54.Timofeeva OA, Plisov S, Evseev AA, Peng S, Jose-Kampfner M, Lovvorn HN, Dome JS, Perantoni AO. 2006. Serine-phosphorylated STAT1 is a prosurvival factor in Wilms' tumor pathogenesis. Oncogene 25:7555–7564. doi: 10.1038/sj.onc.1209742. [DOI] [PubMed] [Google Scholar]
- 55.Zhong Z, Wen Z, Darnell JE Jr. 1994. Stat3 and Stat4: members of the family of signal transducers and activators of transcription. Proc Natl Acad Sci U S A 91:4806–4810. doi: 10.1073/pnas.91.11.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Horvath CM, Wen Z, Darnell JE Jr. 1995. A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes Dev 9:984–994. doi: 10.1101/gad.9.8.984. [DOI] [PubMed] [Google Scholar]
- 57.Besser D, Bromberg JF, Darnell JE Jr, Hanafusa H. 1999. A single amino acid substitution in the v-Eyk intracellular domain results in activation of Stat3 and enhances cellular transformation. Mol Cell Biol 19:1401–1409. doi: 10.1128/MCB.19.2.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nan Y, Wang R, Shen M, Faaberg KS, Samal SK, Zhang YJ. 2012. Induction of type I interferons by a novel porcine reproductive and respiratory syndrome virus isolate. Virology 432:261–270. doi: 10.1016/j.virol.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang YJ, Wang KY, Stein DA, Patel D, Watkins R, Moulton HM, Iversen PL, Matson DO. 2007. Inhibition of replication and transcription activator and latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus by morpholino oligomers. Antiviral Res 73:12–23. doi: 10.1016/j.antiviral.2006.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patel D, Stein DA, Zhang YJ. 2009. Morpholino oligomer-mediated protection of porcine pulmonary alveolar macrophages from arterivirus-induced cell death. Antivir Ther 14:899–909. doi: 10.3851/IMP1409. [DOI] [PubMed] [Google Scholar]