

ABSTRACT
The human cytomegalovirus terminase complex cleaves concatemeric genomic DNA into unit lengths during genome packaging and particle assembly. This process is an attractive drug target because cleavage of concatemeric DNA is not required in mammalian cell DNA replication, indicating that drugs targeting the terminase complex could be safe and selective. One component of the human cytomegalovirus terminase complex, pUL89, provides the endonucleolytic activity for genome cleavage, and the domain responsible is reported to have an RNase H-like fold. We hypothesize that the pUL89 endonuclease activity is inhibited by known RNase H inhibitors. Using a novel enzyme-linked immunosorbent assay (ELISA) format as a screening assay, we found that a hydroxypyridonecarboxylic acid compound, previously reported to be an inhibitor of human immunodeficiency virus RNase H, inhibited pUL89 endonuclease activity at low-micromolar concentrations. Further characterization revealed that this pUL89 endonuclease inhibitor blocked human cytomegalovirus replication at a relatively late time point, similarly to other reported terminase complex inhibitors. Importantly, this inhibitor also prevented the cleavage of viral genomic DNA in infected cells. Taken together, these results substantiate our pharmacophore hypothesis and validate our ligand-based approach toward identifying novel inhibitors of pUL89 endonuclease.
IMPORTANCE Human cytomegalovirus infection in individuals lacking a fully functioning immune system, such as newborns and transplant patients, can have severe and debilitating consequences. The U.S. Food and Drug Administration-approved anti-human cytomegalovirus drugs mainly target the viral polymerase, and resistance to these drugs has appeared. Therefore, anti-human cytomegalovirus drugs from novel targets are needed for use instead of, or in combination with, current polymerase inhibitors. pUL89 is a viral ATPase and endonuclease and is an attractive target for anti-human cytomegalovirus drug development. We identified and characterized an inhibitor of pUL89 endonuclease activity that also inhibits human cytomegalovirus replication in cell culture. pUL89 endonuclease, therefore, should be explored as a potential target for antiviral development against human cytomegalovirus.
KEYWORDS: human cytomegalovirus, terminase, pUL89 inhibitor, hydroxypyridonecarboxylic acid, metal chelation
INTRODUCTION
Human cytomegalovirus (HCMV) infection is a significant cause of morbidity and mortality in immunocompromised patients, including transplant recipients and AIDS patients (reviewed in reference 1). In addition, HCMV infects 1% of all newborns and is a leading cause of brain damage and hearing loss (2). Currently, most of the U.S. Food and Drug Administration (FDA)-approved anti-HCMV drugs target the viral polymerase, including ganciclovir (GCV), valganciclovir, cidofovir, and foscarnet (reviewed in reference 3). Drug-resistant strains of HCMV encoding polymerase mutations have been found for all of these FDA-approved drugs (4). Given that the emergence of robust virus resistance has been reported in the clinic, there is an urgent need to develop new anti-HCMV drugs with new molecular targets.
The HCMV terminase complex plays a critical role in the packaging of viral genomes into assembling viral particles. The genome-packaging mechanism and the role of the terminase complex appear to have been conserved among double-strand DNA (dsDNA) viruses, including herpesviruses (5), adenoviruses (6), and certain bacteriophages (7). In general, the terminase complex binds the viral genomic DNA, cleaves the DNA at a specific sequence, and facilitates genome packaging into the assembling virus particle. The terminase complex is minimally comprised of a large and a small terminase protein. In HCMV, the protein encoded by the UL89 gene, pUL89, is the large terminase subunit, and it contains the ATPase and endonuclease functions. The corresponding HCMV small terminase protein is pUL56. The HCMV terminase complex may also contain another viral protein, pUL51, whose function has yet to be determined (8). pUL89 cleaves a unit-length viral genome from a concatemer to ensure the packaging of an entire genome (9). Mutation of herpes simplex virus 1 (HSV-1) UL15, the homolog of HCMV UL89, results in a virus that is unable to cleave concatemeric DNA into unit-length genomes and abolishes virus replication (10). Therefore, inhibition of pUL89 genome cleavage activity has a high likelihood of blocking viral replication. Several compounds targeting the terminase complex have been identified (e.g., 2-bromo-5,6-dichloro-1-β-d-ribofuranosyl-1H-benzimidazole [BDCRB], BAY 38-4766, and letermovir) (11–13). Letermovir is the most successful terminase inhibitor, having been granted fast-track status by the FDA and orphan-drug status by the European Medicines Agency. Mutations in UL56 confer resistance to letermovir, indicating that pUL56 is the likely target (12). BDCRB is a nucleoside analog, and resistance has been mapped to exon 2 of the UL89 gene (13). BAY 38-4766 is a nonnucleoside inhibitor of HCMV genome cleavage and packaging. Sequence analyses of HCMV strains that are resistant to BAY 38-4766 indicate that pUL89 and pUL104 are its likely targets (14).
The nuclease domain located in the C terminus of pUL89 (pUL89-C) has been reported to have an RNase H/integrase (IN)-like fold (15). Like RNase H, the function of pUL89-C is dependent on metal ions located in the catalytic pocket. The pUL89-C nuclease function was blocked by the human immunodeficiency virus (HIV) IN inhibitor raltegravir (15), suggesting that inhibiting pUL89-C may be possible by using a similar metal-chelating pharmacophore. Recently, compounds with metal-chelating motifs were identified as inhibitors of herpesviruses (16–18) and in a separate study were identified as inhibitors of HSV-1 pUL15C (19). However, the likely target(s) of the inhibitors in infected cells has not been identified. Compounds containing chelating motifs could bind any of several viral or cellular metal-binding proteins or even proteins that may not be metal binding in cells. An example is raltegravir, a weak inhibitor of pUL89-C (15) but a potent pUL15C inhibitor in biochemical assays (19). HSV-1 resistance to raltegravir maps to a viral DNA polymerase accessory factor and not UL15 (20), indicating that raltegravir likely binds the accessory factor in the infected cell. Clearly, it is important to have an understanding of the mechanism of action of these chelating compounds in infected cells prior to pursuing particular chemical scaffolds as terminase inhibitors.
In our previous study, a series of hydroxypyridonecarboxylic acids (HPCAs) was designed to inhibit HIV RNase H via metal chelation (21). We hypothesize here that the chelating triad of the HPCAs could also interact with the two metal cations in the pUL89-C active site to inhibit endonuclease activity. We designed a new enzyme-linked immunosorbent assay (ELISA) format to measure pUL89-C endonuclease activity and identified an HPCA inhibitor. The pUL89-C inhibitor also inhibited HCMV replication and spread in cell culture. We employed both a kinetic block release assay and a time-of-drug-addition assay to demonstrate that this HPCA inhibitor acted late in virus replication. Finally, Southern blot analysis verified that the HPCA pUL89-C inhibitor prevented viral DNA cleavage in infected cells, as would be expected for a compound that targets pUL89. Therefore, we have identified a promising pUL89-C inhibitor scaffold that also shows the characteristics of a pUL89 inhibitor in infected cells.
RESULTS
Establishment of an assay to evaluate anti-pUL89-C activity.
A recombinant form of the pUL89 endonuclease domain, pUL89-C, cleaves a linearized plasmid (pUC18), and this cleavage can be monitored by agarose gel electrophoresis (15). Raltegravir was identified as an inhibitor of pUL89-C nuclease activity by using an agarose gel assay (15). This gel-based assay, however, is low throughput and time-consuming, so we first developed a higher-throughput and more quantitative assay. Toward this end, we noted that pUC18 was degraded almost completely by pUL89-C (15). As pUC18 is an ∼2.7-kb dsDNA, we felt that it was too large to serve as a substrate for the development of a quantitative assay. By using a substrate this large, it is difficult to quantify the extent of pUL89-C cleavage and the inhibition thereof. These difficulties prompted us to identify the minimal size for a pUL89-C DNA substrate because we reasoned that it would be easier and more reliable to monitor one or only a few cleavage events. We began by conducting DpnI restriction enzyme digestion of pUC18, resulting in 15 fragments of different sizes. We then incubated digested pUC18 with pUL89-C for 1 h to identify the smallest degraded fragment. Agarose gel analysis showed that the 153-bp fragment was the smallest degraded fragment (Fig. 1A and B). To determine if fragments smaller than 153 bp could be digested by pUL89-C, we subsequently selected 60-bp and 27-bp fragments derived from the 153-bp fragment for testing. Polyacrylamide gel analysis showed that the 60-bp fragment was cleaved by pUL89-C under normal reaction conditions. However, pUL89-C did not degrade the 27-bp fragment efficiently (Fig. 1C). Therefore, for our purposes, the 60-bp dsDNA from pUC18 was the smallest DNA fragment capable of being cleaved by pUL89-C, and we moved forward with this fragment for the development of a quantitative assay.
FIG 1.

Substrate identification for ELISA to detect pUL89-C activity. (A) pUC18 or pUC18 digested with DpnI was incubated with (+) or without (−) pUL89-C. (B) The band intensity on an agarose gel was analyzed by using ImageJ. A band intensity ratio (absence of pUL89/presence of pUL89) of >1.5 was considered a high cleavage efficiency. (C) Three different sizes of dsDNA fragments incubated with or without pUL89-C. (D) Schematic of the ELISA format for analysis of pUL89-C activity.
We used an ELISA format to detect the cleavage of a small DNA substrate, and a schematic description of this assay is illustrated in Fig. 1D. The 60-bp single-strand DNA (ssDNA) oligonucleotide was labeled with a digoxigenin (DIG) tag at the 5′ end, and a complementary 60-bp ssDNA was labeled with biotin at the 5′ end. Equal moles of ssDNAs were mixed and annealed to obtain the 60-bp dsDNA substrate. The substrate binds to a streptavidin-coated plate through the biotin-streptavidin interaction, and the DIG tag is detected by using alkaline phosphatase (AP)-conjugated anti-DIG antibody. Therefore, UL89-C is added after the substrate is bound in a well, and if cleavage occurs, no DIG-labeled substrate will remain in the well, and no AP activity will be detected after antibody addition. If a compound inhibits pUL89-C cleavage, the anti-DIG-AP antibody will bind the substrate, and the level of the substrate in the well can be quantified by the AP-dependent catalysis of p-nitrophenylphosphate (pNPP) to yellow para-nitrophenol and spectrometry. A reaction time course showed that ∼100% of the substrate was cleaved at 10 min with 3 μM enzyme, whereas it took 60 min to cleave 100% of the substrate when 0.1 μM enzyme was used (Fig. 2A). The Km for the substrate was calculated to be 10 nM (Fig. 2B).
FIG 2.
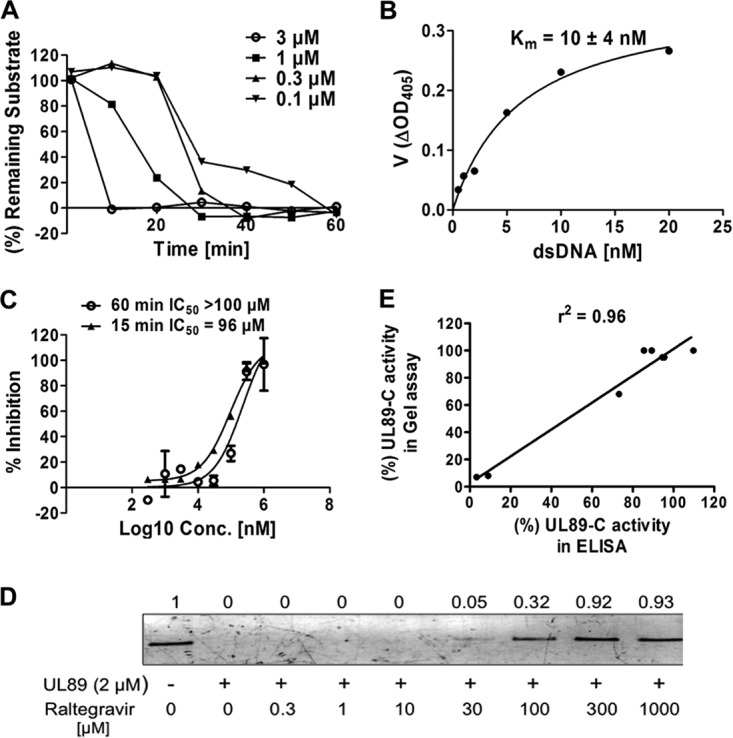
Establishment of an ELISA to evaluate the anti-pUL89-C activity of compounds. (A) Time course for an ELISA using different concentrations of pUL89-C and 10 nM the substrate. (B) Representative graph for the determination of substrate Km values. Experiment were performed two independent times, and mean Km values plus standard deviations are shown. ΔOD405, change in the optical density at 405 nm. (C) Inhibitory effects of raltegravir on pUL89-C activity analyzed by an ELISA. Results included stopping the reaction at 15 or 60 min. (D) Inhibitory effects of raltegravir on pUL89-C activity analyzed by an agarose gel assay. Shown is linearized pUC18 in the absence (lane 1) or presence (lane 2) of pUL89-C. Lanes 3 to 10 show a range of concentrations of raltegravir with pUL89-C. Numbers above bands correspond to the fold change compared with the control. (E) Correlation between the inhibitory response in a gel assay and the inhibitory response in an ELISA (r2 = 0.96). Intensities of gel bands were determined by using ImageJ and used to calculate percent activity in a gel assay. These values were plotted along with percent activity in an ELISA. The best-fit line and r2 values were determined by using linear regression analysis with GraphPad Prism software.
We used raltegravir as a positive control to validate the new ELISA format. The inhibitory effects of raltegravir on pUL89-C nuclease activity were analyzed by both agarose gel electrophoresis and an ELISA. Both assays were performed by using 1 μM pUL89-C for 1 h at 37°C to ensure complete digestion of pUC18 in the gel-based assay and for comparison of data among the two assays and a previously reported assay (15). By agarose gel analysis, raltegravir showed approximately 32%, 92%, and 93% inhibitions at concentrations of 100, 300, and 1,000 μM, respectively (Fig. 2D), when band intensities were quantified by using ImageJ software. In the ELISA, raltegravir inhibited pUL89-C activity with a 50% inhibitory concentration (IC50) of >100 μM (Fig. 2C). A dose-response ELISA using raltegravir was also performed for 15 min at 37°C, a time point prior to complete substrate conversion (Fig. 2A), and an IC50 of 96 μM was obtained (Fig. 2C). There was a strong correlation between the inhibitory response in the gel assay and the inhibitory response in the ELISA (r2 = 0.96) (Fig. 2E), indicating that the ELISA is reliable and can be applied to pUL89-C inhibitor screening.
HPCA as a new chemotype for inhibiting pUL89-C and HCMV.
HPCA compounds inhibit HIV RNase H via metal chelation (21), and we hypothesize that the same HPCA compounds could also inhibit pUL89-C nuclease activity (Fig. 3). To test this, we used an ELISA to examine a series of HPCA compounds for inhibition of pUL89-C activity, and the structure of the most potent HPCA, compound 10k, is shown in Fig. 3. In the pUL89-C ELISA, compound 10k showed an IC50 of 1 μM (6 μM when the assay was extended to 1 h to match the gel assay) (Fig. 4A), which was verified via the pUL89-C gel assay (Fig. 4B). Compound 10k was also tested for its effects on HCMV replication and infectious-virus production (Fig. 4C). Two terminase inhibitors (letermovir and BDCRB) and one polymerase inhibitor (GCV) were selected as positive controls in our anti-HCMV evaluation. We inoculated human fibroblast cultures with ADCREGFP, a green fluorescent protein (GFP)-expressing reporter virus (22) producing a readily detectable GFP signal (Fig. 4C), to quantitate virus spread in cell culture and the production of infectious virus (23–25). HFF cells were inoculated with ADCREGFP at a low multiplicity of infection (MOI) in the presence or absence of potential inhibitors, and 7 days later, the GFP level in the inoculated/treated cells was measured as an indication of the extent of virus spread (Fig. 4D). The titer of the supernatant from the original inoculated/treated cells was determined on fresh HFF cells to measure virus production (Fig. 4E). Letermovir demonstrated low-nanomolar activity for both measures of antiviral activity, with 50% effective concentrations (EC50s) being similar to those reported previously (Fig. 4D and E) (12). Both BDCRB and GCV reduced HCMV replication with EC50s at submicromolar levels. Our HPCA analog, compound 10k, reduced HCMV spread with an EC50 of 4 μM (Fig. 4D) and significantly blocked the production of infectious HCMV with an EC50 of 4 μM (Fig. 4E). To confirm that compound 10k did not affect cell viability at the levels used in our experiments, we determined that the 50% cytotoxic concentration (CC50) value was >200 μM, while the CC50 values for letermovir and GCV were >20 μM, and the CC50 was >100 μM for BDCRB (data not shown). We did not detect a significant reduction in cell viability due to the addition of the compound in any of our cell-based assays for virus replication. We summarize here that compound 10k inhibits pUL89-C endonuclease activity in vitro and also inhibits HCMV replication and infectious-virus production in cell culture.
FIG 3.
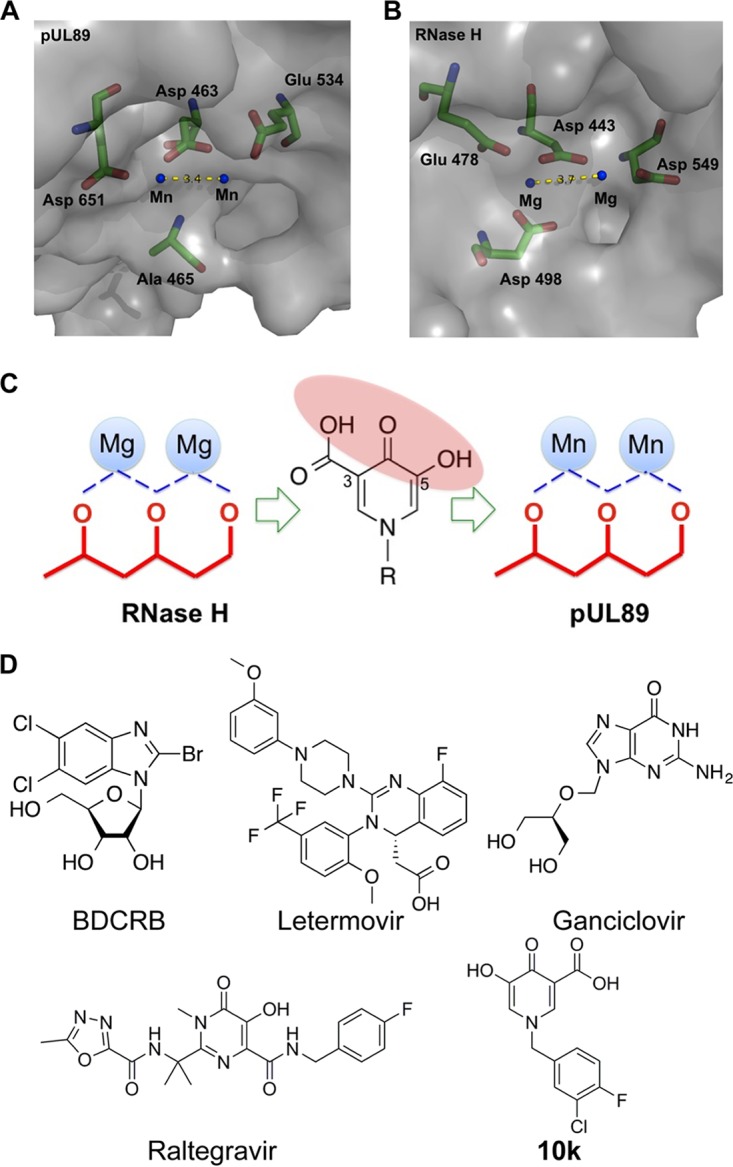
Triad to chelate the two Mn2+ ions for discovery and development of pUL89 inhibitors. (A and B) Molecular structure analysis illustrates the catalytic pocket of HCMV pUL89-C (PDB accession number 3N4Q) (A) and HIV RNase H (PDB accession number 3HYF) (B). The three-dimensional diagrams show the key residues (sticks) (green, C; red, O; blue, N) and the metal ions (blue dots) in the catalytic site. The distance between two metal ions is labeled with yellow dashes. (C) Series of HPCAs designed to inhibit RNase H via metal chelation. We hypothesize that the chelating triad of the HPCAs can also interact with two metal cations in the pUL89 active site to inhibit endonuclease activity. (D) Structure of the HPCA pUL89-C inhibitor 10k and other compounds.
FIG 4.

HPCA compound 10k is a pUL89-C and HCMV inhibitor. (A) Dose-response inhibition of compound 10k for pUL89-C nuclease activity evaluated by an ELISA, stopping the reaction at either 15 or 60 min. (B) Dose-response inhibition of pUL89-C nuclease activity by compound 10k with an agarose gel readout. (C) Schematic representation of the experimental setup. HFF cells were inoculated with HCMV ADCREGFP and treated with serial dilutions of compounds. After 144 h, cell supernatants were collected for a yield reduction assay, and the original inoculated cells were analyzed for GFP expression as a measure of virus replication. For the yield reduction assay, HFF cells were inoculated with the supernatants from the antiviral assay, and GFP expression was measured after 144 h. (D and E) Representative dose-response curves for compound 10k in an HCMV replication assay (D) and a yield reduction assay (E).
Mn2+ ions play a key role in the anti-pUL89-C activity of HPCA.
We predict that the chelating triad of our HPCA analog, compound 10k, targets the pUL89-C catalytic pocket by binding the divalent cations required for endonuclease activity. We performed an initial characterization of the binding of HPCA to pUL89-C using molecular docking analysis. The hydroxyl group at the 5 position of compound 10k interacts with Glu 534 through an H bond, while the carboxyl group at the 3 position forms H bonding with Asp 651 (Fig. 5A). These two hydrogen bond interactions could help position the inhibitor near the metal ions in the active site. To further verify the key role of Mn2+ ions in pUL89 inhibition by compound 10k, we conducted an order-of-addition assay to examine the effect of Mn2+ preincubation conditions on the inhibition of pUL89-C activity. Under our standard biochemical endonuclease reaction conditions, compound 10k was preincubated with pUL89-C and Mn2+ in reaction buffer for 15 min prior to the addition of the substrate to begin the reaction. We reasoned that if compound 10k was interacting with the Mn2+ ions in the pUL89-C active site as a way to bind and inhibit pUL89-C endonuclease activity, then preincubation of compound 10k with pUL89-C and without Mn2+ should result in a reduction of inhibition compared to our standard conditions. The absence of Mn2+ during preincubation of the compound could delay or reduce the binding of compounds containing a chelating triad. To test this, we conducted a biochemical pUL89-C assay using a fixed concentration (IC50 under normal conditions) of an inhibitor with a 15-min pUL89-C preincubation in the presence or absence of Mn2+ prior to the addition of the substrate. For the reaction mixtures without Mn2+ in the preincubation, the dsDNA substrate and Mn2+ were added to the reaction mixture simultaneously. Fifty-percent inhibition of pUL89-C activity was observed by both the ELISA and the gel assay when an inhibitor was preincubated with pUL89-C and Mn2+ (Fig. 5B). The inhibitory activity of compound 10k was significantly reduced in the absence of Mn2+ preincubation (Fig. 5B and C), indicating that the interaction between compound 10k and Mn2+ ions plays a key role in its anti-pUL89-C activity.
FIG 5.

HPCA compound 10k inhibition in the presence or absence of preincubation with Mn2+. (A) Molecular docking analysis illustrates the favorable binding positions of compound 10k with the lowest binding free energy in the inhibitor binding site of pUL89-C (PDB accession number 3N4Q). The three-dimensional diagram shows the interactions of compound 10k (gray sticks) with pUL89-C (gray surface), with labeled amino residues. Chelating and H-bond interactions are depicted as red dashes. (B and C) Cleavage of the substrate under different Mn2+ preincubation or no-preincubation conditions as determined by an ELISA (B) and a gel assay (C).
Reversibility of the anti-HCMV effect of HPCA.
To begin to characterize HPCA's mechanism of inhibition of virus replication, we examined compound 10k using a kinetic block release assay. Compounds targeting the terminase or viral polymerase exhibit different profiles upon withdrawal of the inhibitor from infected cells. Compounds targeting polymerase act relatively early in virus replication (genome production) compared to terminase inhibitors that act relatively late in virus replication (assembly). Upon withdrawal of a polymerase inhibitor from virus-inoculated cells, we would expect a greater lag time to detect the production of infectious virus than we would when we withdraw a terminase inhibitor, as was demonstrated previously (4). If compound 10k targets pUL89 endonuclease activity in infected cells, we should see a relatively rapid recovery of infectious-virus production upon its withdrawal compared to virus polymerase inhibitors like GCV. ADCREGFP-inoculated cells were treated with fixed inhibitory concentrations (∼10× EC50) of compound 10k, BDCRB, letermovir, or GCV to suppress HCMV replication. At 96 h postinoculation (hpi), infected cells were released from the drug block, and the amount of progeny virus in the supernatant was quantified 24 h, 48 h, and 72 h following drug release (Fig. 6A). Upon withdrawal of the two terminase inhibitors and compound 10k, secretion of infectious virus was readily detected in the supernatant starting from 24 h postrelease. In contrast, there was a significant delay in the release of infectious virus after the removal of GCV compared to that after the removal of the terminase inhibitors (Fig. 6B). This result demonstrates that compound 10k inhibits viral replication at a later point than that for the polymerase inhibitor GCV and at a time point similar to that for the late-acting terminase inhibitors.
FIG 6.
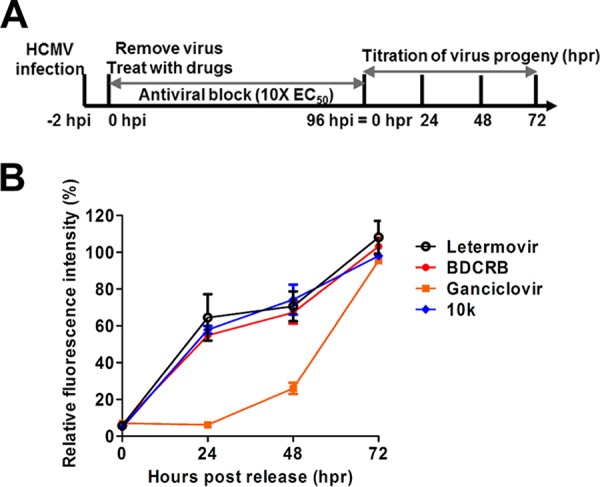
Kinetic block release assay. (A) Schematic representation of the experimental setup. HCMV ADCREGFP-inoculated HFF cells were treated for 96 h with letermovir, BDCRB, GCV, or compound 10k at concentrations 10 times their EC50s (antiviral block step). At 96 hpi, infected cells were released from the drug block, and virus was collected 24 h, 48 h, and 72 h following drug release (hours postrelease). The level of virus produced was determined by titration on fresh HFF cells. (B) Production of progeny virus following block release monitored via virus yield measurements in compound-treated cells compared to the DMSO-treated control.
Time-of-addition studies.
Another assay to characterize the mechanism of action of HPCA in infected cells is a time-of-addition assay. Changing the time of addition of inhibitors after viral infection is a common technique to characterize where in the virus replication process the inhibitor acts (26). Inhibitors are effective only if they are added prior to their target's role in replication. In an HCMV time-of-addition study, GCV retained antiviral activity when added up to 33 hpi, whereas the later-acting terminase inhibitors letermovir and BAY 38-4766 retained antiviral activity when added as late as 57 hpi (4). In order to determine the earliest time point in HCMV replication where compound 10k is active, a time-of-addition study was performed. Fixed inhibitory concentrations (∼5× EC50) of letermovir, BDCRB, GCV, or compound 10k were added to ADCREGFP-inoculated cells at the indicated time points. Virus replication was measured by the detection of GFP expression in infected cells at various time points (Fig. 7A). The inhibitory effect of GCV on HCMV replication was gradually reduced when it was added after 24 hpi. In contrast, the terminase inhibitors BDCRB and letermovir were effective virus replication inhibitors when they were added up to 60 hpi. The efficacy of both drugs drastically decreased when they were added after 60 hpi. Interestingly, the inhibitory effect of compound 10k was slightly reduced when it was added after 48 hpi, with a more drastic loss of anti-HCMV activity being seen when it was added after 60 hpi (Fig. 7B). We are unsure at this time if the slight block at 48 hpi indicates another possible target in addition to pUL89. The time-of-drug-addition studies demonstrate that compound 10k acts at a relatively late step in HCMV replication, similarly to known terminase inhibitors.
FIG 7.
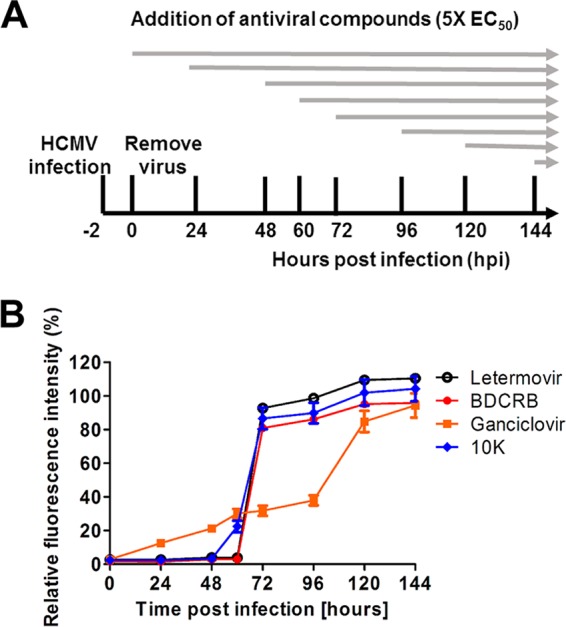
Effect of time-of-addition of inhibitors on HCMV replication. (A) Schematic representation of the experimental setup. HFF cells were inoculated with ADCREGFP and treated with ∼5 times the EC50 of letermovir, BDCRB, GCV, or compound 10k at the indicated time points postinfection (hpi). After 144 h, cells were washed with phosphate-buffered saline and lysed. HCMV replication was measured by measuring relative GFP fluorescence. (B) HCMV replication (relative GFP fluorescence intensity) in compound-treated cells compared to DMSO-treated control.
Compound 10k inhibits viral genomic DNA cleavage in infected cells.
pUL89 is thought to associate with pUL56 and possibly other viral proteins to mediate the cleavage of the viral genomic DNA. pUL56 recognizes the viral cleavage/packaging sequences and may direct pUL89 to cleave the viral DNA at these sites. Consequently, if compound 10k inhibits the endonuclease activity of pUL89 as the major mechanism of anti-HCMV activity, a reduction in genome cleavage should be observed in the presence of compound 10k. Genome cleavage in the presence of compound 10k or letermovir was examined by Southern blotting of infected cells. Viral DNA was isolated from inhibitor-treated and untreated HCMV-inoculated cells and digested with the restriction enzymes EcoRV and MfeI. pUL89 cleavage of viral genomic DNA was detected by using a DIG-labeled DNA probe designed to bind the viral genomic DNA near the cleavage site (Fig. 8A). Analysis of DNA from inoculated cells demonstrates that the specific cleavage of viral DNA (indicated by the 2-kb fragment) was greatly reduced in the presence of letermovir and compound 10k. This result indicates that compound 10k inhibits viral genomic DNA cleavage in HCMV-inoculated cells, providing further evidence that compound 10k is a specific inhibitor of pUL89 in infected cells (Fig. 8B).
FIG 8.
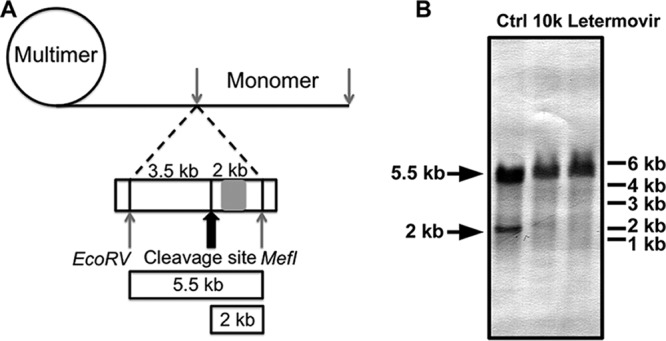
Functional viral DNA cleavage assay. (A) Schematic representation of the experimental setup. The position of the cleavage DNA probe (genome positions 703 to 1524) is indicated by a gray rectangle. Isolated viral DNA was digested, size fractionated by gel electrophoresis, and analyzed by Southern blotting. Concatemeric viral DNA yields an ∼5.5-kb fragment, whereas terminase-cleaved DNA yields an ∼2-kb fragment. The drawing is not to scale. (B) HFF cells were inoculated with HCMV ADCREGFP at an MOI of 0.1 and treated with ∼10× EC50 of letermovir or compound 10k. Ctrl, control (DMSO treatment).
DISCUSSION
To identify new HCMV inhibitors, we implemented a target-based approach taking advantage of a small in-house library of compounds designed to inhibit HIV RNase H, a protein with general structural and functional similarity to the HCMV pUL89 endonuclease domain. HPCAs were found to inhibit pUL89 endonuclease activity by using a novel ELISA format. The HPCAs feature a chelating motif to sequester the Mn2+ ions in the active site and prevent pUL89-C endonuclease activity. However, in infected cells, many cellular and viral proteins bind metal ions, possibly creating selectivity issues for putative pUL89-C inhibitors identified in a biochemical assay. Therefore, we examined the activity and selectivity of the HPCAs in HCMV-infected cells. The most potent HPCA from our screen, compound 10k, inhibited HCMV replication and infectious-virus production, acted late in virus replication, and inhibited genome cleavage. Taken together, these results indicate that compound 10k is an inhibitor of HCMV pUL89 and that HPCA represents a new pUL89 and HCMV inhibitor scaffold. We are currently exploring modifications of this scaffold that increase potency and selectivity, with the ultimate goal of developing a valid preclinical anti-HCMV drug candidate. Our study also indicates that another terminase subunit, the so-called “large subunit” (pUL89 in HCMV), is an attractive new target for antiviral exploration.
Interestingly, pUL89 (27) and pUL89-C (15) have shown nonspecific and unregulated DNA nuclease activity in biochemical assays. Monitoring of pUL89-C cleavage of plasmid DNA proved to be difficult to quantitate and adapt to a higher-throughput scale. To design a more quantitative assay, we investigated the smallest dsDNA substrate that was cleavable by pUL89-C and observed no cleavage of dsDNA substrates of <60 bp. We can more quantitatively measure one or a limited number of cleavage events using a 60-bp dsDNA substrate with the ELISA described here than is possible with larger DNA substrates. It is unclear at this time if smaller substrates are bound by pUL89-C and not cleaved or if they are not bound. How pUL89 recognizes dsDNA in the presence or the absence of pUL56 and other viral proteins remains unknown and warrants further investigation, since our current inhibitor, compound 10k, may be competitive with a substrate. The pUL89-C homolog HSV-1 pUL15C was found to cleave a double-strand DNA substrate as small as 14 to 15 bp (19). The substrates used contained sequence elements designed to mimic aspects of the viral pac sites, sites that contribute to viral genomic cleavage and packaging, whereas our substrate did not. Substrates mimicking pac sites may bind the enzyme more efficiently, and this could explain the difference in minimal substrate sizes.
In our previous study, a series of HPCAs was designed to inhibit RNase H (21). A structure of RNase H in complex with the inhibitor shows that HPCAs bind at the active site, directly chelating the metal cations (21). In view of the structural similarity between the catalytic pocket of pUL89-C and RNase H, we hypothesize that these compounds would bind in a similar way to pUL89-C and inhibit its activity. An analog of HPCA (compound 10k) showed inhibitory effects on pUL89-C endonuclease activity with a low-micromolar IC50. The inhibitory activity increased in the presence of Mn2+ ions, suggesting that metal ion chelation is a mechanism of action. In our molecular docking analysis, the chelating triad of HPCA inhibitors can form H bonds with Glu 534 and Asp 651, respectively. These two hydrogen bond interactions could help position the inhibitor near the key metal ions in the active site. These results support a model whereby certain compounds with chelating motifs can interact with the divalent cations in the pUL89-C active site to inhibit endonuclease activity. However, inhibition of HSV-1 pUL15C endonuclease activity by β-thujaplicinol and related compounds featuring a chelating motif was equivalent regardless of the order of the addition of reagents or preincubation when examined by using a dual-probe fluorescence biochemical assay (19). Differences in compounds, assays, and enzymes can account for these results. The two herpesvirus terminase subunit endonuclease domains are structurally highly related but clearly can display different inhibitor profiles by using the respective assays. Raltegravir was as a weak inhibitor of pUL89-C, with an IC50 of ∼100 μM, by an ELISA and agarose gel readout (Fig. 1) (15), yet it was a potent inhibitor of pUL15C, with an IC50 of 0.39 μM (19). In addition, another HIV IN inhibitor, dolutegravir, did not display detectable inhibition of pUL89-C (data not shown) but inhibited pUL15C with an IC50 of 1.1 μM (19).
Overall, our results indicate that compounds with a chelating motif can be identified to interact with pUL89-C in an Mn2+-dependent manner, inhibit pUL89-C endonuclease activity, and inhibit HCMV replication and genome cleavage, likely by blocking pUL89 endonuclease activity in infected cells.
MATERIALS AND METHODS
Cells and virus.
Human foreskin fibroblasts (HFF) were obtained from Wade Bresnahan (University of Minnesota) and cultured at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and penicillin-streptomycin. The laboratory recombinant HCMV AD169 strain ADCREGFP (22) was obtained from Wade Bresnahan (University of Minnesota). This virus has the UL21.5 gene replaced by a GFP expression cassette driven by the simian virus 40 (SV40) promoter. The UL21.5 gene is not essential for HCMV replication, and ADCREGFP replicates to wild-type levels (22, 28).
Antiviral compounds.
HPCA derivatives were synthesized as previously described (21), as was BDCRB (29). GCV, raltegravir, and letermovir were purchased from MedChemExpress. All compounds were stored as a 20 mM stock solution in dimethyl sulfoxide (DMSO).
Agarose gel in vitro nuclease assay.
pUL89-C was expressed and purified as described previously by others (15). The pUL89-C bacterial expression plasmid was generated by the insertion of the UL89-C sequences into the pET-21a(+) vector using NdeI and NotI restriction enzyme digestion and ligation. The UL89-C insertion was generated by PCR amplification using an HCMV AD169-containing bacterial artificial chromosome (BAC) as the template (provided by Wade Bresnahan, University of Minnesota) and standard procedures. The primers for the PCR were UL89C_NdeI_for (5′-ACTACTACATATGGGCGGTACCAATAAGATC) and UL89C_NotI_rev (5′-AGTAGTGCGGCCGCTTAATGGTGATGGTGATGGTGTCCGCTGACCCTGAAACGGATG). Purified pUL89-C (final concentration, 1 μM) was incubated with 100 ng of the HindIII-digested pUC18 plasmid (2,686 bp) in a reaction buffer (3 mM MnCl2, 30 mM Tris [pH 8], and 50 mM NaCl) for 1 h at 37°C. Compounds or DMSO was preincubated with pUL89-C and reaction buffer for 15 min. To start the reaction, pUC18 was added, and the mixture was incubated for 1 h at 37°C. The reaction was terminated by adding EDTA (final concentration, 30 mM) to the mixture. The samples were analyzed by agarose gel electrophoresis with ethidium bromide staining and imaged by using a Bio-Rad ChemiDoc MP system.
In vitro pUL89 nuclease ELISA.
The 60-bp ssDNA (5′-TAATCGCCTTGCAGCACATCCCCCTTTCGCCAGCTGGCGTAATAGCGAAGAGGCCCGCAC) was labeled with a DIG tag at the 5′ end, and its complementary ssDNA (5′-TCGGTGCGGGCCTCTTCGCTATTACGCCAGCTGGCGAAAGGGGGATGTGCTGCAAGGCGA) was labeled with biotin at the 5′ end. Equal moles of ssDNAs were mixed and annealed to obtain the 60-bp dsDNA. For the standard reaction, purified pUL89-C (final concentration, 1 μM) in reaction buffer (3 mM MnCl2, 30 mM Tris [pH 8], and 50 mM NaCl) was preincubated with the compound in DMSO or DMSO alone for 15 min. The reaction was started by the addition of dsDNA (final concentration, 10 nM), and the mixture was incubated for 15 min to 1 h at 37°C. The reaction was terminated by adding EDTA (final concentration, 30 mM) to the mixture. The samples were transferred to streptavidin-coated plates (Pierce Biotechnology, Rockford, IL), incubated with gentle shaking at room temperature for 30 min, and washed three times with 200 μl of wash buffer (25 mM Tris, 150 mM NaCl, 0.1% bovine serum albumin [BSA], and 0.05% Tween 20 [pH 7.2]). One hundred microliters of the anti-DIG-alkaline phosphatase (AP) conjugate (Roche Applied Sciences, Germany) was added to each well for 30 min at room temperature, washed three times with 200 μl of wash buffer, and incubated with 100 μl of pNPP (Sigma-Aldrich, St. Louis, MO) for 30 min, and the absorbance was determined at 405 nm. For the time course experiment, 0.1 to 3 μM pUL89-C was used, and the reactions were stopped at the times indicated. The percent remaining substrate was calculated by comparing the absorbance reading for the reaction samples to that of control samples, where no cleavage occurs. To study inhibitor preincubation effectiveness in the absence of Mn2+, pUL89-C was preincubated with test compounds in reaction buffer without MnCl2 for 15 min. MnCl2 and substrate DNA were added to the reaction system, and the mixture was incubated for 1 h at 37°C and then treated as described above. For the analysis of substrate Km values, the standard pUL89-C reaction was carried out by using increasing substrate concentrations. The Km was calculated by using nonlinear Michaelis-Menten fitting of activity as a function of the substrate concentration by using GraphPad Prism software.
Molecular docking analysis.
Molecular docking simulation was performed by using the three-dimensional crystal structure of human pUL89 obtained from the Protein Data Bank (PDB) (accession number 3N4Q). The docking simulations were conducted by using AutoDock Vina v.1.0.2. The docking parameters for AutoDock Vina were kept at their default values. The grid box was 16 Å by 14 Å by 16 Å, encompassing the catalytic pocket of pUL89. The binding modes were clustered through the root mean square deviation (RMSD) among the Cartesian coordinates of the ligand atoms. The docking results were ranked by the binding free energy. The binding results were graphically presented by using PyMOL Molecular Graphics System version 1.3 (Schrödinger, LLC).
HCMV replication and virus production assays.
HFF were grown in white-bottom 96-well tissue culture plates to 90% confluence and inoculated with ADCREGFP virus at an MOI of 0.001 in DMEM containing 5% fetal calf serum for 2 h. The inoculated cells were washed with phosphate-buffered saline (PBS) and incubated with 100 μl of DMEM containing 5% fetal calf serum with test compounds or DMSO at 37°C and 5% CO2 for 144 h. After 144 h, cell supernatants were collected for a yield reduction assay (described below), and the infected cells were lysed to measure GFP fluorescence as an indication of the extent of virus replication. For lysis, 200 μl of lysis buffer (25 mM Tris [pH 7.8], 2 mM dithiothreitol [DTT], 2 mM trans-1,2-diaminocyclohexane-N,N,N′,N′-tetraacetic acid, 1% Triton X-100, 10% glycerol) was added to each well, and the mixture was incubated for 10 min at 37°C, followed by a 30-min incubation at room temperature on a shaker. GFP relative fluorescence units were determined at an excitation/emission wavelength of 495/515 nm in a Molecular Devices M5e plate reader. For the yield reduction assay, HFF cells were inoculated for 2 h with the supernatants from the antiviral assay. The supernatants were removed and replaced by culture medium. After 144 h, cells were washed with PBS and lysed. GFP units were determined at an excitation/emission wavelength of 495/515 nm.
Kinetic block release assay.
HFF were cultured in white 96-well tissue culture plates to 90% confluence and inoculated with ADCREGFP at an MOI of 0.001. After virus adsorption, cells were treated with test compounds for 96 h (∼10× EC50) or DMSO as a control. After the 96-h incubation period, cells were washed three times with PBS and then incubated in drug-free medium. Cell culture supernatants of inoculated cells were collected 24, 48, and 72 h after the removal of the antiviral compound and stored at −80°C. Finally, virus titers of each collected cell culture supernatant were quantified via a GFP-based yield reduction assay. Duplicate samples were used for all drug block release studies.
Antiviral time-of-addition assay.
HFF were cultivated in white 96-well tissue culture plates to 90% confluence and inoculated with ADCREGFP virus at an MOI of 0.001. After virus adsorption, cells were treated with test compounds (∼5× EC50) or DMSO at the indicated time points postinfection. After 144 h, cells were washed with PBS and lysed as described above. GFP units were determined at an excitation/emission wavelength of 495/515 nm as described above. Each compound was tested in duplicate at each time point.
Cell viability assay.
HFF cells were plated into 96-well plates at 3.6 × 104 cells/well and incubated with different doses of the compound at 37°C for 72 h. Cellular viability was determined by using the CellTiter 96 Aqueous Non-Radioactive cell proliferation assay (Promega), a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS)-based tetrazolium reduction assay, according to the manufacturer's instructions. Compound doses were evaluated in triplicate, and mean values for triplicate wells were determined and compared to the mean value for the wells that received DMSO alone. The CC50 was determined by comparing cell viabilities for eight serial dilutions of the compound and DMSO-treated cells by using GraphPad Prism software. The CC50 value was defined as the compound concentration resulting in a 50% reduction in viability compared with DMSO.
Viral DNA cleavage assay.
Viral DNA cleavage assays were performed as previously described (11), with a slight modification. Briefly, HFF cells (1 × 107) were inoculated with ADCREGFP at an MOI of 0.1 PFU/cell in a 10-cm tissue culture dish. After virus adsorption, cells were treated for 168 h with ∼10 times the EC50 of a given compound. One sample was treated with DMSO as a control. After the 168-h incubation period, DNA was extracted from infected cells by using a blood and cell culture DNA kit (Qiagen, Hilden, Germany). DNA was digested with 20 U of EcoRV and MfeI for 2 h, size fractionated on a 0.5% agarose gel by electrophoresis, and blotted onto nylon membranes (Millipore, Schwalbach, Germany) via Southern transfer. After UV cross-linking, Southern blot analysis was performed according to standard protocols (30). Terminase-cleaved and uncleaved DNA fragments were visualized by using a DIG-labeled PCR fragment (HCMV genome positions 703 to 1524). Detection was carried out by using an anti-DIG-AP-conjugated antibody according to the manufacturer's instructions (Roche Applied Sciences, Germany).
Statistics.
Statistical significance was determined by using one-way analysis of variance (ANOVA) (GraphPad Software, CA). The results are presented as means ± standard errors of the means (SEM). Significance was accepted when the P value was <0.05. The IC50s or EC50s were calculated by using the inhibitor dose-response function (GraphPad Software, CA).
ACKNOWLEDGMENTS
We thank Wade Bresnahan, University of Minnesota, for providing reagents and Li Qiu and Wenbo Zhou for technical assistance.
This work was supported by the Center for Drug Design at the University of Minnesota.
REFERENCES
- 1.Jean Beltran PM, Cristea IM. 2014. The life cycle and pathogenesis of human cytomegalovirus infection: lessons from proteomics. Expert Rev Proteomics 11:697–711. doi: 10.1586/14789450.2014.971116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morton CC, Nance WE. 2006. Newborn hearing screening—a silent revolution. N Engl J Med 354:2151–2164. doi: 10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- 3.Ahmed A. 2011. Antiviral treatment of cytomegalovirus infection. Infect Disord Drug Targets 11:475–503. doi: 10.2174/187152611797636640. [DOI] [PubMed] [Google Scholar]
- 4.Lischka P, Hewlett G, Wunberg T, Baumeister J, Paulsen D, Goldner T, Ruebsamen-Schaeff H, Zimmermann H. 2010. In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob Agents Chemother 54:1290–1297. doi: 10.1128/AAC.01596-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sigamani SS, Zhao HY, Kamau YN, Baines JD, Tang L. 2013. The structure of the herpes simplex virus DNA-packaging terminase pUL15 nuclease domain suggests an evolutionary lineage among eukaryotic and prokaryotic viruses. J Virol 87:7140–7148. doi: 10.1128/JVI.00311-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ostapchuk P, Almond M, Hearing P. 2011. Characterization of empty adenovirus particles assembled in the absence of a functional adenovirus IVa2 protein. J Virol 85:5524–5531. doi: 10.1128/JVI.02538-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun S, Gao S, Kondabagil K, Xiang Y, Rossmann MG, Rao VB. 2012. Structure and function of the small terminase component of the DNA packaging machine in T4-like bacteriophages. Proc Natl Acad Sci U S A 109:817–822. doi: 10.1073/pnas.1110224109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borst EM, Kleine-Albers J, Gabaev I, Babic M, Wagner K, Binz A, Degenhardt I, Kalesse M, Jonjic S, Bauerfeind R, Messerle M. 2013. The human cytomegalovirus ul51 protein is essential for viral genome cleavage-packaging and interacts with the terminase subunits pUL56 and pUL89. J Virol 87:1720–1732. doi: 10.1128/JVI.01955-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salmon B, Nalwanga D, Fan Y, Baines JD. 1999. Proteolytic cleavage of the amino terminus of the U(L)15 gene product of herpes simplex virus type 1 is coupled with maturation of viral DNA into unit-length genomes. J Virol 73:8338–8348. http://jvi.asm.org/content/73/10/8338.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang K, Wills EG, Baines JD. 2011. A mutation in UL15 of herpes simplex virus 1 that reduces packaging of cleaved genomes. J Virol 85:11972–11980. doi: 10.1128/JVI.00857-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buerger I, Reefschlaeger J, Bender W, Eckenberg P, Popp A, Weber O, Graeper S, Klenk HD, Ruebsamen-Waigmann H, Hallenberger S. 2001. A novel nonnucleoside inhibitor specifically targets cytomegalovirus DNA maturation via the UL89 and UL56 gene products. J Virol 75:9077–9086. doi: 10.1128/JVI.75.19.9077-9086.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldner T, Hewlett G, Ettischer N, Ruebsamen-Schaeff H, Zimmermann H, Lischka P. 2011. The novel anticytomegalovirus compound AIC246 (letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J Virol 85:10884–10893. doi: 10.1128/JVI.05265-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Underwood MR, Harvey RJ, Stanat SC, Hemphill ML, Miller T, Drach JC, Townsend LB, Biron KK. 1998. Inhibition of human cytomegalovirus DNA maturation by a benzimidazole ribonucleoside is mediated through the UL89 gene product. J Virol 72:717–725. http://jvi.asm.org/content/72/1/717.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reefschlaeger J, Bender W, Hallenberger S, Weber O, Eckenberg P, Goldmann S, Haerter M, Buerger I, Trappe J, Herrington JA, Haebich D, Ruebsamen-Waigmann H. 2001. Novel non-nucleoside inhibitors of cytomegaloviruses (BAY 38-4766): in vitro and in vivo antiviral activity and mechanism of action. J Antimicrob Chemother 48:757–767. doi: 10.1093/jac/48.6.757. [DOI] [PubMed] [Google Scholar]
- 15.Nadal M, Mas PJ, Blanco AG, Arnan C, Sola M, Hart DJ, Coll M. 2010. Structure and inhibition of herpesvirus DNA packaging terminase nuclease domain. Proc Natl Acad Sci U S A 107:16078–16083. doi: 10.1073/pnas.1007144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao HY, Lin ZH, Lynn AY, Varnado B, Beutler JA, Murelli RP, Le Grice SFJ, Tang L. 2015. Two distinct modes of metal ion binding in the nuclease active site of a viral DNA-packaging terminase: insight into the two-metal-ion catalytic mechanism. Nucleic Acids Res 43:11003–11016. doi: 10.1093/nar/gkv1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan ZP, Bryant KF, Gregory SM, Angelova M, Dreyfus DH, Zhao XZ, Coen DM, Burke TR, Knipe DM. 2014. HIV integrase inhibitors block replication of alpha-, beta-, and gammaherpesviruses. mBio 5:e01318–14. doi: 10.1128/mBio.01318-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tavis JE, Wang H, Tollefson AE, Ying BL, Korom M, Cheng XH, Cao F, Davis KL, Wold WSM, Morrison LA. 2014. Inhibitors of nucleotidyltransferase superfamily enzymes suppress herpes simplex virus replication. Antimicrob Agents Chemother 58:7451–7461. doi: 10.1128/AAC.03875-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masaoka T, Zhao HY, Hirsch DR, D'Erasmo MP, Meck C, Varnado B, Gupta A, Meyers MJ, Baines J, Beutler JA, Murelli RP, Tang L, Le Grice SFJ. 2016. Characterization of the C-terminal nuclease domain of herpes simplex virus pUL15 as a target of nucleotidyltransferase inhibitors. Biochemistry 55:809–819. doi: 10.1021/acs.biochem.5b01254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou B, Yang K, Wills E, Tang L, Baines JD. 2014. A mutation in the DNA polymerase accessory factor of herpes simplex virus 1 restores viral DNA replication in the presence of raltegravir. J Virol 88:11121–11129. doi: 10.1128/JVI.01540-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kankanala J, Kirby KA, Liu F, Miller L, Nagy E, Wilson DJ, Parniak MA, Sarafianos SG, Wang Z. 2016. Design, synthesis, and biological evaluations of hydroxypyridonecarboxylic acids as inhibitors of HIV reverse transcriptase associated RNase H. J Med Chem 59:5051–5062. doi: 10.1021/acs.jmedchem.6b00465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cantrell SR, Bresnahan WA. 2005. Interaction between the human cytomegalovirus UL82 gene product (pp71) and hDaxx regulates immediate-early gene expression and viral replication. J Virol 79:7792–7802. doi: 10.1128/JVI.79.12.7792-7802.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marschall M, Freitag M, Weiler S, Sorg G, Stamminger T. 2000. Recombinant green fluorescent protein-expressing human cytomegalovirus as a tool for screening antiviral agents. Antimicrob Agents Chemother 44:1588–1597. doi: 10.1128/AAC.44.6.1588-1597.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hutterer C, Eickhoff J, Milbradt J, Korn K, Zeittrager I, Bahsi H, Wagner S, Zischinsky G, Wolf A, Degenhart C, Unger A, Baumann M, Klebl B, Marschall M. 2015. A novel CDK7 inhibitor of the pyrazolotriazine class exerts broad-spectrum antiviral activity at nanomolar concentrations. Antimicrob Agents Chemother 59:2062–2071. doi: 10.1128/AAC.04534-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dal Pozzo F, Andrei G, Daelemans D, Winkler M, Piette J, De Clercq E, Snoeck R. 2008. Fluorescence-based antiviral assay for the evaluation of compounds against vaccinia virus, varicella zoster virus and human cytomegalovirus. J Virol Methods 151:66–73. doi: 10.1016/j.jviromet.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 26.Daelemans D, Pauwels R, De Clercq E, Pannecouque C. 2011. A time-of-drug addition approach to target identification of antiviral compounds. Nat Protoc 6:925–933. doi: 10.1038/nprot.2011.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scheffczik H, Savva CG, Holzenburg A, Kolesnikova L, Bogner E. 2002. The terminase subunits pUL56 and pUL89 of human cytomegalovirus are DNA-metabolizing proteins with toroidal structure. Nucleic Acids Res 30:1695–1703. doi: 10.1093/nar/30.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang D, Bresnahan W, Shenk T. 2004. Human cytomegalovirus encodes a highly specific RANTES decoy receptor. Proc Natl Acad Sci U S A 101:16642–16647. doi: 10.1073/pnas.0407233101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Townsend LB, Devivar RV, Turk SR, Nassiri MR, Drach JC. 1995. Design, synthesis, and antiviral activity of certain 2,5,6-trihalo-1-(beta-d-ribofuranosyl)benzimidazoles. J Med Chem 38:4098–4105. doi: 10.1021/jm00020a025. [DOI] [PubMed] [Google Scholar]
- 30.Gurel F, Ucarli C, Tufan F, Kalaskar DM. 2015. Enhancing T-DNA transfer efficiency in barley (Hordeum vulgare L.) cells using extracellular cellulose and lectin. Appl Biochem Biotechnol 176:1203–1216. doi: 10.1007/s12010-015-1640-0. [DOI] [PubMed] [Google Scholar]