

Abstract
The tendency for genetic architectures to exhibit epistasis among mutations plays a central role in the modern synthesis of evolutionary biology and in theoretical descriptions of many evolutionary processes. Nevertheless, few studies unquestionably show whether, and how, mutations typically interact. Beneficial mutations are especially difficult to identify because of their scarcity. Consequently, epistasis among pairs of this important class of mutations has, to our knowledge, never before been explored. Interactions among genome components should be of special relevance in compacted genomes such as those of RNA viruses. To tackle these issues, we first generated 47 genotypes of vesicular stomatitis virus carrying pairs of nucleotide substitution mutations whose separated and combined deleterious effects on fitness were determined. Several pairs exhibited significant interactions for fitness, including antagonistic and synergistic epistasis. Synthetic lethals represented 50% of the latter. In a second set of experiments, 15 genotypes carrying pairs of beneficial mutations were also created. In this case, all significant interactions were antagonistic. Our results show that the architecture of the fitness depends on complex interactions among genome components.
The nature, amount, and intensity of epistasis among alleles that affect fitness are barely known but are of extreme relevance for many evolutionary theories seeking to explain the origin and maintenance of genetic systems such as sex, recombination, diploidy, genetic canalization, and reproductive isolation; they are also an important component of S. Wright's shifting-balance theory (1). Much effort has gone into finding and characterizing epistasis among genes by using different experimental approaches. Quantitative genetics experiments, in principle, are able to detect epistasis as a component of genetic variance in quantitative traits (2, 3). However, this approach often generates ambiguous results because of low statistical power. Other studies have shown compensatory evolution (4, 5), but these studies do not determine whether epistatic effects are rare or common in absolute terms, because compensatory mutations are favored by selection among a large set of possible mutations. Mutation-accumulation experiments have also been used to show deviations from additive fitness declines (6, 7), but this kind of experiment suffers from two limitations: (i) a lack of knowledge of the number of mutations accumulated and (ii) the fact that deviations from a linear fitness decline may indicate an absence of epistasis or widespread epistasis in which antagonistic and synergistic interactions are equally common (8). So far, the only straightforward approach that avoids all of the above problems relies on constructing genotypes containing exactly the same mutations alone and in combination, then measuring their individual and combined fitness effects and, finally, comparing the results with predictions generated under the null hypothesis of nonepistatic interactions. This approach has been applied to the bacterium Escherichia coli (8) and the fungus Aspergillus niger (9). Both studies reached a similar conclusion: epistases were widespread, but synergistic and antagonistic types were equally common. In addition, all of the above studies explored the interaction among deleterious or quasineutral alleles. However, to our knowledge, no study has yet explored the extent and sign of epistasis among those mutations responsible for adaptive evolution: beneficial mutations.
RNA viruses are characterized as having small compacted genomes, often with overlapping genes and multifunctional components (10). Therefore, epistatic effects on fitness should be especially important for these organisms (10). However, current models of viral evolution do not explicitly incorporate the existence of epistasis (11). Until now, only two studies have sought epistasis for fitness in RNA viruses. The first of them, with foot-and-mouth disease virus, failed to find such epistasis (12). The second, with the segmented bacteriophage φ6, found antagonistic epistasis (13). Nonetheless, these were mutation-accumulation studies, and therefore their conclusions have to be understood with caution for the reasons given above.
Here, we took a direct approach for characterizing the distribution of epistatic effects (8, 9). The starting point for our experiments was a collection of 91 single-nucleotide substitution mutants of vesicular stomatitis virus (VSV) created by site-directed mutagenesis (14). In a first set of experiments, we chose 28 of these genotypes that fulfilled the following two conditions (14): (i) the genomic position to be mutated and the nucleotide incorporated were both randomly chosen, and (ii) mutations had deleterious (although nonlethal) fitness effects. We randomly picked 47 pairs of these mutants and constructed the corresponding double-mutant genotypes. In a second set of experiments, we chose six genotypes for which the mutation incorporated had a beneficial fitness effect (14) and created all of the 15 possible double mutants resulting from combining these single mutations. Table 1, which is published as supporting information on the PNAS web site, contains information about each single and double mutant.
Materials and Methods
Site-Directed Mutagenesis. A full-length infectious VSV cDNA clone was used as template for creating the collection of single and double mutants (15). Site-directed mutageneses were performed as described in the QuikChange XL kit (Stratagene) user's manual. It is important to mention that the method minimizes the chance of the appearance of undesired mutations by using the high-fidelity Pfu DNA polymerase. Sequencing of the cDNAs was done to confirm that each desired mutation was successfully incorporated.
As a first step, we introduced the substitution A-3853→C in the plus strand (Asp-259→Ala substitution in the G surface glycoprotein), which confers the ability of growing in presence of the I1 mAb [mAb-resistant mutant (MARM) phenotype] at a concentration that inhibits wild-type growth (16). This cDNA, named MARM RSV, was used as template for the rest of the mutagenesis.
For the set of randomly chosen mutations, the mutations were distributed in the genome as follows: four mutations were introduced in the nucleocapside N gene (one of them was synonymous); two mutations were generated in the P gene, the small component of the RNA polymerase; one mutant was created in the matrix gene, M; seven mutations were introduced in the envelope glycoprotein G gene (including two synonymous changes); and 12 mutations (including two synonymous substitutions) were introduced in the L gene, the major component of the RNA polymerase. In addition, two mutants were created containing one change in the M-G intercistronic noncoding region. For the set of beneficial mutations, all changes introduced were nonsynonymous: two mutants contained one change in the N gene, three in the M gene, and one in the G gene. More details about each mutant can be found in Table 1.
Virus Recovery from cDNA Clones. Approximately 105 (90–95% confluent) baby hamster kidney (BHK21) cells (obtained from the American Type Culture Collection) were infected with a recombinant vaccinia virus that expressed T7 RNA polymerase. After incubation, cells were cotransfected with the mutant cDNA clone plus three support plasmids encoding the P, L, and N proteins of VSV (15). Transfections were done by using Lipofectamine Plus Reagent (Invitrogen) and adding 25 μg/ml 1-β-d-arabinofuranosylcytosine 6 h postinoculation (hpi) to inhibit vaccinia replication. The supernatant was harvested 96 hpi, and residual vaccinia was removed by filtering the supernatant throughout 0.2-μm membranes (Millipore). Dilutions (100- to 104-fold) were plated on a fresh monolayer with 0.4% agarose in the overlay DMEM (supplemented with 5% calf serum). The presence of plaque-forming units (pfu) 24 hpi indicated the successful recovery of infectious VSV particles. Posttransfection titers, which ranged from 104 to 106 pfu/ml, were equalized to ≈5 × 106 pfu/ml before the competition assays to avoid any possible density effect on fitness (14). Failed transfection experiments were repeated until a positive result was obtained, with a maximum of five trials. The assignation of a mutant as putative lethal was done, taking into account our rate of failure in successfully recovering the wild-type virus, which was in 5 of 28 trials, P = 0.179 (95% confidence interval for the binomial parameter: 0.076 ≤ P ≤ 0.365). After five trials, the probability of not recovering a genotype by chance is as low as 0.1795 = 0.0002 (or, based on the limits of the 95% confidence interval, within the range 2.52 × 10-6 -0.007). In addition, to further minimize the possibility of experimental failure, we repeated the whole process of mutagenesis plus five independent transfection experiments for each putative lethal.
The MARM phenotype of all mutants, as well as the sensitivity of wild type to I1 mAb, was confirmed by plating assays in which the overlay medium was supplemented with 25% (vol/vol) of antibody. A large volume of wild type with a high titer was produced and kept at -80°C. This stock constituted our common competitor for fitness assays.
The study of epistasis among defined pairs of nucleotide substitutions depends strongly on whether each genotype carries only the desired mutations or additional mutations with a fitness effect arising during the early stages of replication and is common to most progeny of a transfection experiment. To rule out this possibility, we took the 2-fold strategy that was explained in detail in ref. 14. In brief, first we ran four independent transfection experiments for five genotypes, and we competed the resulting viruses against our reference wild type (see below). If additional compensatory mutations had accumulated before fitness assays, we would expect to also detect differences among transfection experiments. However, there was no evidence supporting this hypothesis (14). Second, we determined the full-length RNA consensus sequence resulting from one transfection experiment for each of these five genotypes. Not a single unexpected change was observed in three of them. Two of them (originally having single nonsynonymous mutations), however, presented one additional synonymous change with no presumable fitness effect (14). In conclusion, compensatory mutations occurring before competition experiments do not take place at a noticeable rate.
Relative Fitness Assays. The fitness of each mutant relative to the wild type was assessed by seeding ≈2.5 × 103 plaque-forming units (pfu) of each genotype into ≈105 cells. To minimize the probability of fixation of new mutations during competition experiments, they were run for only 12 hpi. Preliminary assays showed that exponential growth occurred during this interval. During competition, the titers of both genotypes were determined by plating the appropriate dilution in the presence and absence of I1 mAb. Intrinsic growth rates, r, were calculated as the slope of the log-titer against time (hpi) during the period of exponential growth. Relative fitness can hence be calculated as ω = exp(rmutant-rwild type). We used this fitness definition because it allows straightforward testing for epistasis in a multiplicative scale (see below). Assays were replicated in 5 independent blocks for random mutations and in 10 blocks when beneficial mutations were combined. Each block contained each pair of singles as well as three replicates of MARM RSV. Doubles were assayed in different blocks that also contained MARM RSV. Fitness estimates of each mutant genotype (W) were adjusted by dividing ω by the grand-mean fitness of MARM RSV estimated in the same block. MARM RSV was effectively neutral relative to the wild type (ω = 0.997 ± 0.009; t59 = 0.405, P = 0.687).
Quantifying the Strength and Direction of Epistasis. Fitness was determined for each double mutant (Wij) as well as for their corresponding single mutants (Wi and Wj). Under the null hypothesis of nonepistatic effects, the expected fitness for the double mutant equals the product of the fitness estimated for each single mutation (i.e., Wij = WiWj). If deleterious mutations were to interact in a synergistic way, then the observed fitness for the double mutant would be lower than expected by multiplying the fitness of both single mutations, and hence the difference between observed and expected fitnesses would became negative. By contrast, if deleterious mutations were to interact in an antagonistic way, then the observed fitness would be larger than expected under the null hypothesis of multiplicative fitness effects, and thus the difference between observed and expected fitnesses would became positive. From this argument, a convenient way of detecting the existence (and sign) of epistasis is by computing the index εij = Wij - WiWj (1). For deleterious alleles (Wi < 1 and Wj < 1), synergistic epistasis is defined by εij < 0, whereas antagonistic epistasis is defined by εij > 0. For beneficial alleles (Wi > 1 and Wj > 1), the signs of εij must be inverted.
Results
Epistasis Among Pairs of Deleterious Mutations. Fig. 1 shows the relationship between observed and expected fitness for each of the 62 double-mutant VSV genotypes. The solid line indicates the result expected under the null hypothesis of nonepistatic interactions. Let us first focus our attention on the 47 randomly chosen pairs of mutations. For each double mutant, we ran t tests assessing the significance of εij (Table 1). Ten cases showed significant antagonistic epistasis, whereas three cases had synergistic epistasis (all P ≤ 0.042). If we apply the Bonferroni method (17) to take into account the multiplicity of tests for interactions, which is a highly conservative approach, only three antagonistic interactions remained significant. Interestingly, in three cases, we were unable to replicate genotypes combining two viable mutations. These can be considered as putative synthetic lethals (18), which represent an extreme form of synergistic interaction. If putative synthetic lethals were removed from the analysis, the distribution of epistatic interactions was effectively symmetrical (Fig. 2; g1 = 0.251 ± 0.357; t43 = 0.703, P = 0.243) and unimodal (Fig. 2; g2 = 0.355 ± 0.702; t43 = 0.506, P = 0.616), and, more interestingly, the average epistatic effect,  , was significantly antagonistic (Fig. 2; t43 = 3.318, P = 0.002).
, was significantly antagonistic (Fig. 2; t43 = 3.318, P = 0.002).
Fig. 1.

Relationship between observed and expected (multiplicative) fitnesses for 65 VSV genotypes carrying pairs of nucleotide substitutions. The solid line represents the null hypothesis of pure multiplicative effects. Deviations from this line are a consequence of the existence of epistatic fitness effects (εij = Wij - WiWj ≠ 0). Filled circles correspond with genotypes carrying two deleterious mutations; open circles correspond with genotypes carrying two beneficial ones.
Fig. 2.

Distribution of the observed minus expected fitness values (εij). The upper distribution was obtained from the 44 mutants carrying pairs of deleterious mutations (after excluding synthetic lethals). The lower distribution was obtained from the 15 mutants that carry two beneficial mutations. The expected values were calculated from the fitness values of the corresponding single mutants assuming no interaction. Antagonistic interactions are more abundant in both cases.
Epistasis Among Pairs of Beneficial Mutations. Let us now move our attention to the 15 genotypes carrying two mutations for which the expected fitness effect was beneficial. The average epistatic effect,  , was also significantly antagonistic (Fig. 2; t14 = 4.522, P < 0.001) and the distribution of epistatic interactions was effectively symmetrical (Fig. 2; g1 = -0.304 ± 0.580; t14 = 0.524, P = 0.608) and unimodal as well (Fig. 2; g2 = -0.576 ± 1.121; t14 = 0.514, P = 0.616). However, the conclusion of a predominance of antagonistic epistasis between pairs of beneficial mutations can be jeopardized by a lack of statistical independence among the 15 genotypes used in this experiment. We created all possible double mutants from six beneficial mutations, but this set of double mutants does not represent a random sample of all possible double mutants on the genome, because several genotypes contain the same mutation. To circumvent this statistical problem, we used a bootstrap approach. One thousand size six random samples were taken from the original dataset, and a
, was also significantly antagonistic (Fig. 2; t14 = 4.522, P < 0.001) and the distribution of epistatic interactions was effectively symmetrical (Fig. 2; g1 = -0.304 ± 0.580; t14 = 0.524, P = 0.608) and unimodal as well (Fig. 2; g2 = -0.576 ± 1.121; t14 = 0.514, P = 0.616). However, the conclusion of a predominance of antagonistic epistasis between pairs of beneficial mutations can be jeopardized by a lack of statistical independence among the 15 genotypes used in this experiment. We created all possible double mutants from six beneficial mutations, but this set of double mutants does not represent a random sample of all possible double mutants on the genome, because several genotypes contain the same mutation. To circumvent this statistical problem, we used a bootstrap approach. One thousand size six random samples were taken from the original dataset, and a 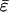 value was computed from each sample. Samples of size six were taken simply because six is the number of independent mutations from which the experiment was initiated. The 95% bootstrap confidence interval for
value was computed from each sample. Samples of size six were taken simply because six is the number of independent mutations from which the experiment was initiated. The 95% bootstrap confidence interval for  ranged between -0.129 and -0.087, slightly more negative than computed above. (The conclusion was completely robust to changes in the sample sizes used during the bootstrap resampling, because 14 of 15 cases had ε < 0.) This result gives even more strength to our conclusion of a predominance of antagonistic epistasis among beneficial mutations.
ranged between -0.129 and -0.087, slightly more negative than computed above. (The conclusion was completely robust to changes in the sample sizes used during the bootstrap resampling, because 14 of 15 cases had ε < 0.) This result gives even more strength to our conclusion of a predominance of antagonistic epistasis among beneficial mutations.
After studying each genotype separately, eight cases showed significant antagonistic epistasis (t tests, P ≤ 0.027) but none showed synergistic epistasis. Six antagonistic cases remained significant even after correcting the significance level with Bonferroni's sequential method (17). Actually, in five of these instances, the fitness of the double mutant was even less than that of either single mutant. This particular case of antagonism between mutational fitness effects is known as decompensatory epistasis (1). Therefore, on average, a viral genotype carrying two beneficial mutations does not get the entire benefit individually associated with each mutation. Indeed, when epistasis is decompensatory, both beneficial alleles involved in the interaction cannot spread to fixation in the population, because the double mutant is less fit than each single mutant (1). As a consequence, lineages bearing alternative beneficial mutations should compete with each other on their way to fixation and, as a consequence of asexuality and clonal interference (19, 20), only the best competitor will eventually become fixed in the population.
Discussion
In this work, the distribution of epistatic interactions on fitness for an RNA virus has been estimated by using explicit pairs of single-nucleotide substitutions, each having a deleterious fitness effect. Among pairs of deleterious mutations, although both synergistic and antagonistic epistases have been detected, interactions were predominantly antagonistic, such that their combined effect is significantly smaller than expected under a multiplicative model. This result is in good agreement with very recent observations made with bacteriophage φ6 (13). A significant fraction of these deleterious, but still viable, mutations create synthetic lethals when combined. We also explored the distribution of epistatic interactions among beneficial mutations. Imaging the results obtained for deleterious mutations, antagonistic epistasis represents the most abundant type of interaction among beneficial mutations, with several cases showing decompensatory epistasis.
Previous studies with E. coli (8) and A. niger (9) found that both synergistic and antagonistic epistases were more or less equally common among deleterious mutations. The results we obtained for deleterious mutations are similar (i.e., both types of interactions exist) but not identical, because antagonistic epistasis are more abundant in VSV. This result might be a reflection of the differences in genome complexity and mutation rate. Evolution under the high mutation rate characteristic of RNA viruses should have been pushing VSV toward regions in sequence space that are more robust to the action of deleterious mutations (21). In this sense, because it involves masking the interaction among deleterious alleles, antagonistic epistasis might be seen as a sort of genetic mutational robustness (22).
The results reported here have two important implications for theories seeking explanations for the evolutionary advantage of recombination and sexual reproduction. First, according to the Fisher–Muller argument, sex and recombination are advantageous because they combine into a common genotype beneficial mutations that arose in different ones, speeding up the rate of adaptation (23, 24). However, if the genetic architecture of RNA viruses determines that, in general, antagonistic epistasis and, in particular, decompensatory epistasis among beneficial mutations is the norm, then recombination would not necessarily imply a benefit in terms of adaptive evolution. Second, sex might still be beneficial for RNA viruses as an efficient mechanism for purging deleterious mutations (25, 26). However, according with the Mutational Deterministic Hypothesis (27), if this is the case, an excess of synergistic epistasis among deleterious mutations is required to compensate the 2-fold advantage of clonal reproduction. Our first data set shows that synergistic interactions among random mutations are neither stronger nor more common than antagonistic interactions. Indeed, the existence of variability among loci in the sign and strength of epistasis, and especially the dominance of antagonistic epistasis, decreases the parameter space over which sex may evolve (28). Taken together, these two observations might explain why VSV, as many other RNA viruses, has evolved asexuality.
Despite the often invoked limitless adaptability of RNA viruses, the existence of abundant antagonistic epistasis among different genome components might impose a strong burden on viral adaptation. As stated by Holmes (10), “Although we can continue to marvel at the adaptability of RNA viruses... rather than thinking about what RNA viruses can do in their evolution, we should concentrate on their limitations. RNA viruses might be more at the mercy of their mutation rates than we think.” The existence of decompensatory epistasis among beneficial mutations might constrain adaptability of RNA viruses.
Finally, we would like to hint that the above findings prompt the necessity of considering nonmultiplicative fitness effects in mathematical descriptions of viral evolution. So far, the only mathematical model put forward to specifically explain viral evolution that explicitly incorporates epistasis (29) considers only the existence of compensatory mutations (i.e., antagonistic epistasis). Although this is a very good starting point, the results we present here suggest that more realistic models must incorporate variance in the type and strength of epistasis among mutations.
Supplementary Material
Acknowledgments
We thank G. T. W. Wertz (University of Alabama, Birmingham) for donating the VSV full-length infectious cDNA as well as the three support plasmids; and A. V. Bordería, C. López-Galíndez, E. Martínez-Salas, and I. S. Novella for invaluable technical advice. This study was supported by the Spanish Ministerio de Educación y Ciencia [Grants BMC2001-3096 (to A.M.) and BMC2003-00066 (to S.F.E.)] and Generalitat Valenciana [Grant GV01-65 (to S.F.E.)]. R.S. received a predoctoral fellowship from the Ministerio de Educación y Ciencia.
Author contributions: R.S. performed research; R.S. and S.F.E. analyzed data; R.S., A.M., and S.F.E. wrote the paper; and A.M. and S.F.E. designed the experiments and supervised R.S.'s Ph.D. thesis.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: hpi, hours postinoculation; MARM, mAb-resistant mutant; VSV, vesicular stomatitis virus.
References
- 1.Wolf, J. B., Brodie, E. D., III, & Wade, M. J. (2000) Epistasis and the Evolutionary Process (Oxford Univ. Press, Oxford).
- 2.Whitlock, M. C., Phillips, P. C., Moore, F. B. G. & Tonsor, S. J. (1995) Annu. Rev. Ecol. Syst. 26, 601-629. [Google Scholar]
- 3.Shaw, A. J., Weir, B. S. & Shaw, F. H. (1997) Evolution (Lawrence, Kans.) 51, 348-353. [DOI] [PubMed] [Google Scholar]
- 4.Lenski, R. E. (1988) Evolution (Lawrence, Kans.) 42, 433-440. [DOI] [PubMed] [Google Scholar]
- 5.Schrag, S. J., Perrot, V. & Levin, B. R. (1997) Proc. R. Soc. London Ser. B 264, 1287-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mukai, T. (1969) Genetics 61, 749-761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Visser, J. A. G. M., Hoekstra, R. F. & van den Ende, H. (1996) Proc. R. Soc. London Ser. B 263, 193-200. [Google Scholar]
- 8.Elena, S. F. & Lenski, R. E. (1997) Nature 390, 395-398. [DOI] [PubMed] [Google Scholar]
- 9.de Visser, J. A. G. M., Hoekstra, R. F. & van den Ende, H. (1997) Evolution (Lawrence, Kans.) 51, 1499-1505. [DOI] [PubMed] [Google Scholar]
- 10.Holmes, E. C. (2003) Trends Microbiol. 11, 543-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eigen, M., McCaskill, J. & Schuster, P. (1988) J. Phys. Chem. 92, 6881-6891. [Google Scholar]
- 12.Elena, S. F. (1999) J. Mol. Evol. 49, 703-707. [DOI] [PubMed] [Google Scholar]
- 13.Burch, C. L. & Chao, L. (2004) Genetics 167, 559-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanjuán, R., Moya, A. & Elena, S. F. (2004) Proc. Natl. Acad. Sci. USA 101, 8396-8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whelan, S. P. J., Ball, L. A., Barr, J. N. & Wertz, G. T. W. (1995) Proc. Natl. Acad. Sci. USA 92, 8388-8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vandepol, S. B., Lefrancois, L. & Holland, J. J. (1986) Virology 148, 312-325. [DOI] [PubMed] [Google Scholar]
- 17.Rice, W. R. (1989) Evolution (Lawrence, Kans.) 43, 223-225. [Google Scholar]
- 18.Phillips, P. C. & Johnson, N. A. (1998) Genetics 150, 449-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerrish, P. J. & Lenski, R. E. (1998) Genetica 102–103, 127-144. [PubMed] [Google Scholar]
- 20.Miralles, R., Gerrish, P. J., Moya, A. & Elena, S. F. (1999) Science 285, 1745-1747. [DOI] [PubMed] [Google Scholar]
- 21.Wilke, C. O. & Adami, C. (2003) Mutat. Res. 522, 3-11. [DOI] [PubMed] [Google Scholar]
- 22.de Visser, J. A. G., Hermisson, J., Wagner, G. P., Ancel Meyers, L., Bagheri-Chaichian, H., Blanchard, J. L., Chao, L., Cheverud, J. M., Elena, S. F., Fontana, W., et al. (2003) Evolution (Lawrence, Kans.) 57, 1959-1972. [DOI] [PubMed] [Google Scholar]
- 23.Fisher, R. A. (1930) The Genetical Theory of Natural Selection. (Oxford Univ. Press, Oxford).
- 24.Muller, H. J. (1932) Am. Nat. 66, 118-138. [Google Scholar]
- 25.Chao, L. (1988) J. Theor. Biol. 133, 99-112. [DOI] [PubMed] [Google Scholar]
- 26.Chao, L. (1991) J. Theor. Biol. 153, 229-246. [DOI] [PubMed] [Google Scholar]
- 27.Kondrashov, A. S. (1988) Nature 336, 435-440. [DOI] [PubMed] [Google Scholar]
- 28.Otto, S. P. & Feldman, M. W. (1997) Theor. Popul. Biol. 51, 134-147. [DOI] [PubMed] [Google Scholar]
- 29.Rouzine, I. M., Wakeley, J. & Coffin, J. M. (2003) Proc. Natl. Acad. Sci. USA 100, 587-592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}