

Abstract
Control of structure and function in membrane proteins remains a formidable challenge. We report here a new design paradigm for the self-assembly of protein components in the context of nonpolar environments of biological membranes. An incrementally staged assembly process relying on the unique properties of fluorinated amino acids was used to drive transmembrane helix–helix interactions. In the first step, hydrophobic peptides partitioned into micellar lipids. Subsequent phase separation of simultaneously hydrophobic and lipophobic fluorinated helical surfaces fueled spontaneous self-assembly of higher order oligomers. The creation of these ordered transmembrane protein ensembles is supported by gel electrophoresis, circular dichroism spectroscopy, equilibrium analytical ultracentrifugation, and fluorescence resonance energy transfer.
The functional properties of proteins are intimately linked to their shape in solution. Proteins spontaneously fold into exquisite three-dimensional structures, representing a delicate balance between protein–protein and protein–solvent interactions. Water-soluble proteins achieve an energetically favorable equilibrium by sequestering nonpolar residues in the interior and placing polar and charged groups on the surface. This simple idea has been exploited as a generalized design paradigm for constructing water-soluble protein architectures (1–4). Indeed, binary patterning with polar and nonpolar amino acids has been successfully used to direct the folding of such proteins (5, 6). Rees et al. (7) have quantitatively analyzed the hydrophobicities of interior versus membrane exposed residues for putative transmembrane helical sequences. They concluded that membrane proteins exhibit a far less pronounced asymmetry in the distribution of polar and nonpolar side chains. In other words, the hydrophobic effect as an organizing force is absent in the long acyl chain region of bilayers. The design of selective protein–protein interfaces in the context of biological membranes is therefore a considerable challenge and remains an unsolved problem in structural biology (8–13).
Naturally occurring protein–protein interfaces either contain elements of polar specificity, for example hydrogen bonding or salt bridges, or are composed of complementary hydrophobic patches containing side chains that maximize van der Waals interactions. DeGrado, Engelman, and colleagues (14–17) have elegantly demonstrated the homomeric association of transmembrane helices by introduction of residues containing polar side chains, including those of Asn, Gln, Asp or Glu (14–17), that can participate in interchain hydrogen bonding. This strategy provides appreciable driving force for oligomerization and has recently been implicated in disease states where a neutral to charged (V232D) mutation within the membrane results in loss of function due to altered assembly and alignment of the cystic fibrosis transmembrane conductance regulator (18). This finding suggests that, in the presence of a number of potential hydrogen-bonding partners in the biological milieu, specificity using this strategy may be difficult to achieve. On the other hand, for proteins embedded within membrane bilayers, tuning the differential van der Waals affinity of protein side chains for one another in the midst of a sea of lipid hydrocarbon tails also proves difficult. What is required from a protein design perspective is an orthogonal hydrophobe that partitions into nonpolar environments away from water but subsequently phase separates from the hydrocarbon lipids.
Highly fluorinated compounds have long been known to have a low propensity to interact with other materials (19). Recent studies have used amino acid side chains incorporating this type of chemical functionality to fashion a new type of protein–protein interaction motif that is simultaneously hydrophobic and lipophobic (20–25). The selectivity in these interfaces is derived from the extra-biological and remarkable properties of highly fluorinated side chains (21). These structures provide an avenue to selectively oligomerize membrane-soluble protein components. We envisioned a two-step assembly for the folding and oligomerization of transmembrane protein segments. First, hydrophobic peptides would partition into micelles. Second, due to phase separation properties of appropriately placed fluorinated amino acids, the peptides would self-assemble within the lipid environment into predetermined structures (Fig. 1). Here, we show that fluorinated surfaces are remarkably effective in mediating helix–helix interactions in transmembrane protein domains.
Fig. 1.

Schematic diagram depicting the two-step self-assembly of membrane-soluble protein segments in micelles. The designed peptides are extremely hydrophobic, are only minimally soluble in water, and partition readily into micelles forming α-helices. One face of the helix exposes a string of hexafluoroleucine residues, shown in space-filling representation that promotes the formation of higher order aggregates. Gray, C; red, O; blue, N; green, F; purple, backbone. Only the backbone (depicted as a helix) and the core packing residues are shown for clarity.
By incorporation of fluorinated side chains, which render selected helical protein surfaces simultaneously hydrophobic and lipophobic, we introduce here a binary patterning scheme suitable for use in biological membranes. We demonstrate the folding and oligomerization of several 29-residue polypeptides in micellar detergents using this strategy. The results described here pave the way for the design of increasingly complex and sophisticated membrane protein architectures involving multiple protein components.
Methods
Synthesis and Purification of Peptides. Peptides were synthesized manually on 4-methylbenzhydrylamine (MBHA) resin by using the in situ neutralization protocol for t-Boc chemistry on a 0.15-mmol scale. For the coupling reactions involving hexafluoroleucine, the reaction time was extended to a minimum of 2 h. The resin-bound, full-length peptides were divided into three; one-third of each was coupled to 4-fluoro-7-nitrobenz-2-oxa-1,3-diazole (NBD-F); one-third was coupled to activated 5-(and- 6)-carboxytetramethylrhodamine (TAMRA); and one-third was left unlabeled. The NBD coupling was carried out between the free terminal amine of the peptide and 3-fold molar excess of NBD-F in the presence of a 20-fold molar excess of N,N-diisopropylethylamine (DIEA). TAMRA was activated with N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) in the presence of a 6-fold molar excess of DIEA. Fluorophore couplings were run for 15 h. The formyl protecting group on tryptophan was removed by treating the resin with 1:10 piperidine in N,N-dimethylformamide (DMF) solution at 0°C for 4 h. Peptides were cleaved from the solid support by using high hydrofluoric acid (HF) conditions (90% anhydrous HF/10% anisole at 0°C for 1.5 h). The lyophilized samples of crude peptides were desalted and purified on reversed-phase HPLC on a C18 column (22 mm × 250 mm, 300 Å, 10–15μm) at 50°C by using linear gradients of water/isopropanol/acetonitrile/0.1% trifluoroacetic acid (TFA).
CD Spectroscopy. CD spectra were obtained on a Jasco (Easton, MD) J-715 spectropolarimeter fitted with a PTC-423S single-position Peltier (Jasco, Easton, MD) temperature controller. The sample conditions were 6 μM peptide, 3 mM SDS, 200 mM NaCl, and 100 mM Tris buffer at pH 8.2 and 25°C. The concentrations of the peptide stock solutions were determined in ethanol by UV absorbance of tryptophan for unlabeled peptides at 280 nm (ε280 nm = 5,690 cm-1·M-1).
SDS/PAGE Analysis. Electrophoresis experiments were performed by using TAMRA-labeled peptides (for ease of visualization without further staining) in 4% stacking and 30% resolving polyacrylamide gel. The running conditions were 0.1% SDS (wt/vol), 200 mM tricine, and 100 mM Tris·HCl buffer at pH 8.2 for 15 h at 90 V.
Analytical Ultracentrifugation. Sedimentation equilibrium experiments were performed with 7-nitrobenz-2-oxa-1,3-diazole (NBD)-labeled peptides in C8E5 or C12E8 micelles at 30,000, 35,000, and 40,000 rpm on a Beckman XL-A ultracentrifuge at 25°C. The density of the buffer was matched to that of the detergent by addition of D2O. Data obtained by UV absorption at 466 nm were analyzed by user-defined nonlinear least squares curve fitting of radial concentration by using a modified Mar-quardt–Levenburg algorithm implemented in igor pro. Data obtained were fit globally to an equation that describes the sedimentation of a homogeneous species.
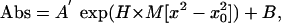 |
[1] |
where Abs = absorbance at radius x, A′ = absorbance at reference radius x0, H = (1 - vρ)ω2/2RT, v = partial specific volume, ρ = density of solvent, ω = angular velocity, M = apparent molecular weight, and B = solvent absorbance. We estimated partial specific volume of the peptides TH1 and TH2 using amino acid composition by the method of Cohn and Edsall (26). Solution densities were calculated by using the program sednterp.
Determination of Partial Specific Volume (v). Density measurements were made by using an Anton Paar (Graza, Austria) DMA5000 densitometer outfitted with a Thermo (Newington, NH) NESLAB RTE-740 digital recirculating bath at 25°C. The instrument was calibrated by using air, H2O, and D2O. Densities of solutions containing H23 and F23 at different concentrations (2–4 mg/ml) were measured.
H23 NH2·QALKKENAQLKWELQALKKELAQ·CONH2
F23 Ac-NH·QALKKENAQLKWELQALKKELAQ·CONH2
An underlined L denotes hexafluoroleucine in F23. The solution densities were then plotted versus peptide concentration in g/ml, and v calculated by using the following expression:
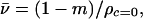 |
where ρc=0 is the solution density of pure solvent and m is the slope of the line. The measured partial specific volume for H23 was consistent with that calculated by using the program sednterp (vH23 = 0.7746 ml/g; calcd = 0.7628 ml/g, and vH23 = 0.7645 ml/g). Normalized values of v adjusted according to the difference in the number of hexafluoroleucine residues in TF1 and TF2 were used for fitting routines used in the analysis of sedimentation equilibrium data.
Fluorescence Resonance Energy Transfer (FRET). Fluorescence spectra were measured on a Varian Cary Eclipse fluorescence spectrophotometer. The concentrations of the peptide stock solutions were determined in ethanol by UV absorbance of tryptophan for unlabeled peptides, of NBD-labeled peptides at 466 nm (ε466 nm = 22,000 cm-1·M-1), and of TAMRA-labeled peptides at 565 nm (ε565 nm = 91,000 cm-1·M-1). Stock solutions in ethanol were dried and then redissolved in buffer. For the FRET experiments, aliquots (125 μl to 1.0 ml) were removed from a starting solution of unlabeled (3 μM) and NBD-labeled peptides (3 μM) and replaced with equivalent volumes of a solution containing TAMRA-labeled peptide (3 μM) and NBD-labeled peptide (3 μM). The net effect was that the total sample volume, concentrations of both the NBD-labeled peptide and total peptide remained constant. Excitation wavelength was set at 460 nm, and emission was monitored at 535 nm. The measured fluorescence at any titration point can be represented in the following way:
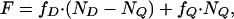 |
[2] |
where fD is molar fluorescence of donor, fQ is the molar fluorescence of quenched donor, ND is the total moles of donor, and NQ is the total moles of quenched donor.
By using a binomial distribution of donors in an oligomeric ensemble of degree n, with the assumption that a single acceptor quenches all donors, the transfer efficiency E (= F/F0) can be described by Eq. 3 for a dimer
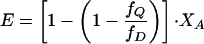 |
[3] |
and by Eq. 4 for a trimeric assembly
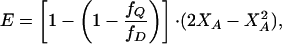 |
[4] |
where fQ/fD denotes the molar efficiency of quenching, XD the mole fraction of the donor peptide, and XA the mole fraction of the acceptor peptide. Dissociation constants were determined by varying the peptide:lipid ratio and globally fitting the data to a monomer–dimer or monomer–trimer equilibrium (17, 27, 28).
Results and Discussion
Membrane proteins can be classified into two broad structural categories, β-barrels and α-helical bundles. We sought to implement our design paradigm into the latter class of protein architectures. The folding and assembly of this class of membrane proteins has been postulated to follow two energetically distinct steps (8), where membrane insertion of independently stable α-helices is followed by intermolecular interactions to give higher order oligomeric structures. The packing interactions within the cellular membrane that promote the second step are van der Waals interactions between neighboring helices, and electrostatic contacts. Whereas soluble extramembranous loops and the binding of ligands also influence packing of individual helices and the overall structure, the primary interhelical interactions within the membrane are essential.
The packing of helices in membrane proteins has been compared to the knobs-into-holes arrangement typically observed in soluble coiled coils (29). Systematic studies have enumerated the preferred side-chain geometries in this protein structural motif and have advanced our general understanding of protein folding and helix–helix interactions (30, 31). Coiled coils typically contain a seven-residue repeat (abcdefg)n, where the a and d residues are predominantly hydrophobic and are the primary components of the packing interface between multiple α-helices. It has been established previously that, for soluble proteins, three to four heptad repeats in the sequence are sufficient to yield a stable dimer (30–32). We designed four peptides with an interaction surface resembling soluble coiled coils to validate our design paradigm. Peptides TH1 and TH2 have identical sequences, except that a single asparagine residue at position 14 in TH1 is substituted with leucine in TH2. All other a and d positions are occupied by leucine (Fig. 2A). Fluorinated peptides TF1 and TF2 are identical in sequence to TH1 and TH2, respectively, except that they contain the nonnatural amino acid 5,5,5,5′,5′,5′-α-S-hexafluoroleucine at all canonical positions constituting the helix-packing interface. The rest of the putative membrane-embedded positions, 20 residues in length and capable of spanning 29.1 Å, were randomly substituted with hydrophobic amino acid residues. On the C-terminal end of the peptides, multiple lysines were included to achieve modest solubility in HPLC solvents to facilitate purification.
Fig. 2.

Peptide sequences, helical wheel diagram, and chemical structures of amino acids and fluorophore labels. (A) Sequences of the peptides used to demonstrate the ability of fluorinated amino acids to promote self-assembly. TH1 and TH2 contain leucine at the a and d positions of the putative micelle-embedded heptad repeats, whereas TF1 and TF2 contain hexafluoroleucine at these positions. (B) Helical wheel diagram depicting the membrane exposed residues (in blue) and the packing interface (green) in a parallel dimer. (C) Structures of the nonproteinogenic amino acids. Peptides labeled with fluorescence donor (NBD) and acceptor (TAMRA) at the N termini were used to assess oligomeric preferences by using FRET. A, Ala; I, Ile; K, Lys; L, Leu; N, Asn; Q, Gln; R, Arg; V, Val; and W, Trp.
Optically pure hexafluoroleucine was prepared according to published procedures (33), and peptides were assembled on 4-methylbenzhydrylamine (MBHA) resin by using the in situ neutralization protocol for t-Boc chemistry. To experimentally examine the preference of the designed peptides to oligomerize, we first tested migration by using gel electrophoresis (Fig. 3A). The mobility of the designed peptides followed the order TH2, TH1, TF2, and TF1 on SDS/PAGE, consistent with higher order oligomeric structures for the latter three peptides. It has previously been shown that peptides similar to TH2 are monomeric and TH1 analogues migrate as a trimeric assembly in SDS/PAGE (14, 17). This result has been ascribed to the tendency of the single polar asparagine residue in TH1 to form interhelical hydrogen bonds. A priori, one expects that TF1 would also participate in interhelical hydrogen bonding, and this prediction is borne out in the retarded mobility during SDS/PAGE. On the other hand, TH2 and TF2 do not contain any side chains capable of forming hydrogen-bonding interactions with partnering helices. However, our design provides TF2 with a fluorinated interaction surface capable of mediating higher order assembly. We anticipated that the lipophobic nature of the supramolecularly organized trifluoromethyl groups would provide the driving force for self-assembly in detergents. The mobility of peptide TF2 is decreased to a much greater extent relative to TH2 during gel electrophoresis, suggesting that interacting fluorinated surfaces alone are, without the aid of polar interactions, sufficient for oligomer formation in membrane environments. We have noted previously that the hydrophobic effect does not contribute significantly to folding in the membrane. Phase separation of fluorinated surfaces is essentially a solvophobic phenomenon, and it seems that mediation of oligomer formation by means of nanophase separation in the cybotactic region (34)§ is energetically viable.
Fig. 3.

Oligomeric states and α-helicity of designed peptides as revealed by gel electrophoresis and CD spectroscopy. (A) Migration of TAMRA-labeled peptides in SDS/PAGE (30% polyacrylamide, 0.1% wt/vol SDS). Irrespective of whether a central asparagine is present or not, the fluorinated peptides show a tendency to form higher order aggregates. (B) CD spectra indicate that all peptides adopt α-helical structures in micellar detergents: shown here are TH2 (blue) and TF2 (red) ([peptide] = 6 μM in 3 mM SDS, 100 mM Tris·HCl, and 200 mM NaCl, pH 8.2).
CD spectroscopy in the presence of micellar detergents suggested that all four peptides adopt α-helical structures, with characteristic minima at 222 and 208 nm (Fig. 3B). To satisfy the stringent demand to neutralize the polar amide backbone through hydrogen bonding in nonpolar environments, all of the designed peptides remain helical, even at elevated temperatures.
The formation of structurally organized higher order aggregates is supported by equilibrium analytical sedimentation. In solutions containing micelles of the neutral lipid dodecyl octa-ethylene glycol ether ([C12E8] = 5 mM), four peptides labeled on the N terminus with NBD were evaluated in the concentration range of 5–30 μM.¶ To cancel out the effect of micellar aggregates on the solution molecular weight of the ensemble, density matching using 18% deuterium oxide was used. This technique effectively matches the solution density to that of C12E8 and eliminates the contribution of micelles to the buoyant molecular weight of the peptide assembly (35). Data obtained from the experiments were fit to an idealized single-species model. As anticipated, the control peptide TH2 sedimented with an apparent molecular weight of a monomer and TH1 was dimeric (Fig. 4 and Table 1). In contrast, both fluorinated peptides were able to form oligomeric ensembles, with apparent molecular weights consistent with a dimeric form for TF2 and a tetrameric form for TF1. Previous solution studies with soluble fluorinated peptides containing an asparagine in the hydrophobic core also indicated a preference for tetramer formation. It is possible that the proper geometry for either hydrogen bonding or anion binding by the asparagine side chains in TF1 is attainable only in the tetramer form (36). Sedimentation experiments in micelles of the lipid octyl pentaethylene glycol ether (C8E5) gave similar results, as did peptides that were devoid of the chromophore label.
Fig. 4.

Analytical equilibrium sedimentation in C12E8 micelles with density matching using 18% D2O. (A) Peptide TH1 sediments with an apparent molecular weight of a dimer. (B) TF1 has an apparent molecular weight that is a little more than a tetramer. (C) Peptide TH2 sediments as a monomer under these conditions. (D) On the other hand, the fluorinated interface in TF2 drives the formation of dimers. Data were fit to an idealized single species model; solid lines calculated for monomer or oligomer assemblies are indicated by labels. All experiments were run at pH 7.0, [peptide] = 10 μM, [C12E8] = 5 mM, 20 mM phosphate buffer, 18% D2O, and 200 mM NaCl, at 25°C and 40,000 rpm.
Table 1. Molecular masses (MM) of peptide ensembles using analytical equilibrium sedimentation.
| Peptide | NBD-TH1 | NBD-TH2 | NBD-TF1 | NBD-TF2 |
|---|---|---|---|---|
| Calculated monomer MM, Da | 3,443 | 3,442 | 4,219 | 4,326 |
| Observed MM, Da | 6,694 ± 109 | 3,535 ± 56 | 17,614 ± 173 | 8,598 ± 103 |
| No. of helices in aggregate | 2 | 1 | 4 | 2 |
Measurements were made as described in Methods, using 10 μM peptide in 5 mM C12E8 (18% D2O) micelles at 25°C at 30,000, 35,000, and 40,000 rpm. Data from multiple speeds were globally fit to an idealized single-species model. Uncertainty represents 2 SD.
FRET was also used to assess the oligomeric state of the peptide ensembles in lipids. Variants of the four peptides equipped with either a donor fluorophore (NBD) or an acceptor fluorophore (TAMRA) attached to the N terminus by means of the agency of a β-alanine linker were used in the FRET experiments. Quenching of the NBD fluorescence by increasing amounts of TAMRA-labeled peptides, in solutions where the donor concentration, total peptide concentration, and peptide/detergent ratio remains constant, offers a convenient method to estimate the oligomer number of the peptide ensembles (Fig. 5). When the relative fluorescence of the quenched donor to that in the absence of quenching is plotted against the mole fraction of the TAMRA-labeled peptide in solutions containing SDS, the magnitude of the quenching indicates the degree of association of donor-acceptor peptides. The results mirror the trend seen in earlier experiments. The hydrocarbon peptide construct TH2 suffers the least quenching upon self-assembly, and TF1 experienced the maximum decrease in donor fluorescence. Peptides TF2 and TH1 show intermediate quenching abilities. By using a model that describes the binomial distribution of donors in an oligomer (27), and by using the frequently invoked assumption that a single acceptor in an oligomer is enough to quench all donors, the average association numbers for the peptides can be calculated. The best fits were obtained for a monomer-dimer equilibrium for TH2 (Kd = 5.75 × 10-3 MF)∥ and TF2 (Kd = 1.12 × 10-3 MF), and for a monomer-trimer equilibrium for TH1 (Kd = 3.42 × 10-6 MF2) and TF1 (Kd = 1.38 × 10-6 MF2). Qualitatively, these results are consistent with the sedimentation data. The slight quantitative deviation of the average association number in the FRET experiments is likely due to influence of the fluorescence labels on the overall thermodynamics of association.
Fig. 5.

FRET between peptides labeled with NBD (donor) and TAMRA (acceptor). An equimolar mixture of unlabeled and donor peptides (3 μM each) in SDS (3 mM) was titrated with an increasing amount of acceptor-labeled peptide in the following way. Aliquots (125 μl to 1.0 ml) were removed and replaced with an equal volume solution containing donor peptide (3 μM) and the acceptor peptide (3 μM). The net effect is that unlabeled peptide is sequentially lowered in concentration and substituted with the TAMRA-labeled peptide, keeping the NBD peptide concentration constant. Theoretical curves (best fit) were generated by using a binomial distribution of donors in oligomeric ensembles and fit to monomer-dimer equilibrium for and TF2, and for a monomer-trimer equilibrium for TH1 and TF1. Plots of relative fluorescence (F/F0) versus mole fraction of the acceptor (XA) reveal that TH2 (▪) suffers the least quenching consistent with the lowest average association number, and TF2 (▾), TH1 (•), and TF1 (▴) show increasing association propensities.
In general, the fluorinated dimerization interface compares favorably with previous studies that used hydrogen bonding as the driving force for oligomer formation (14, 17). In addition, the fluorinated peptides proffer an interface that is orthogonal to side chains found in nature. We envision that this feature will be extremely useful in designing systems that are selective and prevent crossreactivity from other components of biological membranes. Selective and controlled oligomerization of transmembrane peptides provides a powerful method that can be used to modulate biological processes, for example in integrin activation (37), in inhibition of membrane proteases (38), and in signal transduction pathways. With recent advances in methods for incorporation of nonnatural amino acids into proteins, fine control over more sophisticated membrane protein architectures seems within reach (39–41).
Acknowledgments
We thank W. Stafford (Boston Biomedical Research Institute) for help with analytical ultracentrifugation experiments, X. Xing for the synthesis of hexafluoroleucine, and C. Steinem (Universität Regensburg) for helpful discussions. This work was supported in part by National Institutes of Health Grants GM65500 and 1S10RR017948 and by National Science Foundation Grants CHE-0236846 and CHE-0320783. K.K. is a DuPont Young Professor.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: TAMRA, 5-(and-6)-carboxytetramethylrhodamine; FRET, fluorescence resonance energy transfer; NBD, 7-nitrobenz-2-oxa-1,3-diazole.
Footnotes
Cybotactic region has been defined as that part of a solution in the vicinity of a solute molecule in which the ordering of the solvent molecules is modified by the presence of the solute molecule.
Labeled peptides were utilized for sedimentation experiments because the large extinction coefficient of the NBD chromophore allowed greater sensitivity in detection.
Expressed in units of peptide/detergent mole fraction (MF).
References
- 1.Degrado, W. F., Wasserman, Z. R. & Lear, J. D. (1989) Science 243, 622-628. [DOI] [PubMed] [Google Scholar]
- 2.DeGrado, W. F. (1997) Science 278, 80-81. [DOI] [PubMed] [Google Scholar]
- 3.Harbury, P. B., Plecs, J. J., Tidor, B., Alber, T. & Kim, P. S. (1998) Science 282, 1462-1467. [DOI] [PubMed] [Google Scholar]
- 4.Appella, D. H., Christianson, L. A., Klein, D. A., Powell, D. R., Huang, X. L., Barchi, J. J. & Gellman, S. H. (1997) Nature 387, 381-384. [DOI] [PubMed] [Google Scholar]
- 5.Kienker, P. K., Degrado, W. F. & Lear, J. D. (1994) Proc. Natl. Acad. Sci. USA 91, 4859-4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamtekar, S., Schiffer, J. M., Xiong, H. Y., Babik, J. M. & Hecht, M. H. (1993) Science 262, 1680-1685. [DOI] [PubMed] [Google Scholar]
- 7.Rees, D. C., Deantonio, L. & Eisenberg, D. (1989) Science 245, 510-513. [DOI] [PubMed] [Google Scholar]
- 8.Popot, J. L. & Engelman, D. M. (2000) Annu. Rev. Biochem. 69, 881-922. [DOI] [PubMed] [Google Scholar]
- 9.MacKenzie, K. R., Prestegard, J. H. & Engelman, D. M. (1997) Science 276, 131-133. [DOI] [PubMed] [Google Scholar]
- 10.Bowie, J. U. (2001) Curr. Opin. Struct. Biol. 11, 397-402. [DOI] [PubMed] [Google Scholar]
- 11.Ubarretxena-Belandia, I. & Engelman, D. M. (2001) Curr. Opin. Struct. Biol. 11, 370-376. [DOI] [PubMed] [Google Scholar]
- 12.Bowie, J. U. (2004) Proc. Natl. Acad. Sci. USA 101, 3995-3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hong, H. & Tamm, L. K. (2004) Proc. Natl. Acad. Sci. USA 101, 4065-4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou, F. X., Cocco, M. J., Russ, W. P., Brunger, A. T. & Engelman, D. M. (2000) Nat. Struct. Biol. 7, 154-160. [DOI] [PubMed] [Google Scholar]
- 15.Zhou, F. X., Merianos, H. J., Brunger, A. T. & Engelman, D. M. (2001) Proc. Natl. Acad. Sci. USA 98, 2250-2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gratkowski, H., Lear, J. D. & DeGrado, W. F. (2001) Proc. Natl. Acad. Sci. USA 98, 880-885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choma, C., Gratkowski, H., Lear, J. D. & DeGrado, W. F. (2000) Nat. Struct. Biol. 7, 161-166. [DOI] [PubMed] [Google Scholar]
- 18.Therien, A. G., Grant, F. E. M. & Deber, C. M. (2001) Nat. Struct. Biol. 8, 597-601. [DOI] [PubMed] [Google Scholar]
- 19.Horvath, I. T. & Rabai, J. (1994) Science 266, 72-75. [DOI] [PubMed] [Google Scholar]
- 20.Bilgiçer, B., Fichera, A. & Kumar, K. (2001) J. Am. Chem. Soc. 123, 4393-4399. [DOI] [PubMed] [Google Scholar]
- 21.Bilgiçer, B., Xing, X. & Kumar, K. (2001) J. Am. Chem. Soc. 123, 11815-11816. [DOI] [PubMed] [Google Scholar]
- 22.Tang, Y., Ghirlanda, G., Petka, W. A., Nakajima, T., DeGrado, W. F. & Tirrell, D. A. (2001) Angew. Chem. Int. Ed. 40, 1494-1496. [PubMed] [Google Scholar]
- 23.Tang, Y., Ghirlanda, G., Vaidehi, N., Kua, J., Mainz, D. T., Goddard, W. A., DeGrado, W. F. & Tirrell, D. A. (2001) Biochemistry 40, 2790-2796. [DOI] [PubMed] [Google Scholar]
- 24.Tang, Y. & Tirrell, D. A. (2001) J. Am. Chem. Soc. 123, 11089-11090. [DOI] [PubMed] [Google Scholar]
- 25.Yoder, N. C. & Kumar, K. (2002) Chem. Soc. Rev. 31, 335-341. [DOI] [PubMed] [Google Scholar]
- 26.Cohn, E. J. & Edsall, J. T. (1943) Proteins, Amino Acids and Peptides as Ions and Dipolar Ions (Reinhold, New York).
- 27.Li, M., Reddy, L. G., Bennett, R., Silva, N. D., Jones, L. R. & Thomas, D. D. (1999) Biophys. J. 76, 2587-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lear, J. D., Gratkowski, H. & DeGrado, W. F. (2001) Biochem. Soc. Trans. 29, 559-564. [DOI] [PubMed] [Google Scholar]
- 29.Langosch, D. & Heringa, J. (1998) Proteins 31, 150-159. [DOI] [PubMed] [Google Scholar]
- 30.Lupas, A. (1996) Trends Biochem. Sci. 21, 375-382. [PubMed] [Google Scholar]
- 31.Harbury, P. B., Zhang, T., Kim, P. S. & Alber, T. (1993) Science 262, 1401-1407. [DOI] [PubMed] [Google Scholar]
- 32.Lumb, K. J., Carr, C. M. & Kim, P. S. (1994) Biochemistry 33, 7361-7367. [DOI] [PubMed] [Google Scholar]
- 33.Xing, X., Fichera, A. & Kumar, K. (2001) Org. Lett. 3, 1285-1286. [DOI] [PubMed] [Google Scholar]
- 34.McNaught, A. D. & Wilkinson, A. (1997) IUPAC Compendium of Chemical Technology (Blackwell Science, Malden, MA).
- 35.Fleming, K. G. (2000) Methods Enzymol. 323, 63-77. [DOI] [PubMed] [Google Scholar]
- 36.Malashkevich, V. N., Kammerer, R. A., Efimov, V. P., Schulthess, T. & Engel, J. (1996) Science 274, 761-765. [DOI] [PubMed] [Google Scholar]
- 37.Li, R. H., Mitra, N., Gratkowski, H., Vilaire, G., Litvinov, R., Nagasami, C., Weisel, J. W., Lear, J. D., DeGrado, W. F. & Bennett, J. S. (2003) Science 300, 795-798. [DOI] [PubMed] [Google Scholar]
- 38.Das, C., Berezovska, O., Diehl, T. S., Genet, C., Buldyrev, I., Tsai, J. Y., Hyman, B. T. & Wolfe, M. S. (2003) J. Am. Chem. Soc. 125, 11794-11795. [DOI] [PubMed] [Google Scholar]
- 39.Kwon, I., Kirshenbaum, K. & Tirrell, D. A. (2003) J. Am. Chem. Soc. 125, 7512-7513. [DOI] [PubMed] [Google Scholar]
- 40.Wang, L., Brock, A., Herberich, B. & Schultz, P. G. (2001) Science 292, 498-500. [DOI] [PubMed] [Google Scholar]
- 41.Doring, V., Mootz, H. D., Nangle, L. A., Hendrickson, T. L., de Crecy-Lagard, V., Schimmel, P. & Marliere, P. (2001) Science 292, 501-504. [DOI] [PubMed] [Google Scholar]