

Abstract
The first step in the formation of the protease resistant form (PrPSc) of prion proteins involves a conformational transition of the monomeric cellular form of PrPC to a more stable aggregation prone state PrPC*. A search of PDBselect and Escherichia coli and yeast genomes shows that the exact pattern of charges in helix 1 (H1) is rare. Among the 23 fragments in PDBselect with the pattern of charges that match H1, 83% are helical. Mapping of the rarely found (in E. coli and yeast genomes) hydrophobicity patterns in helix 2 (H2) to known secondary structures suggests that the PrPC → PrPC* transition must be accompanied by alterations in conformations in second half of H2. We probe the dynamical instability in H1 and in the combined fragments of H2 and helix 3 (H3) from mPrPC (H2+H3), with intact disulfide bond, using all atom molecular dynamics (MD) simulations totaling 680 ns. In accord with recent experiments, we found that H1 is helical, whereas the double mutant H1[D147A–R151A] is less stable, implying that H1 is stabilized by the (i,i + 4) charged residues. The stability of H1 suggests that it is unlikely to be involved in the PrPC → PrPC* transition. MD simulations of H2+H3 shows that the second half of H2 (residues 184–194) and parts of H3 (residues 200–204 and 215–223) undergo a transition from α-helical conformation to a β and/or random coil state. Simulations using two force fields (optimized potentials for liquid simulations and charmm) give qualitatively similar results. We use the MD results to propose tentative structures for the PrPC* state.
The extracellular globular prion proteins, which are attached to the plasma membrane by a glycosylphosphatidylinositol anchor, have been linked to various transmissible spongiform encephalopathies including bovine spongiform encephalopathy, scrapie disease in sheep, and Creutzfeldt–Jakob disease in humans. The causative agent in these diseases is believed to be the aggregated form (PrPSc) of the cellular prion protein (PrPC) (1). The transition to the scrapie form involves a spectacular conformational change from the mainly α-helical PrPC to the isoform PrPSc that is rich in β-sheet. According to the “protein-only” hypothesis (2), PrPSc serves as a template in inducing conformational transitions in PrPC into a state, PrPC*, that can subsequently be added to PrPSc. The NMR (1, 3, 4) and x-ray (5) structure of PrPC in various species (human, mouse, syrian hamster, bovine, and sheep) shows that the ordered C-terminal part is composed of a short antiparallel β-sheet that contains 8% of the residues in the (90–231) fragment and three helices representing 48% of the secondary structure. Fourier transform infrared spectroscopy measurements (6, 7) indicate that PrPSc(90–231) has 47% β-sheet and 24% α-helical content.
Neither the mechanism of conversion of PrPC into PrPSc nor the tertiary structure of PrPSc is known. Experiments (8) and scenarios of protein aggregation (9) suggest that the earliest event in the conformational transition involves the formation of PrPC* that is more stable than PrPC. The transition from the metastable PrPC to PrPC*, which involves crossing a substantial free energy barrier on the order of 20 kcal/mol (10, 11), results in a state that can nucleate and polymerize to the eventual protease-resistant form. Based on sequence and structural analysis, we proposed (12) that the core of the ordered C terminus of PrPC is frustrated and is susceptible to conformational change. We predicted that the conformational fluctuations in the C-terminal end of helix 2 (H2) and in parts of helix 3 (H3) are involved in the transition to PrPC*. As a result, the transition to PrPC* requires global unfolding of PrPC (13), which explains the origin of the high free energy barrier separating PrPC and PrPC* (12). Recent NMR experiments (8, 14) showed that conformational fluctuations that originate in the C-terminal part of H2 are essential in the formation of PrPC*. Structural and mutational studies have shown that the relatively short helix 1 (H1) is stable over a range of pH values and solvent conditions, and hence is unlikely to undergo conformational change in the transition to PrPC* (15–17).
The required conformational fluctuations in PrPC needed to populate PrPC* suggest that the earliest event involves extensive unfolding of the monomeric PrPC. In this paper, we use results from database search of sequence patterns in helices of PrPC and extensive all-atom molecular dynamics (MD) simulations of helical fragments from the mouse prion protein (mPrPC) to shed light on the nature of instabilities that drive the PrPC → PrPC* transition. Previously, MD simulations have been used to probe other structural aspects of prion proteins, including structures of protofibrils (18). The 10-residue H1, which has an unusual sequence pattern (see below) and is stabilized by salt bridges, remains helical for the duration of the simulation (≈0.09 μs). The double mutant (D147A,R151A), which eliminates one of the three salt bridges in H1, is less stable than the wild type. Multiple MD trajectories of peptides encompassing H2 and H3 (together with their connecting loop) with intact disulfide bond (Cys-179–Cys-214) show that residues in the second half of H2 clustered around positions 187–188 have large conformational flexibility and nonzero preference for β-strand or coil-like structures. Instability in H2 propagates to H3, especially from position 214 onwards. The present study allows us to map the plausible structures of the aggregation prone PrPC*. Despite the limitations (short simulation time and the expected variations of results with different force fields) of all atom simulations, different computational approaches yield qualitatively similar results.
Methods
Sequence Patterns from H1 and H2 in Databases. Because H1 is short (10 residues compared to the average length of 12 residues in a typical α-helix), its stability must arise from the precise positioning of charges along the sequence. To ascertain whether the unusual hydrophilic composition of H1 occurs frequently, we searched the database of structural motifs in proteins (DSMP) for the pattern of charges in H1 and the pattern of hydrophobic residues in H2. Occurrence of low probability of certain patterns might suggest that the particular secondary structure adopted might be hyperstable (H1) or unstable (H2+H3).
We selected from DSMP the set of helices that are at least six residues long, which results in 2,103 helices from the protein structures deposited in the Protein Data Bank (PDB) with <25% sequence similarity. Using the number of pairs, n(+,-), of oppositely charged residues (Arg and Lys are positively charged and Asp and Glu are negatively charged) at positions (i,i + 4), we calculated R(+,-) = LH/n(+,-), where LH is the helix length. From the average 10% of positively charged and 10% of negatively charged residues in a protein (19), we expect that the typical value of R(+,-) ≈ 10.
We also searched the proteins from PDBselect (www.cmbi.kun.nl/gv/pdbsel), the yeast (8,992 proteins) and Escherichia coli (4,289 proteins) genomes for the precise pattern seen in H1 from PrPC, i.e., for finding positions (i,i + 4), (i + 3,i + 7) and (i + 4,i + 8) occupied by oppositely charged residues. The genome-wide search was performed to put the findings from the DSMP into perspective. If the H1 pattern rarely occurs in DSMP, but appears frequently in the genomes, it would mean that this pattern of charges is not unusual and that the protein structures deposited in the PDB are not a representative set of proteins from whole genomes. On the other hand, finding that the H1 pattern occurs infrequently both in DSMP and in whole genomes gives an indication of negative design. We recorded the secondary structure assignment for each H1 charge pattern for protein in PDBselect.
We investigated the sequence pattern of H2 for clues to its potential for conformational instability. Upon clustering the 20 types of amino acids into four classes (H, P, +, and -), the sequence of H2 in mPrP becomes PPHHP-HHPHPH+PPPHPPPP+. The databases mentioned above were searched for the pattern of hydrophobic residues in H2 together with the requirement that the four positions next to the last be a polar amino acid and the last position be a nonhydrophobic amino acid.
Simulation Details. We carried out MD simulations by using a cubic box of appropriate size (depending on the sequence length) with periodic boundary conditions using the moil package (20) in the microcanonical ensemble with the TIP3P model for water molecules. We used the Particle Mesh Ewald method (21) with a tolerance of 10-6 to compute electrostatic interactions (see Table 1).
Table 1. Simulation details.
| System* | Sequence† | System size, Ň | Run time, ns§ | Simulation cutoffs, Ŷ |
|---|---|---|---|---|
| H1 (moil)∥ | 144–153 | 717/30 | 5/85 | Rvdw = 9 Re = 12 |
| H1 (namd) | 144–153 | 717/30 | 2/40 | Rvdw = 12 Re = 12 |
| Rswitch = 10 | ||||
| Rpair = 13.5 | ||||
| H1 [D147A, R151A] | 144–153 | 732/30 | 4/105 | Rvdw = 9 Re = 12 |
| H2 + H3 (moil)** | 172–224 | 1553/40 | 6/165 | Rvdw = 9 Re = 12 |
| H2 + H3 (namd) | 172–224 | 1553/40 | 3/285 | Rvdw = 12 Re = 12 |
| Rswitch = 10 Rpair = 13.5 |
The integration time step is 1 fs. Typical equilibration time ranges from 0.2 to 0.5 ns, and the temperature is 300 K
Numbering in 1ag2 file
Number of water molecules/length of cubic box
Number trajectories/length of simulation
Cut-off distances for nonbonded interactions. Cutoff for van der Waals (Rvdw) and electrostatic (Re) interactions and for switching function (Rswitch) and inclusion of a pair of nonbonded atoms in the pairs list (Rpair) are shown
DWEDRYYREN
QNNFVHDCVNITIKQHTVTTTTKGENFTETDVKMMDRVVEQMCVTQYQKESQA
For H1 from mPrPC (1), the initial conformation was taken from the PDB entry 1ag2 and the ends were capped with the standard charged acetyl and amide groups. To probe the degree of stabilization of the helical conformation caused by the presence of salt-bridges, we also simulated a mutant of H1 in which we replaced Asp-147 and Arg-151 with Ala. The influence of H3 on the dynamics of H2 was investigated by using simulations of the two helices together with the intervening loop (H2+H3) and with intact disulfide bond between Cys-179 and Cys-214. The disulfide bond was constrained by using ES-S = K (D - D0)2, where K = 500 kcal/(mol Å2), D is the distance between the two sulfur atoms in a given conformation, and D0 = 2.05 Å in 1ag2. To probe the dependence of the conformational fluctuations on the details of the force field, we also performed simulations of the dynamics of H1 and H2+H3 by using the namd package (22) with the charmm27 parameter set (23) (Table 1).
Probes of Conformational Fluctuations. To determine the secondary structural (i.e., α-helix or β-strand) conformation of an amino acid, we used the dihedral angles φ and Ψ in conjunction with the procedure given in ref. 24. The average probability for an interior residue of a polypeptide to be in a secondary structure element in a trajectory i is
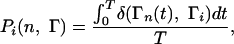 |
[1] |
where Ti is the total simulation time, Γ = H,S,L (helix, strand, loop conformation) is the secondary structure element, and Γn(t) is the secondary structure assignment for residue n at time step t. We measured the average α-helix content (〈H〉), and the average β-strand content (〈S〉) of the fragment as the percentage of residues in an α-helical or β-strand conformation averaged over the length of the trajectory and over the total number of trajectories.
Because of the expectation that the second half of H2 has enhanced conformational flexibility (8, 12), we also probed the global movements of segments that accompany the structural transition. To this end, we monitored the evolution of the torsion angle, Ω(t), between the axes of the two halves of H2 (first half, residues 172–183; second half, residues 184–194). The orientation of the helix axis was obtained based only on the Cα atoms (25). In 1ag2, Ω ≈ 0°. On the other hand, in the human prion protein domain swapped dimer (PDB ID 1i4m; ref. 26), Ω ≈ 58°, which shows that the dimer formation involves a large-scale rotation of the two halves of H2.
Results
Pattern of Charges in H1 Is Rare. The distribution of R(+,-) for the 2,103 helices from the DSMP (Fig. 1a) shows that no other natural sequence has as many (+,-) pairs at positions (i,i + 4) as H1 from PrPC. The search of the entire PDBselect database for the H1 charge pattern shows that this pattern occurs at least once in only 56 (4.6%) sequences, with the total number of patterns being 63. If we restrict the search to be the exact pattern of H1, i.e., i = -, i + 3 = -, i + 4 = +, i + 7 = +, and i + 8 = -, the number of sequences is a mere 23 (or 1.9%). Ziegler et al. (17) arrived at a similar conclusion based on a H1 pattern search in PDB. The 23 rare sequence fragments are either α-helical (83%) or in a random coil state (17%). Analysis of the yeast genome shows that 828 (9.2%) of sequences have the general pattern of H1, with only 253 (2.8%) having the exact pattern. In the E. coli genome, the numbers are 158 (3.7%) for the general charge pattern and 51 (1.2%) for the exact match. These results suggest that the sequence of H1 in PrPC is unusual not only in its high charge content, but also in the positioning of charges along the sequence. More importantly, for the 23 proteins with known 3D structures, the exact charge pattern results overwhelmingly in α-helices. Even more interestingly, analysis of the 19 sequences with mostly α-helical structure reveals that the majority (88%) of (+,-) pairs of residues found at positions (i,i + 4) form salt bridges. These results indicate that the unusual stability of the short helix H1 is possibly associated with its ability to form the highly stabilizing salt bridges involving (i,i + 4) residues.
Fig. 1.

Charge pattern distribution. (a) Distribution of R(+,-) = LH/n(+,-) for 2,103 helices from PDBselect with helix length, LH, ≥6. The arrows show charge patterns for H1 and first helix in 1qp6 (VDDLDKKFKDLWK) that have the highest numbers n(+,-) of (+,-) pairs. (b) Schematic sketch of H1 with dashed lines highlighting the salt bridges between the properly located charged residues.
Pattern of Hydrophobicity in H2 Is Rare. There are very few sequences that share the pattern of hydrophobicity of H2. In PDBAstral40 (27) (proteins in the PDB having at most 40% sequence similarity), there are only 12 (0.2%) such sequences. In the E. coli genome, there are 46 (1%) such sequences, whereas there are 122 (1.4%) in the yeast genome. Inspection of the structures of the 12 proteins from PDBAstral40 shows that the sequence is never entirely helical! For example, the last five residues are found in a helix in only 13% of these proteins. A characteristic pattern seen in H2 from mammalian prion proteins is TTTT (positions 190–193). In the PDBAstral40, this pattern occurs in only 18 proteins, including the prion sequence. In an overwhelming number of these cases (15 of the 18 proteins), the TTTT pattern is found in a strand and/or loop conformation (irrespective of the identity of the flanking amino acids). These results add further support to our proposal (12) that the second half of H2 would be better accommodated in nonhelical conformations.
H1 in mPrP Is Stable. In the five MD trajectories produced with the moil package, the backbone rms deviation (rmsd) with respect to the PDB structure stabilizes around 1.2–1.5 Å after 1 ns. With the exception of residues 150–152, the propensities of the interior residues for α-helical or β-strand conformations (Eq. 1), as averages over the five trajectories, show that the helical structure is overwhelmingly preferred (Fig. 2a). The distribution of distances (Fig. 2b) between residues at positions (i,i + 4) averaged over the five trajectories shows that, with the exception of residues in the second half of H1, the helical structure is preserved. Snapshots of typical conformations at various moments along one of the trajectories show that even the C-terminal end of H1, which becomes disordered after ≈12 ns, returns to the helical conformation toward the end of the run. Small fluctuations in a short helix are unusual because it is known that isolated helices are at best marginally stable (28, 29).
Fig. 2.

moil simulations. (a) Average propensity of residues in H1 in mPrPC for α-helix (PH)or β-strand (PS) conformations averaged over the five trajectories. (b) The distribution of distances between positions (i, i + 4) in the wild-type H1. The averages are calculated by using time and ensemble averages.
To check whether the predicted stability of H1 depends on the force field, we generated two trajectories for a total of 40 ns by using the charmm27 parameter set with the package namd. The backbone rmsd with respect to the PDB structure stabilizes around 2.5–3.0 Å after ≈10 ns. The rmsd for the backbone of the 144–149 fragment of the chain remains close to 0.5 Å for the duration of the run, which is in very good agreement with the previous set of simulations. The difference between these two sets of simulations is in the fraying of the C terminus residues. These results, which are consistent with the moil simulations, also show that the fraction of helix content in H1 is high.
Mutations of Residues in the Second Salt Bridge (D147–R151) Enhance Conformational Fluctuations. The pattern searches suggest that the three (i,i + 4) salt bridges [(Asp-144,Arg-148), (Asp-147,Arg-151), and (Arg-148,Glu-152)] in H1 should stabilize the isolated H1. To probe the importance of the second salt bridge (Asp-147,Arg-151), we simulated the double mutant H1[D147A,R151A]. Replacing D and R by A should not compromise the local helical propensity because Ala is the best helix-former among the amino acids (19). Consequently, any loss of stability in the structure can be attributed largely to the loss of the salt bridge. From an analysis of four trajectories totaling 105 ns for H1[D147A,R151A], we found that the double mutant has increased conformational flexibility compared to the wild-type chain. Most residues, except position 145, have nonzero β-strand propensity.
The larger conformational fluctuations result in extended states with only the first turn of the helix still present. Based on these findings, we conclude that H1[D147A,R151A] populates two basins of attraction: one that is predominantly α-helical with a radius of gyration ≈6 Å, and the other being mostly random coil with a radius of gyration of ≈7.7 Å. Time evolution of distances between (i,i + 4) residues (data not shown) shows that the conformational change starts toward the C-terminal part of the sequence and proceeds in a highly cooperative manner. Our findings are in agreement with recent experiments (17) that showed that the peptide huPrP(140–158)D147A is destabilized compared to wt-huPrP (140–158). The decreased stability of the mutant could result in the efficient conversion of PrPC (90–231) to the protease-resistant form.
By classifying the structures generated in the MD simulations as helical (24), we find that the helical fraction, fH, of the mutant is 0.55, whereas fH for the wild type is 0.64. The value of fH for the wild type is 0.63 when charmm parameters are used. We should emphasize that the absolute values of fH might be overestimated and could depend on the force field. However, meaningful conclusions can be drawn by using the relative values. Using the fH, values we can estimate the free energy of stability by using ΔF = -RT ln(fH/1 - fH). For the wild-type, ΔFWT ≈ -0.37 kcal/mol, whereas for H1[D147A,R151A], ΔFM ≈-0.13 kcal/mol. If fH using the charmm parameter set is used, then ΔFWT ≈ -0.34 kcal/mol. The relative difference ΔΔF = ΔFWT - ΔFM ≈ -0.24 kcal/mol, which arises from the salt bridge formation in wild type. Interestingly, this estimate for free-energy gain caused by salt bridge formation is in the range of the values reported in the literature (30).
Dissecting the Interactions Stabilizing H1. Because (Arg-148,Glu-152) breaks in all five moil trajectories it is likely that it makes a limited contribution to the stability of the helical structure. This finding is consistent with the NMR studies of Liu et al. (15), who showed that, in mPrP(143–58), residues 144–151 strongly favor the helical conformation, whereas there is significantly less helix character in residues 152–154. Our simulations show loss of helical character at 150 and 151, whereas NMR experiments show that they are helical in the longer peptide mPrP(143–158). We propose that Arg-156 leads to favorable electrostatic interaction with the helix macrodipole, which in turn stabilizes the helical conformations at Arg-151 and Glu-152 in mPrP(143–158). This conclusion accords with the recent experiments on peptide fragments from huPrP (17). As shown above, mutations that break up the second salt bridge (Asp-147,Arg-151) are accompanied by destabilization of H1. From the time-dependent variations in the distances between residues in the three salt bridges (Fig. 2a), we conclude that the salt bridge between Asp-144 and Arg-148 is important for the stability of H1. Mutations at these positions could greatly destabilize H1.
Asymmetry in the Stability of N and C Termini of H1. The N-terminal end of H1 in the wild type and mutant is helical for almost the entire duration of our simulations, whereas the C terminus has reduced conformational stability. The observed “asymmetry” in the dynamics of H1 is caused by the greater loss in tertiary contacts by residues in the C terminus compared to those in the N terminus, when H1 is excised from the rest of the protein. The first half of H1 makes 58% of its contacts with other parts of PrPC (especially with H3 and the loop between the first β strand and H1), whereas 75% of the contacts of the second half of H1 are with fragments other than H1 (especially within the loop connecting H1 with the second β strand). This explains, at least in part, the higher degree of conformational instability in the C terminus of H1. In addition, the N-cap residue in H1 is Asp, which, according to ref. 31, is the most preferred type of amino acid at this position in a helix. Also, Glu is the most preferred type of residue at position N2 in a helix (31). Mutations that affect the charge characteristics at the N terminus of helices lead to changes in helical stability (32–34). The observed stability of the N terminus of H1 correlates well with the known stabilizing effects that negatively charged residues have when located at the N-cap and N2 positions in an α-helix. In contrast, the last four positions in H1 are RENM, and none of these residue types is preferred at its location (31). Therefore, the C-terminal end of H1 does not possess an internal source of stabilization similar to that of the N-terminal end.
Second Half of H2 Is Susceptible to Conformational Fluctuations. To assess the origin of structural flexibility in the core of the protein, we generated nine trajectories for H2+H3 (Table 1). In two of the three trajectories, obtained by using the namd package for a total of 185 ns, there is a drastic reduction in the amount of helical structure that is accompanied by an increase in β-strand content (Fig. 3a). The conformational transition starts in the second half of H2 and propagates toward its N terminus, whereas H3 unwinds concomitantly at its two ends. The propensities of residues for α-helical or β-strand conformations show that only positions 178 and 179 (H2) and residues 205–212 (H3) maintain their native α-helical structure (Fig. 3b). The extent of the conformational transition is also reflected in the behavior of the backbone rmsd from the PDB structure (1ag2), which increases monotonically from 3 Å to 6 Å in ≈5 ns and reaches 11 Å in the next 70 ns.
Fig. 3.

NAMD simulations of H2 + H3. (a) Dependence of the average secondary structure contents (calculated by using the method in ref. 24) as a function of time. The dotted line corresponds to helix 〈H(t)〉 and the continuous line is for strand 〈S(t)〉. The averages were performed along the three trajectories generated by using the NAMD package. (b) Position-dependent averages for helix content (PH) and strand content (PS) for H2+H3 by using averages over time and the three namd trajectories. Qualitatively similar results, but with considerably smaller values for PS, are obtained by using the optimized potentials for liquid simulations (OPLS) force field.
The conformational transitions are correlated with an increase in the angle, Ω(t), between the axes of the two halves of H2 (see Methods), which changes from 20° to 90° (in the first 10 ns) followed by rapid oscillations between these values for the remainder of the trajectory. The transition is initiated in the second half of H2, where the distances between (i,i + 4) positions increase from 5 to 14 Å in ≈10 ns. At longer time scales (t ≈ 60 ns), the distances between (i,i + 4) residues in the first half of H2 also increase from 5 to 13 Å. These motions in H2 are correlated with fluctuations in H3, where the distances between the first four (i,i + 4) pairs of residues in H3 and between positions 212 and 218 (with the exception of Cys-214) increase from 5 to 13 Å in ≈10 ns. Almost complete loss of helical structure occurs toward the end of the trajectory (Fig. 4).
Fig. 4.

Schematic representation of PrPC → PrPC* transition, where the conformation for PrPC is taken from the PDB file 1ag2 (yellow). The conformations for PrPC* contain H1 from 1ag2, whereas the residues encompassing H2+H3 are shown in a conformation (red) reached toward the end of our MD simulations using the namd package (b) or the simulations using the moil package (c). The schematic PrPC* structures are representatives from ensembles of fluctuating conformations. In the representative PrPC* structure obtained by using namd simulations, the H1 region, together with the adjacent loops and the β-strands, and residues (205–212) from H3 retain their original conformations and are therefore depicted with same color as in PrPC. In the moil representative PrPC* structure, the H1 region, together with the adjacent loops and the β-strands, residues (175–179), (184–188), and (193, 194) from H2, and residues (203–218) from H3 retain their original conformations and are therefore depicted with same color as in PrPC. The figures are rotated such that the orientation of H1 is the same in all of them. The figures were produced with packages vmd (36) and povray (www.povray.org).
To check the robustness of the results, we also generated six independent trajectories (Table 1) by using the amber and optimized potentials for liquid simulations (OPLS) force fields as implemented in moil. These simulations show that the extent of conformational change is modest (compared to that obtained by using the charmm27 force field) as determined by rmsd fluctuations.
Examination of the position-dependent secondary structural changes shows signs of conformational instability in the second half of H2 and around the C terminus of H3. The positions with the largest probability for nonhelical states are 186–190 (the second half of H2) and positions 215–223 in H3. The unwinding of the second half of H2, which starts from around position 188, is accompanied by a series of rigid body motions, namely, the stretching of the helix around its midpoint and a rotation of the two halves of the helix one with respect to the other.
The angle between the axes of the two halves of H2 fluctuates around 60°, which is similar to the value found in the dimer huPrP structure (1i4m). It appears that the conformational change using the optimized potentials for liquid simulations (OPLS) force field occurs on a much longer time scale than that induced by the use of the charmm27 parameters.
Nevertheless, the qualitative conclusion that residues in the second half of H2 are unstable in their helical states appears not to depend on the force field.
Conformational Instability in the Second Half of H2 Is Independent of the Intact Structure. We analyzed the contact map of H2 and H3 to assess whether the observed instability in the chain is due to the loss of interactions with the rest of the protein. Only 14 of the 51 contacts of H2 (28%) are made with residues from segments not included in simulations. More precisely, 38% of the contacts made by the residues in the first half of H2 and the whole of H3 are with fragments other than H2 or H3, whereas for the second half of H2, this number reduces to only 9%. Because residues in the second half of H2 lose minimum number of contacts by excising the rest of the protein, we would predict that they would undergo minimal conformational fluctuations. The simulations show the exact opposite behavior, which implies that the dynamical instability in this stretch of sequence is due to intrinsic context dependent conformational frustration (9, 12). In other words, the secondary structure preference for residues 188–190 is not a function of the loss of tertiary interactions with the rest of the protein. In addition, because the overall structure of the first half of H2 and first half of H3 is maintained during the simulation, despite their losing more than a third of their contacts, we suggest that the lost contacts do not play a crucial role in the stability of the peptide segment.
The analysis of helix capping motifs (31) in H2 and H3 shows that only the C-cap of H2 is occupied by a stabilizing residue (Lys) and the first three positions in the N terminus of H3 are occupied by slightly stabilizing residues (Glu, Thr, and Asp). Therefore, we would expect to find substantial conformational fluctuations in the N terminus of H2 and the C terminus of H3, and smaller fluctuations in the C terminus of H2 and in the N terminus of H3. This is the observed behavior in the MD simulations, with the notable exception being among the residues in the second half of H2, which are intrinsically dynamically unstable.
Proposed Structures for PrPC*. The present simulations and recent experiments (16, 17) strongly suggest that H1 is unlikely to change conformation in the PrPC → PrPC* transition. The most drastic change occurs in the second half of H2 and parts of H3. Based on the assumption that alterations in the conformation of H2+H3 do not significantly affect the rest of the protein, we have constructed a plausible ensemble of structures for PrPC* (Fig. 4 b and c). In PrPC*(90–231) obtained from the namd trajectories (Fig. 4b), the helical content is ≈20% (a lower bound), and in PrPC*(90–231) reached during the moil simulations (Fig. 4c), the helical content is ≈30% compared to 48% in mPrPC(90–231). The overall characteristics of these structures are consistent with those proposed by James and coworkers (8). It remains to be seen whether formation of PrPC*, with fluctuating regions in H2+H3, is required for oligomerization of PrPC; i.e., whether PrPC* is an on-pathway monomeric intermediate on the route to fibrillization. We should emphasize that the conformation of the prion protein in PrPSc need not coincide with PrPC*.
Conclusions
Sequence pattern matches and long multiple MD simulations of H1 in mPrPC using two force fields show that the stability of H1 is due to the formation of stabilizing internal salt bridges. It follows that multiple mutations that alter the helix macrodipole will be required to greatly decrease the stability of H1. Although the double mutant H1[D147A,R151A] enhances conformational fluctuations it does not destabilize the helical structure entirely. In view of the high propensity of α-helix observed in the isolated H1 in conjunction with supporting experimental results (15–17), it is clear that alterations in the conformation of H1 are unlikely in the PrPC → PrPC* transition.
Using a number of long MD trajectories with two force fields, we have shown that the H2+H3 segment of mPrP presents two regions of high conformational instability, namely, the second half of H2 (more precisely the positions around Thr-188), and the second half of H3 (positions 213–223). The N-terminal end of H3 also shows signs of instability, but on a much more modest scale. These results, whose qualitative aspects do not depend on force fields, are in accord with experimental data and previous theoretical predictions. The predicted tendency for the second half of H2 to be involved in the formation of PrPC* is also consistent with the observation that a number of mutations at 187 and 188 (H187R, T188R, T188K, and T188A) are associated with various prion diseases. In addition, a monoclonal antibody study (35) showed that the peptide 214–226 from huPrP in PrPSc forms a structural epitope with a distinct conformation from the one in PrPC. Based on our findings, we propose that regions 186–190 and 214–226 must play a central role in the initial stages that involve the PrPC → PrPC* transition. The large conformational change is likely to be accompanied by stretching and rotation of the two halves of H2 and by the unwinding of the N-terminal end of H3. The formation of the domain swapped structure in the dimer structure of human PrPC (26) might be facilitated by these large-scale motions.
Acknowledgments
We are grateful to D. K. Klimov for several insightful discussions. This work was supported in part by National Institutes of Health Grant IR01 NS41356-01.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PrP, prion protein; PrPC, cellular PrP; PrPSc, aggregated PrPC; Hn, helix n; mPrPC, mouse PrPC; MD, molecular dynamics; DSMP, database of structural motifs in proteins; PDB, Protein Data Bank; rmsd, rms deviation.
References
- 1.Riek, R., Hornemann, S., Wider, G., Billeter, M., Glockshuber, R. & Wuthrich, K. (1996) Nature 382, 180-182. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner, S. B. (1998) Proc. Natl. Acad. Sci. USA 95, 13363-13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donne, D. G., Viles, J. H., Groth, D., Mehlhorn, I., James, T. L., Cohen, F. E., Prusiner, S. B., Wright, P. E. & Dyson, H. J. (1997) Proc. Natl. Acad. Sci. USA 94, 13452-13457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zahn, R., Liu, A., Luhrs, T., Riek, R., von Schroetter, C., Garcia, F. L., Billeter, M., Calzolai, L., Wilder, G. & Wuthrich, K. (2000) Proc. Natl. Acad. Sci. USA 97, 145-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haire, L. F., Whyte, S. M., Vasisht, N., Gill, A. C., Verma, C., Dodson, E. J., Dodson, G. G. & Bayley, P. M. (2004) J. Mol. Biol. 336, 1175-1183. [DOI] [PubMed] [Google Scholar]
- 6.Caughey, B. W., Dong, A., Bhat, K. S., Ernst, D., Hayes, S. F. & Caughey, W. S. (1991) Biochemistry 30, 7672-7680. [DOI] [PubMed] [Google Scholar]
- 7.Gasset, M., Baldwin, M. A., Fletterick, R. J. & Prusiner, S. B. (1993) Proc. Natl. Acad. Sci. USA 90, 1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuwata, K., Li, H., Yamada, H., Legname, G., Prusiner, S. B., Akasaka, K. & James, T. L. (2002) Biochemistry 41, 12277-12283. [DOI] [PubMed] [Google Scholar]
- 9.Thirumalai, D., Klimov, D. K. & Dima, R. I. (2003) Curr. Opin. Struct. Biol. 13, 146-159. [DOI] [PubMed] [Google Scholar]
- 10.Baskakov, I. V., Legname, G., Prusiner, S. B. & Cohen, F. E. (2001) J. Biol. Chem. 276, 19687-19690. [DOI] [PubMed] [Google Scholar]
- 11.Baskakov, I. V., Legname, G., Baldwin, M. A., Prusiner, S. B. & Cohen, F. E. (2002) J. Biol. Chem. 277, 21140-21148. [DOI] [PubMed] [Google Scholar]
- 12.Dima, R. I & Thirumalai, D. (2002) Biophys. J. 83, 1268-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hosszu, L. P., Baxter, N. J., Jackson, G. S., Power, A., Clarke, A. R., Waltho, J. P., Craven, C. J. & Collinge, J. (1999) Nat. Struct. Biol. 6, 740-743. [DOI] [PubMed] [Google Scholar]
- 14.Kuwata, K. O., Kamatari, K., Akasaka, K. & James, T. L. (2004) Biochemistry 43, 4439-4446. [DOI] [PubMed] [Google Scholar]
- 15.Liu, A., Riek, R., Zahn, R., Hornemann, S., Glockshuber, R. & Wuthrich, K. (1999) Biopolymers 51, 145-152. [DOI] [PubMed] [Google Scholar]
- 16.Speare, J. O., Rush, T. S., III, Bloom, M. E. & Caughey, B. (2003) J. Biol. Chem. 278, 12522-12529. [DOI] [PubMed] [Google Scholar]
- 17.Ziegler, J., Sticht, H., Marx, U. C., Muller, W., Rosch, P. & Schwarzinger, S. (2003) J. Biol. Chem. 278, 50175-50181. [DOI] [PubMed] [Google Scholar]
- 18.DeMarco, M. & Daggett, V. (2004) Proc. Natl. Acad. Sci. USA 101, 2293-2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Creighton, T. E. (1993) Proteins: Structures and Molecular Properties (Freeman, New York).
- 20.Elber, R., Roitberg, A., Simmerling, C., Goldstein, R., Li, H., Verkhiver, G., Keasar, C., Zhang, J. & Ulitsky, A. (1994) Comp. Phys. Comm. 91, 159-189. [Google Scholar]
- 21.Darden, T. A., York, D. M. & Pedersen, L. G. (1993) J. Chem. Phys. 98, 10089-10092. [Google Scholar]
- 22.Kale, L., Skeel, R., Bhandarkar, M., Brunner, R., Gursoy, A., Krawetz, N., Phillips, J., Shinozaki, A., Varadarajan, K. & Schulten, K. (1999) J. Comp. Physiol. 151, 283-312. [Google Scholar]
- 23.MacKerell, A. D., Banavali, N. & Foloppe, N. (2001) Biopolymers 56, 257-265. [DOI] [PubMed] [Google Scholar]
- 24.Klimov, D. K. & Thirumalai, D. (2003) Structure (London) 11, 295-307. [DOI] [PubMed] [Google Scholar]
- 25.Chothia, C., Levitt, M. & Richardson, D. (1981) J. Mol. Biol. 145, 215-250. [DOI] [PubMed] [Google Scholar]
- 26.Knaus, K. J., Morillas, M., Swietnicki, W., Malone, M., Surewicz, W. K. & Yee, V. C. (2001) Nat. Struct. Biol. 8, 770-774. [DOI] [PubMed] [Google Scholar]
- 27.Chandonia, J. M., Walker, N. S., Conte, L. L., Koehl, P., Levitt, M. & Brenner, S. (2002) Nucleic Acids Res. 30, 260-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bundi, A. & Wuthrich, K. (1979) Biopolymers 18, 299-311. [Google Scholar]
- 29.Dyson, H. & Wright, P. E. (1998) Nat. Struct. Biol. 5, 499-503. [DOI] [PubMed] [Google Scholar]
- 30.Makhatadze, G. I., Loladze, V. V., Ermolenko, D. N., Chen, X. & Thomas, S. T. (2003) J. Mol. Biol. 327, 1135-1148. [DOI] [PubMed] [Google Scholar]
- 31.Aurora, R. & Rose, G. D. (1998) Protein Sci. 7, 21-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dao-pin, S., Nicholson, H., Baase, W. A., Zhang, X. J., Wozniak, J. A. & Matthews, B. W. (1991) Ciba Found. Symp. 161, 52-62. [DOI] [PubMed] [Google Scholar]
- 33.Kelly, C. A., Nishiyama, M., Ohnishi, Y., Beppu, T. & Birktoft, J. J. (1993) Biochemistry 32, 3913-3922. [DOI] [PubMed] [Google Scholar]
- 34.Tidor, B. (1994) Proteins 19, 310-323. [DOI] [PubMed] [Google Scholar]
- 35.Serbec, V. C., Bresjanac, M., Popovic, M., Hartman, K. P., Galvani, V., Rupreht, R., Cernilec, M., Vranac, T., Hafner, I. & Jerala, R. (2004) J. Biol. Chem. 279, 3694-3698. [DOI] [PubMed] [Google Scholar]
- 36.Humphrey, W., Dalke, A. & Schulten, K. (1996) J. Mol. Graph. 14, 33-38. [DOI] [PubMed] [Google Scholar]