

Abstract
Highly proliferative tissues such as the gut, skin and bone marrow lose millions of cells each day to normal attrition and challenge from different biological adversities. To achieve a lifespan beyond the longevity of individual cell types, tissue-specific stem cells sustain these tissues throughout the life of a human. For example, the lifespan of erythrocytes is about 100 days and adults make about two million new erythrocytes every second. A small pool of hematopoietic stem cells (HSCs) in the bone marrow is responsible for the lifetime maintenance of these populations. However, there are changes that occur within the HSC pool during aging. Biologically, these changes manifest as blunted immune responses, decreased bone marrow cellularity, and increased risk of myeloid diseases. Understanding the molecular mechanisms underlying dysfunction of aging HSCs is an important focus of biomedical research. With advances in modern health care, the average age of the general population is ever increasing. If molecular or pharmacological interventions could be discovered that rejuvenate aging HSCs, it could reduce the burden of age related immune system compromise as well as open up new opportunities for treatment of hematological disorders with regenerative therapy.
Functional and Molecular Manifestations of HSC Aging
Hematopoietic stem cells are the quiescent, multipotent precursors of the hematopoietic system that reside in specialized niches in the bone marrow and regenerate the hematopoietic system for the life of the organism. HSCs possess two unique functional properties that distinguish them from other cells of the hematopoietic system – the ability to self-renew and generate daughter HSCs which maintain the HSC pool over time, and the capacity for multilineage differentiation to generate all the effector cells of the blood and bone marrow. Over time, the aging process exerts fundamental changes in both of these processes in HSCs. Compared to their young counterparts, old HSCs are characterized by diminished capacity for self-renewal, and a bias in their lineage output towards myeloid lineages1–6.
The diminished self-renewal capacity of old HSCs seems to be partly linked to their replicative history, as HSCs which have undergone more cell divisions (either biologically induced by age or artificially induced by experimentation) have diminished potential in transplantation assays7. But exactly how self-renewal capacity is eroded over time at the molecular level remains to be determined. The observation that old HSCs are more biased towards myeloid cell output can at least be partly explained by the changing dynamics of the stem cell pool over time. Many groups have now experimentally validated that different subtypes of HSCs exist in the bone marrow with inherent biases in lineage output8–13. Aging does not necessarily change the lineage output of individual HSCs, but changes the composition of the HSC pool over time. While lineage-balanced (HSCs that generate blood cell types in the range of that found in an unmanipulated mouse) and lymphoid-biased (generate proportionally more B- and T-cells than myeloid cells) predominate in young bone marrow, the composition of the compartment shifts during age to become dominated by myeloid-biased (or more accurately termed lymphoid-deficient) HSCs2,3,14. Given the observation that these differentiation biases are stable over time, this suggests that at least certain aspects of their behavior may be epigenetically pre-programming.
Epigenetic Modifications Guide HSC Fate Decisions
The term epigenetics refers to changes in gene expression that does not involve changes to the underlying DNA sequence: a change in phenotype without a change in genotype. Epigenetic modifications act like a blueprint to maintain cell identity by informing each specific cell type which genes should be expressed and to what level. Unlike the underlying genetic sequence, epigenetic modifications are malleable and can be altered in response to signals from the environment. Drifting of the epigenetic landscape over time may be responsible for much of the age-related dysfunction of old HSCs.
Epigenetic Modifications to Cytosine Nucleotides
While the protein-coding information in the genome is encoded by the sequence of the four nucleotides, additional epigenetic information can be encoded by chemical modifications to the cytosine base. One such modification occurs when the carbon 5 hydrogen of the cytosine base is replaced by a methyl group, forming 5-methylcytosine (5-mC). 5-mC encodes the same nucleotide sequence as unmodified cytosine and does not affect Watson-Crick base pairing. 5-mC almost always occurs at cytosines in CpG dinucleotides and in mammals, 60–90% of all CpGs are methylated15,16. Normal biological functions of DNA methylation include X-chromosome inactivation, silencing of repetitive elements, and maintenance of parental imprinting patterns17. The pattern of 5-mC distribution can influence binding of regulatory proteins to target sequences in the genome and impact subsequent expression levels of downstream genes. This function of DNA methylation can help orchestrate physiological processes such as stem cell differentiation, and pathologically contribute to oncogenesis.
DNA methylation is established by the DNA methyltransferase enzymes, of which there are three catalytically-active members in mammals. The principle function of Dnmt1 is to reestablish existing DNA methylation patterns during cellular replication by recognizing hemimethylated DNA and copying pre-existing DNA methylation marks from the parental strand to the daughter strand18. Dnmt1 has specific and important functions in HSCs as genetic inactivation leads to nearly immediate and complete loss of HSCs in vivo19. Moreover, HSCs from mice with reduced Dnmt1 activity become restricted to myeloerythroid differentiation due to both impaired silencing of key lineage determinant genes such as Gata1, Id2 and CEBP/α, and an inability to prime master lymphoid regulators such as Ebf1, Pax5 and Il7rα20. DNMT1 is upregulated in some acute myeloid leukemia (AML) patients, and inhibition of Dnmt1 impairs leukemogenesis and inhibits leukemia stem cell function in a mouse model of MLL-AF9 driven AML21. Dnmt3a and Dnmt3b act as de novo DNA methyltransferase enzymes which coordinate the establishment of new DNA methylation patterns during development and stem cell differentiation22. Dnmt3a regulates HSC fate decisions as evidenced by conditional inactivation of Dnmt3a, which skews HSC divisions towards self-renewal divisions at the expense of differentiation. This fate bias of Dnmt3a-null HSCs leads to an accumulation of mutant HSCs in the bone marrow and a relative dearth of their differentiated progeny in the blood23. Over time, the aberrant hematopoiesis resulting from loss of Dnmt3a leads to bone marrow failure and mice succumb to anemia and cytopenias24,25. While deletion of Dnmt3b in adult HSCs seems to be dispensable for HSC function, conditional inactivation of Dnmt3b in a Dnmt3a-null HSC background exacerbates the self-renewal phenotype suggesting Dnmt3b collaborates with Dnmt3a to confer differentiation potential26. Underscoring the importance of these enzymes in hematopoiesis, DNMT3A is one of the most recurrently mutated genes in myeloid malignancies such as AML27,28 and myelodysplastic syndromes (MDS)29. Conversely, DNMT3B is almost never mutated in these diseases, but may also have tumor suppressor function30,31.
HSC aging is associated with changes to the DNA methylome. Old HSCs are characterized by a gradual increase in global DNA methylation levels32, a hypermethylation phenotype that contrasts with aging in post-mitotic somatic cells which show global hypomethylation. The mechanism for HSC hypermethylation during aging has not been defined. As a group, the genes encoding DNA methyltransferase enzymes decrease in expression in old HSCs compared to young32, although this does not account for the expression and function of specific isoforms of Dnmt3a and Dnmt3b. A substantial proportion of differentially methylated regions (DMRs) that change with HSC aging are associated with lineage determinant factors. DMRs that increase in 5-mC content can be localized to genomic loci associated with lymphoid and erythroid lineages33, cell types that decline during aging. This would be consistent with the dogma of DNA methylation acting predominantly as a repressive epigenetic mark. The alterations in DNA methylation at genomic loci in aging HSCs does not correlate with accompanying changes in expression levels of downstream genes. This lack of correlation between DNA methylation and gene expression suggests that alterations in DNA methylation do not necessarily produce functional consequences for the HSCs themselves, but serves as a heritable readout of their lineage potential which ultimately manifests in their downstream progeny.
Cytosine hydroxymethylation (5-hmC) is another important epigenetic modification to the cytosine base. Similar to 5-mC, hydroxymethylation replaces the hydrogen atom at carbon 5 of cytosine with a hydroxymethyl group. 5-hmC is produced by oxidation of 5-mC by the ten-eleven translocation (TET) family of enzymes, and is thought to be an initial step in a potential demethylation pathway34,35. However, the observations that (1) 5-hmC is stable, (2) can be found in relative abundance in certain cell types, and (3) can influence gene expression suggests 5-hmC is a unique commodity and an important part of the regulatory epigenome. While 5-hmC is rare in most somatic cells (<0.2% of all CpGs), it is relatively enriched in pluripotent embryonic stem (ES) cells, and in other adult somatic stem cells34,36,37. Mouse mutant models and patient genome sequencing studies reinforce important roles for the Tet enzymes (Tet1, Tet2, Tet3) in hematopoiesis. Along with DNMT3A, TET2 is one of the most recurrently mutated genes in myeloid malignancies28,38–40. Murine Tet2-null HSCs have increased self-renewal and competitive repopulation capacity compared to wild-type HSCs and increased myeloid cell output41–43. Over the long-term, Tet2-null mice develop a chronic myelomonocytic leukemia (CMML)-like disease, but with long latency and variable penetrance41. However, Tet2-null mice acutely depleted for Tet3 develop a fully-penetrant and highly aggressive myeloid leukemia within a few weeks44. While Tet1 is rarely mutated in human hematopoietic pathologies, conditional deletion in mouse HSCs increases B-cell output and leads to development of B-cell malignancies45. This presents an interesting dichotomy for the epigenetic regulation of B-cell potential in HSCs as Dnmt1 seems necessary for B-lineage priming, while Tet1 may restrict B-cell output.
Corresponding with the global DNA hypermethylation of aging HSCs, there is a concomitant reduction in 5-hmC in old HSCs compared to young32. While all three Tet enzymes are expressed in HSCs, their levels decrease in abundance with age. The role of 5-hmC in gene regulation is still controversial. Although the presence of 5-mC at gene promoters and enhancers is generally associated with gene repression, accumulation of 5-hmC is associated with active genes46–48. While technical advances have facilitated mapping of 5-mC and 5-hmC patterns at unprecedented resolution and permitted correlative observation, our ability to assign functional significance of the modification at specific CpG residues has remained limited. Technological advances such as adaptation of CRISPR/Cas9 systems for site-specific epigenome editing should help address these questions49,50. As abnormal DNA methylation and hydroxymethylation patterns are widely reported in a variety of human blood disorders, assigning functional significance to the placement of these marks is an important priority for biomedical research.
Post-Translational Modifications to Histone Tails
Each human cell has about six feet of linear DNA which is compacted into higher-order structures in the nucleus by wrapping around histone proteins. Covalent post-translational modifications (including methylation, phosphorylation, acetylation, ubiquitylation and sumoylation) to specific amino acid residues of histone tails are another major mechanism of epigenetic regulation. These histone modifications can impact gene expression by altering chromatin structure to make it more/less accessible to transcription factors and the transcriptional machinery, or by recruiting “readers” of the histone code.
Global profiling of histone modifications is performed by chromatin immunoprecipitation (ChIP) followed by next-generation sequencing (ChIP-seq). Although emerging technologies have reduced the scale of material required for such assays51,52, the large cell numbers required to generate sufficient input DNA for ChIP-seq coupled with the limited numbers of long-term HSCs that can be obtained per animal have hindered genome-wide profiling of chromatin marks in HSCs. As such, current genome-wide studies have focused on histone modifications with well-characterized epigenetic roles. Trimethylation of lysine 4 on histone 3 (H3K4me3) is a chromatin landmark that is present at the transcription start sites of protein-coding genes that are either actively transcribed or primed to do so. H3K4me3 protects promoters from DNA methylation, and helps keep the chromatin in an open state for access of the transcriptional machinery. In mammals, at least six genes (predominantly MLL family members) encode for histone methyltransferases that catalyze H3K4me3. Old HSCs exhibit a modest increase in the total number H3K4me3 peaks compared to their young counterparts32. But more interestingly, the breadth of these peaks at existing marks expands considerably32. The spreading of H3K4me3 from existing H3K4me3 peaks could be an important contributing factor to the dysfunction of aging HSCs as they are enriched for genes associated with HSC self-renewal and loss of differentiation capacity. There is a strong correlation between altered H3K4me3 levels and transcriptional activity as some of the genes that increase in expression level most significantly with age (e.g. Selp, Nupr1, Sdpr, Plscr2, HoxB cluster genes) are those which gain the most H3K4me332.
The effect of H3K27me3 is unambiguously and strongly repressive53,54. H3K27me3-associated domains congregate in silenced foci that inhibit transcription by preventing access to RNA polymerase II and other trans-factors55,56. However, this repressive state is both temporally and spatially dynamic. Accordingly, the PRC2 proteins and appropriate regulation of H3K27me3 have been shown to be necessary for differentiation and maintenance of cell type identity in organisms across eukaryotes57,58, and dysregulation of H3K27me3 is implicated in the genesis and progression of cancer59,60. Consistent with the importance of this epigenetic mark in stem cell function, depletion of each of the core PRC2 components (Ezh2, Suz12, Eed) in HSCs severely compromises hematopoiesis. Loss of Eed and Suz12 in HSCs leads to a significant reduction in the number of phenotypically-defined HSCs, compromised function in competitive repopulation assays, and reductions in global H3K27me361–63. While genetic inactivation of Ezh2 has little effect on HSC homeostasis, this may be complicated by compensation by Ezh161. However, over-expression of Ezh2 preserves HSC self-renewal potential over serial transplantation64, and inhibition of both Ezh1 and Ezh2 is catastrophic for HSCs61, showing the importance of this PRC2 component in hematopoiesis. As HSCs age, there appears to be essentially no change in the distribution and total number of H3K27me3 peaks. But the breadth of the peaks increases by ~29%, and the H3K27me3 signal intensity at transcription start sites increases by ~50%32. This pattern may suggest a gradual evolution of this mark over time in HSCs to lock down epigenetic repression of non-HSC genes, or Darwinian selection for HSCs with broader and stronger H3K27me3 peaks during aging.
In pluripotent ES cells and multipotent adult somatic stem cells, many promoters of crucial developmental genes are marked by both the activating H3K4me3 and repressive H3K27me3 histone modifications. Such loci are termed bivalent domains, and are thought to represent a gene poised status ready for rapid activation/repression depending upon the fate choice during lineage commitment57. Accordingly, in the hematopoietic system the frequency of bivalent promoters is highest in HSCs which progressively declines during differentiation as these domains are resolved in lineage-restricted cells65. Genes with a bivalent promoter status are generally not expressed or lowly expressed because of the presence of the repressive H3K27me3 mark. In young HSCs, a large number of H3K27me3-marked loci are also enriched for the H3K4me3 histone modification, and the total number of bivalent domains is comparable to the number found in ES cells (~2000)32. The bivalent domains present in HSCs include many master transcription factors involved in HSC fate determination such as C/EBPα, Ebf1, Pax5, Meis1, Gata2 and Gata3. Over one-third of the bivalent genes are glycoproteins involved in key HSC signaling pathways including Wnt, Hedgehog, BMPs and TGFβ32. As HSCs age, there is a net increase in the number of bivalent domains, which occurs due to gains of both H3K4me3 and H3K27me3 (entirely new bivalent domains), or H3K27me3 only at promoters with existing H3K4me332. However, the mechanisms of this cannot be explained by changing expression levels of the writers and erasers of these modifications, and the functional consequences for HSCs remains to be determined.
Are Epigenome Changes Intrinsic to Each HSC Over Time or Reflect the Reduced Clonal Diversity of the Aging HSC Pool?
HSC aging can be characterized in terms of the underlying epigenetic changes. However, because of technical limitations it remains to be determined if these epigenetic changes represent inherent changes to aging HSCs, or reflect changes in the composition of the HSC pool over time. The aging pool of HSCs in the bone marrow becomes enriched for myeloid-biased HSCs with the highest self-renewal potential2,3. Because all of the assays used to define epigenome features require pools of HSCs, the observed changes in aging epigenetic features may represent different proportions of lineage-biased HSCs in the young/old HSC pools (Figure 1). For a hypothetical example, if Flt3 is marked by H3K27me3 in myeloid-biased HSCs, but less in lymphoid-biased HSCs, then it would appear that H3K27me3 increases at Flt3 as HSCs age. The observation that Flt3 H3K27me3 increases with age, however, could be alternatively explained by the skewing in ratio of myeloid-biased to lymphoid-biased HSCs in the assay sample as lymphoid-biased HSCs may represent up to 50% of the young HSC pool, but less than 10% of the old HSC pool. Even if strict sorting gates are used for flow cytometric purification of HSCs from young and old mice, currently there is little way to avoid this as the trade-off for including more stringent gates for HSC subtypes (e.g. CD150high, CD229, Hoechst low) would mean a final yield of HSCs typically insufficient for the assay.
Figure 1.
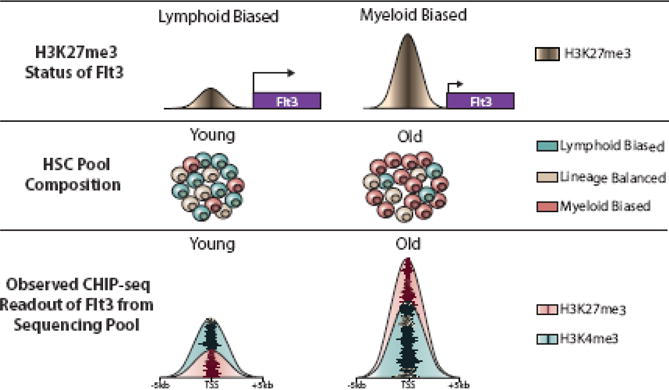
The influence of the composition of the HSC pool in epigenetic comparisons of young and old HSCs. ChIP-seq studies are performed using pools of HSCs, and the outcome can be influenced by variability within distinct HSC subtypes. Using H3K27me3 at the Flt3 gene as an example, there is a low level in lymphoid-biased HSCs and the gene is expressed, but significant H3K27me3 in myeloid-biased HSCs where the gene is repressed. ChIP-seq for H3K27me3 in a pool of young HSCs would show low H3K27me3 at Flt3 because this is the predominant HSC subtype at this developmental stage. The same assay from a pool of old HSCs would produce an observed increase in H3K27me3 at Flt3, but this could be largely due to the over-representation of myeloid-biased HSCs in the pool at this age. Thus, these epigenetic differences over age produced from analyzing pools of cells may in fact reflect the diversity of the cellular composition rather than inherent changes to the HSCs themselves.
To truly resolve the epigenetic changes intrinsic to aging HSCs, single cell resolution is required for epigenome profiling. Methods are currently being adapted for transcriptional profiling of single cells, but such methods still need to be developed for profiling epigenetic marks. However, recent technological advances in ChIP-seq adaptations have reduced the input material to a point (~10,000 cells) that is reasonably achievable for HSC subtype purification51,52. Single cell epigenome profiling may one day be possible with the rate of innovation of next-generation sequencing approaches. Coupling such technology with new strains of fluorescent reporter mice66–68 which isolate more pure populations of functional long-term HSCs should provide the power to answer these important questions.
Is Aging a Byproduct of HSC Epigenome Changes Over Time?
If epigenetic changes are mechanistically shown to be a driver of the aging HSC phenotype, then this raises the possibility that resetting the epigenome to a more youthful state may help restore the functional potential of old HSCs. This premise of this concept has been demonstrated at least indirectly by reprogramming studies. Using transcription-based methods to first reprogram young or old hematopoietic progenitors to induced pluripotent stem (iPS) cells, re-differentiation of these lines back into blood lineages does not show a functional deficit in the potential of re-derived old cells69. This suggests that resetting the old epigenome can overcome the functional deficits of HSC aging. Although global changes to the epigenome occur during aging, it is more likely that the functional decline in aging HSCs is due to specific changes to key regulatory loci. Examining the molecular changes associated with other approaches of rejuvenating old HSCs such as transcription factor-based reprogramming to induced HSCs (iHSCs)70,71, calorie restriction72, or exposure to a young circulatory system through parabiosis73 may identify epigenetic differences at critical loci which enforce the aging phenotype. Reversal of these specific modifications may provide a mechanism to restore self-renewal and lymphoid differentiation potential to old HSCs. Current epigenetic therapies (some of which are used clinically for treatment of hematopoietic malignancies) cause global changes to the epigenome. To more efficaciously reactivate the potential of old HSCs, it is more likely necessary to revert the epigenetic changes at specific age-associated regulatory loci without inducing non-specific global changes. Perhaps most promising for this purpose are modifications to the CRISPR/Cas9 system whereby a catalytically inactive Cas9 (lacks endonuclease activity but can still be recruited by guide RNAs) is fused to the functional domains of epigenetic modifiers (such as DNMT3A for targeted DNA methylation)49,50. Such technologies should provide powerful tools for the study of HSC potential and stem cell biology in general.
Epigenetic Dysfunction as a Precursor to Hematologic Disease in Elderly Individuals
Aging remains the number one risk factor for cancer. The hematopoietic system is no different as age is associated with increased risk of blood cancers (especially malignancies of the myeloid lineage), and abnormal distribution of epigenetic marks is a feature of aging-associated hematopoietic malignancies. One of the most interesting findings from the genomics revolution is the observation that cancer-associated genetic variants can be detected in the blood in a significant proportion of asymptomatic elderly individuals74–76. This phenomenon is known as clonal hematopoiesis, disproportionate blood cell production from a single HSC clone. Studies to date have shown that the presence of such a clone increases an individual’s risk of all-cause mortality, but only marginally increases the hazard ratio for developing a hematopoietic malignancy75. That is, the presence of one of these cancer-associated variants does not mean an individual is destined to develop AML or MDS (although this may just be a matter of limited lifespan at age of detection). More studies are needed to determine what is different between individuals with clonal hematopoiesis who eventually develop a hematopoietic malignancy compared to those who go on to live relatively healthy lives.
In all studies of clonal hematopoiesis across aging humans, the two most commonly variant genes are DNMT3A and TET274–76. Given that murine models of Dnmt3a and Tet2 mutation enhance HSC self-renewal, perhaps this means clonal hematopoiesis is a necessary evil to compensate for the diminished potential of aging HSCs. In other words, DNMT3A and TET2 variants empower old human HSCs with enough self-renewal to extend their lifespan, at the expense of increased risk of malignant transformation. We speculate that this situation may have arisen as a compensatory mechanism over the last two centuries as the rate of stem cell evolution has not kept pace with the extension of human lifespan due to advances in health care technology. Up until relatively recently in human history, the HSC pool was only expected to sustain the blood and bone marrow of an individual for 50 years or so. Over thousands of year, HSCs had evolved to do this very well for this time period. However, average human life expectancy has more than doubled in the last 200 years which has not afforded stem cell evolution the time to adjust to this extended output requirement. One way for HSCs to extend their self-renewal potential beyond their normal lifespan is to select for variants that enhance their self-renewal potential such as those found in DNMT3A and TET2. Thus, there is much to be learned about the relationship between clonal hematopoiesis and risk for hematopoietic transformation, weighed against the overall lifespan of an individual.
Concluding Remarks
Though technological advances have allowed unprecedented access to the HSC epigenome over the last few years, there is still much to be learned about the relationships between different epigenetic marks at distinct regions in the genome and their influence on target gene expression and cellular behavior. Although HSC aging is characterized by changes underlying epigenetic changes, it remains to be determined if the changes in the epigenome over time a cause or a consequence of HSC aging. However, as epigenetic modifications are dynamically malleable, further insights into these processes can pave the way for manipulation of the epigenome to change cell fate, and perhaps restore functional potential to aging HSCs.
Acknowledgments
This work was supported by the following funding sources: National Institutes of Health (NIDDK R01DK102428), the Edward J. Mallinckrodt Jr. Foundation, the American Society of Hematology, Alex’s Lemonade Stand Foundation, the Children’s Discovery Institute, the Sidney Kimmel Foundation and the V Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial disclosures or conflicts of interest – None.
References
- 1.Rossi DJ, Bryder D, Zahn JM, et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. 2005 Jun 28;102(26):9194–9199. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Challen GA, Boles NC, Chambers SM, Goodell MA. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell. 2010 Mar 5;6(3):265–278. doi: 10.1016/j.stem.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beerman I, Bhattacharya D, Zandi S, et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci U S A. 2010 Mar 23;107(12):5465–5470. doi: 10.1073/pnas.1000834107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dykstra B, Olthof S, Schreuder J, Ritsema M, de Haan G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med. 2011 Dec 19;208(13):2691–2703. doi: 10.1084/jem.20111490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. J Exp Med. 2000 Nov 6;192(9):1273–1280. doi: 10.1084/jem.192.9.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007 Aug;5(8):e201. doi: 10.1371/journal.pbio.0050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sawen P, Lang S, Mandal P, Rossi DJ, Soneji S, Bryder D. Mitotic History Reveals Distinct Stem Cell Populations and Their Contributions to Hematopoiesis. Cell reports. 2016 Mar 29;14(12):2809–2818. doi: 10.1016/j.celrep.2016.02.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dykstra B, Kent D, Bowie M, et al. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell. 2007 Aug 16;1(2):218–229. doi: 10.1016/j.stem.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 9.Muller-Sieburg CE, Sieburg HB. Clonal diversity of the stem cell compartment. Curr Opin Hematol. 2006 Jul;13(4):243–248. doi: 10.1097/01.moh.0000231421.00407.65. [DOI] [PubMed] [Google Scholar]
- 10.Muller-Sieburg CE, Sieburg HB, Bernitz JM, Cattarossi G. Stem cell heterogeneity: implications for aging and regenerative medicine. Blood. 2012 Apr 26;119(17):3900–3907. doi: 10.1182/blood-2011-12-376749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller-Sieburg CE, Cho RH, Karlsson L, Huang JF, Sieburg HB. Myeloid-biased hematopoietic stem cells have extensive self-renewal capacity but generate diminished lymphoid progeny with impaired IL-7 responsiveness. Blood. 2004 Jun 1;103(11):4111–4118. doi: 10.1182/blood-2003-10-3448. [DOI] [PubMed] [Google Scholar]
- 12.Muller-Sieburg CE, Cho RH, Thoman M, Adkins B, Sieburg HB. Deterministic regulation of hematopoietic stem cell self-renewal and differentiation. Blood. 2002 Aug 15;100(4):1302–1309. [PubMed] [Google Scholar]
- 13.Morita Y, Ema H, Nakauchi H. Heterogeneity and hierarchy within the most primitive hematopoietic stem cell compartment. J Exp Med. 2010 Jun 7;207(6):1173–1182. doi: 10.1084/jem.20091318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benz C, Copley MR, Kent DG, et al. Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell Stem Cell. 2012 Mar 2;10(3):273–283. doi: 10.1016/j.stem.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 15.Ehrlich M, Gama-Sosa MA, Huang LH, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982 Apr 24;10(8):2709–2721. doi: 10.1093/nar/10.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tucker KL. Methylated cytosine and the brain: a new base for neuroscience. Neuron. 2001 Jun;30(3):649–652. doi: 10.1016/s0896-6273(01)00325-7. [DOI] [PubMed] [Google Scholar]
- 17.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012 Jul;13(7):484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 18.Lei H, Oh SP, Okano M, et al. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 1996 Oct;122(10):3195–3205. doi: 10.1242/dev.122.10.3195. [DOI] [PubMed] [Google Scholar]
- 19.Trowbridge JJ, Snow JW, Kim J, Orkin SH. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell. 2009 Oct 2;5(4):442–449. doi: 10.1016/j.stem.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Broske AM, Vockentanz L, Kharazi S, et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat Genet. 2009 Nov;41(11):1207–1215. doi: 10.1038/ng.463. [DOI] [PubMed] [Google Scholar]
- 21.Trowbridge JJ, Sinha AU, Zhu N, Li M, Armstrong SA, Orkin SH. Haploinsufficiency of Dnmt1 impairs leukemia stem cell function through derepression of bivalent chromatin domains. Genes Dev. 2012 Feb 15;26(4):344–349. doi: 10.1101/gad.184341.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999 Oct 29;99(3):247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 23.Challen GA, Sun D, Jeong M, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012 Jan;44(1):23–31. doi: 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Celik H, Mallaney C, Kothari A, et al. Enforced differentiation of Dnmt3a-null bone marrow leads to failure with c-Kit mutations driving leukemic transformation. Blood. 2015 Jan 22;125(4):619–628. doi: 10.1182/blood-2014-08-594564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayle A, Yang L, Rodriguez B, et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood. 2015 Jan 22;125(4):629–638. doi: 10.1182/blood-2014-08-594648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Challen GA, Sun D, Mayle A, et al. Dnmt3a and Dnmt3b Have Overlapping and Distinct Functions in Hematopoietic Stem Cells. Cell Stem Cell. 2014 Aug 12; doi: 10.1016/j.stem.2014.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010 Dec 16;363(25):2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013 May 30;368(22):2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walter MJ, Ding L, Shen D, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011 Jul;25(7):1153–1158. doi: 10.1038/leu.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schulze I, Rohde C, Scheller-Wendorff M, et al. Increased DNA methylation of Dnmt3b targets impairs leukemogenesis. Blood. 2016 Mar 24;127(12):1575–1586. doi: 10.1182/blood-2015-07-655928. [DOI] [PubMed] [Google Scholar]
- 31.Haney SL, Hlady RA, Opavska J, et al. Methylation-independent repression of Dnmt3b contributes to oncogenic activity of Dnmt3a in mouse MYC-induced T-cell lymphomagenesis. Oncogene. 2015 Oct;34(43):5436–5446. doi: 10.1038/onc.2014.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun D, Luo M, Jeong M, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014 May 1;14(5):673–688. doi: 10.1016/j.stem.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beerman I, Bock C, Garrison BS, et al. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell. 2013 Apr 4;12(4):413–425. doi: 10.1016/j.stem.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009 May 15;324(5929):930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010 Aug 26;466(7310):1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Globisch D, Munzel M, Muller M, et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE. 2010;5(12):e15367. doi: 10.1371/journal.pone.0015367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009 May 15;324(5929):929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009 May 28;360(22):2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 39.Langemeijer SM, Kuiper RP, Berends M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009 Jul;41(7):838–842. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 40.Abdel-Wahab O, Mullally A, Hedvat C, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009 Jul 2;114(1):144–147. doi: 10.1182/blood-2009-03-210039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moran-Crusio K, Reavie L, Shih A, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011 Jul 12;20(1):11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ko M, Bandukwala HS, An J, et al. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U S A. 2011 Aug 30;108(35):14566–14571. doi: 10.1073/pnas.1112317108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shide K, Kameda T, Shimoda H, et al. TET2 is essential for survival and hematopoietic stem cell homeostasis. Leukemia. 2012 Oct;26(10):2216–2223. doi: 10.1038/leu.2012.94. [DOI] [PubMed] [Google Scholar]
- 44.An J, Gonzalez-Avalos E, Chawla A, et al. Acute loss of TET function results in aggressive myeloid cancer in mice. Nature communications. 2015;6:10071. doi: 10.1038/ncomms10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cimmino L, Dawlaty MM, Ndiaye-Lobry D, et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat Immunol. 2015 Jun;16(6):653–662. doi: 10.1038/ni.3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Green BB, Houseman EA, Johnson KC, et al. Hydroxymethylation is uniquely distributed within term placenta, and is associated with gene expression. Faseb J. 2016 Apr 26; doi: 10.1096/fj.201600310R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin SG, Wu X, Li AX, Pfeifer GP. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 2011 Jul;39(12):5015–5024. doi: 10.1093/nar/gkr120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song CX, Szulwach KE, Fu Y, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011 Jan;29(1):68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McDonald JI, Celik H, Rois LE, et al. Reprogrammable CRISPR/Cas9-based system for inducing site-specific DNA methylation. Biology open. 2016 May 11; doi: 10.1242/bio.019067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vojta A, Dobrinic P, Tadic V, et al. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016 Mar 11; doi: 10.1093/nar/gkw159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmidl C, Rendeiro AF, Sheffield NC, Bock C. ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat Methods. 2015 Oct;12(10):963–965. doi: 10.1038/nmeth.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Galen P, Viny AD, Ram O, et al. A Multiplexed System for Quantitative Comparisons of Chromatin Landscapes. Molecular cell. 2016 Jan 7;61(1):170–180. doi: 10.1016/j.molcel.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kharchenko PV, Alekseyenko AA, Schwartz YB, et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011 Mar 24;471(7339):480–485. doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Filion GJ, van Bemmel JG, Braunschweig U, et al. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell. 2010 Oct 15;143(2):212–224. doi: 10.1016/j.cell.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bantignies F, Roure V, Comet I, et al. Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell. 2011 Jan 21;144(2):214–226. doi: 10.1016/j.cell.2010.12.026. [DOI] [PubMed] [Google Scholar]
- 56.Sexton T, Yaffe E, Kenigsberg E, et al. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012 Feb 3;148(3):458–472. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 57.Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006 Apr 21;125(2):315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 58.Feng S, Jacobsen SE. Epigenetic modifications in plants: an evolutionary perspective. Current opinion in plant biology. 2011 Apr;14(2):179–186. doi: 10.1016/j.pbi.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chi P, Allis CD, Wang GG. Covalent histone modifications–miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010 Jul;10(7):457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ellinger J, Kahl P, von der Gathen J, et al. Global histone H3K27 methylation levels are different in localized and metastatic prostate cancer. Cancer investigation. 2012 Feb;30(2):92–97. doi: 10.3109/07357907.2011.636117. [DOI] [PubMed] [Google Scholar]
- 61.Xie H, Xu J, Hsu JH, et al. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell. 2014 Jan 2;14(1):68–80. doi: 10.1016/j.stem.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Majewski IJ, Blewitt ME, de Graaf CA, et al. Polycomb repressive complex 2 (PRC2) restricts hematopoietic stem cell activity. PLoS Biol. 2008 Apr 15;6(4):e93. doi: 10.1371/journal.pbio.0060093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee SC, Miller S, Hyland C, et al. Polycomb repressive complex 2 component Suz12 is required for hematopoietic stem cell function and lymphopoiesis. Blood. 2015 Jul 9;126(2):167–175. doi: 10.1182/blood-2014-12-615898. [DOI] [PubMed] [Google Scholar]
- 64.Herrera-Merchan A, Arranz L, Ligos JM, de Molina A, Dominguez O, Gonzalez S. Ectopic expression of the histone methyltransferase Ezh2 in haematopoietic stem cells causes myeloproliferative disease. Nature communications. 2012;3:623. doi: 10.1038/ncomms1623. [DOI] [PubMed] [Google Scholar]
- 65.Weishaupt H, Sigvardsson M, Attema JL. Epigenetic chromatin states uniquely define the developmental plasticity of murine hematopoietic stem cells. Blood. 2010 Jan 14;115(2):247–256. doi: 10.1182/blood-2009-07-235176. [DOI] [PubMed] [Google Scholar]
- 66.Gazit R, Mandal PK, Ebina W, et al. Fgd5 identifies hematopoietic stem cells in the murine bone marrow. J Exp Med. 2014 Jun 30;211(7):1315–1331. doi: 10.1084/jem.20130428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Acar M, Kocherlakota KS, Murphy MM, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. 2015 Oct 1;526(7571):126–130. doi: 10.1038/nature15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen JY, Miyanishi M, Wang SK, et al. Hoxb5 marks long-term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature. 2016 Feb 11;530(7589):223–227. doi: 10.1038/nature16943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wahlestedt M, Norddahl GL, Sten G, et al. An epigenetic component of hematopoietic stem cell aging amenable to reprogramming into a young state. Blood. 2013 May 23;121(21):4257–4264. doi: 10.1182/blood-2012-11-469080. [DOI] [PubMed] [Google Scholar]
- 70.Riddell J, Gazit R, Garrison BS, et al. Reprogramming committed murine blood cells to induced hematopoietic stem cells with defined factors. Cell. 2014 Apr 24;157(3):549–564. doi: 10.1016/j.cell.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pereira CF, Chang B, Qiu J, et al. Induction of a hemogenic program in mouse fibroblasts. Cell Stem Cell. 2013 Aug 1;13(2):205–218. doi: 10.1016/j.stem.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tang D, Tao S, Chen Z, et al. Dietary restriction improves repopulation but impairs lymphoid differentiation capacity of hematopoietic stem cells in early aging. J Exp Med. 2016 Apr 4;213(4):535–553. doi: 10.1084/jem.20151100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mayack SR, Shadrach JL, Kim FS, Wagers AJ. Systemic signals regulate ageing and rejuvenation of blood stem cell niches. Nature. 2010 Jan 28;463(7280):495–500. doi: 10.1038/nature08749. [DOI] [PubMed] [Google Scholar]
- 74.Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014 Dec 25;371(26):2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014 Dec 25;371(26):2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014 Dec;20(12):1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]