

Abstract
Tubulin and actin exist as monomeric units that polymerize to form either microtubules or filamentous actin. As the polymerization status (monomeric/polymeric ratio) of tubulin and/or actin have been shown to be important in regulating gene expression and phenotype in non‐chondrocyte cells, the objective of this study was to examine the role of cytoskeletal polymerization on the chondrocyte phenotype. We hypothesized that actin and/or tubulin polymerization status modulates the chondrocyte phenotype during monolayer culture as well as in 3D culture during redifferentiation. To test this hypothesis, articular chondrocytes were grown and passaged in 2D monolayer culture. Cell phenotype was investigated by assessing cell morphology (area and circularity), actin/tubulin content, organization and polymerization status, as well as by determination of proliferation, fibroblast and cartilage matrix gene expression with passage number. Bovine chondrocytes became larger, more elongated, and had significantly (P < 0.05) increased gene expression of proliferation‐associated molecules (cyclin D1 and ki67), as well as significantly (P < 0.05) decreased cartilage matrix (type II collagen and aggrecan) and increased fibroblast‐like matrix, type I collagen (COL1), gene expression by passage 2 (P2). Although tubulin polymerization status was not significantly (P > 0.05) modulated, actin polymerization was increased in bovine P2 cells. Actin depolymerization, but not tubulin depolymerization, promoted the chondrocyte phenotype by inducing cell rounding, increasing aggrecan and reducing COL1 expression. Knockdown of actin depolymerization factor, cofilin, in these cells induced further P2 cell actin polymerization and increased COL1 gene expression. To confirm that actin status regulated COL1 gene expression in human P2 chondrocytes, human P2 chondrocytes were exposed to cytochalasin D. Cytochalasin D decreased COL1 gene expression in human passaged chondrocytes. Furthermore, culture of bovine P2 chondrocytes in 3D culture on porous bone substitute resulted in actin depolymerization, which correlated with decreased expression of COL1 and proliferation molecules. In 3D cultures, aggrecan gene expression was increased by cytochalasin D treatment and COL1 was further decreased. These results reveal that actin polymerization status regulates chondrocyte dedifferentiation. Reorganization of the cytoskeleton by actin depolymerization appears to be an active regulatory mechanism for redifferentiation of passaged chondrocytes.
Keywords: actin, cartilage, chondrocyte, cytoskeleton, dedifferentiation, redifferentiation, tubulin
Introduction
Articular chondrocytes grown and passaged in traditional 2D monolayer culture are utilized for cell‐based articular cartilage repair. Although 2D monolayer culture allows for the proliferation required to obtain large cell numbers, the expression of chondrogenic matrix molecules, type II collagen (COL2) and aggrecan (ACAN) is decreased and fibroblastic matrix molecule expression, type I collagen (COL1), is increased following monolayer cell number expansion (Abbott & Holtzer, 1968; Benya et al. 1978). Rather than hyaline cartilage, this phenotype change results in production of fibrocartilage which is incapable of meeting mechanical demands lasting approximately up to 7 years when generated as cartilage repair in vivo (Roberts et al. 2009).
The dedifferentiated phenotype appears to be regulated by cell spreading, as culture on substrates that restricts cell spreading maintained cell production of chondrogenic matrix (Benya et al. 1988; Watt & Dudhia, 1988; Mallein‐Gerin et al. 1991; Cao et al. 2014). The mechanism(s) by which morphology regulates phenotype in chondrocytes is not fully elucidated, but cytoskeletal changes that occur with dedifferentiation may be important. In primary chondrocytes, actin is arranged cortically but when these cells are placed in monolayer culture, actin is rearranged into parallel bundles of actin stress fibers throughout the cytoplasm (Brown & Benya, 1988). Microtubules are organized similarly in both primary and passaged cells but appear longer, more prominent and extensive in passaged chondrocytes (Blain, 2009). Reorganization of either actin or tubulin has been shown to regulate shape and gene expression of several cell types (Fringer & Grinnell, 2001; Hernandez et al. 2013; Liu et al. 2013). Specifically tubulin and actin filaments are polymers made up of monomeric units and there is evidence that gene expression is actively regulated by the proportion of monomeric to polymeric actin and/or tubulin, raising the possibility that cytoskeletal polymerization status may be important in gene regulation in chondrocytes (Rosette & Karin, 1995; Hui et al. 1998; Kuwahara et al. 2005; Rozycki et al. 2016).
The effect of tubulin‐modulating agents on non‐passaged (primary) chondrocytes has provided some insight into the contribution of tubulin to maintenance of chondrocyte phenotype. Depolymerization of polymeric (P‐) into monomeric (M‐) tubulin reduced proliferation (Takigawa et al. 1984) and repressed glycosaminoglycan production by chondrocytes (Jortikka et al. 2000). Induction of tubulin polymerization has been shown to have some beneficial effects on chondrocytes by reducing production of the inflammatory mediator, interleukin (IL)‐1beta, and protease gene expression in primary chondrocytes (Hui et al. 1998). However, to our knowledge the effect of monolayer culture and expansion of chondrocyte numbers on tubulin polymerization status or the effect of tubulin polymerization modulation on the regulation of passaged chondrocyte phenotype has not been elucidated.
The regulation of chondrocyte phenotype by actin has been more thoroughly characterized than tubulin. Monolayer culture of chondrocytes resulted in actin polymerization and a decreased proportion of monomeric to polymerized actin (globular/filamentous; G‐/F‐actin) (Parreno et al. 2014). Depolymerization of actin in passaged chondrocytes favorably affected phenotype. Treatment with the actin depolymerization agent, cytochalasin D, resulted in cell rounding and decreased proliferation. In other cell types, actin depolymerization regulated proliferation by reducing expression of proliferation genes such as cyclin D1 (CCND1) (Fringer & Grinnell, 2001) and Ki‐67 (Ki67) (Salu et al. 2006). CCND1 is of particular interest as it is required for cell cycle G1 to S transition and for chondrocyte proliferation, and is upregulated in chondrocytes by mitogenic factors (Beier et al. 2001). In addition, CCND1 has been shown to regulate cartilage matrix molecule expression by interfering with SOX9 regulation, a transcription factor that regulates ACAN and COL2 expression (Lefebvre et al. 1997; Hwang et al. 2007). This is intriguing as actin depolymerization via cytochalasin D treatment favored proteoglycan deposition (Newman & Watt, 1988) or latrunculin treatment enhanced ACAN gene expression (Parreno et al. 2014). Latrunculin also reduced tenascin C expression in passaged chondrocytes (Parreno et al. 2014). Thus actin polymerization could affect several aspects of the dedifferentiated phenotype including proliferation, fibroblast matrix and/or cartilage matrix molecule gene expression. The studies to date suggest that actin depolymerization plays a role in regulating gene expression during redifferentiation. Thus the hypothesis of this study was that actin and/or tubulin polymerization status modulates chondrocyte phenotype during monolayer culture and passaging as well as redifferentiation.
First, articular chondrocyte dedifferentiation was characterized through examination of cell morphology as well as determination of proliferation, fibroblast and cartilage matrix gene expression with passage number. Phenotypic correlation with actin and tubulin polymerization status in passaged chondrocytes was examined. Although tubulin polymerization status did not change during passaging, actin did. We confirmed that actin depolymerization (increasing G‐/F‐actin) favorably affected the chondrocyte phenotype. We demonstrated that 3D culture, which is used to redifferentiate passaged cells, resulted in actin depolymerization and reversed aspects of the dedifferentiated phenotype, supporting a regulatory role for actin polymerization status in dedifferentiation.
Methods
Cell culture
Bovine full‐thickness articular cartilage was harvested aseptically from 6‐ to 9‐month‐old bovine metacarpophalangeal joints obtained from a local abattoir. Normal‐appearing articular cartilage was obtained from human osteoarthritic joints that were resected with focal cartilage changes of Mankin grades ≥4 following informed patient consent with the approval of the Mount Sinai Hospital Research Ethics Board. Chondrocytes were isolated from articular cartilage by serial enzyme digestion as previously described (Taylor et al. 2015). Briefly, articular cartilage tissue was immersed in 0.5% protease type XIV (Sigma‐Aldrich, Oakville, ON, Canada) in Ham's F12 media and incubated at 37 °C for up to 2 h, followed by 0.1% collagenase A (Roche Diagnostics, Mannheim, Germany) for 18 h at 37 °C and 5% CO2. The digest was filtered using a 40‐μm filter to remove undigested pieces. The cell filtrate was centrifuged and washed twice in media. Chondrocytes were resuspended in Ham's F12 supplemented with 5% fetal bovine serum (FBS; Hyclone; GE Healthcare, Logan, UT, USA) and 1% antibiotics (penicillin, streptomycin, fungizone; Invitrogen).
Chondrocytes were expanded as previously described (Parreno et al. 2014, 2016). Briefly, articular chondrocytes were plated at 1.5 × 103 cells cm−2 in 2D on standard polystyrene flasks (Falcon). Cells were maintained in Ham's F12 supplemented with 5% FBS which was replenished twice weekly, in an incubator at 37 °C, 5% CO2, and at atmospheric O2. At ~70–90% confluency, cells were detached with 0.5% trypsin (5 min), washed in Ham's F12 supplemented with 5% FBS and centrifuged. The remaining cells were re‐plated in monolayer culture at the same density. Primary chondrocytes were designated as ‘P0’ cells. Expanded cells were subsequently identified based on passage number: cells derived after one culture passage were designated ‘P1’ cells; after two passages ‘P2’ cells; after three passages ‘P3’ cells; and after four passages ‘P4’.
For 2D culture studies, either P0 or P2 cells (2.5 × 104 cells cm−2) culture were placed in each well of a six‐well plate and cultured in Ham's F12 supplemented with 5% FBS. For 3D cultures, calcium polyphosphate discs (CPP) were prepared using a previously described method (Waldman et al. 2002). Briefly, approximately 1.6 × 105 cells mm−2 were seeded onto the surface of 4‐mm‐diameter CPP. P2 chondrocytes on CPP were maintained in Ham's F12 supplemented with 5% FBS. After 2 days of culture, the chondrocytes on CPP were either harvested for comparison with cells grown in 2D cultures or were treated with cytoskeletal depolymerization agents in serum‐free Ham's F12 for an additional 24 h depending on the experiment.
Cytoskeletal studies
Following 3 days in 2D or 3D culture, culture media was replaced with serum‐free Ham's F12 media supplemented with either the cytoskeletal disrupting agent(s) or an equivalent amount of vehicle (DMSO) for 24 h. Cytochalasin D (Sigma‐Aldrich), phalloidin (Enzo Life Sciences, Farmingdale, NY, USA), nocodazole (Sigma‐Aldrich), and paclitaxel (Sigma‐Aldrich) were prepared by dissolving agents in DMSO and were diluted in media to obtain working concentrations of 10, 1, 10, and 10 μm, respectively. The cells were then harvested and processed for further studies.
Nucleofection for cofilin knockdown
Non‐targeting SignalSilence® Control siRNA (#6568) or Cofilin siRNA II (#6268; Cell Signaling Techonology Inc., Danvers, MA, USA), which were verified to have bovine cross‐reactivity (communication on 27 March 2014 with Cell Signaling Technology Inc.) were nucleofected into P2 chondrocytes as previously described (Parreno et al. 2016). Briefly, bovine P2 chondrocytes were serum‐starved in Ham's F12 + 0.5% FBS for 2 days. Cells were harvested using trypsin, pelleted, and resuspended in (1 m) nucleofection buffer. Approximately 2 × 106 cells were nucleofected with 100 pmol of siRNA using program U24 of the AMAXA nucleofector (Lonza, Germany). Nucleofected cells were recovered in Ham's F12 supplemented with 20% FBS and plated in monolayer culture at a density of approximately 2.5 × 104 cells cm−2. After 4 h (time at which cells had attached) the media were replaced with Ham's F12 media supplemented with 5% FBS.
Quantification of cell area and spreading
Cells from different passage numbers were seeded at a density of 5 × 103 cells cm−2 on 12‐well polystyrene dishes and grown in Ham's F12 supplemented with 5% FBS for 2 days. This time point and cell density were chosen, as cell boundaries could still be discernible and size as well as shape could be quantified. At this density the cells had similar actin polymerization status to P2 cells seeded at ~2.5 × 104 cells cm−2, which was the cell density utilized for RNA and protein analysis (data not shown). To visualize cells, cells were incubated with live cell dye calcein AM [1 μm in phosphate‐buffered saline (PBS)] at room temperature for 10 min. The cells were then washed with PBS and images acquired using a Leica fluorescent microscope. image J software was utilized to calculate area and circularity as previously outlined (Fardin et al. 2010). Circularity was defined as C = 4π (A/P 2), where π is the mathematical constant ~3.14, P is cell perimeter, and A is cell area. A circularity value of 1.0 indicates perfect circular morphology, whereas a value of 0 would indicate an elongated polygon. A total number of at least 100 cells were evaluated in at least four experiments using cells from different animals on separate occasions.
Confocal microscopy
Cells were cultured at a density of 5 × 103 cells cm−2 on glass coverslips. Following treatments, cells were washed in PBS and fixed in 4% paraformaldehyde at room temperature for 10–15 min. Cells were rinsed in PBS and permeabilized with 0.2% Triton/PBS for 15 min. Following permeabilization, cells were incubated with monoclonal anti‐tubulin antibody (1 : 100 in 0.2% Triton/PBS; Abcam). An Alexa 488‐conjugated anti‐mouse monoclonal antibody (Life Technologies) was utilized as a secondary antibody (1 : 200). A secondary antibody‐alone condition was used as a negative control to ensure specificity of antibody staining. Following immunostaining, cells were stained with Alexa Fluor 488 phalloidin to visualize F‐actin (1 : 20; 0.2% Triton‐PBS; Life Technologies, Burlington, Canada) and DAPI to visualize nuclei. Cells were washed with PBS and then mounted on glass slides using PermaFluor Aqueous Mountant (Fisher Scientific Company, Ottawa, Canada). Confocal images at mid cell level were taken using a Nikon C1si laser scanning confocal microscope at 60× objective with a 1024 × 1024 pixel size.
Total protein extraction
Cells were scraped from culture dishes and pelleted by centrifugation at 800 g for 3 min. Cell pellets were lysed in radioimmunoprecipitation assay buffer [RIPA buffer; 50 mm Tris HCL, 150 mm NaCl, 1% NP‐40, 0.5% sodium deoxycholate, 0.1% SDS, and complete mini protease inhibitor (Roche)]. Debris was cleared by centrifugation at 14 000 g for 30 min. Total protein was quantified using bicinchoninic acid protein assay (Thermo Scientific; Waltham, MA, USA). Approximately 30 μg of protein was prepared for SDS‐PAGE by heating in Laemmli buffer at 98 °C for 10 min.
Actin and tubulin quantification
The Triton X‐100 soluble‐containing fractions and insoluble‐containing fractions were extracted according to previously described protocols (Bershadsky & Gelfand, 1981; McGrath et al. 2000) with slight modifications. Briefly, ~2.5 × 104 cells cm−2 chondrocytes were seeded on polystyrene in Ham's F12 supplemented with 5% FBS (for bovine cells) or DMEM supplemented with 20% FBS (for human cells). After 3 days of culture, cells were treated with cytoskeletal disrupting agent or vehicle (DMSO) for controls in serum‐free Ham's F12 for 24 h. Cells were washed in PBS, scraped from the culture vessels and centrifuged at 800 g for 3 min. The pellet was resuspended in 250 μL of extraction buffer (0.1% Triton X‐100 in PBS and protease inhibitor cocktail tablet) and incubated for 5 min under slight agitation. The samples were then centrifuged at 15 000 g at 4 °C for 5 min. The soluble supernatant (G‐actin and M‐tubulin) were collected. The Triton‐insoluble pellet (predominantly F‐actin and P‐tubulin) was resuspended in 250 μL of RIPA buffer. Both the soluble and insoluble fractions were mixed with Laemmli buffer (5×) with β‐mercaptoethanol, heated at 98 °C and separated by 12% SDS gel electrophoresis and then analyzed further by Western blotting. Equal volumes of the soluble and insoluble fractions were prepared for Western blotting.
Western blotting
Protein fractions were separated by SDS‐PAGE gel and then semi‐dry transferred onto PVDF membranes (program P3 for 7.5 min; iBLOT transfer system; Life Technologies). Membranes were blocked in 5% skim milk for 30 min then incubated with antibody reactive with COL1 (CL50111AP‐1; Cedarlane, Burlington, ON, Canada), alpha‐tubulin (clone B‐5‐1‐2; Sigma) or pan‐actin (clone C4; Millipore) antibody overnight at 4 °C. Membranes were washed in 0.005% Tween/PBS and then incubated at room temperature for 1 h in anti‐rabbit or anti‐mouse HRP secondary antibody (1 : 20 000; Abcam). Chemiluminescent signals were developed using Pierce ECL Western Blot Substrate (Thermo Scientific). Protein bands were assessed through densitometry using image J software.
RNA extraction and real‐time RT‐PCR
Total RNA was extracted from cells using TRIzol reagent and reverse transcribed to cDNA using Superscript II (Invitrogen) according to the manufacturer's instruction. Relative realtime PCR based on SYBR Green (Invitrogen) was performed using primers specific for 18S rRNA, KI67, CCND1, ACAN, COL1, and COL2 (Table 1). Each reaction contained 20 ng of cDNA, 4.8 μL of SYBR Green master mix, and 0.6 μL each of forward and reverse primers (10 mm). Reactions were performed in duplicate. Examination of the melting curve for non‐specific peaks were performed to ensure specificity of PCR reactions and mRNA levels were determined from Ct values according to the the Pfaffl mathematical model for relative real‐time PCR (Pfaffl, 2001) using 18S rRNA for normalization.
Table 1.
List of primers used in study
| Primer | Forward sequence | Reverse sequence | Size | Temp (C) | Species | Reference ID |
|---|---|---|---|---|---|---|
| 18S | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG | 152 | 60 | Bovine, Human | NR_036642.1 |
| COLI | CGGCTCCTGCTCCTCTTAG | CACACGTCTCGGTCATGGTA | 137 | 60 | Bovine, Human | NM 001034039.1; NM_000088.3 |
| COL2 | GTGTCAGGGCCAGGATGTC | GCAGAGGACAGTCCCAGTGT | 127 | 60 | Bovine, Human | NM 001001135.2; NM_001844.4 |
| ACAN | TGGGACTGAAGTTCTTGGAGA | GCGAGTTGTCATGGTCTGAA | 133 | 60 | Bovine, Human | NM 173981.2; NM_001135.3 |
| CCND1 | CCTCTCCTATCACCGCCTGA | TTTGGGGTCCAAGTTCTGCT | 142 | 60 | Bovine | NM_001046273.2 |
| KI67 | GAGACAGCCCAGGACACTTC | CCTGGTTCTCTGCACCATGT | 114 | 60 | Bovine | XM_002698582.2; XM_005225873.1 |
| CCND1 | CACGGACTACAGGGGAGTTT | CTCTGCTGCTCGCTGCTA | 85 | 60 | Human | NM_053056.2 |
| KI67 | ACAGTACCGCAGATGACTCAA | CGTCCAGCATGTTCTGAGGA | 76 | 60 | Human | NM_002417.4 |
| CFL | CCCCTGAGTGTGCACCCCTTAA | CCCCCAGCTTCTCTGCAAGGGT | 150 | 60 | Bovine | NM_001015655.1 |
Statistics
Each experiment was performed at least three times using cells from different animals on different occasions and composed of three to six replicates per experimental condition. Data obtained from separate experiments were pooled. Cell area and circularity are presented as box and whisker plots demonstrating the lower (25%), middle (50%), and upper (75%) quartile values with the whiskers demonstrating the minimum and maximum values. Outlier data were excluded using a Grubbs’ test for detecting outlying observations with an alpha value of 0.05 via graphpad software.
The remainder of the data were expressed as mean ± SD. For gene and protein data, statistical significance (P < 0.05) was determined by analysis of variance followed by Dunnett's post‐hoc tests using graphpad prism5 software.
Results
Passaging of bovine chondrocytes in 2D culture led to larger, more elongated cells with elevated proliferation and type I collagen gene expression, but reduced type II collagen and aggrecan
To investigate the articular chondrocyte dedifferentiation process, bovine cell phenotype at passages 0 (P0) to 4 (P4) underwent analysis of cell shape, size, and gene expression. P0 chondrocytes on polystyrene were small and round and a large majority of cells refracted light as compared to passaged chondrocytes (Fig. 1A). Quantitative analysis of area showed that the P1 cell area was significantly larger (7.1‐fold; P < 0.001) than that of P0 cells (Fig 1B). Cell area remained significantly greater (P < 0.001) than that of P0 cells for later passages but the P2, P3, and P4 cells were smaller than P1 cells: only 6.1‐, 5.1‐, and 3.6‐fold larger, respectively, than P0 cells. In addition, circularity analysis demonstrated that cells elongated with time (Fig. 1C; P < 0.001). P0 cells had an average circularity value of 0.75, whereas cells from P1 to P4 were significantly less circular.
Figure 1.
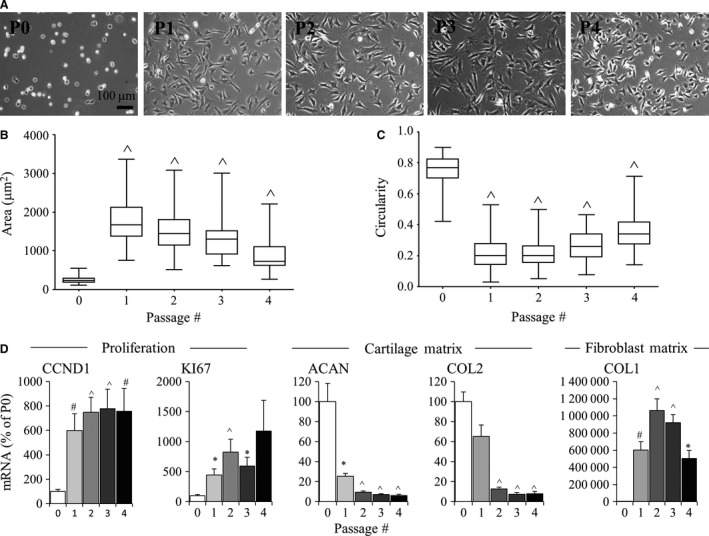
Effect of 2D monolayer passaging on bovine chondrocytes. (A) Light microscopy images of P0 to P4 cells in monolayer culture. Scale bar: 100 μm. (B) Cell area and circularity of P0 to P4 cells in monolayer culture. Box plots represent mean, 25% and 75% quartile, and whiskers represent the 95% confidence intervals. (C) P0 to P4 chondrocytes were evaluated by real‐time relative PCR for proliferation molecules (CCND1, KI67), cartilage matrix (COL2, ACAN) and fibroblast matrix (COL1) genes. PO set at 100. Bars show the mean ± SEM of three to six independent experiments. *P < 0.05; # P < 0.01; ^P < 0.001, vs. P0 chondrocytes.
The gene expression of CCND1 and KI67, which are cell cycle‐associated molecules expressed in proliferating chondrocytes (Muinos‐Lopez et al. 2012; Yu et al. 2013), was determined. CCND1 mRNA levels were significantly elevated in P1, P2, P3 and P4 cells as compared with P0 cells (Fig. 1D; P < 0.01). Similarly, KI67 mRNA levels were significantly elevated (P < 0.05) in P1, P2 and P3 cells as compared with P0 and there was a trend toward elevated KI67 mRNA levels in P4 cells as compared with P0 cells.
To assess the differentiation status of cells, gene expression analysis of specific matrix molecules was carried out. The cartilage matrix molecules ACAN and COL2 decreased with cell passaging; ACAN was significantly downregulated by P1 (P < 0.05) and although COL2 tended to be downregulated by P1, this was not statistically significant (P > 0.05) as compared with P0 chondrocytes. By P2, cells had significantly reduced COL2 levels as compared with P0 chondrocytes. COL1 was significantly upregulated by P1 (P < 0.01) as compared with P0 chondrocytes and peaked at P2 (10 640‐fold greater than P0 chondrocytes; P < 0.001). P2 chondrocytes were used for subsequent experiments, as COL2 and ACAN gene expression was significantly downregulated and COL1 peaked at P2.
P2 chondrocytes had an altered actin but not tubulin polymerization status as compared with P0 chondrocytes
The actin organization and polymerization status in bovine P0 and P2 chondrocytes were determined. Confocal microscopy showed F‐actin was cortically arranged in P0 chondrocytes, whereas P2 cells had developed actin stress fibers (Fig. 2A). Total actin was significantly elevated (P < 0.05) in P2 compared with P0 chondrocytes (Fig. 2B), and the proportion of G‐/F‐actin was significantly lower (P < 0.05) in P2 than in P0 chondrocytes (Fig. 2C).
Figure 2.
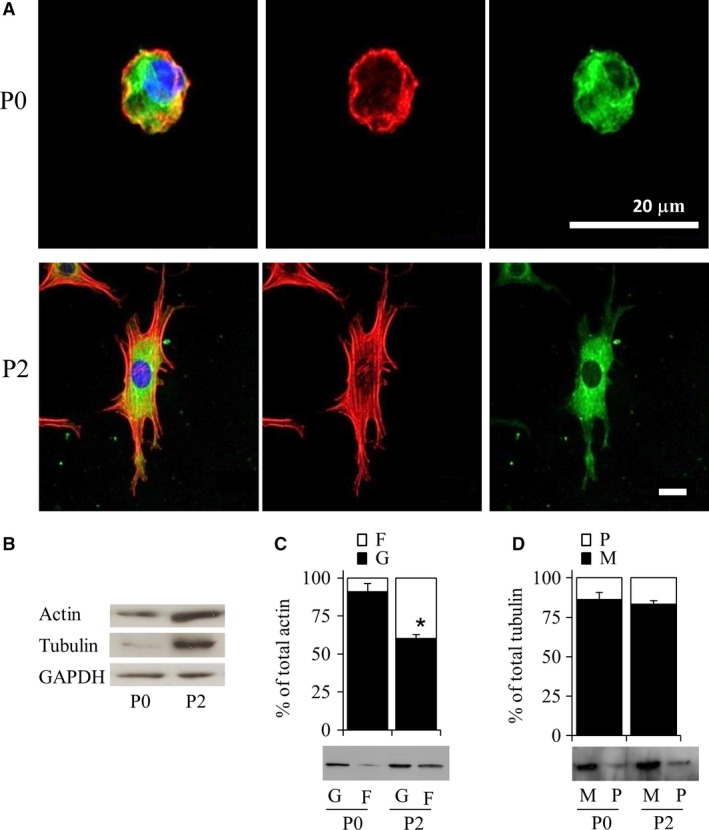
Characterization of actin and tubulin in bovine P0 and P2 chondrocytes after 3 days in monolayer culture. (A) Actin and tubulin organization was examined by confocal microscopy following tubulin (green) antibody or phalloidin (red) staining and DAPI (blue) counterstain. Scale bar: 20 μm. (B) Total actin or tubulin levels as determined by Western blot. (C) G‐/F‐actin and (D) M‐/P‐tubulin levels were quantified by Western blotting. *P < 0.05 vs. P0 chondrocytes.
Tubulin was distributed throughout the cytoplasm in both P0 and P2 cells (Fig. 2A) and no changes in organization were seen. Similar to actin, total tubulin protein levels were also significantly increased (P < 0.05) in P2 cells (Fig. 2B). However, the proportion of M‐/P‐tubulin between P0 and P2 cells was not significantly different (Fig. 2D). In both P0 and P2 cells, M‐tubulin was the predominant form of tubulin.
Exposure of P2 cells to nocodazole results in changes in tubulin and decreased type I collagen mRNA levels
To investigate the influence of tubulin polymerization status on dedifferentiated bovine chondrocytes, cells were exposed to either microtubule depolymerization agent (nocodazole) or microtubule polymerization agent (paclitaxel) (Fig. 3). Nocodazole disrupts microtubules through binding to tubulin and inhibiting polymerization. Paclitaxel both enhances tubulin polymerization and stabilizes microtubules (Grover et al. 1995). Treatment of bovine P2 chondrocytes with nocodazole or paclitaxel resulted in viabilities greater than ~95%, similar to DMSO‐treated controls (data not shown).
Figure 3.
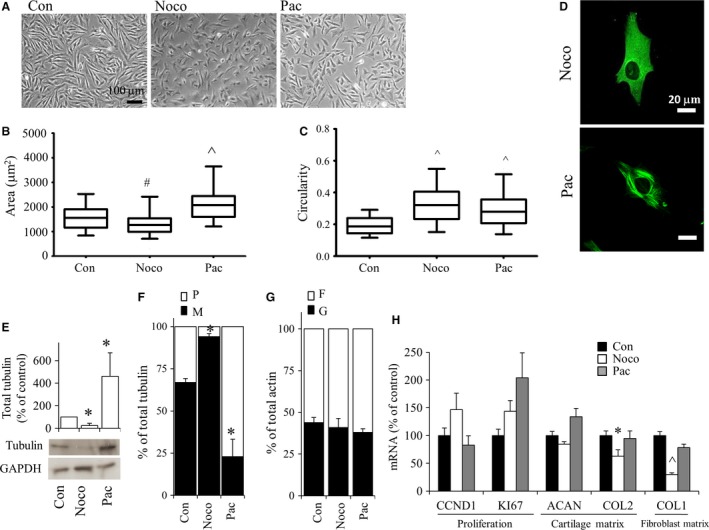
Bovine P2 chondrocytes were cultured in monolayer for 3 days and then exposed to either nocodazole or paclitaxel and evaluated 24 h after treatment. (A) Cell morphology by phase contrast microscopy. Areas (B) as well as circularity (C) of cells were evaluated using image analysis software. Box plots represent mean, 25% and 75% quartile, and whiskers represent the 95% confidence intervals. (D) Tubulin organization was examined by confocal microscopy. Green indicates tubulin. Blue indicates DAPI localized in the nucleus. Scale bar: 20 μm. (E, F) Western blots using anti‐tubulin demonstrated (E) total tubulin levels and (F) M‐ and P‐tubulin fractions separated through Triton solubility. (G) Western blots using anti‐actin demonstrated G‐ and F‐fractions separated through Triton solubility. (H) Real‐time relative PCR evaluation of gene expression normalized to 18S rRNA and expressed as percent of DMSO‐control (vehicle only). Bars show the mean ± SEM of three to six independent experiments. *P < 0.05; # P < 0.01; ^P < 0.001, vs. P2 (control) chondrocytes.
Exposure of P2 cells to nocodazole significantly reduced (P < 0.01) the average cell area (Fig. 3A,B) and increased (P < 0.001) circularity (Fig. 3A,C). Paclitaxel treatment resulted in a significantly larger (P < 0.001) average cell area (Fig. 3B) and increased cell circularity (Fig. 3C).
Nocodazole abolished microtubule organization (Fig. 3D), decreased total tubulin levels (Fig. 3E), and increased the proportion of M‐/P‐tubulin (Fig. 3F). Paclitaxel treatment, on the other hand, led to discrete thick microtubules (Fig. 3D), increased total tubulin (Fig. 3E) and reduced M‐/P‐tubulin (Fig. 3F). Neither nocodazole nor paclitaxel affected G‐/F‐actin in cells (Fig. 3G).
Neither KI67 or CCND1 gene expression were significantly different from controls in cells exposed to either nocodazole or paclitaxel (Fig. 3H). Whereas paclitaxel treatment had no significant effect on COL2 and COL1, nocodazole treatment decreased both COL2 and COL1. ACAN gene expression was not altered following nocodazole or paclitaxel treatment.
Actin depolymerization by cytochalasin D led to small, round cells with altered gene expression
To investigate the effect of modulating actin polymerization status in dedifferentiated cells, bovine P2 chondrocytes were treated with cytochalasin D or phalloidin (Fig. 4). Cytochalasin D binds to the barbed end of actin filaments, inhibiting actin polymerization and promoting a depolymerized state (Carlier et al. 1986), whereas phalloidin stabilizes actin and decreases the rate of actin dissociation from filament ends (Low et al. 1976) and has been shown to decrease G‐/F‐actin in rat kidney cells and human marrow stromal cells (Nickola & Frimmer, 1986; Chen et al. 2015). Treatment of bovine P2 chondrocytes with cytochalasin D or phalloidin resulted in viabilities greater than ~95% and there was no apparent difference compared to vehicle (DMSO)‐treated cells (data not shown).
Figure 4.
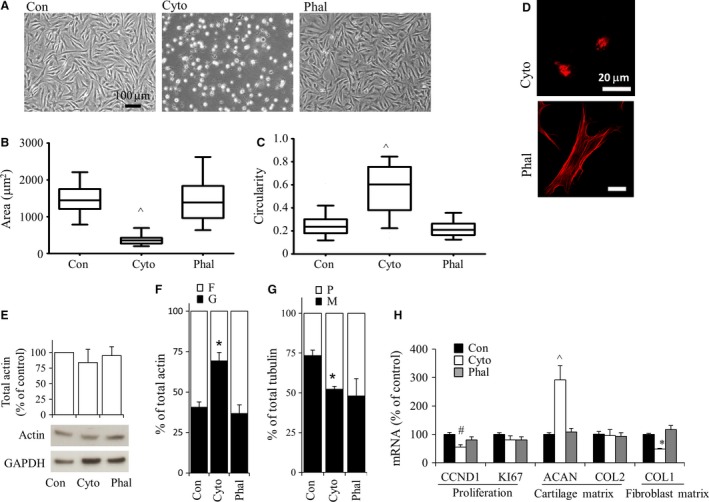
Bovine P2 chondrocytes were cultured in monolayer for 3 days, then exposed to either 10 μm cytochalasin D or 1 μm phalloidin, and evaluated 24 h after treatment. (A) Cell morphology was examined using phase contrast microscopy. Areas (B) as well as circularity (C) of cells were evaluated using imageJ. Box plots represent mean, 25% and 75% quartile, and whiskers represent the 95% confidence intervals. (D) Actin organization was examined by confocal microscopy. Red indicates F‐actin. Blue indicates DAPI. Scale bar: 20 μm. (E) Western demonstrating total actin levels using anti‐pan actin antibody. (F) G‐ and F‐actin fractions which were separated through Triton solubility followed by centrifugation (representative Western blots shown in Supporting Information Fig. S3). (G) Quantification of M‐ and P‐tubulin fractions separated through Triton solubility followed by Western blots. (H) Real‐time relative PCR evaluation of gene expression normalized to 18S rRNA and expressed as percent of control (DMSO vehicle only). Bars show the mean ± SEM of three to six independent experiments. *P < 0.05; # P < 0.01; ^P < 0.001, vs. vehicle‐treated P2 (control) chondrocytes.
Cytochalasin D treatment of P2 chondrocytes resulted in cells that were significantly smaller (P < 0.001; Fig. 4A,B) and rounder (P < 0.001), and refracted light more than non‐treated cells (Fig. 4A,C). The magnitude of shape change was greater than that attained by nocodazole treatment (Fig. 3A–C). Cytochalasin D treatment resulted in loss of actin stress fibers. F‐actin was still visible as punctate staining throughout the cytoplasm (Fig. 4D). Exposure of cells to cytochalasin D did not affect total actin levels (Fig. 4E) but resulted in an increased proportion of G‐/F‐actin (Fig. 4F) as well as a decreased proportion of M‐/P‐tubulin (Fig. 4G). Cells exposed to phalloidin were not statistically different (P > 0.05) in terms of average cell area or circularity as compared with control cells. F‐actin was present as stress fibers in cells exposed to phalloidin (Fig. 4D), and total actin levels (Fig. 4E) as well as G‐/F‐actin (Fig. 4F) were not significantly altered. Whereas phalloidin appeared to decrease M‐/P‐tubulin (Fig. 4G) this change was not statistically significant (P > 0.05).
As previous studies revealed that cytoskeletal integrity is required for the mitogenic capacity of cells other than chondrocytes (Bohmer et al. 1996), proliferation genes were examined in P2 chondrocytes. CCND1 mRNA levels were significantly reduced in cells treated with cytochalasin D but not with phalloidin (Fig. 4H). KI67 mRNA levels were not significantly changed with either cytochalasin D or phalloidin treatment.
Exposure of P2 cells to phalloidin treatment did not significantly affect gene expression. However, cytochalasin D treatment resulted in increased ACAN but unchanged COL2 gene expression. Bovine P2 COL1 mRNA levels were repressed by cytochalasin D treatment.
Cofilin knockdown increases actin polymerization status and COL1 gene expression in bovine passaged chondrocytes
Although treatment of P2 chondrocytes with phalloidin resulted in a trend toward decreased G‐/F‐actin, this was not statistically significant (P > 0.05). Thus we next investigated cofilin (CFL), an actin‐binding molecule that promotes F‐actin depolymerization in mammalian non‐muscle cells (Hotulainen et al. 2005) and was elevated in P2 chondrocytes (Fig. 5A). To investigate whether CFL could decrease G/F‐actin, we knocked down CFL.
Figure 5.
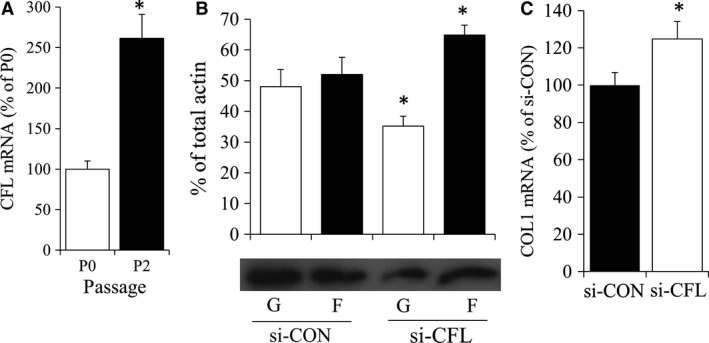
(A) P0 and P2 chondrocytes were evaluated by real‐time relative PCR for actin depolymerization molecule cofilin (CFL). Bars show the mean ± SEM of three to six independent experiments. *P < 0.05 vs. P0 chondrocytes. Bovine P2 chondrocytes were nucleofected with either non‐targeting (si‐CON) or cfl (si‐CFL) siRNA. (B) Bar graph and representative Western blot of G‐ and F actin fractions 2 days following nucleofection. (C) COL1 gene expression in nucleofected P2 chondrocytes. *P < 0.05 vs. si‐con chondrocytes.
Nucleofection was used to knockdown CFL, which we previously have shown to result in a 98% transfection efficiency of siRNA into P2 chondrocytes (Parreno et al. 2016). As we have found that nucleofection alone, even in the absence of siRNA, can affect gene expression, our control P2 cells were also nucleofected with scrambled, non‐targeting siRNA. siRNA‐mediated knockdown of CFL decreased G‐/F‐actin in P2 chondrocytes (Fig. 5B). This resulted in increased COL1 mRNA levels (Fig. 5C). CFL knockdown did not significantly affect the expression of the ACAN or COL2 (data not shown).
Effect of cytoskeletal depolymerization on human chondrocyte gene expression
To examine whether cytoskeletal depolymerization affected human passaged chondrocyte gene expression in a similar manner to bovine P2 chondrocytes, human P2 chondrocytes were treated with cytochalasin D or nocodazole. Similar to bovine chondrocytes, treatment of human chondrocytes with cytochalasin D or nocodazole resulted in smaller, rounder cells which refracted light (Fig. 6A). In human P2 chondrocytes, COL1 was also significantly downregulated (P < 0.01) by cytochalasin D (Fig. 6B). No other genes tested were significantly downregulated by cytochalasin D in human P2 chondrocytes. P2 human chondrocyte gene expression was unaltered by nocodazole treatment.
Figure 6.

Effect of cytochalasin D or nocodazole on human P2 chondrocytes in 2D monolayer culture. (A) Phase contrast images. Scale bar: 100 um. (B) Real‐time relative PCR evaluation of gene expression normalized to 18S rRNA and expressed as percent compared with control cells (DMSO exposed). ^P < 0.00, vs. control chondrocytes.
Effect of cytoskeletal depolymerization on type I collagen protein levels
Modulation of actin polymerization in P2 chondrocytes in monolayer culture correlated with COL1 gene expression. To confirm that cytochalasin D resulted in changes in COL1 protein levels, COL1 levels were examined by Western blot analysis. Cytochalasin D caused significant reductions in COL1A1 (5.5‐fold; P < 0.01) and COL1A2 (2.9‐fold; P < 0.05) protein levels in bovine P2 chondrocytes (Fig. 7). Although COL1 gene expression was also repressed in bovine P2 chondrocytes (Fig. 3H) by nocodazole, there were no significant differences in either the COL1A1 or COL1A2 protein.
Figure 7.
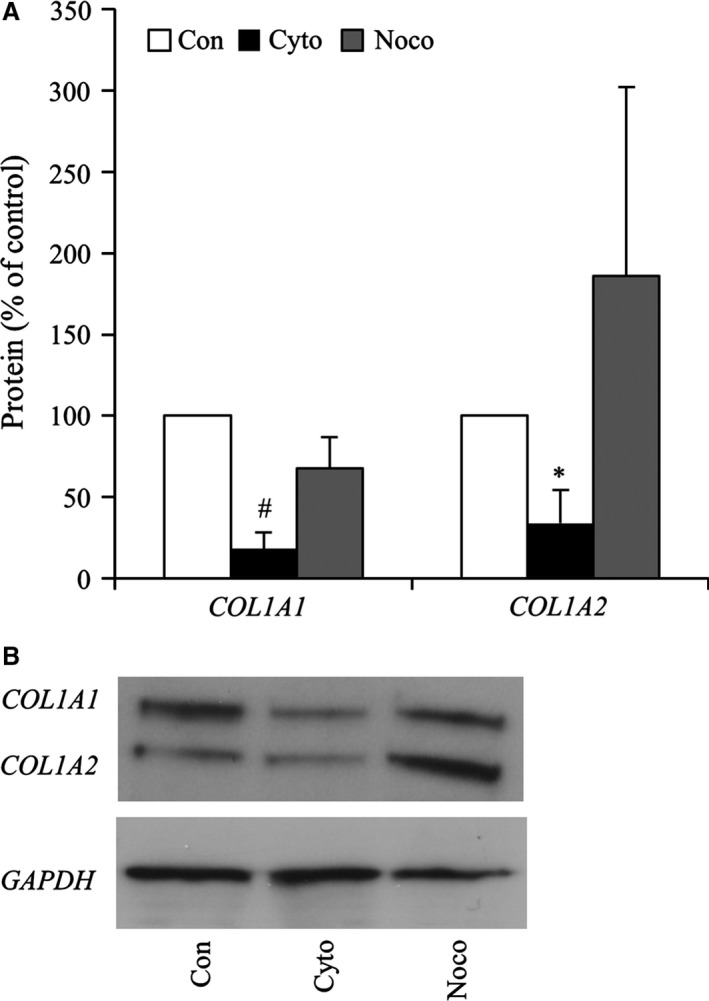
(A) Evaluation of COL1A1 and COL1A2 protein expression densitometry normalized to GAPDH and expressed as percent compared with control (DMSO exposed). *P < 0.05; # P < 0.01, vs. control chondrocytes. (B) Representative Western blot of COL1A1, COL1A2, GAPDH proteins following treatment of bovine P2 cells with cytochalasin D or nocodazole.
3D culture of P2 chondrocytes modulates actin polymerization, which correlates with decreased COL1 expression
It has been shown previously that 3D culture supports passaged chondrocyte redifferentiation of both bovine and human passaged chondrocytes (Caron et al. 2012; Ahmed et al. 2014). To examine the effect of 3D culture on cells, bovine P2 chondrocytes were cultured at high density on a porous ceramic substrate (Fig. 8A). The cells had greater G‐/F‐actin than chondrocytes in 2D culture (Fig. 8B). Culture of P2 cells in 3D resulted in repressed expression of CCND1, KI67, and COL1. ACAN and COL2 expression were not significantly different in 3D than in 2D culture at these early time points. We next investigated whether treatment of P2 chondrocytes in 3D culture with cytochalasin D would further enhance these changes in gene expression. Cytochalasin D resulted in a diffuse distribution of actin, confirming it could change actin in these cells (Fig. 8D). This was associated with increased ACAN and further decreased COL1 gene expression (Fig. 8E). COL2 was not significantly modulated by cytochalasin D treatment of these cells. Treatment of P2 chondrocytes with phalloidin, nocodazole or paclitaxel did not have any additional effects on ACAN, COL, or COL1 gene expression in 3D culture (data not shown).
Figure 8.
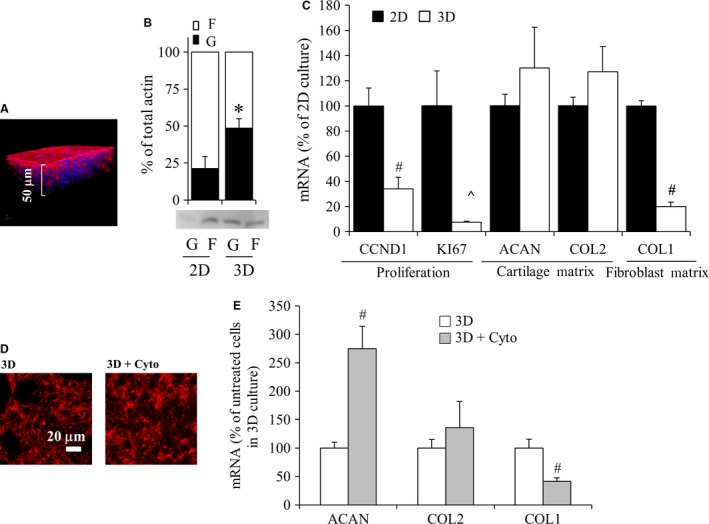
Effect of 3D culture on P2 cells. (A) Z‐stacked confocal image demonstrating that high‐density seeding of P2 chondrocytes resulted in a 3D culture after 2 days. Red indicates F‐actin; blue indicates DAPI‐stained nuclei. Scale bar: 50 μm. (B) Globular and filamentous actin from bovine P2 chondrocytes cultured either in 2D monolayer or 3D for 2 h. (C) Real‐time relative PCR evaluation of P2 chondrocyte gene expression when cultured in 3D compared with 2D. (D) Characterization of actin in P2 cells in 3D on calcium polyphosphate substrate following exposure to cytochalasin D as compared with untreated (3D on CPP) controls. Actin organization was examined by confocal microscopy. Red indicates F‐actin. Scale bar: 20 μm. (E) Real‐time relative PCR evaluation of gene expression in P2 chondrocytes in 3D culture treated with cytochalasin D as percent of untreated P2 chondrocytes in 3D culture. Gene results were normalized to 18S rRNA and are expressed as the percent of controls; either P2 chondrocytes in 2D (C) or untreated P2 chondrocyte in 3D (E). Control values were set to 100%. Bars show the mean ± SEM of three to six independent experiments. *P < 0.05; # P < 0.01; ^P < 0.001, vs. control chondrocytes.
Discussion
This study showed that dedifferentiation is a progressive process which appears to be complete after two passages and that actin polymerization status may be involved in regulating this process. It was observed that actin polymerization status changed, as there was reduced G‐/F‐actin in passaged compared to unpassaged cells. A regulatory role for actin polymerization status during dedifferentiation was supported by the finding that increasing the G‐/F‐actin ratio by cytochalasin D treatment of P2 cells in 2D culture or by culturing P2 cells in 3D culture promoted the chondrocyte phenotype by decreasing COL1 expression. Additionally, knockdown of CFL, to decrease G‐/F‐actin, led to increased COL1 expression, further supporting the involvement of actin polymerization status. Interestingly tubulin polymerization status did not change during passaging, suggesting that it may not be involved directly in regulating dedifferentiation. However, pharmacological modulation of tubulin polymerization was associated with selected gene changes (Fig. 3), raising the possibility that it is involved in other ways.
The present study is in keeping with previous findings from Lefebvre et al. (1990) which demonstrated that dedifferentiation is a progressive process. They also showed that fibroblast matrix and proliferation genes were modulated by P1, whereas COL2 was not significantly modulated until P2. In this study, as compared with P2 chondrocytes, cells from later passages (P3 and P4) chondrocytes were smaller, rounder, and had further decreased levels of ACAN and COL2. This supports previous findings that shape does not always correlate with the differentiated chondrogenic phenotype (Benya et al. 1988; Watt & Dudhia, 1988; Mallein‐Gerin et al. 1991). Others have also shown that extensive passaging results in further changes to chondrocytes, including higher rates of senescence (Lin et al. 2007 and apoptosis, as well as a reduced ability to redifferentiate (Kang et al. 2007). For this study, P2 chondrocytes were utilized to investigate the role of actin and tubulin polymerization in gene regulation based on the findings that P2 chondrocytes were fully dedifferentiated [in terms of significantly downregulated (P < 0.05) COL2 and ACAN as well as peaked COL1 gene expression and that P2 chondrocytes are still capable of redifferentiation (Ahmed et al. 2014).
Tubulin polymerization status did not appear to be involved in regulating chondrocyte phenotype, as demonstrated by two observations: first, tubulin polymerization status was not different between P0 and P2 chondrocytes (Fig. 2D); secondly, tubulin modulation through nocodazole or paclitaxel treatment did not specifically promote redifferentiation (decreased COL2 with nocodazole treatment and no gene differences with paclitaxel; Fig. 3H), which was not unexpected, as there were no changes in actin polymerization status. Although tubulin polymerization status did not appear to be involved in promoting the chondrocyte phenotype, there were several changes such as a reduction in P2 cell area and increased circularity, as well as decreased COL1 gene expression with nocodazole treatment in bovine cells (Fig. 3). This further supports the notion that cell shape/size is not the critical regulator of chondrogenic gene expression, in keeping with observations by Benya et al. (1988). Interestingly, nocodazole treatment decreased bovine COL2 gene expression. The effect nocodazole had on both COL1 and COL2 may be a result of nocodazole activation of nuclear factor (NF)‐kB (Rosette & Karin, 1995), a transcription factor that has been shown to regulate COL1 expression in other cell types (Rippe et al. 1999) as well as COL2 in chondrocytes (Fan et al. 2006).
The effects of actin depolymerization on the chondrogenic phenotype has been characterized previously by several groups (Benya et al. 1988; Watt & Dudhia, 1988; Mallein‐Gerin et al. 1991; Woods et al. 2005; Woods & Beier, 2006). The work by Woods et al. predominantly dealt with the effect of actin on the differentiation of cartilage anlagen cells (including growth plate) that eventually form bone. The present study is different as we investigated the role of actin polymerization status on dedifferentiated articular chondrocytes obtained from mature articular cartilage. Our investigations support the work by three other groups who showed that actin disassembly favorably affected chondrocyte phenotype (Benya et al. 1988; Watt & Dudhia, 1988; Mallein‐Gerin et al. 1991), even though we utilized passaged chondrocytes. We also quantified actin polymerization status under different conditions and utilized 3D cultures (Fig. 8) and CFL knockdown (Fig. 5) to support the correlation of actin polymerization status with COL1 expression levels.
Intriguingly CFL, an actin‐severing molecule, is upregulated in passaged chondrocytes which have an increased proportion of F‐actin (decreased G‐/F‐actin). The role of CFL in passaged chondrocytes has not been clearly defined; however, CFL‐severing in other cell types results in the creation of free barbed ends on actin, making F‐actin susceptible to further elongation and/or nucleation of new filaments in conjunction with nucleating molecule actin‐related protein 2/3 (McGough et al. 1997; DesMarais et al. 2004). Thus increased CFL in P2 chondrocytes could contribute to the decreased G/F‐actin that was observed in these cells. Additional studies are required to examine this as well as the contribution of other actin‐binding proteins in primary chondrocytes which promote high G/F‐actin.
In keeping with our previous study (Parreno et al. 2014), the current findings demonstrated that increasing G‐/F‐actin ratio favors the chondrocyte phenotype. This was demonstrated by treatment of cells with cytochalasin D or by 3D culture of cells. Although cytochalasin D and latrunculin B (which we utilized previously; Parreno et al. 2014) both led to actin depolymerization, they have been shown to do this through different mechanisms. Cytochalasin binds to the barbed ends of actin filaments (Brown & Spudich, 1981), whereas latrunculin binds in a 1 : 1 ratio with G‐actin to prevent polymerization (Coue et al. 1987). This is intriguing, as the differential binding sites of these drugs to actin lead to opposite effects on myocardin‐related transcription factor‐a (MRTF) localization, a transcription factor we have shown previously to regulate COL1 gene expression in P2 chondrocytes (Parreno et al. 2014). Latrunculin is an MRTF deactivator as it decreases nuclear localization in passaged chondrocytes. Conversely, cytochalasin D is an activator of MRTF and increases its nuclear localization. Intriguingly, both latrunculin and cytochalasin D treatment led to decreased COL1 gene levels, suggesting that other actin‐regulated pathways independent of MRTF are involved in modulating COL1 expression. In addition to regulation of COL1, cytochalasin D decreased CCND1. CCND1 expression is of interest as not only is it responsible for regulating proliferation in chondrocytes (Beier et al. 2001), but it also interferes with the ability of SOX9 to bind to promoter regions of target genes (Hwang et al. 2007). ACAN has been shown to be regulated by SOX9, thus CCND1 is a potential way that cytochalasin D may regulate ACAN expression levels.
Previous studies have demonstrated 3D culture to contribute to redifferentiation of passaged chondrocytes (Caron et al. 2012; Schuh et al. 2012; Ahmed et al. 2014) and here we show that 3D culture of passaged chondrocytes resulted in increased G‐/F‐actin. While actin depolymerization by 3D culture had similar effects on gene expression as cytochalasin D treatment of cells in 2D culture, 3D culture had additional effects, such as decreasing KI67 expression. This supports the finding that 3D culture suppresses proliferation, as demonstrated in fibroblasts (Mio et al. 1996), and also demonstrates that the regulation of KI67 gene expression may be independent of actin, as cytochalasin D treatment of cells did not affect KI67 gene expression. This signaling mechanism requires further elucidation. Furthermore, the extent of actin depolymerization by 3D culture was insufficient on its own to induce chondrogenic gene expression. These results are in contrast to previous studies by us and others which have shown increased levels of either ACAN or COL2 in 3D culture systems (Murphy & Sambanis, 2001; Tallheden et al. 2004; Martinez et al. 2008; Bernstein et al. 2009; Yu et al. 2010; Ahmed et al. 2014; Rottmar et al. 2014). There are other factors implicit in such systems such as the extent of passaging/dedifferentiation in culture (Bernstein et al. 2009), substrates/scaffolds used (Rottmar et al. 2014), oxygen tension (Murphy & Sambanis, 2001; Martinez et al. 2008), and media supplements (Yu et al. 2010; Ahmed et al. 2014) which have been shown to affect the chondrogenic differentiation state of cells and may explain the discrepant results. However, we did find that further actin depolymerization by treatment of cells in 3D culture with cytochalasin D enhanced ACAN expression. This contradicts findings from Woods et al. where treatment of cells in 3D with cytochalasin D reduced ACAN levels (Woods & Beier, 2006). These differences may be attributed to differences in the cell type utilized, as P2 chondrocytes were used in our studies in contrast to the embryonic day 11.5 mouse limb bud cells used by Woods & Beier.
Regulation of chondrocyte phenotype is complex and is both dependent on and independent of actin. Although actin polymerization status may actively regulate certain genes (for instance COL1, CCND1, and ACAN) in bovine P2 chondrocytes, it did not regulate others (KI67, COL2). Other mechanism(s) in addition to actin polymerization status are required to restore chondrocyte phenotype completely. Cortical actin has been shown to be essential for redifferentiation (Park et al. 2008) and the inability of these cells to attain a cortical distribution of actin even in 3D culture may explain the incomplete redifferentiation. In a separate study, we have shown that P2 chondrocytes grown in serum‐free DMEM supplemented with insulin and dexamethasone in 3D culture recovers COL2 expression over time (Ahmed et al. 2014). Insulin has been shown to promote cortical actin formation and it may therefore provide the necessary signal(s) to recover cortical actin and complete redifferentiation. Alternatively, insulin‐induced COL2 expression may be independent of actin and may be due to SOX9 activation (Ahmed et al. 2014). Further study is required to determine what signaling mechanisms, in addition to actin polymerization, and/or organization are required for complete redifferentiation.
In this study we examined whether cytoskeletal perturbation could also modulate gene expression in human passaged chondrocytes from osteoarthritic cartilage, a cell source utilized clinically for cartilage repair (Brittberg et al. 1994). There was a limited effect of either actin or tubulin modulation on human chondrocyte gene expression which contrasts the findings in bovine chondrocytes (Fig. 6). This may be due to the intrinsic differences between species, age and/or health of the cells. The human chondrocytes were from older adults with osteoarthritis as compared with the bovine chondrocytes, which were from young, healthy calves. Donor age may explain the lack of response in human chondrocytes, as it has been shown, for example, that the ability to synthesize proteoglycans in response to chondrogenic stimuli is compromised in chondrocytes from older individuals (Martin et al. 1997). In addition, although human chondrocytes were isolated from what appeared to be non‐damaged regions of human articular cartilage, donors had osteoarthritis (Mankin grades ≥ 4) and it is unclear whether the chondrocytes were affected by the disease state. The use of bovine cells could have affected the response to cytoskeletal agents and is a potential limitation of this study. Nevertheless, both bovine and human passaged chondrocytes in this study showed decreased levels of COL1 expression following actin depolymerization with cytochalasin D. A similar finding has been shown following treatment of human passaged chondrocytes with bistratene A, which disrupts actin, also decreased COL1 expression (Gargiulo et al. 2002).
A major issue faced in cell‐based therapies for cartilage repair is the production of fibrocartilage by cells, which is characterized by increased COL1 production (Roberts et al. 2009). In this study COL1 expression was most sensitive to changes in G‐/F‐actin. This is supported by the observation that COL1 was downregulated by actin depolymerization (cytochalasin D treatment and/or 3D culture) and upregulated by actin polymerization through CFL knockdown. Thus understanding the molecular links between the actin cytoskeleton and gene expression may identify a methodology to more effectively redifferentiate chondrocytes to allow for a useable source of chondrocytes that produce hyaline rather than fibrocartilage matrix after cell number expansion in monolayer culture. This would facilitate translation of cartilage repair methods into clinical use.
Author contributions
Study conception and design – J.P., M.N., R.K. Acquisition of data – J.P., M.N., K.A., A.J., P.W. Analysis and interpretation of data – J.P., M.N., K.A., A.J., P.W., R.K. Drafting of manuscript – J.P., R.K. Critical revision – J.P., M.N., K.A., A.J., P.W., R.K. Approval of submission – J.P., M.N., K.A., A.J., P.W., R.K.
Conflict of interest
None.
Acknowledgements
We thank Dr. Pilliar for providing the CPP discs. J.P. was supported by a University of Toronto Fellowship, a Canadian Arthritis Network – Ontario Graduate Studentship in Science and Technology and an NSERC – Alexander Graham Bell Canada Graduate Scholarship. This work was supported by Canadian Institutes of Health Research (CIHR) grant MOP12611 and NSERC Discovery Grant RGPIN‐2016‐06088.
References
- Abbott J, Holtzer H (1968) The loss of phenotypic traits by differentiated cells, V. The effect of 5‐bromodeoxyuridine on cloned chondrocytes. Proc Natl Acad Sci U S A 59, 1144–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed N, Iu J, Brown CE, et al. (2014) Serum‐ and growth‐factor‐free three‐dimensional culture system supports cartilage tissue formation by promoting collagen synthesis via Sox9‐Col2a1 interaction. Tissue Eng Part A 20, 2224–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier F, Ali Z, Mok D, et al. (2001) TGFbeta and PTHrP control chondrocyte proliferation by activating cyclin D1 expression. Mol Biol Cell 12, 3852–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benya PD, Padilla SR, Nimni ME (1978) Independent regulation of collagen types by chondrocytes during the loss of differentiated function in culture. Cell 15, 1313–1321. [DOI] [PubMed] [Google Scholar]
- Benya PD, Brown PD, Padilla SR (1988) Microfilament modification by dihydrocytochalasin B causes retinoic acid‐modulated chondrocytes to reexpress the differentiated collagen phenotype without a change in shape. J Cell Biol 106, 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein P, Dong M, Corbeil D, et al. (2009) Pellet culture elicits superior chondrogenic redifferentiation than alginate‐based systems. Biotechnol Prog 25, 1146–1152. [DOI] [PubMed] [Google Scholar]
- Bershadsky AD, Gelfand VI (1981) ATP‐dependent regulation of cytoplasmic microtubule disassembly. Proc Natl Acad Sci U S A 78, 3610–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blain EJ (2009) Involvement of the cytoskeletal elements in articular cartilage homeostasis and pathology. Int J Exp Pathol 90, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohmer RM, Scharf E, Assoian RK (1996) Cytoskeletal integrity is required throughout the mitogen stimulation phase of the cell cycle and mediates the anchorage‐dependent expression of cyclin D1. Mol Biol Cell 7, 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittberg M, Lindahl A, Nilsson A, et al. (1994) Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. N Engl J Med 331, 889–895. [DOI] [PubMed] [Google Scholar]
- Brown PD, Benya PD (1988) Alterations in chondrocyte cytoskeletal architecture during phenotypic modulation by retinoic acid and dihydrocytochalasin B‐induced reexpression. J Cell Biol 106, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SS, Spudich JA (1981) Mechanism of action of cytochalasin: evidence that it binds to actin filament ends. J Cell Biol 88, 487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B, Peng R, Li Z, et al. (2014) Effects of spreading areas and aspect ratios of single cells on dedifferentiation of chondrocytes. Biomaterials 35, 6871–6881. [DOI] [PubMed] [Google Scholar]
- Carlier MF, Criquet P, Pantaloni D, et al. (1986) Interaction of cytochalasin D with actin filaments in the presence of ADP and ATP. J Biol Chem 261, 2041–2050. [PubMed] [Google Scholar]
- Caron MM, Emans PJ, Coolsen MM, et al. (2012) Redifferentiation of dedifferentiated human articular chondrocytes: comparison of 2D and 3D cultures. Osteoarthritis Cartilage 20, 1170–1178. [DOI] [PubMed] [Google Scholar]
- Chen L, Shi K, Frary CE, et al. (2015) Inhibiting actin depolymerization enhances osteoblast differentiation and bone formation in human stromal stem cells. Stem Cell Res 15, 281–289. [DOI] [PubMed] [Google Scholar]
- Coue M, Brenner SL, Spector I, et al. (1987) Inhibition of actin polymerization by latrunculin A. FEBS Lett 213, 316–318. [DOI] [PubMed] [Google Scholar]
- DesMarais V, Macaluso F, Condeelis J, et al. (2004) Synergistic interaction between the Arp2/3 complex and cofilin drives stimulated lamellipod extension. J Cell Sci 117, 3499–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Yang H, Bau B, et al. (2006) Role of mitogen‐activated protein kinases and NFκB on IL‐1β‐induced effects on collagen type II, MMP‐1 and 13 mRNA expression in normal articular human chondrocytes. Rheumatol Int 26, 900–903. [DOI] [PubMed] [Google Scholar]
- Fardin MA, Rossier OM, Rangamani P, et al. (2010) Cell spreading as a hydrodynamic process. Soft Matter 6, 4788–4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fringer J, Grinnell F (2001) Fibroblast quiescence in floating or released collagen matrices: contribution of the ERK signaling pathway and actin cytoskeletal organization. J Biol Chem 276, 31047–31052. [DOI] [PubMed] [Google Scholar]
- Gargiulo BJ, Cragg P, Richardson JB, et al. (2002) Phenotypic modulation of human articular chondrocytes by bistratene A. Eur Cell Mater 3, 9–18. [DOI] [PubMed] [Google Scholar]
- Grover S, Rimoldi JM, Molinero AA, et al. (1995) Differential effects of paclitaxel (Taxol) analogs modified at positions C‐2, C‐7, and C‐3′ on tubulin polymerization and polymer stabilization: identification of a hyperactive paclitaxel derivative. Biochemistry 34, 3927–3934. [DOI] [PubMed] [Google Scholar]
- Hernandez F, Garcia‐Garcia E, Avila J (2013) Microtubule depolymerization and tau phosphorylation. J Alzheimer's Dis 37, 507–513. [DOI] [PubMed] [Google Scholar]
- Hotulainen P, Paunola E, Vartiainen MK, et al. (2005) Actin‐depolymerizing factor and cofilin‐1 play overlapping roles in promoting rapid F‐actin depolymerization in mammalian nonmuscle cells. Mol Biol Cell 16, 649–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui A, Min WX, Tang J, et al. (1998) Inhibition of activator protein 1 activity by paclitaxel suppresses interleukin‐1‐induced collagenase and stromelysin expression by bovine chondrocytes. Arthritis Rheum 41, 869–876. [DOI] [PubMed] [Google Scholar]
- Hwang SG, Song SM, Kim JR, et al. (2007) Regulation of type II collagen expression by cyclin‐dependent kinase 6, cyclin D1, and p21 in articular chondrocytes. IUBMB Life 59, 90–98. [DOI] [PubMed] [Google Scholar]
- Jortikka MO, Parkkinen JJ, Inkinen RI, et al. (2000) The role of microtubules in the regulation of proteoglycan synthesis in chondrocytes under hydrostatic pressure. Arch Biochem Biophys 374, 172–180. [DOI] [PubMed] [Google Scholar]
- Kang SW, Yoo SP, Kim BS (2007) Effect of chondrocyte passage number on histological aspects of tissue‐engineered cartilage. Bio‐Med Mater Eng 17, 269–276. [PubMed] [Google Scholar]
- Kuwahara K, Barrientos T, Pipes GC, et al. (2005) Muscle‐specific signaling mechanism that links actin dynamics to serum response factor. Mol Cell Biol 25, 3173–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre V, Peeters‐Joris C, Vaes G (1990) Production of collagens, collagenase and collagenase inhibitor during the dedifferentiation of articular chondrocytes by serial subcultures. Biochim Biophys Acta 1051, 266–275. [DOI] [PubMed] [Google Scholar]
- Lefebvre V, Huang W, Harley VR, et al. (1997) SOX9 is a potent activator of the chondrocyte‐specific enhancer of the pro alpha1(II) collagen gene. Mol Cell Biol 17, 2336–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Chen X, Deng L (2007) Observation of replicative senescence of rat chondrocytes in vitro. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi 21, 1228–1232. [PubMed] [Google Scholar]
- Liu YR, Huang SY, Chen JY, et al. (2013) Microtubule depolymerization activates the Epstein‐Barr virus lytic cycle through protein kinase C pathways in nasopharyngeal carcinoma cells. J Gen Virol 94, 2750–2758. [DOI] [PubMed] [Google Scholar]
- Low I, Dancker P, Wieland T (1976) Stabilization of actin polymer structure by phalloidin: ATPase activity of actin induced by phalloidin at low pH. FEBS Lett 65, 358–360. [DOI] [PubMed] [Google Scholar]
- Mallein‐Gerin F, Garrone R, van der Rest M (1991) Proteoglycan and collagen synthesis are correlated with actin organization in dedifferentiating chondrocytes. Eur J Cell Biol 56, 364–373. [PubMed] [Google Scholar]
- Martin JA, Ellerbroek SM, Buckwalter JA (1997) Age‐related decline in chondrocyte response to insulin‐like growth factor‐I: the role of growth factor binding proteins. J Orthop Res 15, 491–498. [DOI] [PubMed] [Google Scholar]
- Martinez I, Elvenes J, Olsen R, et al. (2008) Redifferentiation of in vitro expanded adult articular chondrocytes by combining the hanging‐drop cultivation method with hypoxic environment. Cell Transplant 17, 987–996. [DOI] [PubMed] [Google Scholar]
- McGough A, Pope B, Chiu W, et al. (1997) Cofilin changes the twist of F‐actin: implications for actin filament dynamics and cellular function. J Cell Biol 138, 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JL, Osborn EA, Tardy YS, et al. (2000) Regulation of the actin cycle in vivo by actin filament severing. Proc Natl Acad Sci U S A 97, 6532–6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mio T, Adachi Y, Romberger DJ, et al. (1996) Regulation of fibroblast proliferation in three‐dimensional collagen gel matrix. . In vitro Cell Dev Biol ‐ Animal 32, 427–433. [DOI] [PubMed] [Google Scholar]
- Muinos‐Lopez E, Rendal‐Vazquez ME, Hermida‐Gomez T, et al. (2012) Cryopreservation effect on proliferative and chondrogenic potential of human chondrocytes isolated from superficial and deep cartilage. Open Orthop J 6, 150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy CL, Sambanis A (2001) Effect of oxygen tension and alginate encapsulation on restoration of the differentiated phenotype of passaged chondrocytes. Tissue Eng 7, 791–803. [DOI] [PubMed] [Google Scholar]
- Newman P, Watt FM (1988) Influence of cytochalasin D‐induced changes in cell shape on proteoglycan synthesis by cultured articular chondrocytes. Exp Cell Res 178, 199–210. [DOI] [PubMed] [Google Scholar]
- Nickola I, Frimmer M (1986) Effects of phalloidin and cytochalasin B on cytoskeletal structures in cultured rat hepatocytes. Cell Tissue Res 245, 635–641. [DOI] [PubMed] [Google Scholar]
- Park EH, Kang SS, Lee YS, et al. (2008) Integrity of the cortical actin ring is required for activation of the PI3K/Akt and p38 MAPK signaling pathways in redifferentiation of chondrocytes on chitosan. Cell Biol Int 32, 1272–1278. [DOI] [PubMed] [Google Scholar]
- Parreno J, Raju S, Niaki MN, et al. (2014) Expression of type I collagen and tenascin C is regulated by actin polymerization through MRTF in dedifferentiated chondrocytes. FEBS Lett 588, 3677–3684. [DOI] [PubMed] [Google Scholar]
- Parreno J, Delve E, Andrejevic K, et al. (2016) Efficient, low‐cost nucleofection of passaged chondrocytes. Cartilage 7, 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW (2001) A new mathematical model for relative quantification in real‐time RT‐PCR. Nucleic Acids Res 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rippe RA, Schrum LW, Stefanovic B, et al. (1999) NF‐kappaB inhibits expression of the alpha1(I) collagen gene. DNA Cell Biol 18, 751–761. [DOI] [PubMed] [Google Scholar]
- Roberts S, Menage J, Sandell LJ, et al. (2009) Immunohistochemical study of collagen types I and II and procollagen IIA in human cartilage repair tissue following autologous chondrocyte implantation. Knee 16, 398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosette C, Karin M (1995) Cytoskeletal control of gene expression: depolymerization of microtubules activates NF‐kappa B. J Cell Biol 128, 1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottmar M, Mhanna R, Guimond‐Lischer S, et al. (2014) Interference with the contractile machinery of the fibroblastic chondrocyte cytoskeleton induces re‐expression of the cartilage phenotype through involvement of PI3K, PKC and MAPKs. Exp Cell Res 320, 175–187. [DOI] [PubMed] [Google Scholar]
- Rozycki M, Bialik JF, Speight P, et al. (2016) Myocardin‐related transcription factor regulates Nox4 protein expression: linking cytoskeletal organization to redox state. J Biol Chem 291, 227–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salu KJ, Bosmans JM, Huang Y, et al. (2006) Effects of cytochalasin D‐eluting stents on intimal hyperplasia in a porcine coronary artery model. Cardiovasc Res 69, 536–544. [DOI] [PubMed] [Google Scholar]
- Schuh E, Hofmann S, Stok K, et al. (2012) Chondrocyte redifferentiation in 3D: the effect of adhesion site density and substrate elasticity. J Biomed Mater Res, Part A 100, 38–47. [DOI] [PubMed] [Google Scholar]
- Takigawa M, Takano T, Shirai E, et al. (1984) Cytoskeleton and differentiation: effects of cytochalasin B and colchicine on expression of the differentiated phenotype of rabbit costal chondrocytes in culture. Cell Differ 14, 197–204. [DOI] [PubMed] [Google Scholar]
- Tallheden T, Karlsson C, Brunner A, et al. (2004) Gene expression during redifferentiation of human articular chondrocytes. Osteoarthritis Cartilage 12, 525–535. [DOI] [PubMed] [Google Scholar]
- Taylor DW, Ahmed N, Parreno J, et al. (2015) Collagen type XII and versican are present in the early stages of cartilage tissue formation by both redifferentating passaged and primary chondrocytes. Tissue Eng Part A 21, 683–693. [DOI] [PubMed] [Google Scholar]
- Waldman SD, Grynpas MD, Pilliar RM, et al. (2002) Characterization of cartilagenous tissue formed on calcium polyphosphate substrates in vitro. J Biomed Mater Res 62, 323–330. [DOI] [PubMed] [Google Scholar]
- Watt FM, Dudhia J (1988) Prolonged expression of differentiated phenotype by chondrocytes cultured at low density on a composite substrate of collagen and agarose that restricts cell spreading. Differentiation 38, 140–147. [DOI] [PubMed] [Google Scholar]
- Woods A, Beier F (2006) RhoA/ROCK signaling regulates chondrogenesis in a context‐dependent manner. J Biol Chem 281, 13134–13140. [DOI] [PubMed] [Google Scholar]
- Woods A, Wang G, Beier F (2005) RhoA/ROCK signaling regulates Sox9 expression and actin organization during chondrogenesis. J Biol Chem 280, 11626–11634. [DOI] [PubMed] [Google Scholar]
- Yu SM, Kim HA, Kim SJ (2010) 2‐Deoxy‐D‐glucose regulates dedifferentiation through beta‐catenin pathway in rabbit articular chondrocytes. Exp Mol Med 42, 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Li X, Cai L, et al. (2013) Achyranthes bidentata polysaccharides induce chondrocyte proliferation via the promotion of the G1/S cell cycle transition. Mol Med Rep 7, 935–940. [DOI] [PubMed] [Google Scholar]