

Abstract
In favor of their outgrowth, cancer cells must resist immune surveillance and edit the immune response. Cancer immunoediting is characterized by fundamental changes in the cellular composition and the inflammatory cytokine profiles in the microenvironment of the primary tumor and metastatic niches, with an ever increasing complexity of interactions between tumor cells and the immune system. Recent data suggest that genetic instability and immunoediting are not necessarily disparate processes. Increasing mutational load may be associated with multiple neoepitopes expressed by the tumor cells and thus increased chances for the immune system to recognize and combat these cells. At the same time the immune system is more and more suppressed and exhausted by this process. Consequently, immune checkpoint modulation may have the potential to be most successful in genetically highly altered and usually extremely unfavorable types of cancer. Moreover, the fact that epitopes recognized by the immune system are preferentially encoded by passenger gene mutations opens windows of synergy in targeting cancer-specific signaling pathways by small molecules simultaneously with antibodies modifying T-cell activation or exhaustion.
This review covers some aspects of the current understanding of the immunological basis necessary to understand the rapidly developing therapeutic endeavours in cancer treatment, the clinical achievements made, and raises some burning questions for translational research in this field.
Keywords: Checkpoint inhibitor, Cancer, Immunoediting, Exhaustion, Mutational load, T cell repertoire
Background
Tumor immunotherapy has a long-standing history. Starting with the work of William Coley in the 1890s [1], some progress in the treatment of malignancies was achieved with the introduction of interferons, interleukin-2 given either systemically or used for in vitro expansion of T-cells and reinfusion of lymphokine-activated killer cells. At the price of substantial side effects, success was seen in hematological cancers such as multiple myeloma, follicular lymphoma and myeloproliferative disorders, including chronic myeloid leukemia and polycythemia vera (interferons) and acute myeloid leukemia (AML) post allogeneic stem cell transplantation (interleukin-2) [2, 3]. In solid cancers, including melanoma and renal cell cancer, some long-term survivors and even cures were observed with extremely high-dose immuno- or chemoimmunotherapy approaches with interferons or interleukin-2 in the metastatic setting, but toxicities were severe [4, 5].
Substantial progress has later been made with the introduction of monoclonal antibodies (MAb) inducing apoptosis and/or eliciting antibody- or complement-dependent cytotoxicity after binding to tumor antigens. Just to name a few, anti-CD20- [6], anti-Her2- [7], anti-epidermal growth factor receptor (EGFR)- [8] and anti-CD38-MAb [9] are highly efficient in the clinics. Antibodies armed with toxins (eg brentuximab vedotin [10], gemtuzumab ozogamicin [11], trastuzumab emtansine [12], rovalpituzumab tesirine [13], denileukin diftitox [14]) have also proven to be successful.
The most exciting recent progress in the treatment of cancers, however, is derived from the better understanding of how tumor cells escape immune recognition [15] and how they exhaust, suppress and even kill immunocompetent T-cells directed against the tumor [16–20]. T-cell exhaustion is thereby induced by consistent antigen exposure leading to altered T-cell differentiation, loss of effector functions and anergy as well as upregulation and coexpression of inhibitory receptors that are also used as exhaustion markers (eg programmed death 1 (PD1)), and alterations of other key characteristics (for reviews see [21–23]). In addition, cancer cells cleverly expand regulatory T-cells (Tregs) [24] and further B-, natural killer- and dendritic-regulatory cells (for review see [25]) in order to prime the microenvironment towards a tumor supportive milieu. Under normal conditions, immune checkpoint molecules serve to regulate T-cell responses, which is necessary to avoid uncontrolled expansion resulting in organ destruction and fatal outcomes. Tumor cells use these intrinsic ‘brakes’ of the immune system as immune escape mechanisms by inducing functionally exhausted T-cells [15, 25].
The generality of these mechanisms across most -if not all- cancer types implies a yet unexploited applicability of drugs targeting immune suppression in a wide range of tumor entities. In fact, antibodies counteracting suppression of the T-cell receptor (TCR) signaling via CD28/cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) (eg ipilimumab), or interfering with T-cell exhaustion via the PD1/PD ligand 1 (PDL1) axis (eg nivolumab, pembrolizumab, atezolizumab, durvalumab etc.) display impressive therapeutic efficacy in melanoma [26–32], squamous [33] and non-squamous non-small-cell lung cancer (NSCLC) [34], squamous cell cancer of the head and neck [35], renal [36], urothelial cancers [37] and Hodgkin’s disase [38, 39]. Anticancer drugs targeting these so-called ‘immune checkpoints’ on T-cells have been termed ‘checkpoint inhibitors’. The opposite side of the coin, however, is the relevant side effect profile of checkpoint inhibitors, with some patients developing autoimmunity against various organs including hypophysis, adrenal glands, beta cells of the pancreas, thyroid, lungs, liver, gut and nerves. In fact, knockout of PD1 [40] or CTLA-4 [41] resulted in severe and lethal autoimmune diseases in murine models. In humans, treatment with anti-CTLA-4 or anti-PD1 MAbs led to significant autoimmune phenomena and the number of patients with treatment-related grade 3–4 side effects increased up to 55% when both drugs were combined [26].
There is need to systematically clarify the potential exploitation of targeting individual receptors expressed by T-cells, with the aim of circumventing the immunosuppressive effects cleverly used by cancer cells to evade host anti-tumor immune responses. In brief, T-cells exhibit various activating and inhibitory ‘checkpoint’ receptors or molecules (Fig. 1a).
Fig. 1.
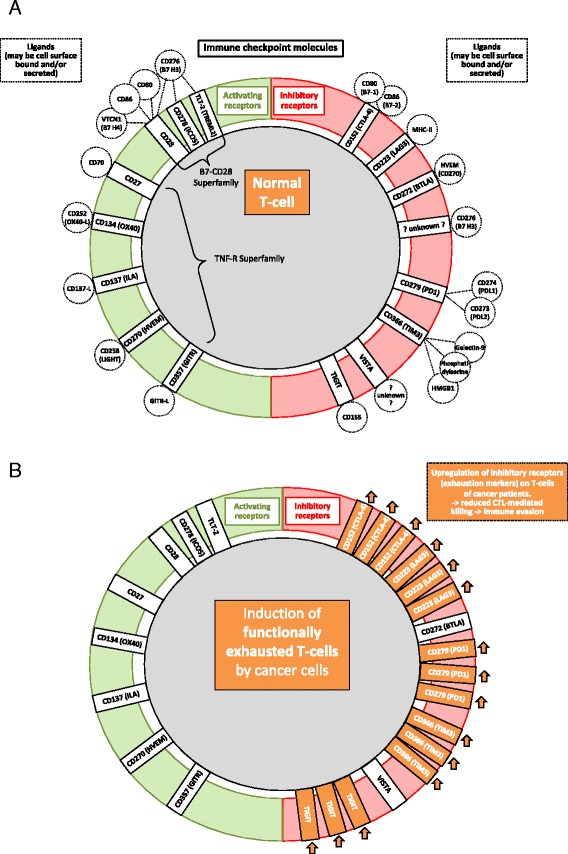
Checkpoint receptors on T cells. Figure 1 a shows negative checkpoint receptors (NCRs) on the right, and activating (costimulatory) checkpoint receptors (ACRs) of a normal T-cell on the left. The balance between the expression of these receptors, and the ligation with respective ligands, determines the functional status of the T-cell during varying physiological processes. Figure 1 b schematically shows how cancer cells may modulate T-cells, to prefentretially express and upregulate NCRs. Therefore, T-cells of cancer patients often become exhausted, anergic, and/or incapable of efficiently attacking and killing the cancer cells. This is one of the mechanisms by which the malignant cells induce tumor imune escape
Activating costimulatory immune checkpoint molecules expressed by T-cells include (i) the B7-CD28 superfamily, which encompasses CD28 (the receptor for CD80 and CD86), CD278 (inducible T-cell costimulator (ICOS) and TREML-2/TLT-2 (Trem-like transcript 2 protein), and (ii) members of the tumor necrosis factor receptor (TNFR) superfamily such as CD27, CD134 (OX40), CD137 (induced by lymphocyte activation (ILA)), CD270 (herpesvirus entry mediator (HVEM)) and CD357 (glucocorticoid-induced TNFR family related gene (GITR)) (reviewed eg in [42]).
Inhibitory checkpoint molecules found on T-cells include CD152 (CTLA-4), CD223 (lymphocyte activation gene 3 (LAG3)), CD272 (B- and T-lymphocyte attenuator (BTLA)), CD279 (PD1) and CD366 (T-cell immunoglobulin and mucin protein 3 (TIM3)), V-domain Ig suppressor of T-cell activation (VISTA), as well as the newly discovered T-cell immunoreceptor with Ig and ITIM domains (TIGIT).
These checkpoint molecules are extremely important, as they help the body to discriminate between ‘foreign’ and ‘self’ as well as help restrain immune responses against foreign targets, while sparing ‘self’. They are often deregulated in cancer, eg by expression or upregulation of inhibitory molecules by the cancer cells themselves, and/or by production of soluble factors by the cancer cells that result in downregulation or blockage of activating receptors, or in ligation and/or upregulation of inhibitory receptors on T-cells, respectively (Fig. 1b). In addition, the phenomenon of T-cell exhaustion can be induced by cancer, thereby hindering anti-tumor immune control (reviewed in [21, 22]).
Numerous drugs have been developed to intercept the malignant control of the immune system by specifically targeting these checkpoint molecules on T-cells. Activating checkpoint molecules can be therapeutically targeted with agonistic molecules, whereas inhibitory immune checkpoint molecules expressed by T-cells can be targeted with blocking antibodies, and the respective drugs that are currently tested and/or have been approved are listed in Tables 1, 2, 3 and 4.
Table 1.
Current status of agents targeting negative checkpoint receptors (NCRs)
| Drug Target, (Synonym) |
Effect on T-cells and immune system | Compound (Synonym) |
Drug company (Trade name) |
Drug type (application) |
Status | Indication |
|---|---|---|---|---|---|---|
| CD152 (CTLA4) | Blocking of CTLA4, activation of T-cells and anti-tumor immune response | Ipilimumab (MDX-010) |
BMS (Yervoy) |
Human MAb, IgG1ĸ (i.v., Q3W) | FDA 25.03.2011 EMA 25.07.2011 Investigational |
MEL (metastatic and adjuvant) MEL NSCLC, SCLC |
| Tremelimumab (CP-675,206) |
AstraZeneca | Human MAb, IgG2ĸ (i.v., Q4-12 W) | Phase II | Mesothelioma, MEL | ||
| CD223 (LAG3) | Blocking of LAG3-mediated immune down-regulation; Activation and expansion of tumor antigen-specific T-effector cells; activation of antigen presenting cells to remove tumor debris; | BMS-986016 | BMS | MAb, (i.v.) | Phase I | Relapsed hematologic malignancies |
| LAG-525 | Novartis | Humanized MAb, (i.v.) | Phase I | Advanced solid tumors | ||
| IMP-701 | Novartis | MAb, (i.v.) | Preclinical | No data available; Cancer | ||
| IMP-321 | Prima BioMed | Recombinant human soluble LAG3 MAb fragment (fusion protein), (i.v.) | Phase I/II | Pancreatic cancer, MEL, MRCC, metastatic breast cancer | ||
| CD223 (LAG3) | Blocks LAG3; Depletion of activated autoaggressive T-cells | GSK-2831781 (initially IMP-731) |
GSK | Humanized MAb, (i.v.) | Phase I | Psoriasis, autoimmune diseases |
| CD272 (BTLA) | - | - | - | - | Preclinical | In vitro, murine models |
| CD279 (PD1) | Blocking of PD1; Activation of anti-tumor immune response via prevention of CD8+ T-cell exhaustion | Nivolumab (BMS-936558) (MDX-1106) |
BMS (Opdivo) |
Human MAb, IgG4ĸ (i.v.) | FDA 22.12.2014 EMA 16.07.2015 |
MEL, NSCLC, RCC, M. Hodgkin MEL, NSCLC, RCC |
| Pembrolizumab (Lambrolizumab) (MK-3475) |
Merck (Keytruda) |
Humanized MAb, IgG4ĸ (i.v., Q3W) | FDA 04.09.2015 EMA 30.07.2015 |
MEL, NSCLC MEL |
||
| BGB-A317 | BeiGene | Humanized MAb, (i.v. Q2-3 W) | Phase I | Advanced solid tumors, B-cell malignancies | ||
| TSR-042 | Tesaro, AnaptysBio | Humanized MAb, (i.v.) | Phase I | Advanced or metastatic solid tumors | ||
| PDR-001 | Novartis | Humanized MAb IgG4ĸ, (i.v.) | Phase I/II | Advanced malignancies, rec./metastatic nasopharyngeal carcinoma | ||
| Pidilizumab (CT-011) (MDV9300) |
Medivation | Humanized MAb IgG1ĸ, (i.v.) | Phase II | DLBCL, relapsed myeloma, follicular lymphoma | ||
| AMP-224 (AFC-111CL) |
Creative Biolabs | ADCC enhanced MAb. (i.v.) | Phase I | Advanced, refractory cancer, metastatic CRC | ||
| MEDI-0680 (AMP-514) |
MedImmune | Humanized MAb IgG4ĸ, (i.v.) | Phase I | Advanced solid tumors | ||
| CD366 (TIM3) | Blocking of TIM3; Activation of anti-tumor immune response via prevention of CD8+ T-cell exhaustion | TSR-022 | Tesaro, AnaptysBio | Humanized MAb IgG4, (i.v.) | Phase I | Advanced solid tumors |
| MBG-453 | Novartis | MAb, (i.v.) | Phase I | Advanced malignancies | ||
| VISTA CD274 (PDL1) |
Blocking of VISTA and PDL1; Activation of T-cell proliferation & cytokine production | CA-170 (AUPM-170) |
Curis, Aurigene | Small molecule antagonist (p.o.) |
Phase I | Advanced solid tumors or lymphomas, nonresponsive to available therapies |
| TIGIT | - | - | - | - | Preclinical | Murine cancer models |
MAb indicates monoclonal antibody, i.v. intravenous, p.o. per os, FDA Agency for Food and Drug Administraton, EMA European Medicines Agency, MEL metastatic melanoma, NSCLC non small cell lung cancer, SCLC small cell lung cancer, MRCC metastatic renal cell carcinoma, DLBCL diffuse large B-cell lymphoma, CRC colorectal cancer
Table 2.
Current status of therapeutic agents targeting ligands of negative checkpoint receptor (NCR)
| Drug Target, (Synonym) | Effect on T-cells and immune system | Compound (Synonym) | Drug company (Trade name) | Drug type (application) | Status | Indication |
|---|---|---|---|---|---|---|
| CD270 (HVEM) | Activation/Inhibition | - | - | - | Preclinical | - |
| CD274 (PDL1) | Blocks PD1/PDL1 ligation; Activation of anti-tumor immune response via prevention of CD8+ T-cell exhaustion | Durvalumab (MEDI-4736) |
AstraZeneca | Human MAb IgG1ĸ (i.v., Q2-3 W) | FDA 17.02.2016 BTD Phase III Phase I/II |
Metastatic urothelial cancer NSCLC Head and neck, gastric cancer, pancreatic cancer, hematologic malignancies, myelodysplastic syndromes |
| Atezolizumab (MPDL-3280A) (RG7446) |
Roche Genentech (Tecentriq) |
Humanized MAb IgG1ĸ, (i.v., Q3W) | FDA 18.05.2016 Phase III |
Metastatic urothelial cancer NSCLC |
||
| Avelumab (MSB-0010718C) |
Merck & Pfizer | Human MAb IgG1ĸ, (i.v., Q2W) | Phase III | Advanced malignancies, bladder cancer, ovarian cancer, MRCC, head and neck, NSCLC, gastric cancer, Merkel cell carcinoma, M. Hodgkin | ||
| BMS-936559 (MDX-505) |
BMS | Human MAb IgG4 ĸ, (i.v., Q2W) | Phase I | MEL, advanced refractory solid tumors and hematologic malignancies | ||
| CD274 (PDL1) VISTA |
Blocking of PDL1 and VISTA - > indirect activation of T-cell proliferation & cytokine production | CA-170 (AUPM-170) |
Curis, Aurigene | Small molecule antagonist (p.o., BID Q4D) |
Phase I | Advanced solid tumors or lymphomas, non-responsive to available therapies |
| HGMB1 | Sensitizes MDR AML cells to chemotherapy, significant decrease in AML cell proliferation | miR181b | - | Inhibits HMGB1 expression | Preclinical | In vitro |
MAb indicates monoclonal antibody, i.v. intravenous; p.o. per os, FDA Agency for Food and Drug Administraton, BTD Breakthrough Therapy Designation, NSCLC non small cell lung cancer, MRCC metastatic renal cell carcinoma, MEL metastatic melanoma, p.o. per os, MDR multi-drug resistant, AML Acute myeloid leukemia
Table 3.
Current status of therapeutic agents targeting activating checkpoint receptors
| Drug Target (Synonym) |
Effect on T-cells and immune system | Compound (Synonym) |
Drug company (Trade name) |
Drug type (application) |
Status | Indication |
|---|---|---|---|---|---|---|
| CD27 | Binds and activates CD27 - > activation of anti-tumor immune response; Treg depletion; Direct targeting of CD27 expressing tumors | Varlilumab (CDX-1127) |
Celldex | Human mAb IgG1ĸ, (i.v., D1,8,15,22 Q8W) | Phase I/II | CRC, NSCLC, RCC, MEL, ovarian cancer, head and neck squamous cell carcinoma |
| CD28 | Immunosuppression via preferential activation of Tregs | TAB-08 (TGN-1412) |
TheraMab (TeGenero) | Humanized MAb IgG4ĸ, (i.v., Q1W) | Phase I/II | Rheumatoid arthritis, Psoriasis, SLE |
| CD134 (OX40) | Binds and activates OX40 - > increases T-cell proliferation and cytokine secretion - > activation of dormant anti-tumor immune response + reduction and suppression of Tregs | PF-04518600 (PF-8600) |
Pfizer | Human MAb IgG2, (i.v.) | Phase I | Neoplasms |
| MOXR-0916 | Genentech | Humanized MAb IgG1, (i.v.) | Phase I | Advanced or metastatic tumors | ||
| MEDI-0562 | MedImmune | Humanized MAb (i.v.) | Phase I | Advanced or metastatic tumors | ||
| MEDI-6469 | MedImmune | Humanized MAb (i.v.) | Phase I | Metastatic CRC, locoregionally advanced head and neck cancer | ||
| CD137 (ILA, 4-IBB) |
Activation and expansion of T-cells and anti-tumor immune response | Urelumab (BMS-663513) |
BMS | Human MAb IgG4ĸ (i.v.) | Phase I | Recurrent glioblastoma, advanced solid tumors, hematologic neoplasms |
| Utomilumab (PF-05082566) |
Pfizer | Human MAb IgG2ĸ (i.v.) | Phase I | Avanced Solid Tumors | ||
| CD270 (HVEM) | Activation/Inhibition | - | - | - | Preclinical | - |
| CD278 (ICOS) | Activation | - | - | - | Preclinical | - |
| CD357 (GITR) | Binds and activates GITR - > activates tumor tumor antigen specific T-effector cells, suppresses Tregs - > activates anti-tumor immune response | MK-4166 | Merck | MAb, (i.v.) | Phase I | Advanced Solid Tumors |
| INCAGN-01876 | Agenus | MAb, (i.v.) | Phase I | Advanced or Metastatic Solid Tumors | ||
| TRX-518 | Leap Therapeutics | Humanized MAb, (i.v.) | Phase I | Advanced Solid Tumors, MEL | ||
| GWN-323 | Novartis | MAb, (i.v.) | Phase I/II | Advanced solid tumors, lymphomas | ||
| TLT-2, TREML | Activation | - | - | - | Preclinical | - |
MAb indicates monoclonal antibody, i.v. intravenous, CRC colorectal cancer, NSCLC non small cell lung cancer, RCC metastatic renal cell carcinoma, MEL metastatic melanoma, SLE systemic lupus erythematodes
Table 4.
Current status of therapeutic agents targeting ligands activating checkpoint receptors
| Drug Target (Synonym) |
Effect on T-cells and immune system | Compound (Synonym) |
Compound (Trade name) |
Drug type (application) |
Status | Indication |
|---|---|---|---|---|---|---|
| CD80 (B7-1) CD86 (B7-2) |
T-cell costimulation blocker, inhibits T-cell proliferation and production of cytokines | Belatacept (LEA29Y) |
BMS (Nulojix) |
CTLA4 fusion-Ig, costimulation blocker | FDA approval 15.11.2011 EMA approval 07.07.2011 |
Prophylaxis of renal graft rejection Prophylaxis of renal graft rejection |
| Abatacept | BMS (Orencia) |
CTLA4 fusion-Ig, costimulation blocker |
FDA approval 31.07.2011 EMA approval 25.01.2010 |
Rhematoid arthritis; Polyarticular juvenile idiopathic arthritis | ||
| CD80 | Activates ADCC on B-NHL cells with upregulated CD80 | Galiximab (IDEC-114) |
Biogen | Humanized MAb IgG1, (i.v.) | Phase I/II Phase II |
Untreated follicular lymphoma Relapsed/refractory M. Hodgkin |
| CD80 DNMT1 |
Upregulation of CD80 on cancer cells - > costimulatory activation of T-cells | Decitabine | Janssen-Cilag (Dacogen) | Deoxycytidine analogue, i.v. (D1-5, Q6W) | FDA approval 02.05.2006 EMA approval 28.09.2012 |
Myelodysplastic syndromes Acute myeloid leukemia |
| CD86 HDAC |
Upregulation of CD86 on cancer cells, HDAC-I - > costimulatory activation of T-cells | Romidepsin (FR901228) |
Gloucester Pharmaceuticals, Celgene (Istodax) | Depsipeptide, (i.v. Q1W) | FDA approval 05.11.2009 EMA refused approval 09.07.2012 |
Cutaneous T-NHL, PTCL |
| CD137-L | Induction of differentiation of AML cell samples in vitro | - | - | Recombinant CD137-L protein | Preclinical | In vitro |
| CD252 (OX40-L) | Binds and activates Ox40 by mimicking Ox40-L - > proliferation of TAA specific T-effector cells - > activation of anti-tumor immune response | MEDI-6383 | MedImmune | Fusion protein | Phase I | Advanced solid tumors |
| CD252 (OX40-L) | Binds and inhibits Ox40L and the interaction with Ox40 - > inhibition of allergen induced immune response | Oxelumab (huMAb OX40L) (R4930) |
Genentech | Human MAb IgG1, (i.v.) | Phase II | Mild allergic asthma |
| CD276 (B7-H3) | Inhibits CD276 | Enoblituzumab (MGA-271) |
MacroGenics | Humanized MAb IgG1ĸ, (i.v.) | Phase I | B7-H3 expressing refractory solid tumors |
| CD276 (B7-H3) CD3 |
Redirection of T-cells to kill B7-H3 overexpressing tumor cells | MGD-009 | MacroGenics | Dual affinity retargeting protein | Phase I | B7-H3 expressing unresectable or metastatic solid tumors |
MAb indicates monoclonal antibody, i.v. intravenous, FDA Agency for Food and Drug Administraton, EMA European Medicines Agency, ADCC antibody dependent cellular cytotoxicity, NHL non-Hodgkin lymphoma, PTCL peripheral T-cell lymphoma, TAA tumor associated antigen
Predicting response to checkpoint blockade
The degree of efficacy of checkpoint inhibitors is highly divergent between different tumor types. This phenomenon may be attributed to differences in PDL1 expression on neoplastic or microenvironmental cells, suggesting that this marker should be quantified ahead of therapy. However, reported thresholds for PDL1 expression to predict the probability of response towards anti-PD1 MAbs vary between ≤ vs. >1% (for nivolumab) or ≤ vs. >50% (for pembrolizumab) and ≤ vs. >1%, 5% or 10% (for atezolizumab) with many reasons suggested, but none being really convincing in explaining these differences [43, 44]. Given the relevant side effect profiles of immune checkpoint inhibitors and their exceptionally high costs, novel and better predictors of response are therefore needed.
Notably, the mutational burden defined as the number of mutations per megabase, may correlate with -and thus predict the occurrence of- tumor-specific (neo)antigens (TSA) which are expressed on the tumor cell surface and presented to T-cells. T-cells exposed to TSA can learn to specifically target and eliminate (ie kill) tumor cells. In contrast to tumor-associated antigens (TAA), which are in essence massively overexpressed ‘normal antigens’ that also occur on normal, non-cancerous tissues of the body, TSA are true neoantigens that cannot be found on any non-malignant cell. TAA are much more common than TSA, and strategies targeting TAA molcules include eg the clinically widely successful targeting of CD20 in lymphomas and CD33 in AML, as briefly mentioned above. Targeting of TSA would in theory eliminate the bystander killing of normal cells, which also bear these molecules to a lesser extent (eg normal B-cells or myeloid cells for the two molecules mentioned above).
The mutational burden varies substantially over a range of 3–4 logs in different tumor entities [45] and even within the same tumor considerable interpatient variability may be observed. Provided that in tumors with high neoantigen frequency T cells are more prone to recognize TSA, but are exhausted by specific ligands during immunoediting, strategies aimed at re-instating T cell functions could be particularly effective in these patients [23]. This view is supported by the following facts:
The response and efficacy of checkpoint inhibitors seems to be highest in tumor types with the highest mutational burden (eg melanoma, NSCLC) [46]. This is especially relevant in the light of the fact that patients with high numbers of mutations are usually weakly responsive to chemotherapy and/or rapidly develop chemo-resistance.
In NSCLC patients treated with the PD1-inhibitor pembrolizumab, progression-free survival massively differed according to mutational burden in an as yet unseen manner [47]. This has also been observed in urothelial cancers treated with the anti-PDL1 antibody atezolizumab [48].
Most patients with colon cancer usually do not respond to checkpoint inhibitors [49], however, in a small subset of advanced colorectal cancer patients high microsatellite instability due to deficient DNA mismatch repair [50] was observed, the occurrence of which has been associated with a high number of mutations, potentially resulting in an elevated expression of TSA on the tumor cell surface [51]. Treatment of these patients with PD1-inhibitors resulted in a response rate of nearly 40%, as compared to only 11% stable disease in those with microsatellite stability [51].
APOBEC family members, mutational burden, the role of the immune system and its use as a predictor of response to checkpoint inhibitors
On average, 2–4 oncogenic driver gene mutations are present in various tumors [52, 53]. It is not clear yet whether the extent of driver gene mutations correlates with overall mutational burden (including passenger mutations) and how this impacts on checkpoint molecule expression on T-cells.
The AID (activation-induced cytidine deaminase)/APOBEC (apolipoprotein B mRNA editing enzyme catalytic polypeptide-like) gene family members are cytidine deaminases causing alterations in DNA and mRNA sequences by cytidine-to-uracil (C-U) transitions -with subsequent conversion of U to thymine (T) during DNA replication- a process called DNA/mRNA-editing that results in protection from parasitic viruses as well as protein and antibody diversity. Loss of cellular control of APOBEC activities results in DNA hypermutations, promiscuous RNA editing, and ultimately genetic instability and tumorigenesis (for recent review see [54, 55]). One of the main functions of AID is to regulate mutations in immunoglobulin (Ig) heavy and light chain genes during B-cell development in lymph nodes, thereby creating antibody diversity. AID also alters gene regulation by interfering with epigenetic DNA modification. However, AID is to a certain degree ‘leaky’ and may induce off-target gene mutations and/or translocations of oncogenes towards Ig genes, thereby promoting leukemogenesis and/or lymphomagenesis [56–58]. Similarly, other APOBEC family members, whose canonical function is to induce showers of mutations in cDNA intermediates of RNA viruses, contribute to tumor induction and progression in many types of neoplasias, including -but not limited to- chronic lymphocytic leukemia (CLL) and breast cancer [57–61]. Various members of the AID/APOBEC family may differ up to tenfold in their mutagenic capacity [62] and APOBEC enzymes may significantly drive subclonal evolution and tumor heterogeneity [63]. It is therefore not surprising that APOBEC family mutational signatures characterize patients with poor prognosis (eg in multiple myeloma), mostly via their involvement in generating translocations, which are often associated with adverse outcome [64]. In breast cancer, APOBEC-3B expression is associated with unfavorable clinicopathological features and poor outcome [65]. In line with these observations, APOBEC-3B expression has been associated with mutations of p53, as well as of the catalytic subunit of phosphatidylinositol 3-kinase [66].
On the other hand, activated members of the APOBEC family might increase the number of neoantigens, cancer-specific T-cell clones and may induce a broader TCR repertoire. Thus, APOBEC family member expression, function, or mutational pattern could serve as a biomarker for the response to checkpoint inhibitors and other immunomodulatory drugs. Initial evidence supporting this hypothesis includes:
In non-invasive early urothelial cancers APOBEC-related mutational signatures were predominantly seen in high-risk tumors [67].
In another small series of urothelial tumors, the expression of certain APOBEC family members (A3A, A3D and A3H) was associated with PDL1 positive mononuclear cells infiltrating the tumor and increased expression of the variants A3F_a and A3F_b correlated with upregulated expression of PDL1 on tumor cells [68], indicating that PDL1 may serve as a therapeutic target. As a side note, increased expression of A3D and A3H was associated with a better overall survival (OS) in this study, which seems paradoxical, or at least cannot be explained yet. Thus, further investigations concerning APOBEC expression patterns and response to checkpoint inhibitors are warranted.
In high grade serous ovarian carcinomas APOBEC3 expression was significantly associated with T-cell infiltration and -seemingly paradoxically- with improved clinical outcome [69].
Furthermore, breast cancer developed more commonly in women with germline APOBEC3B (A3B) deleting polymorphisms, but these women were not subject to unfavorable risk profiles or worse outcome [65], suggesting potential value for the determination of A3B deletion status in predicting response to checkpoint inhibitors.
All of the above data encourage deeper analyses of the correlation (and presumed interaction) between (i) APOBEC family member expression profiles, splice variants and/or polymorphisms and (ii) mutational burden, clonal evolution, and effects on expression profiles of immunomodulatory molecules and their function. This might lead to a better understanding and fine-tuning of immunotherapies in cancer.
TCR repertoire and T-cell diversity in predicting response to immunotherapy
Often, driver gene mutations and associated atypical proteins remain immunologically silent. In fact, over 90% of cancer cell mutations recognized by CD4+ and CD8+ T-cells occur in passenger genes [45]. This suggests that the TCR repertoire broadens with increasing numbers of (passenger) gene mutations, resulting in a broader pool of T-cell clones capable of fighting cancer cells. This might be exploited with therapeutic strategies aimed at reactivating or boosting the host anti-tumor immune response. Therefore, although the presence of high mutational burden is generally acknowledged to be an adverse predictor of outcome across all tumor entitites, it may predict TCR diversity and thus good response to checkpoint inhibitors and/or activating immunotherapies.
Indeed, TCR diversity was associated with good clinical outcomes following treatment with the MAb ipilimumab targeting CTLA-4 in a small series of melanoma patients [70]. This was confirmed in conference papers by others, who show that a TCR diversity score higher than 20% is necessary for good outcome of melanoma patients receiving anti-CTLA-4 antibody treatment [71]. Interestingly, an inverse constellation was found for treatment with anti-PD1 antibodies [71]. However, as CTLA-4 blockade itself can broaden the TCR repertoire [72], this may partly explain the seeming discrepancies. Given the high number of immunomodulatory ligand/receptor pairs modifying cancer/T-cell interactions, a thorough investigation of these issues, ideally in prospective clinical trials, is warranted.
Peripheral blood instead of tissue examinations of biomarkers
Most analyses of biomarkers thought to predict response to checkpoint inhibitors are currently performed in primary samples of tumor tissue. However, tumor biopsies are sometimes difficult or even impossible to obtain and, depending on the location of the tumor, may be associated with relevant side effects such as an increased risk of bleeding, organ perforation and/or infection, as well as high medical costs for the procedure itself. In addition, tissue biopsies usually cannot be performed continuously during the course of the disease due to these potential risks. Therefore, the actual status of tumor clone evolution and expansion or reduction of T-cell clones capable of actively combating the tumor, remains obscure in patients treated with checkpoint inhibitors. The provision of a rationale for therapeutic decision making and the choice of the optimal immunomodulatory drug most suitable to fight malignant subclones, would ideally require the serial analysis of (i) representative tumor DNA from tissue biopsies, and (ii) various T-cell subsets from peripheral blood.
Recently, the detection of circulating tumor DNA (ctDNA) has shown very high identification rates of mutations that were also found in primary tumors using a deep-coverage (15,000x) next-generation sequencing test of 70 genes [73].
Nevertheless, it has been shown that 9/10 gastrointestinal cancer patients had CD4+ and/or CD8+ tumor infiltrating lymphocytes that recognized 1–3 neo-epitopes from somatic mutations occurring in the respective cancers [74]. Moreover, very recently it has been demonstrated that circulating PD1+ lymphocytes from cancer patients were enriched in naturally occurring tumor-reactive and mutation-specific cells [75]. Exhaustion of T-cells has mostly, if not exclusively, been examined on tumor-infiltrating lymphocytes. Thus, longitudinal analyses and functional examination of T-cells during the natural disease course and/or during various treatment phases are lacking. In fact, it remains unknown at present, whether there is a correlation between ctDNA (ie type and frequency of mutations) and peripheral blood T-cell exhaustion profiles. This underlines the necessity to characterize peripheral blood T-cells within the frame of clinical trials that aim to aid the immune system to adapt to clonal tumor evolution via therapeutic immunoediting.
Synergistic immunotherapeutic opportunities
Interaction between various members of checkpoint inhibitors or immune-activators
The approach of repressing multiple pathways, or of combining repressive with immunostimulatory antibodies seems particulary exciting and is currently investigated in numerous trials (Table 5). In preclinical studies synergy for such approaches (eg inhibition of PD1 and activation of CD137, or combined inhibition of inhibitory checkpoint molecules) has been demonstrated [76–78]. Early phase I data support the view that such combinations of repressive with activating MAbs are feasible ([78]; NCT00803374, NCT02253992, NCT00351325). However, these therapeutic approaches must be viewed with caution and be closely monitored, given the overwhelming activation of autoimmunity which could arise.
Table 5.
Clinical trials testing combined targeting strategies of more than 1 checkpoint receptor
| Drug target (Synonym) |
Compound (Synonym) |
Status | Indication | ClinicalTrials.gov identifier |
|---|---|---|---|---|
| CD134 (OX40) CD137 (ILA, 4-IBB) |
PF-04518600 Utomilumab (PF-05082566) |
Phase I | Neoplasms | NCT02315066 |
| CD134 (OX40) CD137 (ILA, 4-IBB) CD274 (PDL1) |
PF-04518600 Utomilumab (PF-05082566) Avelumab (MSB-0010718C) |
Phase I | Neoplasms | NCT02554812 |
| CD137 (ILA, 4-IBB) CD279 (PD1) |
PF-05082566 MK-3475 |
Phase I | Advanced solid tumors | NCT02179918 |
| CD123 (OX40) CD274 (PDL1) VEGF |
MOXR0916 Atezolizumab (MPDL-3280A) Bevacizumab |
Phase I | Advanced or metastatic solid tumors | NCT02410512 |
| CD134 (OX40) CD152 (CTLA4) CD274 (PDL1) |
MEDI-0562 Tremelimumab (CP-675,206) Durvalumab (MEDI-4736) |
Phase I | Advanced or metastatic solid tumors | NCT02705482 |
| CD134 (OX40) CD152 (CTLA4) CD274 (PDL1) |
MEDI-6469 Tremelimumab (CP-675,206) Durvalumab (MEDI-4736) |
Phase I/II | Advanced solid tumors or DLBCL | NCT02205333 |
| CD252 (OX40L) CD274 (PDL1) |
MEDI-6383 Durvalumab (MEDI-4736) |
Phase I | Recurrent or metastatic solid tumors | NCT02221960 |
| CD276 (B7-H3) CD152 (CTLA4) |
Enoblituzumab (MGA-271) Ipilimumab |
Phase I | B7-H3 expressing solid tumors | NCT02381314 |
| CD276 (B7-H3) CD279 (PD1) |
Enoblituzumab (MGA-271) Pembrolizumab |
Phase I | B7-H3 expressing solid tumors | NCT02475213 |
| VISTA CD274 (PDL1) |
CA-170 (AUPM-170) |
Phase I | Advanced solid tumors or lymphomas, non-responsive to available therapies | NCT02812875 |
| CD152 (CTLA4) CD274 (PDL1) Cytotoxic |
Tremelimumab (CP-675,206) Durvalumab (MEDI-4736) Chemotherapy |
Phase I Phase I Phase I |
Resectable CRC with liver metastases Solid malignancies Multiple Myeloma |
NCT02754856 NCT02537418 NCT02716805 |
| CD152 (CTLA4) CD274 (PDL1) Cytotoxic |
Tremelimumab (CP-675,206) Durvalumab (MEDI-4736) Radiotherapy |
Phase I Phase II Phase II |
Unresectable pancreatic cancer NSCLC, CRC Relapsed SCLC |
NCT02311361 NCT02888743 NCT02701400 |
| CD152 (CTLA4) CD274 (PDL1) |
Tremelimumab (CP-675,206) Durvalumab (MEDI-4736) |
Phase I Phase II Phase II Phase II Phase III |
Advanced solid tumors Unresectable hepatocellular carcinoma Metastatic Her2 negative breast cancer Head and neck cancer NSCLC |
NCT02261220 NCT02519348 NCT02536794 NCT02319044 NCT02453282 |
| CD274 (PDL1) CD279 (PD1) |
Durvalumab (MEDI-4736) MEDI-0680 |
Phase I/II | Advanced malignancies | NCT02118337 |
| CD137 (ILA, 4-IBB) CD279 (PD1) |
Urelumab Nivolumab (BMS-936558) |
Phase I Phase II |
Recurrent glioblastoma Cisplatin-ineligible bladder carcinoma |
NCT02658981 NCT02845323 |
| CD152 (CTLA4) CD137 (ILA, 4-IBB) |
Ipilimumab BMS-663513 |
Phase I | Advanced melanoma | NCT00803374 |
| CD223 (LAG3) CD279 (PD1) |
BMS-986016 Nivolumab (BMS-936558) |
Phase I | Relapsed hematologic malignancies | NCT02061761 |
| CD366 (TIM3) CD279 (PD1) |
MBG-453 PDR-001 |
Phase I/II | Advanced malignancies | NCT02608268 |
| CD279 (PD1) CD357 (GITR) |
PDR-001 GWN-323 |
Phase I/II | Advanced malignancies and lymphomas | NCT02740270 |
| CD279 (PD1) CD223 (LAG3) |
PDR-001 LAG-525 |
Phase I/II | Advanced malignancies | NCT02460224 |
| CD357 (GITR) CD279 (PD1) |
MK-4166 Pembrolizumab (MK-3475) |
Phase I | Advanced solid tumors | NCT02132754 |
DLBCLindicates diffuse large B-cell lymphoma, CRC colorectal cancer, NSCLC non small cell lung cancer, SCLC small cell lung cancer, MRCC metastatic renal cell carcinoma
Data in melanoma have shown that nivolumab outcompetes ipilimumab and that the combination of both is superior over single treatment strategies [28]. In this regard it is interesting to note that in a murine model acquired resistance against anti-PD1 antibodies was accompanied by an upregulation of another exhaustion marker, TIM3. The resistance could be broken by inhibition of TIM3 with a blocking antibody and these preclinical mouse data were supported by in vivo findings in two lung cancer patients [79]. Therefore the combination -or sequential application- of eg anti-PD1 or anti-PDL1 antibodies with anti-TIM3 antibodies is an approach that should be further evaluated in controlled clinical trials.
Notably, TIM3 is expressed on tumor-infiltrating Tregs (CD4+, CD25+, Foxp3+), which suppress CD8+ cytotoxic T-cells (CTLs) [80]. Blocking of TIM3 would thus reduce the Treg mediated suppression of (tumor-specific) CTLs and allow them to target the tumor. However, the degree to which such an effect might be offset by TIM3 expression on CD4+ [81] and CD8+ [82] effector T cells remains to be determined, particularly as TIM3 expression was also associated with improved survival under certain conditions [83]. Clearly, a systematic serial analysis of changes in the expression profiles of immunodulatory molecules during immunoediting in carcinogenesis, progression of disease as well as during (effective) treatment needs to be carried out in individual tumor entities in order to dissect optimal time points and types of immunologic interventions.
Synergistic opportunities with other therapies
Off-target effects of small molecules on T-cells
Kinase inhibitors might synergize with immunotherapy in combating cancer, even without direct interaction of the molecular targets. In fact, phase I clinical trials have shown a synergy between gefitinib, which targets EGFR with the PD1 checkpoint inhibitor durvalumab in EGFR mutated NSCLC patients with ~80% response rates [84]. In addition, drugs targeting the proteins of mutated driver genes might directly increase the re-activation of the specific immune system exerted by checkpoint inhibitors. In part, these effects may be caused by interference of some kinase inhibitors with signaling pathways essential for T-cell function, activation, survival and proliferation. Indeed, it has recently been shown that the mitogen-activated protein kinase (MAPK) kinase (MEK) inhibitor cobimetinib increased major histocomaptibility complex (MHC) class I molecule expression on cancer cells and induced a 17% response rate in colorectal cancer patients treated with the anti-PDL1 mAb atezolizumab [85]. Midostaurin, an flt-3 inhibitor with a broad kinase inhibition dendrogram, increases OS in AML patients (when added to daunorubicin and cytarabine) [86], yet does not hamper TCR signaling or T-cell activation [87]. Its effect on expression and function of checkpoint molecules on the cell surface of T-cells of AML patients has not yet been analyzed, although the combination of flt-3 inhibition with checkpoint inhibitors is currently being tested in Phase I to III trials in this disease.
The Bruton’s tryosine kinase (BTK) inhibitor ibrutinib binds covalently to BTK, thus inhibiting B-cell-receptor-mediated proliferation, inducing apoptosis and migration of neoplastic B-cells out of the protective micromilieu of lymph nodes. The drug has shown impressive efficacy in CLL [88, 89], particularly in patients with p53 mutations or deletions. Moreover, ibrutinib also binds to and inhibits interleukin-2-inducible T-cell kinase (ITK), thereby leading to a T-helper (Th) cell 1 polarization in vitro and in vivo, which aids in inducing an anti-tumor immune response [90]. When mice carrying aggressive lymphomas, breast or colon cancers, which all were insensitive towards ibrutinib treatment, were treated with anti-PDL1 MAb or a combination of anti-PDL1 MAb and ibrutinib, the combination showed significantly enhanced efficacy over anti-PDL1 mAb monotherapy [91]. In addition, murine and human myeloid-derived suppressor cells, which play a relevant role in suppressing an efficient anti-tumor immune reaction, express BTK and ibrutinib has been shown to eliminate these cells in vivo [92].
In addition, PDL1-exposed lymphocytes cocultured with melanoma cell lines showed downregulation of MAPK signaling which could be reverted by the B-Raf inhibitor vemurafenib in vitro [93]. In murine (transplantation) models for hepatocellular cancer, tumor shrinkage was induced by sorafenib which was linked to a downregulation of PD1+/CD8+ and Treg cells in the tumor microenvironment [94]. In addition, in murine B-Raf wild-type syngeneic transplantable tumors Raf-kinase inhibitors paradoxically induced hyperactivation of extracellular-signal regulated kinase (ERK) signaling and thus increased T-cell activation and signaling [95]. This may serve as an explanation for increased anti-tumor activity of the combination of CTLA-4-and Raf-kinase inhibitors in preclinical models. Little has been done to systematically analyze these interactions of Raf-kinase with checkpoint inhibitors on a broader, preclinical level.
Other kinase inhibitors have been shown to increase tumor cell infiltration by T-cells, as detected in core biopsies of patients, which predicts a more favorable spontaneous clinical course and better response to neoadjuvant Her2-targeting agents in breast cancer [96, 97]. These effects predominantly seem to be reflected by the CD8+/Treg ratio within the tumor tissue [98].
Likewise, janus kinase 2 (Jak2) mRNA expression in breast cancer cells was associated with increased numbers of tumor infiltrating leukocytes and better prognosis [99]. However, Jak2-inhibitors, which aim to suppress the growth supporting function of this kinase in tumor cells, are currently tested in clinical trials, but since the detailed role of Jak2 inhibitors on T-cell activation, exhaustion and tumor recognition has not yet been fully addressed, a potential unfavorable effect of Jak2-inhibitors cannot be excluded [99].
Combination of cytotoxic drugs and checkpoint inhibitors – novel aspects
It is clear that the current results achieved with checkpoint inhibitors in clinical practice are exciting, but far from being good enough. Therefore, various combinations with chemotherapy, radiotherapy or endocrine therapy are currently being tested in clinical trials. This approach was initially followed only reluctantly due to the widespread view that these chemotherapeutic drugs suppress the immune system [100]. However, it is becoming increasingly clear that conventional chemotherapeutics may induce the expression of neoantigens, induce Th1-differentiation and/or suppress Tregs. These drugs have thus been termed ‘immunogenic chemotherapy’ [101], and may ultimately sensitize tumor cells to checkpoint inhibitors [101–104].
In line with this hypothesis, it was demonstrated in a systemically progressive melanoma patient that local radioation therapy induced upregulation of the tumor antigen NY-ESO-1 and resulted in consecutive systemic resensitization towards ipilimumab [105]. This observation was later confirmed in a larger number of patients [106].
Other drugs such as hypomethylating agents may also show additive immunomodulatory effects with checkpoint inhibitors, via upregulation of MHC-I on the neoplastic myeloid-derived suppressor cells (for review see [107]). In this regard, the combination of anti-PD1 with anti-CTLA-4 antibodies and 5-azacytidine as well as a histon-deacetylase inhibitor completely eradicated murine breast and colorectal cancer cells in vivo [107].
The influence of the gut microbiota on the immune response
Gut microbiota, their development during treatment with chemotherapy and immunomodulators and their influence on the effect of checkpoint inhibitors
Gut microbiota comprise several trillions of microorganisms with a weight of 2 kg (reviewed in [108]). These microorganisms include bacteria, archaea, eukarya and viruses, with the main phyla being firmicutes, bacteroidetes and actinobacteria [108]. Of note, significant interindividual differences in the species and functional composition of the human enterotypes may result from long-term dietary habits [109, 110]. More importantly, gut colonization essentially influences the development of the immune system [111, 112], as seen in inoculation experiments using germ-free mouse models [113–115], and gut microbiota have been reported to be centrally involved in carcinogenesis [116, 117], eg in colorectal cancer (for reviews see [118, 119]). Alterations in the composition of gut microbiota have also been shown to exert systemic effects by modulating estrogen metabolism, thereby affecting women’s risk of developing postmenopausal estrogen receptor-positive breast cancer (reviewed in [120]). In addition, certain gut microbiota can induce DNA double-strand breaks and thus adversely influence the genomic stability of intestinal epithelial cells in vitro (eukaryotic cell lines) [121] and in vivo (mouse model) [122]. In addition, gut microbiota may also exert an influence on epigenetic modifications, and can thus influence inflammatory and immunological reactions (reviewed in [108]), and also directly modulate endogenous T-cell immune responses in mice [123].
Gut microbiota also seem to be involved in the regulation of extrathymic differentiation of Tregs in vitro and in vivo [124] and Th1 infiltration into cancer tissues following treatment with cyclophosphamide. Antibiotic treatment –by subsequent changes in gut microbiota– may turn down the effect of immunostimulation exerted by these drugs. In turn, microbiotic constitution within the gut has been shown to be modified in number and class distribution by cytotoxic drugs, including irinotecan, 5-fluorouracil (reviewed in [125]), and others (reviewed in [108]). Vice versa, the microbiota may also be indispensible for the in vivo anti-tumoral effects of certain cytotoxic drugs such as cyclophosphamide [126] or platinum salts [127] as observed in mouse models. For example, gut microbiota have been shown to be involved in the metabolization of cytotoxic drugs (reviewed in [108]) and in modifying local toxicity of anticancer drugs in vivo [128–130].
Recently it was also reported that PD1−/− mice have altered composition of the gut microbiota [131], and that the efficacy of anti-CTLA-4 treatment in animals and patients with metastatic melanoma and NSCLC may depend on the constitution of gut bacteria [132]. Studying the interactions between gut microbiota and (i) the efficacy of conventional cytotoxic anticancer drugs, and (ii) immune cells capable of targeting the tumor, are expected to increase our understanding of how one might best therapeutically modulate antitumor immune responses.
Conclusion
In summary, despite the clinical benefit observed in a relevant proportion of patients by targeted immune checkpoint modulation, this field of research is still in its infancy. However, our increasing understanding of tumor immunology in general, and the immunoediting process exerted by cancer cells in particular, opens a wide window of opportunities to improve therapeutic immunomodulatory approaches against cancer, making translational science in this exciting field more important than ever.
Burning questions for translational research
Which factors could serve as predictors of response to checkpoint mediators?
At which time points and from which source(s) (ie peripheral blood or tissue biopsy) should the analysis of potential predictors/biomarkers be performed?
Which combinations of checkpoint mediators with other therapies seem promising?
Are the best effects of checkpoint mediators achieved using combinational or sequential approaches?
What are the optimal time points for which type of immunologic intervention(s)?
Which biological interactions with the tumor microenvironment might affect the response to checkpoint mediators?
Acknowledgements
The authors would like to acknowledge and thank: Keri Davies from FireKite, an Ashfield company, part of UDG Healthcare plc, editing of the figures to the required journal format; the Province of Salzburg and the Austrian Science fund (FWF): W 1213 and P 24100 for funding.
Funding
Austrian Science fund (FWF) W 1213 and P 24100 to RG and W 1213 to TNH.
Availability of data and materials
Not applicable.
Authors’ contributions
All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- A3B
APOBEC3B
- AID
Activation-induced cytidine deaminase
- AML
Acute myeloid leukemia
- APOBEC
Apolipoprotein B mRNA editing enzyme catalytic polypeptide-like
- BTK
Bruton’s tryosine kinase
- BTLA
B- and T-lymphocyte attenuator
- CLL
Chronic lymphocytic leukemia
- ctDNA
Circulating tumor DNA
- CTLA-4
Cytotoxic T-lymphocyte-associated protein 4
- CTLs
Cytotoxic T-cells
- EGFR
Epidermal growth factor receptor
- ERK
Extracellular-signal regulated kinase
- GITR
Glucocorticoid-induced TNFR family related gene
- HVEM
Herpesvirus entry mediator
- ICOS
Inducible T-cell costimulator
- ILA
Induced by lymphocyte activation
- ITK
Interleukin-2-inducible T-cell kinase
- Jak2
Janus kinase 2
- LAG3
Lymphocyte activation gene 3
- MAb
Monoclonal antibody/antibodies
- MAPK
Mitogen-activated protein kinase
- MEK
Mitogen-activated protein kinase kinase
- MHC
Major histocomaptibility complex
- NSCLC
Non-small-cell lung cancer
- OS
Overall survival
- PD1
Programmed death 1
- PDL1
PD1 ligand
- TAA
Tumor-associated antigens
- TCR
T-cell receptor
- Th
T-helper
- TIGIT
T-cell immunoreceptor with Ig and ITIM domains
- TIM3
T-cell immunoglobulin and mucin protein 3
- TNFR
Tumor necrosis factor receptor
- Tregs
Regulatory T-cells
- TREML-2
Trem-like transcript 2 protein
- TSA
Tumor-specific (neo)antigens
- VISTA
V-domain Ig suppressor of T-cell activation
Contributor Information
Richard Greil, Phone: +43 57 255025801, Email: r.greil@salk.at.
Evelyn Hutterer, Email: e.hutterer@salk.at.
Tanja Nicole Hartmann, Email: t.hartmann@salk.at.
Lisa Pleyer, Email: l.pleyer@salk.at.
References
- 1.Coley WB., II Contribution to the knowledge of Sarcoma1. Ann Surg. 1891;14(3):199–220. doi: 10.1097/00000658-189112000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galvani DW, Cawley JC. The current status of interferon alpha in haemic malignancy. Blood Rev. 1990;4(3):175–80. doi: 10.1016/0268-960X(90)90045-T. [DOI] [PubMed] [Google Scholar]
- 3.Toren A, et al. Role of interleukin-2 in human hematological malignancies. Med Oncol. 1995;12(3):177–86. doi: 10.1007/BF01571195. [DOI] [PubMed] [Google Scholar]
- 4.Kruit WH, et al. High-dose regimen of interleukin-2 and interferon-alpha in combination with lymphokine-activated killer cells in patients with metastatic renal cell cancer. J Immunother. 1997;20(4):312–20. doi: 10.1097/00002371-199707000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Atkins MB, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105–16. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 6.Maloney DG. Anti-CD20 antibody therapy for B-cell lymphomas. N Engl J Med. 2012;366(21):2008–16. doi: 10.1056/NEJMct1114348. [DOI] [PubMed] [Google Scholar]
- 7.Maximiano S, et al. Trastuzumab in the treatment of breast cancer. BioDrugs. 2016;30(2):75–86. doi: 10.1007/s40259-016-0162-9. [DOI] [PubMed] [Google Scholar]
- 8.Goldstein DA, et al. Metastatic colorectal cancer: a systematic review of the value of current therapies. Clin Colorectal Cancer. 2016;15(1):1–6. doi: 10.1016/j.clcc.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lonial S, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet. 2016;387(10027):1551–60. doi: 10.1016/S0140-6736(15)01120-4. [DOI] [PubMed] [Google Scholar]
- 10.Deng C, Pan B, O’Connor OA. Brentuximab vedotin. Clin Cancer Res. 2013;19(1):22–7. doi: 10.1158/1078-0432.CCR-12-0290. [DOI] [PubMed] [Google Scholar]
- 11.Loke J, et al. Mylotarg has potent anti-leukaemic effect: a systematic review and meta-analysis of anti-CD33 antibody treatment in acute myeloid leukaemia. Ann Hematol. 2015;94(3):361–73. doi: 10.1007/s00277-014-2218-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verma S, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367(19):1783–91. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.ADC Shows Effectiveness in SCLC. Cancer Discov. 2015;5(12):OF4. 10.1158/2159-8290.CD-NB2015-144. [DOI] [PubMed]
- 14.Kadin ME, Vonderheid EC. Targeted therapies: Denileukin diftitox--a step towards a ‘magic bullet’ for CTCL. Nat Rev Clin Oncol. 2010;7(8):430–2. doi: 10.1038/nrclinonc.2010.105. [DOI] [PubMed] [Google Scholar]
- 15.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 16.Greil R, et al. Tuning the rheostat of the myelopoietic system via Fas and TRAIL. Crit Rev Immunol. 2003;23(4):301–22. doi: 10.1615/CritRevImmunol.v23.i4.30. [DOI] [PubMed] [Google Scholar]
- 17.Greil R, et al. Tracking death dealing by Fas and TRAIL in lymphatic neoplastic disorders: pathways, targets, and therapeutic tools. J Leukoc Biol. 2003;74(3):311–30. doi: 10.1189/jlb.0802416. [DOI] [PubMed] [Google Scholar]
- 18.Tinhofer I, et al. Differential sensitivity of CD4+ and CD8+ T lymphocytes to the killing efficacy of Fas (Apo-1/CD95) ligand + tumor cells in B chronic lymphocytic leukemia. Blood. 1998;91(11):4273–81. [PubMed] [Google Scholar]
- 19.Villunger A, et al. Drug-induced apoptosis is associated with enhanced Fas (Apo-1/CD95) ligand expression but occurs independently of Fas (Apo-1/CD95) signaling in human T-acute lymphatic leukemia cells. Cancer Res. 1997;57(16):3331–4. [PubMed] [Google Scholar]
- 20.Villunger A, et al. Constitutive expression of Fas (Apo-1/CD95) ligand on multiple myeloma cells: a potential mechanism of tumor-induced suppression of immune surveillance. Blood. 1997;90(1):12–20. [PubMed] [Google Scholar]
- 21.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–9. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 23.Topalian SL, et al. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16(5):275–87. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pleyer L, Valent P, Greil R. Mesenchymal Stem and Progenitor Cells in Normal and Dysplastic Hematopoiesis-Masters of Survival and Clonality? Int J Mol Sci. 2016;17(7):1009. doi: 10.3390/ijms17071009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eggermont AM, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2015;16(5):522–30. doi: 10.1016/S1470-2045(15)70122-1. [DOI] [PubMed] [Google Scholar]
- 27.Postow MA, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006–17. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larkin J, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373(13):1270–1. doi: 10.1056/NEJMc1509660. [DOI] [PubMed] [Google Scholar]
- 29.Weber JS, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16(4):375–84. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 30.Robert C, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–30. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 31.Robert C, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521–32. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 32.Hamid O, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brahmer J, et al. Nivolumab versus docetaxel in advanced squamous-cell Non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–35. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Borghaei H, et al. Nivolumab versus docetaxel in advanced nonsquamous Non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–39. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gillison ML, et al. Abstract CT099: Nivolumab (nivo) vs investigator’s choice (IC) for recurrent or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC): CheckMate-141. 2016. [Google Scholar]
- 36.Motzer RJ, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373(19):1803–13. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosenberg, J.E. et al. PD-L1 expression, Cancer Genome Atlas (TCGA) subtype, and mutational load as independent predictors of response to atezolizumab (atezo) in metastatic urothelial carcinoma (mUC; IMvigor210). ASCO Meeting Abstracts, 2016. 34(15_suppl): p. 104.
- 38.Ansell SM, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–9. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Armand P. et al. Programmed Death-1 Blockade With Pembrolizumab in Patients With Classical Hodgkin Lymphoma After Brentuximab Vedotin Failure. J Clin Oncol. 2016;34(31):3733–39. [DOI] [PMC free article] [PubMed]
- 40.Nishimura H, et al. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11(2):141–51. doi: 10.1016/S1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 41.Tai X, et al. Induction of autoimmune disease in CTLA-4−/− mice depends on a specific CD28 motif that is required for in vivo costimulation. Proc Natl Acad Sci U S A. 2007;104(34):13756–61. doi: 10.1073/pnas.0706509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009;9(4):271–85. doi: 10.1038/nri2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.To PD-L1 or Not to PD-L1: That Is the Question | Cancer Commons. 2016; Available from: https://www.cancercommons.org/2015/09/09/to-pd-l1-or-not-to-pd-l1-that-is-the-question/. Accessed 11 Oct 2016.
- 44.Herbst RS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–7. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 46.George T.J. et al. Tumor mutational burden as a potential biomarker for PD1/PD-L1 therapy in colorectal cancer. ASCO Meeting Abstracts, 2016. 34(15_suppl): p. 3587.
- 47.Rizvi NA, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosenberg JE, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909–20. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh PP, et al. Gastroenterol Rep (Oxf) 2015. Immune checkpoints and immunotherapy for colorectal cancer; pp. 289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koopman M, et al. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer. 2009;100(2):266–273. doi: 10.1038/sj.bjc.6604867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Le DT, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372(26):2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ali SM, et al. Prospective comprehensive genomic profiling of advanced gastric carcinoma cases reveals frequent clinically relevant genomic alterations and new routes for targeted therapies. Oncologist. 2015;20(5):499–507. doi: 10.1634/theoncologist.2014-0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Papaemmanuil E, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–21. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salter JD, Bennett RP, Smith HC. The APOBEC protein family: united by structure, divergent in function. Trends Biochem Sci. 2016;41(7):578–94. doi: 10.1016/j.tibs.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knisbacher BA, Gerber D, Levanon EY. DNA editing by APOBECs: a genomic preserver and transformer. Trends Genet. 2016;32(1):16–28. doi: 10.1016/j.tig.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 56.Robbiani DF, Nussenzweig MC. Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Annu Rev Pathol. 2013;8:79–103. doi: 10.1146/annurev-pathol-020712-164004. [DOI] [PubMed] [Google Scholar]
- 57.Rebhandl S, et al. APOBEC3 signature mutations in chronic lymphocytic leukemia. Leukemia. 2014;28(9):1929–32. doi: 10.1038/leu.2014.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rebhandl S, et al. Alternative splice variants of AID are not stoichiometrically present at the protein level in chronic lymphocytic leukemia. Eur J Immunol. 2014;44(7):2175–87. doi: 10.1002/eji.201343853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roberts SA, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45(9):970–6. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet. 2013;45(9):977–83. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morganella S, et al. The topography of mutational processes in breast cancer genomes. Nat Commun. 2016;7:11383. doi: 10.1038/ncomms11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chan K, et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat Genet. 2015;47(9):1067–72. doi: 10.1038/ng.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Swanton C, et al. APOBEC enzymes: mutagenic fuel for cancer evolution and heterogeneity. Cancer Discov. 2015;5(7):704–12. doi: 10.1158/2159-8290.CD-15-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Walker BA, et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun. 2015;6:6997. doi: 10.1038/ncomms7997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cescon DW, Haibe-Kains B, Mak TW. APOBEC3B expression in breast cancer reflects cellular proliferation, while a deletion polymorphism is associated with immune activation. Proc Natl Acad Sci U S A. 2015;112(9):2841–6. doi: 10.1073/pnas.1424869112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Henderson S, et al. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 2014;7(6):1833–41. doi: 10.1016/j.celrep.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 67.Hedegaard J, et al. Comprehensive transcriptional analysis of early-stage urothelial carcinoma. Cancer Cell. 2016;30(1):27–42. doi: 10.1016/j.ccell.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 68.Mullane SA, et al. Correlation of apobec mrna expression with overall survival and pd-l1 expression in urothelial carcinoma. Sci Rep. 2016;6:27702. doi: 10.1038/srep27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leonard B, et al. APOBEC3G expression correlates with T-cell infiltration and improved clinical outcomes in high-grade serous ovarian carcinoma. Clin Cancer Res. 2016;22(18):4746–55. doi: 10.1158/1078-0432.CCR-15-2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Postow MA, et al. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J Immunother Cancer. 2015;3:23. doi: 10.1186/s40425-015-0070-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goldinger S.M. et al. The peripheral blood TCR repertoire to facilitate patient stratification for immune checkpoint blockade inhibition in metastatic melanoma. ASCO Meeting Abstracts, 2016. 34(15_suppl): p. 3026.
- 72.Robert L, et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zill, O.A. et al. Somatic genomic landscape of over 15,000 patients with advanced-stage cancer from clinical next-generation sequencing analysis of circulating tumor DNA. ASCO Meeting Abstracts, 2016. 34(18_suppl): p. LBA11501.
- 74.Tran E, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350(6266):1387–90. doi: 10.1126/science.aad1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gros A, et al. Selection of circulating PD-1+ lymphocytes from cancer patients enriches for tumor-reactive and mutation-specific lymphocytes. J Immunother Cancer. 2015;3(Suppl 2):O2. doi: 10.1186/2051-1426-3-S2-O2. [DOI] [Google Scholar]
- 76.Fourcade J, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207(10):2175–86. doi: 10.1084/jem.20100637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Woo SR, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–27. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Márquez-Rodas I, et al. Immune checkpoint inhibitors: therapeutic advances in melanoma. Ann Transl Med. 2015;3(18):267. doi: 10.3978/j.issn.2305-5839.2015.10.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Koyama S, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bu M, et al. Ovarian carcinoma-infiltrating regulatory T cells were more potent suppressors of CD8(+) T cell inflammation than their peripheral counterparts, a function dependent on TIM3 expression. Tumour Biol. 2016;37(3):3949–56. doi: 10.1007/s13277-015-4237-x. [DOI] [PubMed] [Google Scholar]
- 81.Moorman JP, et al. Tim-3 pathway controls regulatory and effector T cell balance during hepatitis C virus infection. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sakuishi K, et al. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–94. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Farren MR, et al. Systemic immune activity predicts overall survival in treatment-naive patients with metastatic pancreatic cancer. Clin Cancer Res. 2016;22(10):2565–74. doi: 10.1158/1078-0432.CCR-15-1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gibbons DL, et al. 57O Efficacy, safety and tolerability of MEDI4736 (durvalumab [D]), a human IgG1 anti-programmed cell death-ligand-1 (PD-L1) antibody, combined with gefitinib (G): A phase I expansion in TKI-naive patients (pts) with EGFR mutant NSCLC. J Thorac Oncol. 2016;11(4 Suppl):S79. doi: 10.1016/S1556-0864(16)30171-X. [DOI] [Google Scholar]
- 85.Bendell J.C. et al. Clinical activity and safety of cobimetinib (cobi) and atezolizumab in colorectal cancer (CRC). ASCO Meeting Abstracts, 2016. 34(15_suppl): p. 3502.
- 86.Stone RM et al. The Multi-Kinase Inhibitor Midostaurin (M) Prolongs Survival Compared with Placebo (P) in Combination with Daunorubicin (D)/Cytarabine (C) Induction (ind), High-Dose C Consolidation (consol), and As Maintenance (maint) Therapy in Newly Diagnosed Acute Myeloid Leukemia (AML) Patients (pts) Age 18–60 with FLT3 Mutations (muts): An International Prospective Randomized (rand) P-Controlled Double-Blind Trial (CALGB 10603/RATIFY [Alliance]). 2015.
- 87.Wolleschak D, et al. Clinically relevant doses of FLT3-kinase inhibitors quizartinib and midostaurin do not impair T-cell reactivity and function. Haematologica. 2014;99(6):e90–3. doi: 10.3324/haematol.2014.104331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Burger JA, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373(25):2425–37. doi: 10.1056/NEJMoa1509388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Byrd JC, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213–23. doi: 10.1056/NEJMoa1400376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dubovsky JA, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122(15):2539–49. doi: 10.1182/blood-2013-06-507947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sagiv-Barfi I, et al. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci U S A. 2015;112(9):E966–72. doi: 10.1073/pnas.1500712112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stiff A, et al. Myeloid-Derived Suppressor Cells Express Bruton’s Tyrosine Kinase and Can Be Depleted in Tumor-Bearing Hosts by Ibrutinib Treatment. Cancer Res. 2016;76(8):2125–36. doi: 10.1158/0008-5472.CAN-15-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Atefi M, et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res. 2014;20(13):3446–57. doi: 10.1158/1078-0432.CCR-13-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen ML, et al. Sorafenib relieves cell-intrinsic and cell-extrinsic inhibitions of effector T cells in tumor microenvironment to augment antitumor immunity. Int J Cancer. 2014;134(2):319–31. doi: 10.1002/ijc.28362. [DOI] [PubMed] [Google Scholar]
- 95.Callahan MK, et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol Res. 2014;2(1):70–9. doi: 10.1158/2326-6066.CIR-13-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Salgado R, et al. Tumor-Infiltrating Lymphocytes and Associations With Pathological Complete Response and Event-Free Survival in HER2-Positive Early-Stage Breast Cancer Treated With Lapatinib and Trastuzumab: A Secondary Analysis of the NeoALTTO Trial. JAMA Oncol. 2015;1(4):448–54. doi: 10.1001/jamaoncol.2015.0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Denkert C, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol. 2015;33(9):983–91. doi: 10.1200/JCO.2014.58.1967. [DOI] [PubMed] [Google Scholar]
- 98.Ladoire S, Martin F, Ghiringhelli F. Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: the paradox of colorectal cancer. Cancer Immunol Immunother. 2011;60(7):909–18. doi: 10.1007/s00262-011-1046-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Miller CP, et al. JAK2 expression is associated with tumor-infiltrating lymphocytes and improved breast cancer outcomes: implications for evaluating JAK2 inhibitors. Cancer Immunol Res. 2014;2(4):301–6. doi: 10.1158/2326-6066.CIR-13-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zitvogel L, et al. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8(1):59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
- 101.Pfirschke C, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. 2016;44(2):343–54. doi: 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zitvogel L, et al. The anticancer immune response: indispensable for therapeutic success? J Clin Invest. 2008;118(6):1991–2001. doi: 10.1172/JCI35180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Galluzzi L, et al. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. 2015;28(6):690–714. doi: 10.1016/j.ccell.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 104.Melero I, et al. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer. 2015;15(8):457–72. doi: 10.1038/nrc3973. [DOI] [PubMed] [Google Scholar]
- 105.Postow MA, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366(10):925–31. doi: 10.1056/NEJMoa1112824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chandra RA, et al. A systematic evaluation of abscopal responses following radiotherapy in patients with metastatic melanoma treated with ipilimumab. Oncoimmunology. 2015;4(11):e1046028. doi: 10.1080/2162402X.2015.1046028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kim K, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. 2014;111(32):11774–9. doi: 10.1073/pnas.1410626111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Viaud S, et al. Gut microbiome and anticancer immune response: really hot Sh*t! Cell Death Differ. 2015;22(2):199–214. doi: 10.1038/cdd.2014.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Arumugam M, et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–80. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gophna U. Microbiology. The guts of dietary habits. Science. 2011;334(6052):45–6. doi: 10.1126/science.1213799. [DOI] [PubMed] [Google Scholar]
- 111.Dimmitt RA, et al. The role of postnatal acquisition of the intestinal microbiome in the early development of immune function. J Pediatr Gastroenterol Nutr. 2010;51(3):262–73. doi: 10.1097/MPG.0b013e3181e1a114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kau AL, et al. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474:327–336. doi: 10.1038/nature10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Neumann E, et al. Monoassociation with Lactobacillus acidophilus UFV-H2b20 stimulates the immune defense mechanisms of germfree mice. Braz J Med Biol Res. 1998;31(12):1565–73. doi: 10.1590/S0100-879X1998001200009. [DOI] [PubMed] [Google Scholar]
- 114.Duarte R, et al. Influence of normal microbiota on some aspects of the immune response during experimental infection with Trypanosoma cruzi in mice. J Med Microbiol. 2004;53(Pt 8):741–8. doi: 10.1099/jmm.0.45657-0. [DOI] [PubMed] [Google Scholar]
- 115.Oliveira MR, et al. Germ-free mice produce high levels of interferon-gamma in response to infection with Leishmania major but fail to heal lesions. Parasitology. 2005;131(Pt 4):477–88. doi: 10.1017/S0031182005008073. [DOI] [PubMed] [Google Scholar]
- 116.Round JL, Mazmanian SK. The gut microbiome shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9(5):313–23. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Russo E, et al. The interplay between the microbiome and the adaptive immune response in cancer development. Therap Adv Gastroenterol. 2016;9(4):594–605. doi: 10.1177/1756283X16635082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tjalsma H, et al. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol. 2012;10(8):575–82. doi: 10.1038/nrmicro2819. [DOI] [PubMed] [Google Scholar]
- 119.Boleij A, Tjalsma H. Gut bacteria in health and disease: a survey on the interface between intestinal microbiology and colorectal cancer. Biol Rev Camb Philos Soc. 2012;87(3):701–30. doi: 10.1111/j.1469-185X.2012.00218.x. [DOI] [PubMed] [Google Scholar]
- 120.Kwa M et al. The Intestinal Microbiome and Estrogen Receptor-Positive Female Breast Cancer. J Natl Cancer Inst. 2016;108(8). doi:10.1093/jnci/djw029. [DOI] [PMC free article] [PubMed]
- 121.Nougayrede JP, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313(5788):848–51. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 122.Mangerich A, et al. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc Natl Acad Sci U S A. 2012;109(27):E1820–9. doi: 10.1073/pnas.1207829109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wu S, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15(9):1016–22. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Arpaia N, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504(7480):451–5. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zitvogel L, et al. Cancer and the gut microbiota: an unexpected link. Sci Transl Med. 2015;7(271):271ps1. doi: 10.1126/scitranslmed.3010473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Viaud S, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342(6161):971–6. doi: 10.1126/science.1240537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Iida N, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342(6161):967–70. doi: 10.1126/science.1240527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Stringer AM, et al. Faecal microflora and beta-glucuronidase expression are altered in an irinotecan-induced diarrhea model in rats. Cancer Biol Ther. 2008;7(12):1919–25. doi: 10.4161/cbt.7.12.6940. [DOI] [PubMed] [Google Scholar]
- 129.Gupta E, et al. Metabolic fate of irinotecan in humans: correlation of glucuronidation with diarrhea. Cancer Res. 1994;54(14):3723–5. [PubMed] [Google Scholar]
- 130.Takasuna K, et al. Involvement of beta-glucuronidase in intestinal microflora in the intestinal toxicity of the antitumor camptothecin derivative irinotecan hydrochloride (CPT-11) in rats. Cancer Res. 1996;56(16):3752–7. [PubMed] [Google Scholar]
- 131.Maruya M, et al. Impaired selection of IgA and intestinal dysbiosis associated with PD-1-deficiency. Gut Microbes. 2013;4(2):165–71. doi: 10.4161/gmic.23595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Vétizou M, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350(6264):1079–84. doi: 10.1126/science.aad1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.