

Introduction
Hepatocellular carcinoma (HCC) is a frequent and deadly human disease (1). Several lines of evidence indicate that the gradual accumulation of genomic alterations, leading to progressive deregulation of different signaling pathways, induces the progressive evolution of initiated liver cells to dysplastic nodules and malignant lesions (1). Molecular events leading to cell cycle deregulation in HCC include up-regulation of RAS/ERK, PI3K/AKT, IKK/NF-kB, WNT, TGF-β, NOTCH, HEDGEHOG, and HIPPO signaling pathways, and genes involved in DNA repair process (2). Better understanding of the molecular mechanisms underlying hepatocarcinogenesis may hasten the identification of novel molecular HCC progression markers and development of new diagnostic and therapeutic strategies.
Heparan-sulfate glycosaminoglycans (HSGAGs)
The HSGAGs are linear polysaccharides constituted by 50–200 disaccharide repeats, with regions of glucuronic acid-N-acetylglucosamine and regions of 2-O-sulfated uronic acid/N-glucosamine sulfated at 3-O and 6-O positions interspaced by transition areas in which both sulfoglucosamine and N-acetylglucosamine are present. The N-position of glucosamine can be sulfated, acetylated, or unmodified (3). The polysaccharides, covalently attached to a polypeptide core, form heparan sulfate proteoglycans (HSPGs) located at the cell surface (Figure 1A) and also present in the extracellular matrix.
Figure 1.
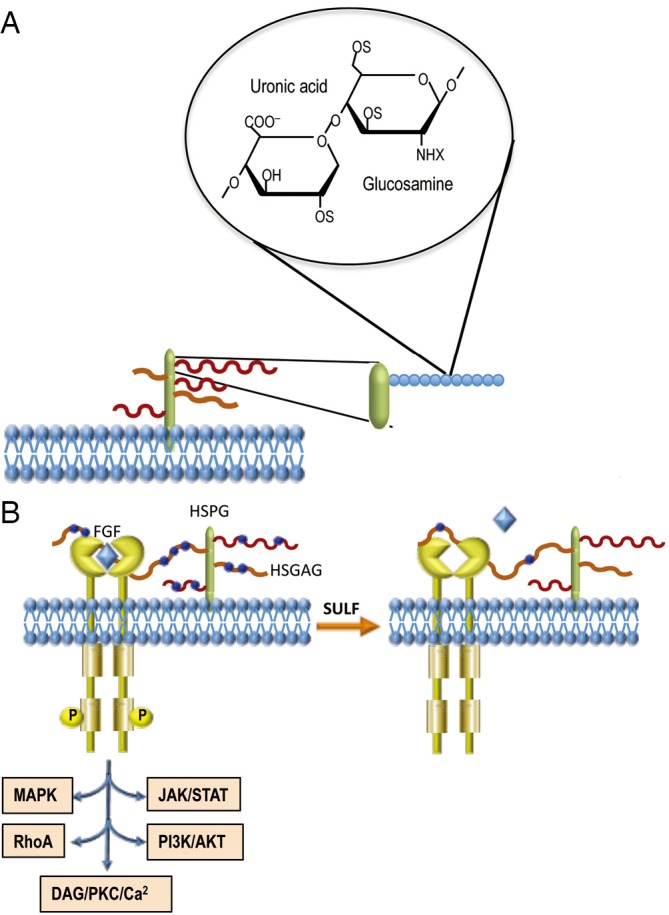
Structure of HSGAGs and proposed model of SULFs in heparin bindings growth factors. (A) Heparan sulfate proteoglycans on cell surface showing heparan-sulfate glycosaminoglycans (HSGAGs) attached to a protein core. Enlarged detail: 2-O-sulfated uronic acid-N-glucosamine sulfated at the 3-O and 6-O positions. NHX on glucosamine: N position sulfated, acetylated, or unchanged; (B) sulfate residues on HSGAG (blue circles) can favor ligand bridging (i.e., FGF) to its receptor, and signaling pathways activation. HSGAG desulfation by sulfatases can impede the link of ligands to their receptors.
HSGAGs bind and interact with chemokines, enzymes, and growth factors involved in tumor development. The latter include fibroblast growth factors (FGF1, FGF2), vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), transforming growth factor-β (TGF-β), and platelet-derived growth factor (PDGF) (4). Predominantly, extracellular proteins such as FGFs bind to a tetra- or hexasaccharide motif within an HSGAG chain (5). Long oligosaccharide sequences can bridge protein-protein complexes, thus favoring the homo-oligomerization and/or bridging a ligand to its receptor (Figure 1B). Thus, HSGAG chains of proteoglycans function as binding sites for signaling molecules. The sequence and conformational plasticity of the HSGAG polymer determine the binding specificity with consequent regulation of different biological processes.
HSGAGs may play important roles in cancer initiation and progression by modulating tumor cell growth, invasiveness, and metastatic potential (6). Different observations indicate that changes in expression level and oligosaccharide sequence of cell-surface HSPGs might contribute to cell transformation (7-9).
Heparin-degrading sulfatases
The heparin sulfate 6-O-endosulfatases 1 and 2, designated as sulfatase 1 (SULF1) and sulfatase 2 (SULF2), respectively, hydrolyze the sulfate ester bonds of HSGAGs. SULF1 and SULF2 show the same in vitro specificity to trisulfated disaccharides, but the two sulfatases display structural and functional differences in mice and humans (10). Lai and coworkers (11) reported that SULF1 desulfates HSPGs on cells surface and inhibits HCC tumor cell growth in vitro and in nude mice, partially through effects on gene expression mediated through histone H4 acetylation. SULF1 is downregulated in most HCC cell lines and approximately 30% of primary HCCs. SULF1 transfection in HCC cells reduces proliferation rate by suppressing heparin-binding to growth factor signaling. In contrast, SULF2 promotes hepatocarcinogenesis in nude mice and its expression is associated with more rapid recurrence and shorter survival of HCC patients, after surgical resection (11). Furthermore, Shire and coworkers (12) showed SULF1 mRNA down-regulation in 9/11 HCC cell lines and in only 6/10 primary tumors. They found that SULF1 promoter acquires a silenced chromatin state in low SULF1-expressing cells, through an increase in di/trimethyl-K9H3 and trimethyl-K27H3 and a concomitant loss of activating acetyl K9, K14H3. Restoration of SULF1 mRNA expression by 5-Aza-dC sensitized HCC cells to drug-induced apoptosis.
The oncosuppressor effect of SULF1 was confirmed by the recent observation that microRNA-21, a suppressor of PTEN and hSulf-1 expression, promotes HCC progression through the AKT/ERK pathways (13). It was hypothesized that the inhibition of histone deacetylase by SULF1 induces a rise in acetylated histone H4, which leads to inhibition of RAS/ERK and PI3K/AKT signaling (13). A recent contribution (14) provides a hypothetical mechanism whereby cancer cells could evade SULF1 suppressor action. The removal by sulfatases of the sulfate moiety from 6-O of heparan sulfate on HSPGs, should result in decreased FGF binding sites on HSPG that should disfavor FGF bridging to its receptor (Figure 1B). However, HIF-1α stabilization, under low oxygen conditions prevailing in solid tumors, shuts down the transcription of sulfatases, which may results in sulfation of 6-O of heparan sulfate on HSPGs. This would favor FGF signaling, cell migration, and invasion (14).
In apparent contradiction with above reports, gene expression analysis of human HCCs showed that high SULF1 overexpression is associated with poor survival, suggesting that SULF1 is oncogenic in most HCC in vivo (15). This conclusion is supported by Dhanasekaran and coworkers, who in a recent study (16) provided convincing data in support of the oncogenic role of SULF1 in hepatocarcinogenesis and unraveled some of the molecular mechanisms involved. These authors used a transgenic mouse model overexpressing SULF1 (Sulf1-Tg) to evaluate the effects of SULF1 on the diethylnitrosamine (DENA) model of hepatocarcinogenesis. They showed a higher incidence of large and multifocal HCCs in DENA-treated Sulf1-Tg mice, compared to wild-type (WT) mice. They also found that lung metastases were present in 75% of Sulf1-Tg mice but not in WT mice. These in vivo experiments clearly indicated that SULF1 overexpression enhances liver tumor progression and strongly support a tumor promoter role for SULF1.
In order to identify the molecular players responsible for the oncogenic role of SULF1, Dhanasekaran and coworkers (16) evaluated by transcriptome analysis of non-DENA treated liver tissues the pathways and biological processes activated in Sulf1-Tg compared to WT mice. They observed the up-regulation of biological processes involving cytoskeletal remodeling, cell adhesion, and muscle development in Sulf1-Tg mice. Noticeably, they also found the preferential activation of the epithelial mesenchymal transition (EMT) process in Sulf1-Tg mice. EMT is a process implicated in tumor progression and development of metastases (17) and its activation is in line with the presence of larger tumors and lung metastases in Sulf1-Tg mice.
TGF-β is known to be implicated in EMT of cancer cells (18,19). The analysis by Dhanasekaran and coworkers (16) of genes involved in TGF-β signaling showed higher expression of Smad2 and Smad6 in Sulf1-Tg than in WT mice. Also, an increase in phosphorylation of Smad2/3 in peritumoral liver tissues and, at a higher extent, in tumors of Sulf1-Tg mice than WT mice occurred. These changes were associated with a lower expression of the epithelial marker E-cadherin, and an increase in the mesenchymal markers N-cadherin and vimentin in tumors of Sulf1-Tg mice when compared to HCC of WT mice. Further, the authors provided the immunohistochemical evidence of cytoplasmic expression of the mesenchymal proteins in tumor cells of Sulf1-Tg mice suggesting the acquisition of the mesenchymal traits by the epithelial cells (Figure 2).
Figure 2.
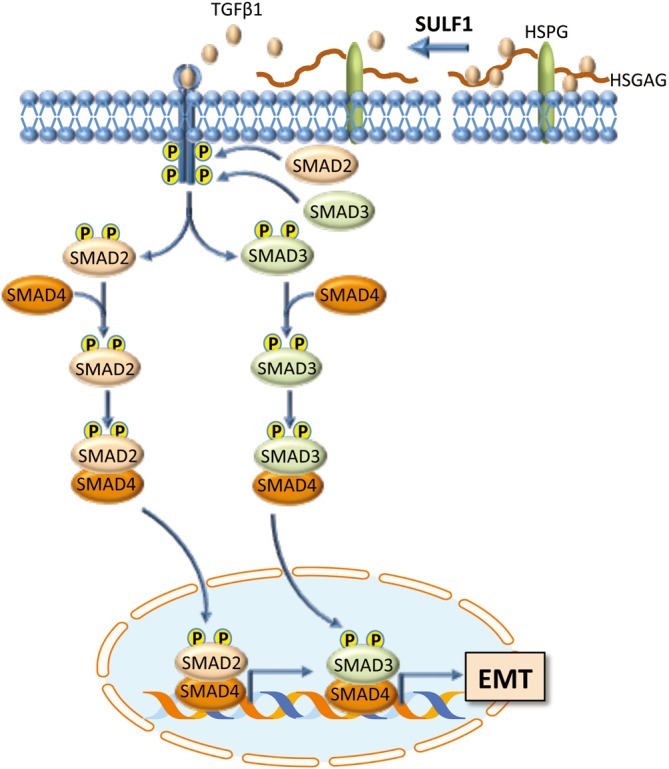
A possible mechanism responsible for the oncogenic role of SULF1. TGF-β1, bound to the sulfate moiety from 6-O of heparan sulfate on HSPGs, is released following the desulfation of 6-O of heparan sulfate on HSPGs by SULF1. This allows TGF-β1 binding to the receptor system. After binding and phosphorylation, the receptor activates by phosphorylation the SMAD2 and SMAD3 effectors. This is followed by the formation of heteromeric complexes of SMAD2 and SMAD3 with SMAD4, which then translocate to the nucleus and activate specific DNA sequences involved in EMT.
The results in mice were confirmed in a series of experiments with various human HCC cell lines. In particular, it was observed that the overexpression of SULF1 increased the phosphorylation of both SMAD2 and SMAD3, whereas the suppression of SULF1 expression led to the opposite effects. Moreover, it was clearly shown a link between SULF1 up-regulation and EMT: SULF1-transfected cells, treated with TGF-β1, exhibited a decrease in the tight junction protein Zona occludens protein 1 (Zo-1) and in E-cadherin, as well as an increase in the mesenchymal markers N-cadherin, vimentin, and α-smooth muscle actin (αSMA). Opposite changes occurred when SULF1 expression was suppressed.
Some elegant experiments were devoted to the analysis of the role of SULF1 catalytic activity. For this aim, a SULF1 mutant with loss of catalytic activity was created. It was thus demonstrated that the stimulation of cell migration and invasiveness and SMAD2/3 phosphorylation, present in cell transfected with SULF1 and treated with TGF-β1, was lost in cells transfected with the mutant SULF1 devoid of sulfatase activity.
The classification of HCC patients into two prognostic clusters, according the microarray expression profile, showed that the majority of patients with high SULF1 expression (76%) belonged to the poor prognosis cluster and gene expression correlation analysis confirmed the association between SULF1, TGF-β activation, EMT and five EMT driver genes (vimentin, SNAI1, COL1A2, TGF-β1, SPARC) in human HCC. The association between high SULF1 and high phospho-SMAD2/3 expression, decreased expression of E-cadherin and increased that of vimentin and αSMA was confirmed in the tumor specimens.
Importantly, the authors presented evidence that Hep3B and PLC/PRF5PRF/5 cell lines, used in the SULF1 experiments, do not express SULF2. Thus, it is unlike that the observed results are a consequence of overexpression of the SULF2 gene, which is known to possess an oncogenic activity.
Conclusions
According to the results of Dhanasekaran and coworkers, SULF1 is a potential biomarker of tumor progression and thus a novel target for drug development. At present, there are no sufficient elements to understand the conflictual results about the role of SULF1 in tumorigenesis. TGF-β is known to behave as an oncogene or an oncosuppressor gene in cancer (20). The effect and function of genes can be opposite and adaptable in cells with different genomes or in different contexts and the response to the same protein could be cellular genetic/context-dependent (20). However, a link between SULF1 overexpression and the oncosuppressor role of TGF-β has not yet been demonstrated. Further research is needed to confirm a possible antagonistic role of SULF1 in liver carcinogenesis and solve this dilemma.
Acknowledgements
None.
Footnotes
Provenance: This is a Guest Commentary commissioned by Guest Editor Haitao Zhao, MD, PhD (Department of Liver Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China).
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Frau M, Biasi F, Feo F, et al. Prognostic markers and putative therapeutic targets for hepatocellular carcinoma. Mol Aspects Med 2010;31:179-93. 10.1016/j.mam.2010.02.007 [DOI] [PubMed] [Google Scholar]
- 2.Calvisi DF, Frau M, Tomasi ML, et al. Deregulation of signalling pathways in prognostic subtypes of hepatocellular carcinoma: novel insights from interspecies comparison. Biochim Biophys Acta 2012;1826:215-37. [DOI] [PubMed] [Google Scholar]
- 3.Kreuger J, Spillmann D, Li JP, et al. Interactions between heparan sulfate and proteins: the concept of specificity. J Cell Biol 2006;174:323-7. 10.1083/jcb.200604035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forsten-Williams K, Chu CL, Fannon M, et al. Control of growth factor networks by heparan sulfate proteoglycans. Ann Biomed Eng 2008;36:2134-48. 10.1007/s10439-008-9575-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morimoto-Tomita M, Uchimura K, Werb Z, et al. Cloning and characterization of two extracellular heparin-degrading endosulfatases in mice and humans. J Biol Chem 2002;277:49175-85. 10.1074/jbc.M205131200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Z, Fan JQ, Li J, et al. Promoter hypermethylation correlates with the Hsulf-1 silencing in human breast and gastric cancer. Int J Cancer 2009;124:739-44. 10.1002/ijc.23960 [DOI] [PubMed] [Google Scholar]
- 7.Ai X, Do AT, Lozynska O, et al. QSulf1 remodels the 6-O sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J Cell Biol 2003;162:341-51. 10.1083/jcb.200212083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delehedde M, Lyon M, Gallagher JT, et al. Fibroblast growth factor-2 binds to small heparin-derived oligosaccharides and stimulates a sustained phosphorylation of p42/44 mitogen-activated protein kinase and proliferation of rat mammary fibroblasts. Biochem J 2002;366:235-44. 10.1042/bj20011718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang S, Ai X, Freeman SD, et al. QSulf1, a heparan sulfate 6-O-endosulfatase, inhibits fibroblast growth factor signaling in mesoderm induction and angiogenesis. Proc Natl Acad Sci U S A 2004;101:4833-8. 10.1073/pnas.0401028101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei G, Bai X, Gabb MM, et al. Location of the glucuronosyltransferase domain in the heparan sulfate copolymerase EXT1 by analysis of Chinese hamster ovary cell mutants. J Biol Chem 2000;275:27733-40. [DOI] [PubMed] [Google Scholar]
- 11.Lai JP, Thompson JR, Sandhu DS, et al. Heparin-degrading sulfatases in hepatocellular carcinoma: roles in pathogenesis and therapy targets. Future Oncol 2008;4:803-14. 10.2217/14796694.4.6.803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shire A, Lomberk G, Lai JP, et al. Restoration of epigenetically silenced SULF1 expression by 5-aza-2-deoxycytidine sensitizes hepatocellular carcinoma cells to chemotherapy-induced apoptosis. Med Epigenet 2015;3:1-18. 10.1159/000375461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bao L, Yan Y, Xu C, et al. MicroRNA-21 suppresses PTEN and hSulf-1 expression and promotes hepatocellular carcinoma progression through AKT/ERK pathways. Cancer Lett 2013;337:226-36. 10.1016/j.canlet.2013.05.007 [DOI] [PubMed] [Google Scholar]
- 14.Khurana A, Beleford D, He X, et al. Role of heparan sulfatases in ovarian and breast cancer. Am J Cancer Res 2013;3:34-45. [PMC free article] [PubMed] [Google Scholar]
- 15.Yang JD, Sun Z, Hu C, et al. Sulfatase 1 and sulfatase 2 in hepatocellular carcinoma: associated signaling pathways, tumor phenotypes, and survival. Genes Chromosomes Cancer 2011;50:122-35. 10.1002/gcc.20838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dhanasekaran R, Nakamura I, Hu C, et al. Activation of the transforming growth factor-β/SMAD transcriptional pathway underlies a novel tumor-promoting role of sulfatase 1 in hepatocellular carcinoma. Hepatology 2015;61:1269-83. 10.1002/hep.27658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Ma J, Qian X, et al. Regulation of EMT by Notch signaling pathway in tumor progression. Curr Cancer Drug Targets 2013;13:957-62. 10.2174/15680096113136660101 [DOI] [PubMed] [Google Scholar]
- 18.Morrison CD, Parvani JG, Schiemann WP. The relevance of the TGF-β Paradox to EMT-MET programs. Cancer Lett 2013;341:30-40. 10.1016/j.canlet.2013.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014;15:178-96. 10.1038/nrm3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stepanenko AA, Vassetzky YS, Kavsan VM. Antagonistic functional duality of cancer genes. Gene 2013;529:199-207. 10.1016/j.gene.2013.07.047 [DOI] [PubMed] [Google Scholar]