

Abstract
Endothelial progenitor cells (EPCs) have an important role in maintaining endothelial homeostasis. Previous studies reported that smoking has detrimental effects on EPCs; however, recent studies revealed that short-term nicotine exposure may benefit EPCs. As most smokers are exposed to nicotine over an extended time period, the present study aimed to investigate the long-term effects of nicotine on EPCs. Mice were administered nicotine orally for 1, 3 or 6 months. The mice exposed to nicotine for 1 month demonstrated increased EPC counts and telomerase activity and reduced cell senescence compared with control mice, consistent with previous reports. However, long-term nicotine exposure resulted in opposing effects on EPCs, causing decreased counts, functional impairment and reduced telomerase activity. Furthermore, the effects of nicotine exposure were correlated with changes in sirtuins type 1 (SIRT1) protein expression. The current study indicated that long-term nicotine exposure induces dysfunction and senescence of EPCs, which may be associated with impairment of telomerase activity through SIRT1 downregulation. The present results emphasize the necessity of smoking cessation to prevent dysfunction of EPCs.
Keywords: endothelial progenitor cells, nicotine, senescence, telomerase, SIRT1
Introduction
Endothelial progenitor cells (EPCs) are important in maintaining endothelial homeostasis (1,2). In a variety of pathological processes such as atherosclerosis, acute myocardial infarction and percutaneous coronary intervention, EPCs migrate to the site of endothelial injury or tissue ischemia, and promote endothelial repair and neovascularization (3). Numerous previous studies have reported a number of factors, including smoking, hypertension and diabetes mellitus, that reduce the number of circulating EPCs and impair their function (4,5).
Smoking is a factor that demonstrates a marked correlation with cardiovascular diseases and associated mortality. In 2000, >1 in 10 cardiovascular deaths were attributable to smoking (6). However, multiple previous studies focused on the association between smoking and endothelial cells, reporting that smoking markedly impaired endothelial function (4) and integrity (7). More recently, reports documented that smoking has detrimental effects on EPCs: In 2001, Vase et al (8) identified that smoking was an important risk factor for a reduction in circuiting EPC number. Subsequent studies demonstrated that the number of circulating EPCs was reduced in chronic smokers, and that reduced EPC levels are restored following smoking cessation (9,10).
Notably, nicotine may have benefits for EPCs. Wang et al (11) reported that low concentrations of nicotine increase the EPC count and their proliferative, migratory, adhesive and vasculogenic capacity. Furthermore, nicotine administration improved the blood flow in a rat ischemic hindlimb model by enhancing the function of transplanted EPCs (12). A hypothesized explanation of these effects was that nicotine reduced EPC senescence by activating telomerase through the PI3K/Akt pathway (13). However, these conclusions were made from short-term experiments (>4 weeks), and the effect of long-term nicotine administration on EPCs remains unclear. As most smokers are long-term consumers of tobacco and cigarettes, the present study aimed to determine the long-term effects of nicotine on EPCs.
Materials and methods
Animal experiments
All animal experiments conformed to the Guide for the Care and Use of Laboratory Animals (14) and the protocol was approved by the Experimental Animal and Ethics Committee of the Third Military Medical University (Chongqing, China).
A total of 120 C57BL/6 male mice (age, 8 weeks; weight, 28±1.32 g) were purchased from the Animal Centre at the Medical Department of Beijing University (Beijing, China) and maintained in 22°C with a 12 h light/dark cycle, specific pathogen-free conditions and ad libitum access to water. Prior to experimental treatment, all mice were maintained on a normal chow diet for 1 week. Mice were randomly allocated into 6 groups (n=20 mice per group): Control group mice sacrified at 1, 3 or 6 months (abbreviated as C-1 M, C-3 M and C-6 M, respectively); and nicotine-treated groups sacrificed at 1, 3 or 6 months (abbreviated as N-1 M, N-3 M and N-6 M, respectively). In nicotine-treated groups, 100 ng/ml nicotine (Jingkehuaxue, Shanghai, China) diluted in 2% sucrose water was administered orally, on a daily basis. The control groups received 2% sucrose water.
Murine EPC culture and identification
Murine EPCs were isolated and cultured as previously described (15). Briefly, mice were sacrificed by cervical dislocation following anesthetization with an overdose of sodium pentobarbitone (intraperitoneal injection, 60 mg/kg). Bone marrow were obtained by flushing mice femurs, then the flushing fluid was subjected to density gradient centrifugation in order to isolate mononuclear cells. The cells were resuspended in endothelial cell growth medium (EGM-2MV BulletKit medium; Lonza, Basel, Switzerland) containing 5% fetal bovine serum (FBS) and supplemented with recombinant human (rh) epidermal growth factor, rh fibroblast growth factor-B, rh insulin-like growth factor-1, rh vascular endothelial cell growth factor (VEGF), ascorbic acid and heparin. Equal cell densities (4.106 cells/cm2) were seeded in fibronectin-coated culture plates and cultured in an incubator at 37°C and 5% CO2. The culture medium was changed after 48 h following seeding, and every 3 days subsequent to this. In order to identify EPCs, cells were subjected to fluorescent labeling with acetylated low density lipoprotein (DiI-ac-LDL) and fluorescein isothiocyanate-conjugated Ulex europaeus agglutinin (FITC-UEA-I); the double-positive cells were identified as EPCs.
EPC number and proliferation assay
After culturing for 7 days, cells were stained with DiI-ac-LDL and FITC-UEA-I, and the number of double-positive cells was counted in six randomly selected fields of view. FITC-labelled bromodeoxyuridine (BrdU) incorporation was used to assess EPC proliferation activity according to the manufacturer's instructions. Briefly, 10 mM BrdU was added to cells cultured in 96-well plates, and these were incubated for 24 h. The cells were fixed with 4% paraformaldehyde and DAPI stained. Fluorescent cells were observed with a fluorescence microscope, and images were analyzed using Image J software (version 1.50i; imagej.nih.gov/ij/). The proliferative ratio was calculated using the equation: Proliferative ratio (%) = (BrdU-positive cells/DAPI positive cells) × 100.
EPC migration assay
The migration of EPCs was assessed within a modified Boyden chamber assay. In brief, after culturing for 7 days, EPCs were resuspended in serum-free EGM-2 medium, and 5×104 cells were placed in the upper chamber, whilst the lower chamber was filled with medium containing 5% FBS and 20 ng/ml VEGF. After incubation for 12 h, the upper side of the membrane was wiped with a cotton swab, and the membranes were collected and fixed with 4% paraformaldehyde, which was followed by crystal violet staining. The stained cells were counted in six random fields of view using a light microscope.
EPC senescence assay
EPC senescence was assessed using a Senescence β-galactosidase Staining kit (Beyotime Institute of Biotechnology, Haimen, China) according to the manufacturer's instructions. Briefly, cells were cultured in 6-well plates for 14 days, were washed with phosphate-buffered saline, fixed with 4% paraformaldehyde, then cells were incubated overnight with the kit staining mixture at 37°C. The positive (blue) cells were observed under a light microscope.
Telomerase activity assay
Quantitative polymerase chain reaction (qPCR) of telomerase reverse transcriptase (TERT) expression was used to assess telomerase activity. Total RNA was extracted and purified using RNAiso Plus (cat. no. 9018; Takara Biotechnology Co., Ltd., Dalian, China). Single cDNAs were synthesized from RNA using a PrimeScript™ RT reagent Kit (TaKaRa, China, Cat number: RR037A) according to the manufacturer's instruction A qPCR kit to detect mouse TERT (KeyGen Biotech. Co. Ltd., Nanjing, China) was used according to the manufacturer's instructions. The specific primers for TERT and GAPDH (as the reference gene) were supplied in the kit. The thermal cycling conditions were as follows: Initial denaturation at 95°C for 10 sec, 40 cycles of denaturation at 95°C for 15 sec, then annealing at 60°C for 60 sec. The expression level of TERT mRNA was calculated by the ΔCq method (16).
Furthermore, the telomerase activity was also assessed by the telomeric repeat amplification protocol assay (17). The telomerase reaction product was amplified by PCR using the TeloTAGGG Telomerase PCR ELISAPLUS kit (cat. no. 11854666910; Roche Diagnostics, Basel, Switzerland) according to the manufacturer's instructions.
Western blot analysis
EPCs were solubilized using a lysis buffer (Beyotime Institute of Biotechnology), supplemented with 0.5 mM phenylmethylsulfonyl fluoride and centrifuged at 12,000 x g for 15 min. Protein concentrations were assayed using a bicinchoninic acid assay kit (Beyotime Institute of Biotechnology), 30 µg total protein from each group was separated by sodium dodecyl sulfate-polyacrylamide gel (10% for nAChR and 5% for SIRT1) electrophoresis and these proteins were transferred onto polyvinylidene fluoride membranes. The membranes were blocked with 5% bovine serum albumin, and probed with antibodies against sirtuin 1 (SIRT1; dilution, 1:200; cat. no. sc-15404; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), alpha-7 nicotinic receptor (nAChR-α7; dilution, 1:500; cat. no. AB15332; Merck Millipore, Darmstadt, Germany) and β-actin (dilution, 1:1,000; cat. no. AA128; Beyotime Institute of Biotechnology) at 4°C overnight. Following this, horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibodies (dilution, 1:1,000; Beyotime Institute of Biotechnology) were incubated at 37°C for 1 h, and bands were detected using an enhanced chemiluminescence kit (cat. no. 32109; Thermo Fisher Scientific, Inc., Waltham, MA, USA). Densiometry signals were quantified by ImageQuant TL software (GE Healthcare Life Sciences, Chalfont, UK).
Statistical analysis
Data is presented as the mean ± standard error. Intergroup comparisons were performed by one-way analysis of variance test accompanied by post-hoc least significant difference test. P-values <0.05 were considered to indicate a statistically significant difference.
Results
Long-term nicotine exposure reduces EPC number
EPCs derived from mice in the different groups were cultured in the same conditions for 7 days. At this stage, the percentage of cells that double positive for the uptake of DiI-ac-LDL and binding of UEA-1 were 85.3±3.4, 84.2±2.6, 82.9±3.8, 86.1±4.3, 79.6±3.5 and 82.3±5.9% in C-1M, N-1M, C-3M, N-3M, C-6M and N-6M groups, respectively (Fig. 1A-C). There was no significant difference in the number of EPCs between the control groups at the different time points. Short-term nicotine exposure (1 month) significantly increased the EPC number; however, as duration of nicotine exposure increased, the EPC number was gradually reduced compared with the corresponding control group (Fig. 1D).
Figure 1.

Effect of nicotine on EPC number. EPCs positive for (A) acetylated low density lipoprotein; (B) fluorescein isothiocyanate-conjugated Ulex europaeus agglutinin, and (C) double-positive cells. (D) Statistical analysis of EPC number. *P<0.05 vs. C-1 M group; **P<0.05 vs. C-3 M group; ***P<0.05 vs. C-6 M group. Cells were extracted from 3 mice. EPCs, endothelial progenitor cells; C, control; M, month(s); N, nicotine-treated.
Long-term nicotine exposure reduces EPC proliferation and migration
The effect of nicotine on EPC proliferation was evaluated by BrdU incorporation assay. Short-term nicotine exposure non-significantly increased EPC proliferation compared with the matched control group. However, EPC proliferative activity was significantly reduced compared with the corresponding control group following exposure to nicotine for 6 months (Fig. 2A). Next, migratory activity of EPCs was determined using a modified Boyden chamber assay. There was no significant difference between the number of migrating cells in control groups, however, long-term nicotine exposure (6 months) significantly reduced EPC migration (Fig. 2B).
Figure 2.
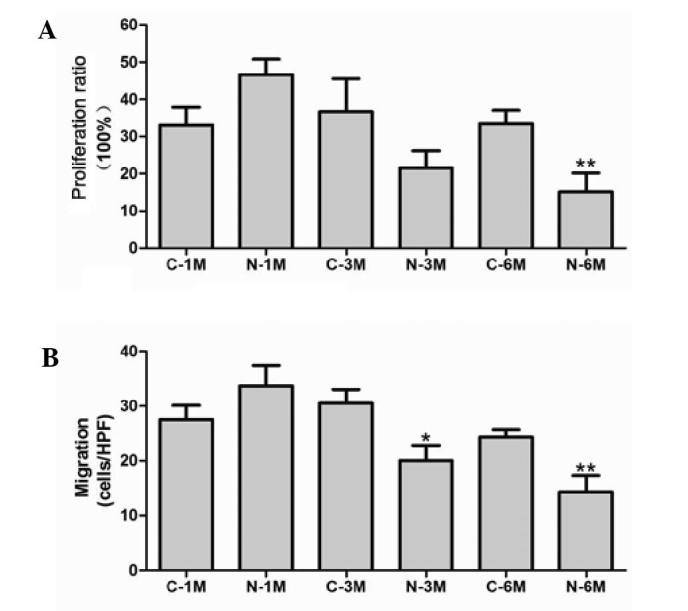
Effect of nicotine on (A) proliferation and (B) migration of endothelial progenitor cells. *P<0.05 vs. C-3 M group; **P<0.05 vs. C-6 M group. Cells were extracted from 3 mice. C, control; HPF, high-power field (x400); M, month(s); N, nicotine-treated..
Long-term nicotine exposure promotes EPC senescence
To assess EPC senescence, cells were cultured for 14 days and β-galactosidase was detected to determine the onset of cellular senescence. Short-term nicotine exposure significantly decreased the number of β-gal-positive, senescent cells compared with the control group; however, long-term exposure to nicotine significantly increased the number of β-gal-positive, senescent cells (Fig. 3).
Figure 3.

Effect of nicotine on endothelial progenitor cell senescence, determined by β-gal-positive cells in the (A) C-1, (B) N-1, (C) C-6 and (D) N-6 groups. (E) Statistical analysis of β-gal-positive cells *P<0.05 vs. C-1 M group; **P<0.05 vs. C-6 M group. Cells were extracted from 3 mice. C, control; M, month(s); N, nicotine-treated.
Long-term nicotine exposure reduces EPC telomerase activity
In the present study, the mRNA level of TERT was used to evaluate telomerase activity. As reported in Fig. 4A, expression of TERT was similar across the control groups. Short-term exposure to nicotine significantly increased TERT mRNA expression, but long-term exposure decreased this compared with corresponding control groups. Furthermore, telomerase activity was also determined, using a TeloTAGGG Telomerase PCR ELISA kit. As demonstrated in Fig. 4B, short-term exposure to nicotine significantly increased the telomerase activity but long-term exposure decreased this when compared with matched control groups. These results indicate that long-term nicotine exposure reduces EPC telomerase activity.
Figure 4.
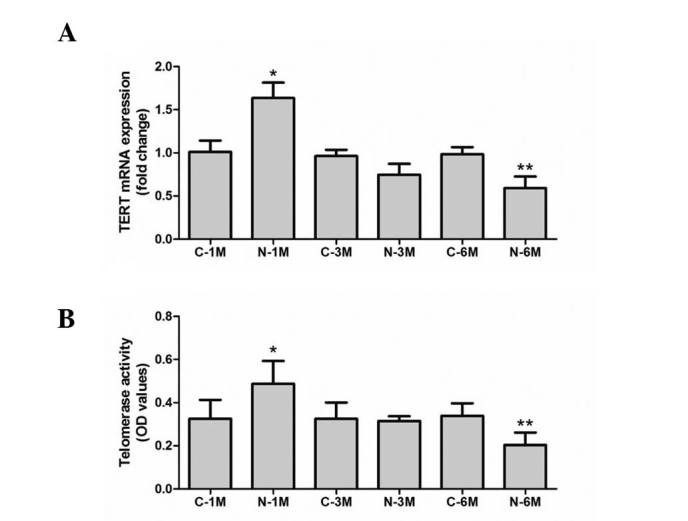
Effect of nicotine on endothelial progenitor cell telomerase activity. Telomerase activity was assessed by (A) TERT mRNA expression level and (B) a telomeric repeat amplification protocol assay. *P<0.05 vs. C-1 M group; **P<0.05 vs. C-6 M group. Cells were extracted from 3 mice. C, control; M, month(s); N, nicotine-treated; TERT, telomerase reverse transcriptase; OD, optical density.
Long-term nicotine exposure reduces the expression of nAChRs
The effect of nicotine is reliant on nAChR activity. In the current study, the expression of nAChR-α7 was detected by western blotting in EPCs. As demonstrated in Fig. 5, short-term nicotine exposure increased the expression of nAChR-α7 compared with the corresponding control group. However, following long-term exposure to nicotine, the expression of nAChR-α7 decreased compared with expression in control group mice.
Figure 5.
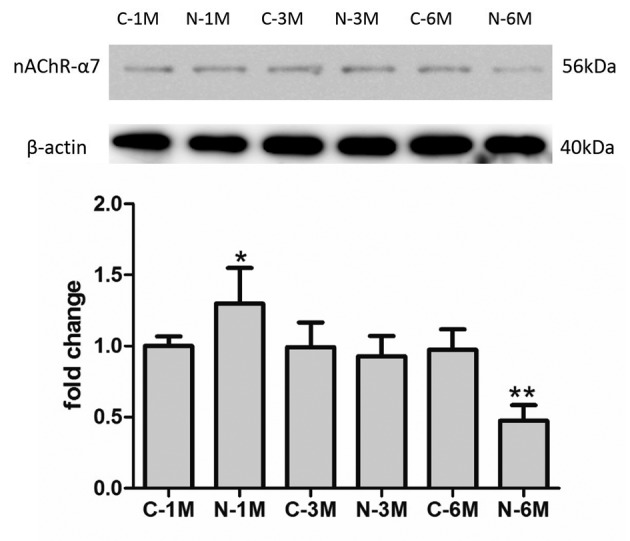
Effect of nicotine on nAChR expression. *P<0.05 vs. C-1 M group; **P<0.05 vs. C-6 M group. Cells were extracted from 3 mice. C, control; M, month(s); N, nicotine-treated; nAChR-α7, alpha-7 nicotinic receptor.
Long-term nicotine exposure reduced expression of SIRT1 protein
SIRT1 protein exerts protective effects against EPC senescence (18). In the present study, the effects of nicotine exposure on SIRT1 expression were examined. As indicated in Fig. 6, the expression of SIRT1 increased compared with that of control group mice following short-term nicotine exposure; however, subsequent to increased duration of nicotine exposure, the expression of SIRT1 decreased compared with the matched control group.
Figure 6.
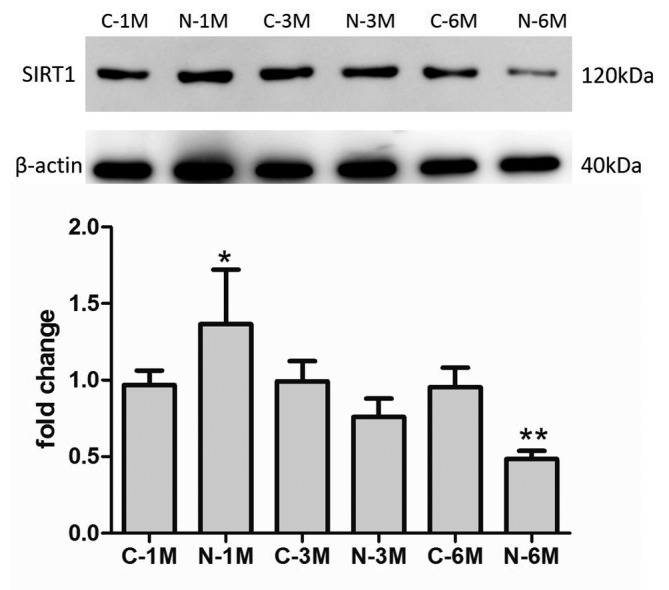
Effect of nicotine on SIRT1 protein expression.*P<0.05 vs. C-1 M group; **P<0.05 vs. C-6 M group. Cells were extracted from 3 mice. C, control; M, month(s); N, nicotine-treated; SIRT1, sirtuin 1.
Discussion
EPC dysfunction is common in chronic smokers, but increasing evidence suggests that short-term nicotine exposure presents benefits to EPCs (12). In the present study, the long-term effects of nicotine on EPCs were investigated. Short-term nicotine exposure was revealed to increase EPC number and to inhibit EPC senescence. However, long-term exposure to nicotine reduced EPC number and impaired EPC proliferation and migration, in addition to promoting EPC senescence. These effects appear to correlate with the change in SIRT1 protein expression.
Cigarette smoke is a complex mixture of >4,000 chemical constituents. The most impactful constituents of these are nicotine, carbon monoxide, aldehydes and sulfur compounds (19). Nicotine was previously demonstrated to have complex pharmacological effects on endothelial cells, and most of these effects were deleterious. For example, a previous study investigating coronary heart disease identified smoking as one of the five key risk factors in development of atherosclerosis (20). In animal models, chronic nicotine treatment also increased the rate of development of atherosclerosis in arteries (21). Furthermore, nicotine in similar concentrations to those experienced by smokers impaired the structure and function of vascular smooth muscle and endothelial cells (22,23). Notably, a previous study also reported that short-term exposure to nicotine enhanced EPC function. In the present study, it was similarly demonstrated that short-term nicotine exposure increased EPC number, consistent with the previous report. However, short-term nicotine exposure did not affect the proliferation and migration of these cells. Contrastingly, long-term nicotine exposure significantly decreased EPC number and impaired the proliferation and migration of EPCs.
The exposure time difference may explain the conflicting effects of nicotine on EPCs. Short-term nicotine exposure may act as a stimulus, promoting EPC functions such as migration, chemoattraction and proliferation (11). These effects have been observed in some pathological conditions; for example, patients undergoing percutaneous coronary intervention demonstrated increases in circulating EPC number (24,25). In the present study, it was contrastingly demonstrated that long-term nicotine exposure impaired EPC function. This impairment was similar to that observed in relation to other risk factors to EPCs, such as cholesterol (26), proinflammatory factors and reactive oxygen species, which result in EPC dysfunction through their chronic effects on these cells (27).
Besides the effects on the proliferative and migratory capacity of EPCs, long-term nicotine exposure also accelerated EPC senescence. Telomerase length and activity is a recognized marker of the cellular life span (28). Factors such as oxidization of low density lipoprotein, angiotensin II activity and high glucose are established to be involved in promoting EPC senescence by inhibiting telomerase activity (29–31). These findings are consistent with the results in the present study: Short-term nicotine exposure increased the telomerase activity and resulted in decreased numbers of senescent cells, whilst long-term nicotine exposure resulted in the opposite effect. These results suggested that the mechanism by which nicotine exposure affects EPCs senescence appears to involve telomerase activity.
It is established that the effects of nicotine depend on the signaling via nAChRs. Therefore, the current study evaluated the expression of nAChR-α7 in EPCs, revealing that short-term nicotine exposure increases the expression of nAChR-α7, but that long-term exposure decreased expression of these receptors. It is hypothesized that this may be a negative feedback mechanism to alleviate the detrimental effects of nicotine. However, additional research is required to determine the specific association between nicotine exposure and the expression of nAChR-α7 and other nAChRs.
In order to investigate the mechanisms by which nicotine affected telomerase activity, the expression of SIRT1 was evaluated in this study. Previous studies have demonstrated that SIRT1 exerts protective effects following stress-induced endothelial cell (32–34) and EPC (18,35) senescence. In the present study, nicotine exposure similarly affected the expression of SIRT1 protein, which was correlated and consistent with alterations to telomerase activity and EPC senescence. These results suggested that the effects of nicotine exposure on telomerase activity was mediated, at least in part, by regulation of SIRT1 expression.
In conclusion, the results of the present study suggest that long-term nicotine exposure induces dysfunction and senescence of EPCs, which may be associated with impairment of telomerase activity through the downregulation of SIRT1. These results additionally emphasize the necessity of smoking cessation in order to prevent EPC dysfunction and other tobacco-associated disease.
References
- 1.Urbich C, Dimmeler S. Endothelial progenitor cells: Characterization and role in vascular biology. Circ Res. 2004;95:343–353. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- 2.Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, Böhm M, Nickenig G. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353:999–1007. doi: 10.1056/NEJMoa043814. [DOI] [PubMed] [Google Scholar]
- 3.Aicher A, Zeiher AM, Dimmeler S. Mobilizing endothelial progenitor cells. Hypertension. 2005;45:321–325. doi: 10.1161/01.HYP.0000154789.28695.ea. [DOI] [PubMed] [Google Scholar]
- 4.Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, Zeiher AM, Dimmeler S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001;89:E1–E7. doi: 10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- 5.Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR, Levine JP, Gurtner GC. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106:2781–2786. doi: 10.1161/01.CIR.0000039526.42991.93. [DOI] [PubMed] [Google Scholar]
- 6.Ezzati M, Henley SJ, Thun MJ, Lopez AD. Role of smoking in global and regional cardiovascular mortality. Circulation. 2005;112:489–497. doi: 10.1161/CIRCULATIONAHA.104.521708. [DOI] [PubMed] [Google Scholar]
- 7.Powell JT. Vascular damage from smoking: Disease mechanisms at the arterial wall. Vasc Med. 1998;3:21–28. doi: 10.1177/1358836X9800300105. [DOI] [PubMed] [Google Scholar]
- 8.Bondjers G, Hansson G, Olsson G, Pettersson K. Smoking, catecholamines and their effects on endothelial cell integrity. Adv Exp Med Biol. 1990;273:51–59. doi: 10.1007/978-1-4684-5829-9_6. [DOI] [PubMed] [Google Scholar]
- 9.Kondo T, Hayashi M, Takeshita K, Numaguchi Y, Kobayashi K, Iino S, Inden Y, Murohara T. Smoking cessation rapidly increases circulating progenitor cells in peripheral blood in chronic smokers. Arterioscler Thromb Vasc Biol. 2004;24:1442–1447. doi: 10.1161/01.ATV.0000135655.52088.c5. [DOI] [PubMed] [Google Scholar]
- 10.Puls M, Schroeter MR, Steier J, Stijohann L, Hasenfuss G, Konstantinides S, Schäfer K. Effect of smoking cessation on the number and adhesive properties of early outgrowth endothelial progenitor cells. Int J Cardiol. 2011;152:61–69. doi: 10.1016/j.ijcard.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 11.Wang X, Zhu J, Chen J, Shang Y. Effects of nicotine on the number and activity of circulating endothelial progenitor cells. J Clin Pharmacol. 2004;44:881–889. doi: 10.1177/0091270004267593. [DOI] [PubMed] [Google Scholar]
- 12.Sugimoto A, Masuda H, Eguchi M, Iwaguro H, Tanabe T, Asahara T. Nicotine enlivenment of blood flow recovery following endothelial progenitor cell transplantation into ischemic hindlimb. Stem Cells Dev. 2007;16:649–656. doi: 10.1089/scd.2007.9991. [DOI] [PubMed] [Google Scholar]
- 13.Junhui Z, Xiaojing H, Binquan Z, Xudong X, Junzhu C, Guosheng F. Nicotine-reduced endothelial progenitor cell senescence through augmentation of telomerase activity via the PI3K/Akt pathway. Cytotherapy. 2009;11:485–491. doi: 10.1080/14653240902887267. [DOI] [PubMed] [Google Scholar]
- 14.National Institutes of Health (NIH), corp-author Guide for the Care and Use of Laboratory Animals. 7th. NIH; Bethesda, MD, USA: 1996. Publication No. 85–23. [Google Scholar]
- 15.Li W, Wang H, Kuang CY, Zhu JK, Yu Y, Qin ZX, Liu J, Huang L. An essential role for the Id1/PI3K/Akt/NFkB/survivin signalling pathway in promoting the proliferation of endothelial progenitor cells in vitro. Mol Cell Biochem. 2012;363:135–145. doi: 10.1007/s11010-011-1166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 17.Reed J, Gunaratnam M, Beltran M, Reszka AP, Vilar R, Neidle S. TRAP-LIG, a modified telomere repeat amplification protocol assay to quantitate telomerase inhibition by small molecules. Anal Biochem. 2008;380:99–105. doi: 10.1016/j.ab.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 18.Vassallo PF, Simoncini S, Ligi I, Chateau AL, Bachelier R, Robert S, Morere J, Fernandez S, Guillet B, Marcelli M, et al. Accelerated senescence of cord blood endothelial progenitor cells in premature neonates is driven by SIRT1 decreased expression. Blood. 2014;123:2116–2126. doi: 10.1182/blood-2013-02-484956. [DOI] [PubMed] [Google Scholar]
- 19.Stedman RL. The chemical composition of tobacco and tobacco smoke. Chem Rev. 1968;68:153–207. doi: 10.1021/cr60252a002. [DOI] [PubMed] [Google Scholar]
- 20.Kannel WB, McGee D, Gordon T. A general cardiovascular risk profile: The Framingham Study. Am J Cardiol. 1976;38:46–51. doi: 10.1016/0002-9149(76)90061-8. [DOI] [PubMed] [Google Scholar]
- 21.Strohschneider T, Oberhoff M, Hanke H, Hannekum A, Karsch KR. Effect of chronic nicotine delivery on the proliferation rate of endothelial and smooth muscle cells in experimentally induced vascular wall plaques. Clin Investig. 1994;72:908–912. doi: 10.1007/BF00190750. [DOI] [PubMed] [Google Scholar]
- 22.Leake DS, Bowyer DE. Quantitative studies of pinocytosis by arterial endothelial and smooth muscle cells in culture. Exp Mol Pathol. 1981;35:84–97. doi: 10.1016/0014-4800(81)90009-5. [DOI] [PubMed] [Google Scholar]
- 23.Zimmerman M, McGeachie J. The effect of nicotine on aortic endothelial cell turnover. An autoradiographic study. Atherosclerosis. 1985;58:39–47. doi: 10.1016/0021-9150(85)90054-1. [DOI] [PubMed] [Google Scholar]
- 24.Banerjee S, Brilakis E, Zhang S, Roesle M, Lindsey J, Philips B, Blewett CG, Terada LS. Endothelial progenitor cell mobilization after percutaneous coronary intervention. Atherosclerosis. 2006;189:70–75. doi: 10.1016/j.atherosclerosis.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 25.Hristov M, Zernecke A, Liehn EA, Weber C. Regulation of endothelial progenitor cell homing after arterial injury. Thromb Haemost. 2007;98:274–277. [PubMed] [Google Scholar]
- 26.Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 27.Lin CP, Lin FY, Huang PH, Chen YL, Chen WC, Chen HY, Huang YC, Liao WL, Huang HC, Liu PL, Chen YH. Endothelial progenitor cell dysfunction in cardiovascular diseases: Role of reactive oxygen species and inflammation. Biomed Res Int. 2013;2013:845037. doi: 10.1155/2013/845037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Counter CM. The roles of telomeres and telomerase in cell life span. Mutat Res. 1996;366:45–63. doi: 10.1016/S0165-1110(96)90006-8. [DOI] [PubMed] [Google Scholar]
- 29.Imanishi T, Hano T, Sawamura T, Nishio I. Oxidized low-density lipoprotein induces endothelial progenitor cell senescence, leading to cellular dysfunction. Clin Exp Pharmacol Physiol. 2004;31:407–413. doi: 10.1111/j.1440-1681.2004.04022.x. [DOI] [PubMed] [Google Scholar]
- 30.Imanishi T, Hano T, Nishio I. Angiotensin II accelerates endothelial progenitor cell senescence through induction of oxidative stress. J Hypertens. 2005;23:97–104. doi: 10.1097/00004872-200501000-00018. [DOI] [PubMed] [Google Scholar]
- 31.Kuki S, Imanishi T, Kobayashi K, Matsuo Y, Obana M, Akasaka T. Hyperglycemia accelerated endothelial progenitor cell senescence via the activation of p38 mitogen-activated protein kinase. Circ J. 2006;70:1076–1081. doi: 10.1253/circj.70.1076. [DOI] [PubMed] [Google Scholar]
- 32.Ota H, Akishita M, Eto M, Iijima K, Kaneki M, Ouchi Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J Mol Cell Cardiol. 2007;43:571–579. doi: 10.1016/j.yjmcc.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 33.Zu Y, Liu L, Lee MY, Xu C, Liang Y, Man RY, Vanhoutte PM, Wang Y. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ Res. 2010;106:1384–1393. doi: 10.1161/CIRCRESAHA.109.215483. [DOI] [PubMed] [Google Scholar]
- 34.Wan YZ, Gao P, Zhou S, Zhang ZQ, Hao DL, Lian LS, Li YJ, Chen HZ, Liu DP. SIRT1-mediated epigenetic downregulation of plasminogen activator inhibitor-1 prevents vascular endothelial replicative senescence. Aging Cell. 2014;13:890–899. doi: 10.1111/acel.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lemarié CA, Shbat L, Marchesi C, Angulo OJ, Deschênes ME, Blostein MD, Paradis P, Schiffrin EL. Mthfr deficiency induces endothelial progenitor cell senescence via uncoupling of eNOS and downregulation of SIRT1. Am J Physiol Heart Circ Physiol. 2011;300:H745–H753. doi: 10.1152/ajpheart.00321.2010. [DOI] [PubMed] [Google Scholar]