

Abstract
Mycobacterium tuberculosis is a human respiratory pathogen that causes the deadly disease tuberculosis. The rapid global spread of antibiotic‐resistant M. tuberculosis makes tuberculosis infections difficult to treat. To overcome this problem new effective antimicrobial strategies are urgently needed. One promising target for new therapeutic approaches is PonA1, a class A penicillin‐binding protein, which is required for maintaining physiological cell wall synthesis and cell shape during growth in mycobacteria. Here, crystal structures of the transpeptidase domain, the enzymatic domain responsible for penicillin binding, of PonA1 from M. tuberculosis in the inhibitor‐free form and in complex with penicillin V are reported. We used site‐directed mutagenesis, antibiotic profiling experiments, and fluorescence thermal shift assays to measure PonA1's sensitivity to different classes of β‐lactams. Structural comparison of the PonA1 apo‐form and the antibiotic‐bound form shows that binding of penicillin V induces conformational changes in the position of the loop β4′‐α3 surrounding the penicillin‐binding site. We have also found that binding of different antibiotics including penicillin V positively impacts protein stability, while other tested β‐lactams such as clavulanate or meropenem resulted in destabilization of PonA1. Our antibiotic profiling experiments indicate that the transpeptidase activity of PonA1 in both M. tuberculosis and M. smegmatis mediates tolerance to specific cell wall‐targeting antibiotics, particularly to penicillin V and meropenem. Because M. tuberculosis is an important human pathogen, these structural data provide a template to design novel transpeptidase inhibitors to treat tuberculosis infections.
Database
Structural data are available in the PDB database under the accession numbers 5CRF and 5CXW.
Keywords: β‐lactams, antibiotic resistance, PBP, PonA1, transpeptidase domain, tuberculosis
Abbreviations
- FTS
fluorescence thermal shift
- PBP
penicillin‐binding protein
- TB
tuberculosis
- TG‐
transglycosylase domain mutant
- TP‐
transpeptidase domain mutant
Introduction
Mycobacterium tuberculosis is a bacterial pathogen of the human respiratory system that primarily infects lungs but could also infect other parts of the body including kidney, spine, and brain. Tuberculosis (TB) infection is fatal for ~ 1.5 million people worldwide each year according to the World Health Organization and remains silent within 90% of the infected population 1. Emergence of antibiotic‐resistant M. tuberculosis bacteria has become a serious health problem during the last 40 years. In order to control and treat TB disease, the development of new effective drugs is urgently needed.
Mycobacterium tuberculosis has a complicated cell wall architecture compared to other antibiotic resistant bacteria 2, 3, 4. The complexity of the cell wall is considered to be one of the reasons for the bacterium's natural tolerance to antibiotics. One major cell wall component is peptidoglycan. The mutations of proteins involved in peptidoglycan synthesis in M. tuberculosis consequently lead to antibiotic resistance 5. The peptidoglycan layer is formed by glycan chains of β‐(1,4) linked N‐acetylglucosamine and N‐acetylmuramic acid which are additionally crosslinked by 3,3 or 4,3 transpeptide bonds between short amino acid fragments of alanine, glutamate, and diaminopimelic acid residues. In mycobacteria, the 3,3 crosslinks are generated in equal abundance in all phases of bacterial growth by the l,d‐transpeptidases 6, 7. The classical 4,3 crosslinks are formed during the exponential phase of growth by the d,d‐transpeptidase activity of penicillin‐binding proteins (PBPs) 8. The 4,3 transpeptidase activity could be easily inhibited by penicillin or other β‐lactam antibiotics by forming a stable covalent complex with the serine at the active site of the PBP 9. PBPs have not traditionally been chosen as primary drug targets for M. tuberculosis because β‐lactams are very susceptible to degradation by endogenous bacterial β‐lactamases; however, recent reports indicate some level of efficacy against drug‐sensitive and even drug‐resistant M. tuberculosis strains 10. A more recent study in Escherichia coli indicates that β‐lactams do not only inhibit transpeptidase activity of PBPs but also induce degradation of the newly formed peptidoglycan chains, leading to systemic toxicity 11. Therefore, investigating PBPs from M. tuberculosis may provide novel opportunities for the development of new antibiotics that would be resistant to β‐lactamase cleavage and would simultaneously target multiple cellular proteins involved in peptidoglycan biosynthesis.
Mycobacterium tuberculosis possesses only two class A PBPs, PonA1 and PonA2, that contain both a transglycosylase domain involved in polymerization of the polyglycan chains and a transpeptidase domain, the enzymatic domain responsible for binding of penicillin 12, 13. Different studies indicate that PonA1 and PonA2 play a complex role in bacterial physiology and have an unusual hypersusceptibility to β‐lactam antibiotics, suggesting that novel therapeutic development could optimally target these enzymes 14, 15, 16, 17. To understand the role of class A PBPs in mycobacterial peptidoglycan biology, we examined PonA1 from the M. tuberculosis strain H37Rv. PonA1 is a high molecular weight 71 kDa, two‐domain protein that contains a noncleavable signal peptide at the N terminus. The N‐terminal signal peptide of PonA1 is phosphorylated and was identified as a substrate for the serine–threonine protein kinase PknB 15, 18. The peptidoglycan transglycosylase domain is homologous to PBP1 from E. coli 19 and catalyzes the ligation of N‐glycolyl muramic acid from the polyglycan sacculus to N‐acetyl glucosamine from amphipathic peptidoglycan precursor molecules (lipid II). The C‐terminal domain of PonA1 crosslinks the penultimate d‐alanine to the d‐meso‐diaminopimelic acid between parallel peptidoglycan strands. PonA1 localizes to the poles and septa of M. smegmatis and is required for maintaining normal cell length in both M. tuberculosis and M. smegmatis 15, 20, 21. Changes to the level of PonA1 protein result in a structurally abnormal cell shape, suggesting that PonA1 is critical for maintaining physiological cell wall synthesis and cell shape during growth. PonA1 regulates essential RipA peptidoglycan hydrolase activity and binds to the RipA endopeptidase domain at the end of PonA1's C‐terminal transpeptidase domain 20. Additionally, recent work shows that PonA1 activity impacts antibiotic tolerance in mycobacteria 5. Altering PonA1 transpeptidase or phosphorylation activity affects bacterial growth in the presence of the antibiotic teicoplanin, which targets peptidoglycan biosynthesis 15.
In this study, we describe the high‐resolution crystal structure of the PonA1 transpeptidase domain from the M. tuberculosis virulent strain H37Rv in ligand‐free form and in complex with penicillin V. We characterize the structural details and penicillin‐binding site. Furthermore, using site‐directed mutagenesis and antibiotic profiling, we provide evidence that PonA1's transpeptidase activity in both M. tuberculosis and M. smegmatis mediates tolerance to different classes of β‐lactams. Loss of PonA1's transpeptidase activity renders cells more susceptible to β‐lactams, especially to meropenem and penicillin V. Additionally, we tested the stability of PonA1 in the presence of different classes of β‐lactams by fluorescence thermal shift (FTS) assays. We find that different antibiotics have different impact on the folding of PonA1. FTS data show that binding and formation of the acyl‐enzyme by compounds like carbenicillin or penicillin V result in positive Tm shifts, while others like clavulanate or meropenem give negative Tm shifts, indicating that they induce a more destabilized conformation of PonA1. The structure of PonA1's transpeptidase domain in complex with penicillin V provides a starting point to understand the mechanism underlying the development of antibiotic resistance in M. tuberculosis and to design novel transpeptidase inhibitors, which hold great potential for the treatment of TB disease.
Results
Overall structure of PonA1's transpeptidase domain
Four PonA1 molecules in the asymmetric unit of the inhibitor‐free form share a similar structure with an average root‐mean‐square deviation (RMSD) value of 0.2 Å calculated between the Cα atoms of their main chains. Gel filtration chromatography and analysis by the PISA server 22 suggest that the biological unit of PonA1 is a monomer (data not shown). Crystallized PonA1 molecules (residues 249–643) contain the transpeptidase domain and one small adjacent domain at the N terminus of the transpeptidase domain (Figs 1A and 2). The first 156 residues that form part of the N‐terminal glycosyltransferase domain and 33 residues at the C terminus of PonA1 were not observed in the protein structure. These modifications could be due to protein degradation and/or structural disorder. In the structure, the PonA1 molecule has a unique unstructured C terminus that contains a proline‐rich region. This region forms an exposed long hydrophobic tail (Fig. 1B), suggesting that it may be involved in the protein–protein interactions that have been suggested by previous studies 23. The small adjacent N‐terminal segment of the transpeptidase domain comprises two α‐helices [α1n (250–268) and α2n (270–276)] and a three‐stranded [β1n (280–284), β2n (480–486), and β3n (490–495)] antiparallel β‐sheet (Figs 1A and 2). The PonA1 transpeptidase (or penicillin‐binding domain) resembles other PBPs secondary structure: PBP1 of class A from Staphylococcus pneumonia (RMSD of 1.9 Å) 24, 25, the transpeptidase domain of PBP2 from Neisseria gonorroeae (RMSD of 2.00 Å) 26, class A PBP1 from Pseudomonas aeruginosa (RMSD of 1.65 Å) 27, 28, and PBP4 from Listeria monocytogenes (RMSD of 2.01 Å) 29. A cleft in the middle of the domain contains the active site. The cleft divides the transpeptidase domain of PonA1 into two subdomains (Figs 1A and 2). One subdomain has α/β fold and contains an antiparallel β‐sheet surrounded by α‐helices in a following order: α1 (286–302), β1 (307–316), β2 (321–326), β1′ (341–343), β5′ (473–475), β6′ (503–505), α8 (520–525), β3 (537–545), β4 (550–560), β5 (563–572), and α9 (590–605). The second subdomain has an α‐helical structure with six α‐helices: α2 (349–358), α3 (391–397), α4 (399–410), α5 (413–425), α6 (458–471), and α7 (507–519). Additionally, the second subdomain contains a long loop with three short β strands, β2′ (364–368), β3′ (371–373), and β4′ (376–378), that forms part of the inhibitor‐binding pocket. The conformation of this loop is held by disulfide bond between Cys386 and Cys389 (Fig. 1A).
Figure 1.
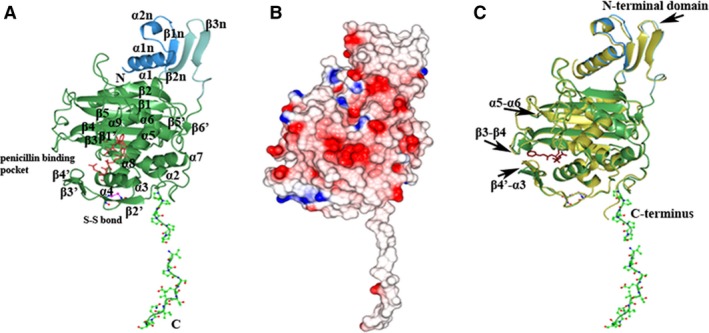
Structure of the transpeptidase domain of PonA1. (A) Ribbon diagram of the C‐terminal transpeptidase domain of PonA1 monomer from Mycobacterium tuberculosis. The small adjacent N‐terminal segment of the PonA1 transpeptidase domain is colored in blue and cyan. The PonA1 penicillin‐binding domain is colored in green. Residues of the hydrophobic C‐terminal segment and cysteine residues are shown as a ball‐and‐stick model, where carbon atoms are in green (or blue for cysteines), nitrogen atoms in blue, oxygen atoms in red, and sulfur atoms in pink. Residues composing conserved motifs in the penicillin‐binding pocket of PBPs are shown as a cylinder model in crimson. The secondary structure elements are labeled. (B) Electrostatic surface representation. The surface is colored by surface potential charge from negative in red to positive in blue. (C) Comparison of PonA1 structures with (gold) and without penicillin V (green). Penicillin V is shown as a cylinder model in dark purple. The disposition of the loops in superimposed structures is marked with arrows.
Figure 2.

PonA1 domain composition and protein secondary structure assignment. (A) Functional domains of PonA1 protein. The analysis of PonA1 sequence was performed on the Pfam database (http://pfam.xfam.org/). (B) PonA1 sequence of the crystallized transpeptidase domain. The secondary structure elements are shown above the sequence. The stars below the sequence indicate the first and last residue that was observed in the structure of the crystallized PonA1 domain.
Fluorescence thermal shift assays
To investigate whether certain drugs may impact the stability of the protein, we determined thermal denaturation curves using FTS assays with different derivatives of β‐lactam antibiotics. Effects of 23 antibiotics on the denaturation temperature of PonA1 were determined (Fig. 3). The data show that hetacillin (ΔT m = 3.0 °C), carbenicillin (ΔT m = 2.2 °C), dicloxacillin (ΔT m = 1.9 °C), penicillin V (ΔT m = 1.6 °C), or cefotaxime (ΔT m = 1.6 °C) shifts the T m of PonA1 by more than 1° in the positive direction, while nafcillin (ΔT m = −2.0 °C), clavulanate (ΔT m = −2.7 °C), and meropenem (ΔT m = −3.8 °C) cause significant T m shifts in the negative direction. The data shown are for PonA1 at 2.8 μm and the antibiotics at 5 μm.
Figure 3.
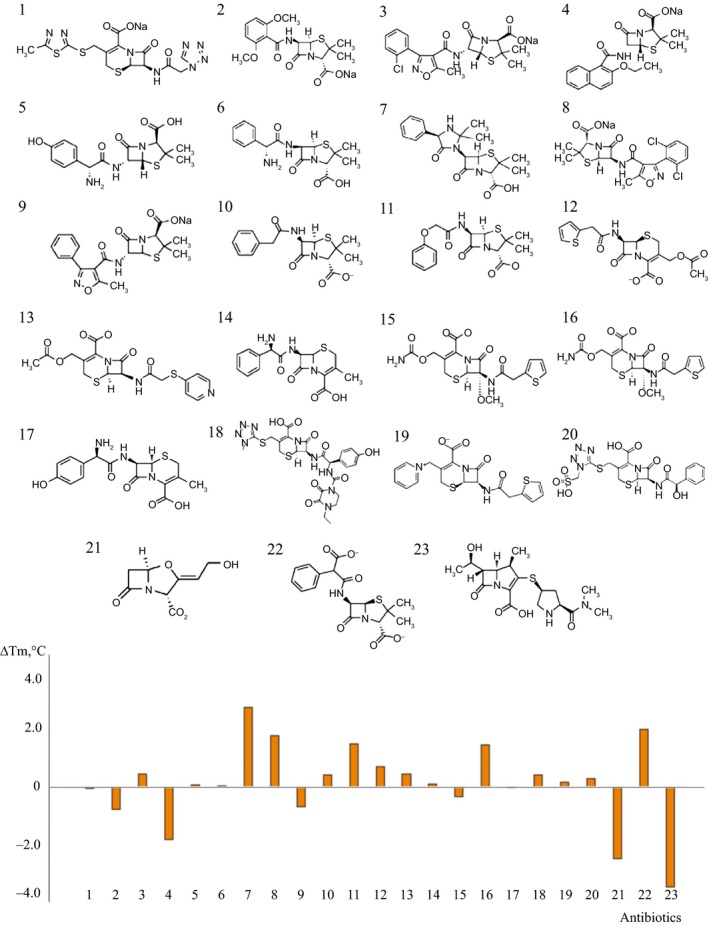
Modulation of thermal stabilities of PonA1 by different classes of antibiotics as indicated by FTS determined ΔT m. The ordinate is calculated as the difference between the T m values of PonA1 at presence and absence of antibiotics: cefazolin (1), methicillin (2), cloxacillin (3), nafcillin (4), amoxicillin (5), ampicillin (6), hetacillin (7), dicloxacillin (8), oxacillin (9), penicillin G (10), penicillin V (11), cephalothin (12), cephapirin (13), cephalexin (14), cefoxitin (15), cefotaxime (16), cefadroxil (17), cefoperazone (18), cefhaloridine (19), cefonicid (20), clavulanate (21), carbenicillin (22), meropenem (23). The data shown are for PonA1 at 2.8 μm and the antibiotics at 5 μm. Of the 23 derivatives of β‐lactam antibiotics, nafcillin, clavulanate, and meropenem give rise to significant negative T m shift indicating that these compounds make PonA1 less stable. In contrast, hetacillin, carbenicillin, dicloxacillin, and penicillin V induces the T m shift of PonA1 in the positive direction as observed for other PBP proteins.
The significant transition temperature shift at this low drug to protein ratio (1.8–1) indicates specific binding of these antibiotics to PonA1. Furthermore, carbenicillin gave rise to a similar positive T m shift as penicillin V, cefotaxime, and dicloxacillin, while compounds such as clavulanate, meropenem, and nafcillin exhibited negative T m shifts, indicating that these compounds make PonA1 less stable in these conditions. While β‐lactam compounds induced ~ 10 °C T m shift for other PBP proteins we studied (data not shown), the largest shift we observed for PonA1 was < 3.5 °C.
Catalytic site and PonA1–penicillin V interactions
Similar to all known β‐lactam‐binding proteins, the PonA1 transpeptidase domain contains three conserved sequence motifs: SxxK (which contains the catalytic serine), SxN, and KTG at the penicillin‐binding cleft 8, 9. The SxxK motif comprising residues Ser345, Ser346, Phe347, and Lys348 is located at the N terminus of the helix α2 and contains two important residues for catalysis in PBPs: Ser345 and Lys348. Ser345 is acylated by the substrate and situated at the center of the catalytic site. Lys348 is thought to enhance the nucleophilicity of the serine. The SxN motif comprising residues Ser398, Leu399, and Asn400 is located on the loop between helices α3 and α4. The KTG motif comprising residues Lys539, Thr540, and Gly541 is situated on the strand β3 of the PonA1 transpeptidase domain. In the inhibitor‐free PonA1 structure, the Ser345 hydrogen bonds with Lys348, Glu382, and Thr540 and through a water molecule with Ser398. At the penicillin‐binding site of the PonA1 structure, Lys348 hydrogen bonds with Asn400 from the SxN motif. We identify another hydrogen bond between Lys539 and Ser398 from the SxN motif. Despite the similarity in the position of the three conserved motifs within the active site, the rate of acylation of β‐lactams varies considerably in different PBPs 30, 31, 32, 33. This suggests that the architecture of the antibiotic‐binding pocket and adjacent loops plays an important role in the activity of different PBPs and likely influences PonA1's interaction with inhibitors.
The high‐resolution crystal structure of PonA1 in the penicillin V acylated form was determined and contained one protein molecule (residues 249–628) in the asymmetric unit. The part of the N‐terminal glycosyltransferase domain (156 residues) and C terminus (51 residues) were missing in the structure due to protein degradation and/or disorder. We calculated RMSD values between the Cα atoms of PonA1 monomers in the apo‐form and PonA1 monomer in the ligand‐bound structure. The average RMSD value was 0.5 Å. Conformational changes with RMSD > 0.8 Å were observed in the position of the loops between β4′‐α3 (loop with disulfide bridge), α5‐α6, β3‐β4, β5‐α9 (in monomer C of the apo‐form), and β3n‐β6′ (in monomer A of the apo‐form) as well as in the position of the small adjacent domain at the N terminus of the transpeptidase domain (Fig. 1C). Another change that was observed was in the position of the C‐terminal proline‐rich tail in all monomers of PonA1 (Fig. 1C). To investigate if packing interactions between adjacent monomers in the crystal lattice were responsible for conformational differences in the structures, we analyzed their crystal contacts. In the crystal lattice, PonA1 monomers were very tightly packed with Matthews coefficients Vm = 1.81 and Vm = 2.17 for the inhibitor‐free form and penicillin V‐bound form, respectively. The contacts between the PonA1 monomers and symmetry‐related monomers in ligand‐free and penicillin V‐bound structures with Van der Waals interactions < 4.6 Å were calculated with the program contact (ccp4 support program 34). The analysis indicated that all monomers have different contact areas with symmetry‐related monomers. Most of the conformational changes including the N‐terminal adjacent domain and C‐terminal proline‐rich tail have contacts with symmetry equivalent monomers in one or both structures. However, the position of the loop between β4′‐α3 surrounding the active site was not associated with crystal packing and was most likely affected by reaction with the penicillin V (Fig. 4A). The disposition of the loop β4′‐α3 in the PonA1 monomers has the largest RMSD value ~ 3 Å (calculated average value). In two monomers (B and D) of the apo‐form structure, this loop is partially disordered due to its mobility.
Figure 4.
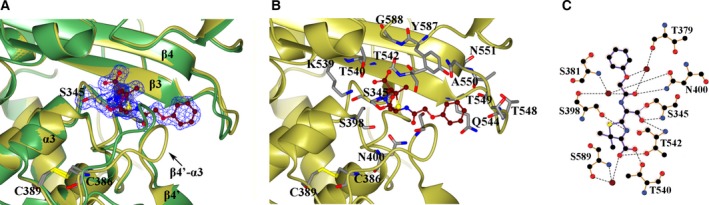
Penicillin V‐binding site. (A and B) Comparison of PonA1 active site with (gold) and without (green) penicillin V. Penicillin V covalently bound to Ser345 is presented as a ball‐and‐stick model. Residues of the penicillin‐binding site are shown as a cylinder model, where carbon atoms are in gray, nitrogen atoms in blue, oxygen atoms in red, and sulfur atoms in yellow. At the protein active site of the liganded structure, penicillin V is shown in the electron density omit map. (C) LigPlot diagram of penicillin V‐binding site. Hydrogen bonds for antibiotic on the LigPlot+ are shown as black broken lines. In the LigPlot+ diagram, water molecules are colored in brown, oxygen atoms in red, carbon atoms in black, nitrogen atoms in blue, and sulfur in yellow.
At the binding cleft, penicillin V forms an acyl–enzyme complex through the covalent bond between the OG atom of Ser345 and the C6 atom of the β‐lactam ring (Fig. 4A and 4B). The carboxyl group of the thiazolidine ring forms hydrogen bonds with OG1 atoms of Thr540 and Thr542 located on the β3 strand, and through a water molecule with the nitrogen of the main chain and the OG atom of the side chain of Ser589 on the loop β5‐α9 (Fig. 4C). The N7 atom of the thiazolidine ring of penicillin V hydrogen bonds with the OG atom of Ser398 located on helix α3. The N4 and O18 atoms of the acyl side chain of penicillin V interact with the O atom of the main chain of Thr542 of β3 and the ND2 atom of the side chain of Asn400 of α4, respectively. Other interactions occur between the O1 and O18 atoms of penicillin V and Thr379 of β4′, Ser381 of β4′, and Ser398 of α4 residues through water molecules (Fig. 4C). Therefore, structural analysis of PonA1 in an antibiotic‐bound and free form suggests that positioning of the R group of penicillin V at the active site is accompanied by conformational change of the loop β4′‐α3 that widened the penicillin‐binding pocket (Fig. 4A).
Antibiotic profiling experiments
PonA1's interaction with small molecules occurs in the inhibitor cleft, which includes its active site serine. Because of the importance of PonA1's active site in cell wall biosynthesis and cell growth, we hypothesized that PonA1's active site serine plays a key role in the protein's interactions with small molecules during cell growth. To test the importance of PonA1's active site for interaction with antibiotics, we generated M. tuberculosis and M. smegmatis mutants of the transpeptidase active site. Coupling these genetic mutants with several derivatives of β‐lactams, we assessed the efficacy of particular PBP‐targeting antibiotics in M. tuberculosis and M. smegmatis (Fig. 5A). For both M. tuberculosis and M. smegmatis, loss of PonA1's transpeptidase activity rendered cells more susceptible to β‐lactams, specifically to carbenicillin and meropenem (Fig. 5A). It is possible that the observed difference in antibiotic tolerance may be due to altered effective antibiotic concentration for the remaining PBPs in the cell (because transpeptidase mutant PonA1 no longer covalently binds transpeptidase inhibitors). However, our previous work demonstrated that M. tuberculosis cells that lack PonA1 and PonA2 exhibit differential susceptibility to cell wall‐targeting antibiotics, including transpeptidase inhibitors. Thus, although these proteins are homologous, their distinct physiological roles in mycobacterial cell wall biology result in different susceptibility to cell wall‐active drugs 35.
Figure 5.
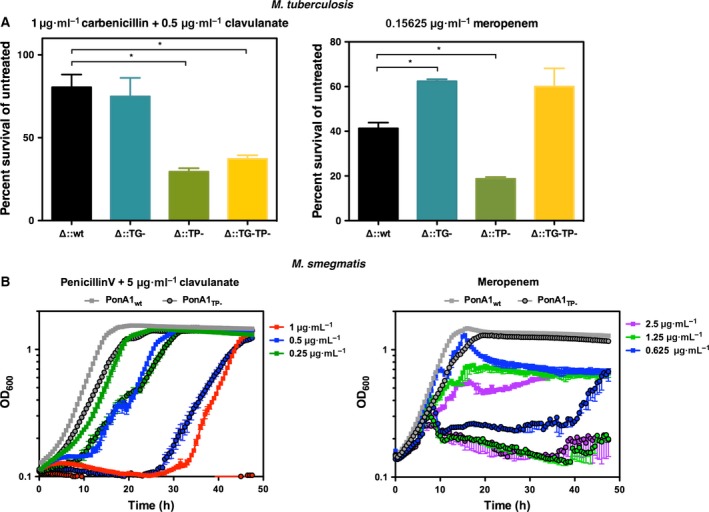
In vitro antibiotic efficacy experiments. (A) The indicated Mycobacterium tuberculosis H37Rv strains were grown in the presence of carbenicillin and clavulanate (left panel) or meropenem (right panel) at the indicated concentrations. Bacterial survival in the presence of antibiotic was calculated in comparison to untreated bacterial cells. Wild‐type PonA1, wt; TG‐mutant PonA1, TG‐; TP‐mutant PonA1, TP‐; double TG‐ and TP‐mutant PonA1, TG‐TP‐. Data were collected in duplicate for each strain and are representative of two experiments. Error bars represent SD. The asterisk indicates P‐value of < 0.05 and was determined by an unpaired, two‐tailed t‐test. (B) M. smegmatis mc2155 strains expressing wild‐type PonA1 (wt, nonoutlined squares) or transpeptidase mutant PonA1 (TP‐, outlined circles) were grown in the presence of penicillin V and clavulanate (left panel) or meropenem (right panel) at the indicated concentrations. Untreated control cells are shown in gray for both strains. Data were collected in triplicate (for carbenicillin data) or duplicate (for penicillin V data) for each strain and are representative of two experiments. Error bars represent SEM.
Interestingly, cell growth plateaued at different times when M. smegmatis cells were treated with penicillin V compared with meropenem. The PonA1 transpeptidase‐inactivated cells exhibit a substantial delay in growth at a range of penicillin V concentrations when compared to wild‐type control (Fig. 5B). At 1 μg·mL−1, the TP cells never recover. These data suggest that cells lacking PonA1's transpeptidase activity grow much less robustly in the presence of penicillin V or meropenem.
We observed that when we treat cells that encode an allele of ponA1 wherein both enzymatic domains are inactivated (Fig. 5A, TG‐TP‐) with meropenem, the survival rate is almost the same as the single TG‐mutant. This may correlate with other cell biology data that show the expression of a TP allele is more toxic than a TG‐allele or a TG‐TP‐allele (and that the latter phenocopy each other) if the cells also undergo inhibition of crosslinking 11, 15. One model in E. coli explains that this may be due to cells experiencing a buildup of uncrosslinked glycan strands 15. However, the carbenicillin data do not fit this model; the TG‐TP‐cells phenocopy the TP‐cells. That could be related to the frequency of 4,3 and 3,3 crosslinks in mycobacterial peptidoglycan and how these 4,3 and 3,3 crosslinking inhibitors work in mycobacteria. Because mycobacteria have more 3,3 crosslinks than 4,3 crosslinks, we may observe this glycan strand toxicity more in the presence of 3,3 inhibitors (such as meropenem), unlike the situation in E. coli.
Discussion
In this study, we characterize the crystal structure of the transpeptidase domain of PonA1, which affects antibiotic tolerance in mycobacteria. The structural comparison of inhibitor‐free and inhibitor‐bound states indicates that binding of penicillin V induces conformational changes of the loop β4′‐α3 leading to a widening of the penicillin‐binding pocket. In the structure, penicillin V forms a network of interactions with PonA1 residues of highly conserved motifs (SxxK, SxN, and KTG) in known β‐lactam PBPs. These findings highlight why penicillins are such effective inhibitors of this class of enzymes.
Our antibiotic profiling experiments suggest that PonA1's transpeptidase activity is important for normal cellular tolerance of transpeptidase inhibitors, especially to penicillin V and meropenem. We also observed that cells that lack PonA1's transpeptidase activity grown in the presence of penicillin V exhibit a substantial delay in growth and develop resistance in 25 h. This may suggest that homologous peptidoglycan synthesizing enzymes more insensitive to penicillin V take over PonA1's function to promote new peptidoglycan synthesis or that mutations occurred that alter the PBP binding affinity of penicillin V. Moreover, our data indicate that meropenem and penicillin V affect cell growth differently. This may be correlated with the ability of these classes of compounds to target 4,3 crosslinking enzymes vs. 3,3 crosslinking enzymes. For example, meropenem is thought to target both the enzyme groups, whereas carbenicillin or penicillin V should target just the 4,3 crosslinking enzymes.
In addition, the FTS assays show that different derivatives of β‐lactam antibiotics have different effects on PonA1 protein stability. The presence of meropenem, clavulanate, or nafcillin induces unstable conformations of the protein, while carbenicillin, penicillin V, hetacillin, dicloxacillin have positive effects on protein stability. There is a possibility that binding of meropenem, clavulanate, or nafcillin increases more disordered conformations of the protein. The initial noncovalent complexes should have favorable interactions. When the covalent complex is formed and the serine is acylated, the interactions with product change. The covalent adducts with the acylated serine make more unfavorable interactions at the protein active site that destabilize the structure. We also suggest that, when compared to acylation with penicillin V, acylation with meropenem may affect the formation of the disulfide bond, which could lead to a more unstable conformation of the PonA1 transpeptidase domain. We found that binding of penicillin V induced conformational changes in the loop β4′‐α3 with the two cysteines (C386 and C389). The changes in the transpeptidase domain consequently may disturb contacts between PonA1 and its interactors, such as the endopeptidase RipA, which are important for normal peptidoglycan metabolism 23. It has been shown that the C‐terminal 259 residues of the transpeptidase domain of PonA1, where the last 55 residues form an unstructured proline‐rich tail, are required for interactions with the 25 C‐terminal residues of RipA thus contributing to peptidoglycan remodeling processes 20. Our combined data suggest that meropenem could be a useful model for future drug design to target mycobacterial PBPs. Alternatively, TB treatment could be served by further optimization of meropenem to improve its efficacy and render it even less susceptible to beta‐lactamase cleavage 10.
In conclusion, because M. tuberculosis is an important human pathogen, these data provide clear molecular details toward understanding the mechanism underlying the development of penicillin resistance. These structural data provide a template to design novel transpeptidase inhibitors and improve already known drugs to treat TB disease.
Experimental procedures
Cloning
The gene encoding PonA1 (NCBI accession code YP_177687.1) containing residues 92–676 excluding N‐terminal signal peptide was amplified from the genomic DNA of M. tuberculosis strain H37Rv using PCR forward and reverse primers: 5′‐TACTTCCAATCCAATGCCAACAACCTGTTCGGCGGCGAT and 5′‐TTATCCACTTCCAATGTTACGGCGTGGGAGTCGCAG, respectively. The product was cloned into the pMCSG73 vector, which incorporates a T7 promoter, TVMV‐cleavable N‐terminal NusA tag, TEV‐cleavable N‐terminal hexa‐histidine tag, and StrepII tag developed at Midwest Center of Structural Genomics (www.mcsg.anl.gov). The vector was transformed into E. coli BL21(DE3) Magic cells.
Expression and purification
PonA1 fusion protein with NusA was expressed in 4 L of High Yield selenomethionine M9 medium (Medicilon Inc., Shanghai, China) containing 100 mg·L−1 ampicillin and 50 mg·L−1 kanamycin. BL21‐Magic cells were overgrown at 37 °C until OD600 reached 0.6–0.8 and were induced with 5 mm isopropyl β‐D‐thiogalactoside (IPTG) overnight at 22 °C. Cells were harvested by centrifugation at 10 876 g for 10 min and resuspended in lysis buffer pH 8.3 containing 0.25 m sodium chloride, 0.1 m ammonium sulfate, 43.6 mm sodium phosphate dibasic, 3.25 mm citric acid, 10% glycerol, 1 mm tris‐(2‐chloroethyl)‐phosphate (TCEP), 5 mm imidazole, and 0.08% n‐dodecyl β‐D‐maltoside (DDM). The cells were then sonicated and the cleared lysate was prepared by centrifugation at 36 000 g for 40 min at 4 °C. The NusA fusion protein in pMCSG73 constructs is removed before purification by TVMV protease cleavage. TVMV protease was expressed the same way as PonA1 fusion protein and cleared lysate was obtained. The two cleared lysates were mixed together in 5 : 1 ratio (PonA1–NusA:TVMV protease), 5 mm β‐mercaptoethanol was added, and the mixture was incubated overnight at 4 °C. The protein was purified by Ni‐affinity chromatography followed by gel filtration. Loading buffers (10 mm Tris/HCl pH 8.3, 0.5 m sodium chloride, 1 mm TCEP) containing 25 mm and 0.5 m imidazole, respectively, were used for washing and elution. The purity of acquired protein was examined with SDS/PAGE and Coomassie blue staining. SDS/PAGE revealed that the protein showed a tendency to degrade. An additional chromatographic step was performed to improve purity. Protein was loaded on a Ni column, washed with loading buffer containing 0.1 mm DDM, 1 m sodium chloride, 1 mm TCEP, and eluted with 0.5 m imidazole in loading buffer with 0.5 m sodium chloride. It was then dialyzed against buffer containing 10 mm Tris/HCl pH 8.3, 0.5 m sodium chloride, and 1 mm TCEP. The hexa‐histidine tag was cleaved overnight at 4 °C by recombinant TEV protease having a noncleavable histidine tag. PonA1 was separated from TEV protease by passing through the Ni column. Purified PonA1 was concentrated to 16 mg·mL−1 in the loading buffer defined above for crystallization.
Crystallization and data collection
Crystallization trays of selenomethionine‐labeled PonA1 were set up on Corning 96‐well sitting‐drop plates using the sitting‐drop vapor‐diffusion method at 19 °C. One microlitre of protein solution (concentration 8 mg·mL−1) was mixed with 1 μL of reservoir solution from the crystal screens Classics, Classics II, JCSG+, PACT, PEGs, and PEGs II (Qiagen Inc., Valencia, CA, USA). Prior to cocrystallization with antibiotic, PonA1 was incubated with 5 mm penicillin V for 30 min. Crystals of PonA1 in inhibitor‐free form were obtained with a reservoir solution containing 0.2 m lithium chloride, 0.1 m HEPES buffer pH 7.0, and 20% (w/v) PEG6000. The cocrystals of PonA1 with penicillin V were obtained with a reservoir solution containing 0.1 m HEPES buffer pH 7.5 and 25% (w/v) PEG3350. Before data collection, cocrystals of PonA1 with antibiotic were soaked with 10 mm penicillin V for 10 min in 5 μL of well solution. All crystals were flash cooled in liquid nitrogen using reservoir solution as a cryo‐protectant. Low temperature (100 K) X‐ray diffraction data were collected at LS‐CAT 21ID‐G and 21ID‐F beamlines of the Advanced Photon Source (Argonne National Laboratory, Argonne, IL, USA) from crystals of PonA1 in inhibitor‐free form and in the presence of penicillin V at 1.8 and 1.75 Å resolutions, respectively. The crystal space groups and cell parameters are summarized in Table 1.
Table 1.
Data collection, structure determination, and refinement statisticsa
| Inhibitor‐free form | Penicillin V‐bound form | |
|---|---|---|
| Crystal parameters | ||
| Resolution (Å) | 30.0–1.8 (1.83–1.8) | 30.0–1.75 (1.78–1.75) |
| Space group | P21 | P212121 |
| Unit cell parameters | ||
| a, b, c (Å) | 46.0, 333.3, 47.5 | 47.3, 60.6, 133.9 |
| α, β, γ (°) | 90, 108.4, 90 | 90, 90, 90 |
| Matthews coefficient (Å3/Da) | 1.81 | 2.17 |
| Solvent content (%) | 31.9 | 43.2 |
| Data collection | ||
| Completeness (%) | 96.8 (97.6) | 99.5 (99) |
| No. of unique reflections | 120754 | 39656 |
| I /σ(I) | 30.3 (2.4) | 23.5 (3.1) |
| R merge (%) | 0.07 (0.5) | 0.09 (0.6) |
| CC1/2 | 0.99 (0.79) | 0.99 (0.87) |
| Redundancy | 4.9 (4.1) | 7.2 (7.1) |
| Wilson B‐factor (Å2) | 43.9 | 16.3 |
| Refinement | ||
| R (%)/R free (%) | 16.1/20.5 | 14.7/18.9 |
| RMSD bond length (Å) | 0.014 | 0.016 |
| RMSD bond angle (°) | 1.6 | 1.7 |
| Average B value (Å2) | 31.9 | 20.1 |
| Number of molecules in AU | 4 | 1 |
| No. of atoms | ||
| Protein | 11251 | 2834 |
| Inhibitor/other ligands | –/25 | 24/21 |
| Water molecules | 964 | 458 |
| Ramachandran analysisb | ||
| Favored (%)/n | 95.4/ | 97.2/ |
| Allowed (%)/n | 4.2/43 | 2.8/28 |
| Outlier (%)/n | – | – |
Data for the highest resolution shell are given in parentheses. The abbreviations RMSD and AU stand for root‐mean‐square deviation and asymmetric unit, respectively.
Defined by validation program molprobity.
Structure determination and refinement
The structure of the PonA1 inhibitor‐free form was determined using the single wavelength anomalous dispersion method (SAD) and the AutoSol Wizard from the phenix program suite 36. AutoSol contains hyss 37, solve/resolve 38, xtriage, and phenix.refine scripts to determine Se positions, generate experimental phases, build and refine model structure. The structure of PonA1 in complex with penicillin V was determined by molecular replacement using PHASER 39. The structure of the inhibitor‐free form was taken as the starting model. The structure of the PonA1 inhibitor‐bound form was processed in HKL‐3000 40. Structures were refined in refmac 41 and manually built in coot 42. Water molecules were placed using arp/warp 43 and manually checked in coot 42. The quality of structures was checked using the PDB ADIT validation server (http://deposit.pdb.org/validate/) and molprobity 44. Refinement and validation statistics are summarized in Table 1. The figures were generated with pymol 45, ccp4mg 46, ligplot + 47, and espript 3.0 48.
PDB accession codes
Atomic coordinates and structure factors of PonA1 in inhibitor‐free form and in penicillin V‐bound form are deposited into the Protein Data Bank 49 with accession codes 5CRF and 5CXW, respectively.
Fluorescence thermal shift assay
Fluorescence thermal shift assays were conducted in 384‐well plate format with an assay volume of 10 μL. Concentrated PonA1 protein sample and 5000× Sypro Orange (Invitrogen, Carlsbad, CA, USA) were diluted and mixed in a Hepes buffer (100 mm HEPES, 150 mm NaCl, pH7.5), resulting in protein assay concentration of 2.8 μm and 5× Sypro Orange. After adding the protein Sypro Orange mix to a PCR plate, antibiotics from 10 mm DMSO stocks of the spectrum collection (MicroSource Discovery Systems, Gaylordsville, CT, USA) were added in nanoliter volumes using a Labcyte Echo550 acoustic liquid transfer robot for testing. The assay plates were mixed with a plate shaker, sealed with optical clear plastic seal, and centrifuged. Thermal scanning coupled with fluorescence detection was performed on a Bio‐Rad CFX384 qPCR machine at 1.5 °C·min −1 from 10 °C to 80 °C. Data analysis was performed using the in‐house software excelFTS which uses IDBS XLfit for fitting the fluorescence data to a Boltzmann function to determine the melting temperature T m and other thermal transition parameters. The experiments were performed in triplicate.
Site‐directed mutagenesis
Transpeptidase active site mutants were generated as previously described 15. The M. smegmatis mutant is S468/469A (using the appropriate start site for the enzyme, see 15)—both serines in the canonical SSxK motif were mutated to alanine to ensure that activity would be abolished. For M. tuberculosis, the mutations are S487/488A. The mutations were generated in single copy with vectors that integrate into the L5 phage site in the genome (in M. smegmatis, pMC1s TetO MSMEG_6900‐FLAG; or in M. tuberculosis pMC1s TetO Rv0050‐FLAG) in background strains where endogenous ponA1 was deleted. The FLAG epitope does not compromise PonA1 function 15, as this form of the protein fully complements bacterial growth by OD assay.
Antibiotic profiling experiments
The single copy mutant strains described above were used for all experiments. Isogenic wild‐type controls (wherein wild‐type ponA1 is integrated at the same L5 phage site and endogenous ponA1 is deleted) were included for all experiments, along with untreated cells. For M. tuberculosis experiments, log phase M. tuberculosis H37Rv strains were back‐diluted to a calculated OD600 of 0.01 and grown in 7H9 + OADC complete medium ± antibiotic for 6 days in 24‐well plates. Cells were grown at 37 °C with shaking. Each strain was cultured in triplicate. Control wells contained no antibiotic. Optical density of each well was measured after 6 days. Triplicate wells treated with antibiotic were averaged and then normalized to the average of triplicate untreated wells to calculate the percent survival in the presence of antibiotic. Carbenicillin (1 μg·mL −1) was cotreated with clavulanate (0.5 μg·mL −1). Meropenem was used at 0.15625 μg·mL −1. For M. smegmatis experiments, log phase M. smegmatis mc2155 strains were back‐diluted to a calculated OD600 of 0.01 and grown in 7H9 + ADC complete medium ± antibiotic for 48 h in 96‐well plates. Cells were grown at 37 °C with shaking. Control wells contained no antibiotic. Optical density of each well was measured every 30 min. All data were plotted without normalization to untreated control strains. Cells were treated with penicillin V at 1, 0.5, and 0.25 μg·mL −1 in the presence of 5 μg·mL −1 clavulanate (cultured in triplicate) or meropenem at 2.5, 1.25, and 0.625 μg·mL −1 (cultured in duplicate).
Conflict of interest
The authors have no conflict of interest to declare.
Author contributions
EVF, KJK, CHL, EJR, and WFA planned experiments; EVF, KJK, CHL, ZW, and OK performed experiments; EVF, KJK, CHL, EJR, and WFA analyzed data; EVF, KJK, CHL, OK, EJR, and WFA wrote the paper.
Acknowledgements
The data collection was performed at the LS‐CAT at the Advanced Photon Source supported by the Argonne National Laboratory, which is operated by University of Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE‐AC02‐06CH11357. The LS‐CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri‐Corridor (Grant 085P1000817). The project was supported by NIH PSI grant GM074942 to the Midwest Center for Structural Genomics. KJK was supported by a National Science Foundation Graduate Research Fellowship (DGE1144152 and DGE0946799). This project has been funded in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contracts No. HHSN272200700058C and HHSN272201200026C (WFA), and the National Science Foundation grant MCB 1024945 (MAB).
References
- 1. World Health Organization (2014) Global Tuberculosis Report 2014. World Health Organization, Geneva. [Google Scholar]
- 2. Brennan PJ (2003) Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis . Tuberculosis 83, 91–97. [DOI] [PubMed] [Google Scholar]
- 3. Mahapatra S, Crick DC, McNeil MR & Brennan PJ (2008) Unique structural features of the peptidoglycan of Mycobacterium leprae . J Bacteriol 190, 655–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kieser KJ & Rubin EJ (2014) How sisters grow apart: mycobacterial growth and division. Nat Rev Microbiol 12, 550–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Farhat MR, Shapiro BJ, Kieser KJ, Sultana R, Jacobson KR, Victor TC, Warren RM, Streicher EM, Calver A, Sloutsky A et al (2013) Genomic analysis identifies targets of convergent positive selection in drug‐resistant Mycobacterium tuberculosis . Nat Genet 45, 1183–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kumar P, Arora K, Lloyd JR, Lee IY, Nair V, Fischer E, Boshoff HI & Barry CE 3rd (2012) Meropenem inhibits D, D‐carboxypeptidase activity in Mycobacterium tuberculosis . Mol Microbiol 86, 367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Correale S, Ruggiero A, Capparelli R, Pedone E & Berisio R (2013) Structures of free and inhibited forms of the L, D‐transpeptidase LdtMt1 from Mycobacterium tuberculosis . Acta Crystallogr D Biol Crystallogr 69, 1697–1706. [DOI] [PubMed] [Google Scholar]
- 8. Sauvage E, Kerff F, Terrak M, Ayala JA & Charlier P (2008) The penicillin‐binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32, 234–258. [DOI] [PubMed] [Google Scholar]
- 9. Zapun A, Contreras‐Martel C & Vernet T (2008) Penicillin‐binding proteins and beta‐lactam resistance. FEMS Microbiol Rev 32, 361–385. [DOI] [PubMed] [Google Scholar]
- 10. Hugonnet JE, Tremblay LW, Boshoff HI, Barry CE 3rd & Blanchard JS (2009) Meropenem‐clavulanate is effective against extensively drug‐resistant Mycobacterium tuberculosis . Science 323, 1215–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cho H, Uehara T & Bernhardt TG (2014) Beta‐lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell 159, 1300–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE 3rd et al (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. [DOI] [PubMed] [Google Scholar]
- 13. Goffin C & Ghuysen JM (2002) Biochemistry and comparative genomics of SxxK superfamily acyltransferases offer a clue to the mycobacterial paradox: presence of penicillin‐susceptible target proteins versus lack of efficiency of penicillin as therapeutic agent. Microbiol Mol Biol Rev 66, 702–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Patru MM & Pavelka MS (2010) A role for the class A penicillin‐binding protein PonA2 in the survival of Mycobacterium smegmatis under conditions of nonreplication. J Bacteriol 192, 3043–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kieser KJ, Boutte CC, Kester JC, Baer CE, Barczak AK, Meniche X, Chao MC, Rego EH, Sassetti CM, Fortune SM et al (2015) Phosphorylation of the peptidoglycan synthase PonA1 governs the rate of polar elongation in mycobacteria. PLoS Pathog 11, e1005010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Billman‐Jacobe H, Haites RE & Coppel RL (1999) Characterization of a Mycobacterium smegmatis mutant lacking penicillin binding protein 1. Antimicrob Agents Chemother 43, 3011–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Flores AR, Parsons LM & Pavelka MS (2005) Characterization of novel Mycobacterium tuberculosis and Mycobacterium smegmatis mutants hypersusceptible to beta‐lactam antibiotics. J Bacteriol 187, 1892–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Prisic S, Dankwa S, Schwartz D, Chou MF, Locasale JW, Kang CM, Bemis G, Church GM, Steen H & Husson RN (2010) Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc Natl Acad Sci USA 107, 7521–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sung MT, Lai YT, Huang CY, Chou LY, Shih HW, Cheng WC, Wong CH & Ma C (2009) Crystal structure of the membrane‐bound bifunctional transglycosylase PBP1b from Escherichia coli . Proc Natl Acad Sci USA 106, 8824–8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hett EC, Chao MC & Rubin EJ (2010) Interaction and modulation of two antagonistic cell wall enzymes of mycobacteria. PLoS Pathog 6, e1001020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chao MC, Kieser KJ, Minami S, Mavrici D, Aldridge BB, Fortune SM, Alber T & Rubin EJ (2013) Protein complexes and proteolytic activation of the cell wall hydrolase RipA regulate septal resolution in mycobacteria. PLoS Pathog 9, e1003197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krissinel E & Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- 23. Hett EC, Chao MC, Deng LL & Rubin EJ (2008) A mycobacterial enzyme essential for cell division synergizes with resuscitation‐promoting factor. PLoS Pathog 4, e1000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Conteras‐Martel C, Job V, Di Guilmi AM, Vernet T, Dideberg O & Dessen A (2006) Crystal structure of penicillin‐binding protein 1a (PBP1a) reveals a mutational hotspot implicated in beta‐lactam resistance in Streptococcus pneumoniae. J Mol Biol 355, 684–696. [DOI] [PubMed] [Google Scholar]
- 25. Yamada M, Watanabe T, Baba N, Takeuchi Y, Ohsawa F & Gomi S (2008) Crystal structures of biapenem and tebipenem complexed with penicillin‐binding proteins 2X and 1A from Streptococcus pneumoniae . Antimicrob Agents Chemother 52, 2053–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Powell AJ, Tomberg J, Deacon AM, Nicholas RA & Davies C (2009) Crystal structures of penicillin‐binding protein 2 from penicillin‐susceptible and ‐resistant strains of Neisseria gonorrhoeae reveal an unexpectedly subtle mechanism for antibiotic resistance. J Biol Chem 284, 1202–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sainsbury S, Bird L, Rao V, Shepherd SM, Stuart DI, Hunter WN, Owens RJ & Ren J (2011) Crystal structures of penicillin‐binding protein 3 from Pseudomonas aeruginosa: comparison of native and antibiotic‐bound forms. J Mol Biol 405, 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Han S, Zaniewski RP, Marr ES, Lacey BM, Tomaras AP, Evdokimov A, Miller JR & Shanmugasundaram V (2010) Structural basis for effectiveness of siderophore‐conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa . Proc Natl Acad Sci USA 107, 22002–22007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jeong JH, Kim YS, Rojviriya C, Ha SC, Kang BS & Kim YG (2013) Crystal structures of bifunctional penicillin‐binding protein 4 from Listeria monocytogenes . Antimicrob Agents Chemother 57, 3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mouz N, Di Guilmi AM, Gordon E, Hakenbeck R, Dideberg O & Vernet T (1999) Mutations in the active site of penicillin‐binding protein PBP2x from Streptococcus pneumoniae. Role in the specificity for beta‐lactam antibiotics. J Biol Chem 274, 19175–19180. [DOI] [PubMed] [Google Scholar]
- 31. Graves‐Woodward K & Pratt RF (1998) Reaction of soluble penicillin‐binding protein 2a of methicillin‐resistant Staphylococcus aureus with beta‐lactams and acyclic substrates: kinetics in homogeneous solution. Biochem J 332, 755–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fuda C, Suvorov M, Vakulenko SB & Mobashery S (2004) The basis for resistance to β‐lactam antibiotics by penicillin binding protein 2a of methicillin‐resistant Staphylococcus aureus . J Biol Chem 279, 40802–40806. [DOI] [PubMed] [Google Scholar]
- 33. Nicholas RA, Krings S, Tomberg J, Nicola G & Davies C (2003) Crystal structure of wild‐type penicillin‐binding protein 5 from Escherichia coli: implications for deacylation of the acyl‐enzyme complex. J Biol Chem 278, 52826–52833. [DOI] [PubMed] [Google Scholar]
- 34. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A et al (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kieser KJ, Baranowski C, Chao MC, Long JE, Sassetti CM, Waldor MK, Sacchettini JC, Ioerger TR & Rubin EJ (2015) Peptidoglycan synthesis in Mycobacterium tuberculosis is organized into networks with varying drug suscentibility. Proc Natl Acad Sci USA 112, 13087–13092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW et al (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Grosse‐Kunstleve RW & Brunger AT (1999) A highly automated heavy‐atom search procedure for macromolecular structures. Acta Crystallogr D Biol Crystallogr 55, 1568–1577. [DOI] [PubMed] [Google Scholar]
- 38. Terwilliger TC (2003) SOLVE and RESOLVE: automated structure solution and density modification. Methods Enzymol 374, 22–37. [DOI] [PubMed] [Google Scholar]
- 39. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC & Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Minor W, Cymborowski M, Otwinowski Z & Chruszcz M (2006) HKL‐3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62, 859–866. [DOI] [PubMed] [Google Scholar]
- 41. Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F & Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emsley P & Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 43. Langer GG, Hazledine S, Wiegels T, Carolan C & Lamzin VS (2013) Visual automated macromolecular model building. Acta Crystallogr D Biol Crystallogr 69, 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Davis IW, Leaver‐Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB 3rd, Snoeyink J, Richardson JS et al (2007) MolProbity: all‐atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35, W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Delano WL (2002) The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA. [Google Scholar]
- 46. McNicholas S, Potterton E, Wilson KS & Noble ME (2011) Presenting your structures: the CCP4mg molecular‐graphics software. Acta Crystallogr D Biol Crystallogr 67, 386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wallace AC, Laskowski RA & Thornton JM (1996) LIGPLOT: a program to generate schematic diagrams of protein‐ligand interactions. Protein Eng 8, 127–134. [DOI] [PubMed] [Google Scholar]
- 48. Robert X & Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42 (W1), 320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, Feng Z, Gilliland GL, Iype L, Jain S et al (2002) The Protein Data Bank. Acta Crystallogr D Biol Crystallogr 58, 899–907. [DOI] [PubMed] [Google Scholar]