

Abstract
The definition of sepsis has been recently modified to accommodate emerging knowledge in the field, while at the same time being recognized as challenging, if not impossible, to define. Here we seek to clarify the current understanding of sepsis as one that has been typically framed as a disorder of inflammation to one in which the competing interests of the microbiota, pathobiota and host immune cells leads to loss of resilience and non-resolving organ dysfunction. Here we challenge the existence of the idea of non-infectious sepsis given that critically ill humans never exist in a germ free state. Finally, we propose a new vision of the pathophysiology of sepsis that includes the invariable loss of the host’s microbiome with the emergence of a pathobiome consisting of both healthcare acquired and healthcare adapted pathobiota. Under this framework, the critically ill patient is viewed as a host colonized by pathobiota dynamically expressing emergent properties which drive, and are driven by, a pathoadaptive immune response.
Introduction
“Every judgment in science stands on the edge of error, and is personal” Jacob Bronowski (1)
While there is general agreement that the term “sepsis” is difficult, if not impossible to define, framing it as a disorder of inflammation that can be treated with pathway-blocking agents remains a pervasive line of inquiry. Virtually every report on sepsis begins with the declaration that the problem is increasingly affecting more than 750,000 patients each year and that more than 200,000 patients in the US die each year of sepsis (2). Grants and review articles continue to claim that “sepsis” and its associated mortality rate is escalating at an alarming rate. Like cancer, there is now a war on sepsis, and, with proper funding, the cure is right around the corner.
Yet an often overlooked fact of sepsis is that in the overwhelming majority of cases, the actual cause of death in a patient with sepsis remains ill-defined and obscured by the term itself. The main reason for this is threefold: 1. the clinical syndrome of sepsis cannot be precisely defined in any meaningful clinical or biologic context despite numerous and recent attempts (3). 2. the sepsis continuum has been dichotomously split into “non-infection related sepsis” and “infection-related sepsis” without any clear defined confirmation of the involvement, or lack of involvement, of pathogenic bacteria in one circumstance versus the other (4) and 3. the underlying disorder of the patient and its survivability (end stage cancer, ruptured aortic aneurysm with irreversible shock, 90% burn injury) is regularly decoupled from sepsis-related causes of mortality whereby an individual is declared to have died from “sepsis” and “multiple organ failure” and not, for example, from end-stage metastatic cancer or irreversible shock (5). Finally, it is now widely recognized that most septic patients today who die, die as a result of discontinuation of care when it is deemed to be futile. Today, it is extremely rare for a patient to die within 48 hours of presenting with the diagnosis of “sepsis.” Even then, death is often attributed to failure to diagnose/rescue, inappropriate resuscitation (6), insufficient/inappropriate antibiotic coverage or delay in achieving source control (7). While frailty, major co-morbid conditions, or an unsurvivable injury are well known factors that contribute to so-called “sepsis-related mortality” (8), these factors are rarely, if ever, properly accounted for in the final tally. In order to more precisely understand the pathobiology of sepsis in the context of today’s critically ill patients, deaths due to sepsis must be properly adjudicated by analyzing and documenting the most proximate and plausible cause of death. Today, most patients die late in the course of their critically ill state in association with colonization by highly virulent multi-drug resistant pathogens (9). Late deaths of this sort are regularly attributed to immunoparalysis or immunosuppression with the pathogens acting simply as opportunistic secondary actors (10). We must also come to grips with the fact that virtually every patient with severe sepsis is being administered multiple antibiotics whether an infectious source is identified or not (11). While more recent drafts of the definition of sepsis include “suspected infections,” if we are to move the field forward, we must explain and justify why dying critically ill patients with sterile cultures and normal imaging studies invariably remain on antibiotics (3, 12). This practice likely continues for two reasons: clinicians are using antibiotics as a desperate last resort measure or they actually suspect that a pathogen or pathogen community itself is driving the septic process despite its source remaining unidentified. We suspect most experienced intensivists would agree it is the latter. Until which time patients can be rendered germ-free, it is not appropriate to claim that there is such a thing as “non-infectious sepsis.”
Recently, our laboratory presented findings in a mouse model of lethal gut-derived sepsis to the committee on immunology at the University of Chicago. In these experiments, mice underwent a recoverable surgical procedure (30% hepatectomy) to mimic the physiologic stress of injury, followed by direct intestinal inoculation (via cecal puncture) of a highly resistant human four-pathogen community. The pathogen community was isolated from the stool of a critically ill patient who developed multiple organ dysfunction following a liver transplant, remained in the intensive care unit for a prolonged period and eventually died (13). The pathogen community consisted of Candida albicans, multi-drug resistant Enterococcus faecalis, Klebsiella oxytoca, and Serratia marcescens. All of the pathogen-inoculated animals appeared sick by 24 hours and 60% went on to die within 72 hours, while the remaining 40% recovered fully (14). An immunologist in the audience asked “what was the cause of death in the mice that died? Did they die of disseminated bacteremia?” We responded by stating that death in this model was not necessarily predicted by the presence or concentration of disseminated bacteria (i.e in the blood, liver, lung, etc). Both, the dying mice and recovered mice, showed evidence of bacterial dissemination, supporting the well-documented observation that most patients do not die of bacteremia, they die with it (15). So then why did the mice die?
Both clinically and experimentally, it is not yet clear whether patients or experimentally manipulated mice actually die of bacteremia or with it, despite the bias that the presence of bacteremia itself must confer a greater mortality rate. Results from experiments spanning 15 years using our gut-derived sepsis models, have demonstrated that neither bacteremia nor fungemia discriminates between those mice that live versus those that die within a treatment group- only between treatments (control versus septic). Why is this? All of the pathogen-inoculated animals look septic at 24 hours postoperatively; however, some go on to survive while others die. Bacterial translocation to the lymph nodes, liver, or spleen, per se also does not discriminate between progression to lethality versus recovery. The ensuing discussion consisted of using a variety of immune knockout approaches to identify the immune pathway/unit responsible for the mortality in this model. The invariable direction of this line of inquiry is a product of the pervasive immunocentric view of sepsis research – an immune/inflammatory unit/pathway must be identified that is required for mortality in any model of sepsis, because - in terms of ultimate causality- mortality is due to the response and not to the inciting pathogen (s). But isn’t it both?
Here are questions the audience did NOT ask- “What were the life-histories and genetic ancestry of the four organisms you injected? What antibiotics were these organisms exposed to when they were in their original habitat (i.e dirt, chickens, cows, or the septic patient’s intestine)? What were the characteristics of the previous hosts and host environments to which they were exposed? Were comparative pathogenomics performed between these strains and standard laboratory pairs? Did you determine if the disseminated pathogens expressed different virulence genes between the dying and surviving mice? The reason none of these questions are even contemplated in experimental sepsis research is because it is assumed that the mortality from sepsis is due to an immunologic disorder, where the inciting test pathogen (usually a laboratory strain) is considered to be irrelevant as the immune/inflammatory disorder takes over (16). Once the pathogen can no longer be recovered (in blood, sputum, urine or abscess), it is no longer considered to contribute to the sepsis continuum. This bias also occurs on the part of the infectious disease specialist whose primary interest is identifying the causative pathogen and implementing a broad-based kill strategy (17).
Perhaps much of the distraction in sepsis research away from the pathogens’ involvement is a result of observations generated in acute sepsis models such as cecal ligation and puncture and endotoxin administration. Investigators in the field have been traditionally anchored to the notion that these models are sufficient to inform the path forward for drug development in human sepsis. Evidence for this can be easily obtained by searching the NIH website “Grantome” using sepsis as the search word. With no exception, every funded grant is based on the immunocentric theory of sepsis and almost every grant has a promissory note that blockade of a pathway or molecule will inform a strategy to improve the outcome of human sepsis. Implicit in each of these proposals is the practice of dismissing any ongoing involvement of the inciting pathogen or any role for the ecological collapse of the normal microbiota (microbiome) in the sepsis process. Finally, in order for the immunocentric view to prevail, the cause of death from sepsis must be believed to be due to the response itself. This means that in the majority of patients dying in intensive care units today, patients are not dying of an infection from a virulent pathogen, they are dying of the response to the pathogen. For decades, when patients were dying of sepsis in the first 24–48 hours, sepsis was considered to be a result of “overexuberant inflammation.” Today, as late onset sepsis predominates, it has been framed as a problem of immunoparalysis (18). Here we assert, that while immunologic interrogation is critically important and needs to be understood along the entire continuum of sepsis, it needs to be balanced with more causal inference from the pathogens that are the primary drivers of the disorder itself. Finally, the important role that the core microbiome plays in driving a recovery-directed immune response when exposure to highly pathogenic bacteria occurs is just coming to light and needs to be accounted for in mouse models in which survival is not anticipated.
Cecal Ligation and Puncture and endotoxin administration- necessary evils in the war against sepsis
Before mounting an all-out assault on animal models of sepsis, to paraphrase Box (19), it is important to keep in mind that while most animal models are not clinically relevant, they are nonetheless useful. In many cases of basic scientific discovery, investigators need to have the freedom to perform experiments that are solution agnostic. This type of freedom to explore is critical for the process of discovery. It is true that regardless of the clinical relevance of a given animal model, if it informs basic biology, then it adds value on some level. However, at the same time it is important to recognize that the cecal ligation and puncture (CLP) model of sepsis, as performed today and prior, is at best, nothing more than a model of gross medical negligence and surgical malpractice. Surgery is performed to create a necrotic perforated cecum with localized peritonitis that is left completely untreated until which time animals die. Somehow this model has been considered to mimic the mechanism by which patients die in an intensive care unit. CLP represents a completely remedial surgical problem, where were adequate volume resuscitation to take place, appropriate surgical intervention and administration of antibiotics provided, most, if not all animals would survive. Although both cecal excision and antibiotic administration following CLP have been shown to improve mortality, that we are aware, there are no reports applying both treatments to the CLP model (20, 21). Furthermore, volume resuscitation was not standard (5% dextrose only) in these reports. As such my laboratory performed a full source control treatment with saline resuscitation as depicted in figure 1 and as would be standard for a patient with a perforated viscus. We used the more severe form of the CLP model where a long segment of cecum is ligated and an 18 gauge needle used for puncture. This more severe model is known to have a high and rapid developing mortality rate (22). As can be seen, with proper medical care (i.e antibiotics, fluid resuscitation) and surgical care (excision of necrotic tissue and abscess drainage), high grade CLP is a completely survivable injury.
Figure 1.
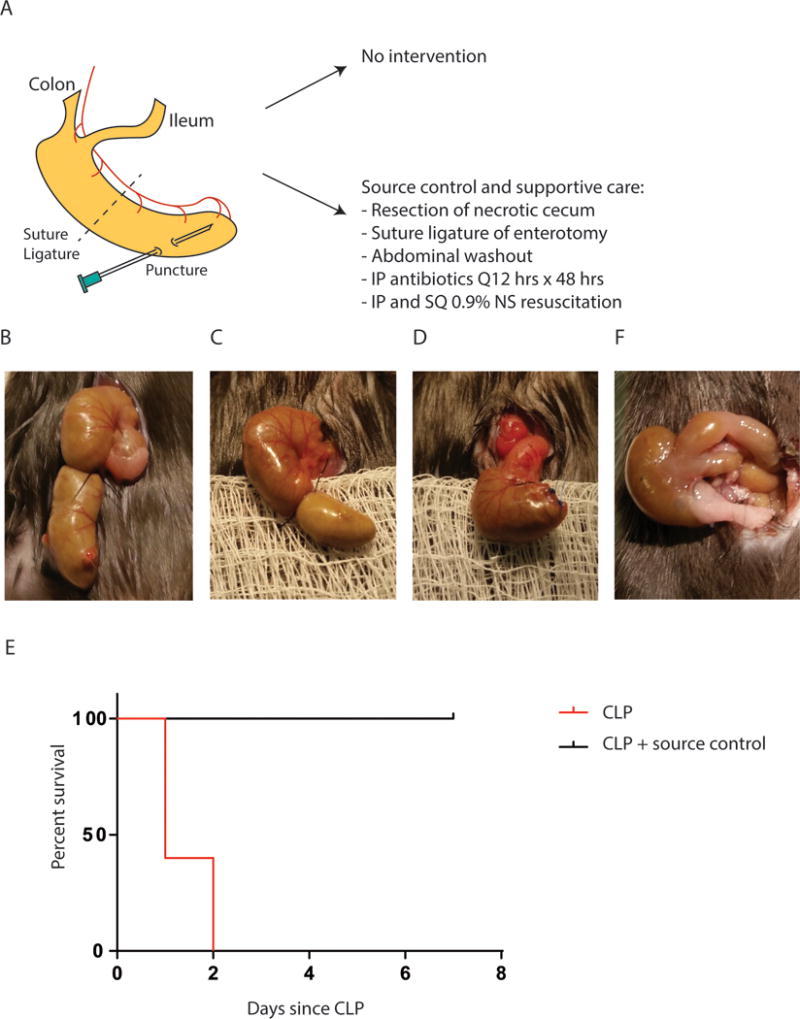
Mouse sepsis due to cecal ligation and puncture (CLP) can be reversed with proper source control. (A) Experimental design of mouse model of high grade CLP known to result in 100% mortality within 48–72 hrs (75). Following CLP, mice were divided into two groups: (1) no intervention and (2) operative source control, antibiotic treatment, and fluid resuscitation at 6 hours following CLP when first signs of sepsis are noted. (B) Appearance of cecum immediately following CLP. (C) Appearance of cecum 6 hours following CLP. (D) Appearance of cecum following surgical source control, i.e. resection of necrotic cecum and suture ligature of the enterotomy. (E) Mortality is 100% at day 2 following CLP. Source control and supportive therapy result in 0% mortality and all mice recover completely by day 7 (p=0.0023, n=5/group). (F) Appearance of cecum on day 7 following successful operative source control and supportive therapy.
In many decades previously, ostensibly the CLP model was developed to mimic a perforated viscus that went untreated, representing then a more common cause of death. Today however, in the overwhelming majority of patients treated in advanced care systems, survival from a perforated viscus approaches 100%. Again if such patients do die, it is usually attributed to failure to diagnose, rescue or implement the proper source control and antibiotics. While delayed treatments of appendicitis and other perforated viscera undoubtedly do cause deaths in modern ICUs, such cases do not represent the majority of causes of sepsis and multiple organ failure seen in the ICU today (23). Yet in support of the continued use of the CLP model is the claim that if we can understand the early phases of this response, we can prevent the later stages of sepsis. Yet, if we unpack the logic behind this assertion, it is untenable. Untreated peritonitis with normal rodent flora, without the exposure to the hospital environment and the selective pressure of antibiotics on the colonizing microbiota/pathobiota and without the surgical intervention that removes the disease process itself, the CLP model is not likely to inform the pathogenesis of sepsis, as it occurs today when patients are maximally treated in an ICU with the appropriate care.
It should be recognized that indeed some patients can present with rapidly evolving pneumonia due to highly aggressive pathogens such as Pseudomonas aeruginosa and can suddenly die due to vascular collapse. P aeruginosa can express exotoxin A, an ADP ribosylotransferase that can open endothelial tight junctions and cause rapid and untreatable vascular collapse (24). Yet today patients rarely, if ever, die this type of “rapidly evolving sepsis” when treated appropriately. Unfortunately animal models have not changed in decades to accommodate the evolving demographics of sepsis where patients die late in the course of infection or injury. A similar line of reasoning could be applied to animal models of sepsis in which a near lethal dose of endotoxin is administered (25). Again, it is important to recognize that much biology has been elucidated using endotoxin shock model, and such models should, on some level, continue. However development of an agent that prevents death in mice administered a lethal dose of endotoxin is unlikely to save a critically ill patient colonized by healthcare adapted pathogens, whose core protective microbiome has been eliminated by antibiotics and whose hospital course is characterized by dialysis and ventilator dependence for one month with multiple organ failure. Again, such models seem to dismiss the chronicity of critical illness, its attendant adaptive and counteradaptive responses to aggressive and invasive medical care and the influence of all of these selective pressures on the emerging pathobiome, which itself can directly cause immunosuppression (26).
It should also be recognized that the most common cause of mortality following sepsis among hospitalized patients is pneumonia. Most patient who develop life-threatening pneumonia and sepsis are immunocompromised, elderly, have COPD, are on ventilators and have received multiple antibiotics both prior to, and concurrent with their disease. Among the most aggressive pathogens associated with lethal pneumonia is Pseudomonas aeruginosa. So who dies of Pseudomonas pneumonia and why? Again there are compelling reasons to model and understand the pathobiology of P. aeruginosa infection in the lung at the most basic level. However, when studying why patients die of sepsis due to Pseudomonas lung infection, animal models need to move beyond intratracheal injection of a massive inoculum of the pathogen. Pseudomonas will eventually colonize the gut following lung inoculation and can express a wide variety of antibiotics that normally kill off competing bacteria, thus dramatically altering the microbiome (27). The microbiome itself plays a key role in regulating the local, lung and systemic immune system (28, 29). Fecal transplants are able to rescue mice from lethal pneumonias and patients routinely receive broad spectrum antibiotics when being treated for pneumonia which will necessarily alter the gut microbiota (30). If a patient with P. aeruginosa pneumonia dies 30 days after admission to the hospital, did they die of frailty (immune exhaustion), failure to rescue (inappropriate medical care), or simply from a highly evolved pathogen or pathogen community (pathobiome) capable of eliminating the competing microbiota and subverting the immune system? Perhaps it is more honest to declare that the patient simply died of Pseudomonas pneumonia. To claim this death is due to sepsis implies that endotoxin models and CLP models of sepsis will help define the path forward to drug development in Pseudomonas pneumonia because converging lines of evidence will define the “final common pathway” in which pharmacologic interference will stop the insidious inflammatory disorder common to all “sepsis.” The many failures of clinical trials informed by this approach are testament to the failed thinking of this mechanistic framework. Perhaps some pathogens are simply short sighted and make the fundamental tradeoff to kill the very host upon whom their survival may depend (31). In some cases this occurs rapidly, in others slowly. As one author declared “medicine needs evolution” and this is certainly the case with sepsis research (32).
Intestinal permeability, bacterial translocation, intestinal epithelial apoptosis: unpacking the confusion
The leaky gut hypothesis of sepsis syndrome leading to multiple organ failure claims that critical illness creates a primary disorder of the gut epithelial barrier in which there is loss of intestinal permselectivity to proinflammatory microbial exoproducts (i.e endotoxins) that results in counterproductive inflammation (33). At its best, this neatly packed oversimplified story has advanced our understanding of the biology of tight junctional regulation as it relates to physiologic stress such as intestinal ischemia and traumatic injury (34). At its worst, it has led investigators down a path in which they are studying the consequences of the post-injury response, not its cause. Intestinal permeability defects have been linked to bacterial translocation events and the term “bacterial translocation” has been inappropriately used to describe how the intestinal microbiota drive the immunopathology of sepsis (35). The fallacy implied in the term bacterial translocation is that the event itself, i.e. the mere relocation of any luminal bacterium to any extraintestinal site, is, in and of itself, pathologic. The same can be said of intestinal permeability defects. While there is evidence that both these events may contribute to inflammation following injury, ischemia or infection, they alone do not discriminate between those subjects (animal or human) that develop inflammation or not, develop sepsis or not, or who survive sepsis or not (36). Many of the models point to intestinal ischemia-reperfusion (I/R) injury, a common occurrence following any sudden insult, as the inciting event. These studies often begin with the declaration that intestinal I/R following shock causes altered barrier function that leads to sepsis and multiple organ failure. Yet, despite numerous attempts to document this, the fact remains that most humans that suffer profound shock (cardiac arrest, massive hemorrhage, etc), rarely develop intestinal ischemia reperfusion injury that can be linked to multiple organ failure (37, 38). Furthermore when sepsis syndrome and multiple organ failure develop post arrest, there is no evidence that they are causally linked to intestinal barrier failure (39). Although experimentally, elimination of the microbiota during intestinal ischemia reperfusion attenuates the permeability defect (40) and the presence of pathobiota exacerbate it (41), the extent to which the defined permeability defect is causative to the subsequent immunopathology and organ failure observed remains unknown.
Some of the best work in the field on the role of intestinal permeability and sepsis has come from the Coopersmith laboratory. Work from this group has provided some of the most compelling evidence that indeed the intestinal permeability defect observed during infection and injury may contribute to the sepsis continuum. Using a targeted genetic approach, Coopersmith and colleagues created an animal model of forced expression of the BCLR-2 gene in the gut holding intestinal apoptosis constant. Septic mice did not develop intestinal apoptosis and demonstrated enhanced survival despite the presence of bacterial translocation (42). These and other studies using IL-15 and epidermal growth factor administration demonstrate that attempts to control hyperpermeability over the course of physiologic or infective stress do indeed improve survival in animals (43). As such understanding the causes and consequences of the intestinal permeability defect and its contribution to multiple organ failure is critically important. Yet because the intestinal microbiome and the emerging pathobiome are so intimately involved in how the intestine regulates is permselectivity, this group has recently proposed a more holistic framework suggesting that changes in the microbiome and bacterial virulence remain yet-to be-accounted for factors in the gut hypothesis of sepsis (44, 45).
Clarification is needed in field of intestinal permeability and bacterial translocation to define the gain and loss of specific microbes and their phenotypic characterization as principle regulators of the barrier disruption (mucus, apoptosis, etc) and hyperpermeability process. While our group was the first to show that that stress-induced intestinal permeability defects are dependent on the presence and phenotype of intestinal microbiota (46, 47), and is now confirmed by others (48), the community structure and phenotype of the intestinal microbiota that initiate and drive the permeability defect remain unknown.
Basic science has demonstrated that certain bacteria express both structural appendages and soluble exoproducts that bind pathogen recognition receptors on intestinal epithelial cells, which then transduce a number of downstream pathways that not only alter tight junctional permselectivity, but also alter local and systemic immune function (49). Bacteria need not translocate to transduce these downstream pathways as both epithelial receptors and antigen presenting cell receptors are readily accessible to luminal pathogens at the epithelial surface itself. Opportunism for such events develops when there is critical loss of the protective microbiota, which can develop in response to physiologic stress itself. As soon as 6 hours after a sudden insult, 90% of the normal anaerobic flora is lost in the gut (50). The mechanisms of this effect remains unexplored and thus unknown. As a consequence there is a “bloom” in select pathogens that can then express proteases which break down the intestinal epithelial mucus layer (loss of barrier function). Once mucus is eliminated, spatial recognition mechanisms by microbes induces the expression of adhesins facilitating adherence to pathogen recognition receptors on epithelial cells. Ligation of the bacterial appendage/exoproduct then initiates the cascade of downstream transduction events which have been traditionally interpreted as primary events (intestinal hyperpermeability, loss of mucus, bacterial translocation). Yet in virtually every model of survival from injury (intestinal I/R, CLP, etc), intestinal decontamination of the intestinal microbiota not only reverses the secondary effects (loss of mucus, hyperpermeability, bacterial translocation) but also improves survival (51, 52). Alternatively introduction of pathogenic bacteria in these models, which alone can kill off competing microbiota (i.e Pseudomonas), worsens survival (41). For example, intestinal I/R causes loss of the overlying mucus as well as an intestinal permeability defect due to alterations in tight junctional proteins (53). Intestinal mucus production is directly dependent on the presence of the microbiota (54, 55). Healthy unstressed mice decontaminated of their intestinal microbiota with antibiotics display an increase in tight junctional protein expression and a decrease in intestinal permeability (55). Therefore, while it is well described that microbes control the epithelial barrier and are the primary drivers of the gut-origin sepsis paradigm, much, if not all, of the work in the field is focused on epithelial biology and immunology as primary events in the process. This has been promulgated by the ease with which their functional parameters (i.e transepithelial electrical resistance, flux of probes across ex vivo tissues, serum cytokines, etc.) can be measured. Yet the actual bacterial ligands that are responsible for the proposed gut-derived sepsis in a given patient, their pathogen(s) of origin, the molecular details by which the pathogen of origin is “cued” in vivo to secrete a given ligand and the molecular targets of secreted ligands, remain uncharacterized. As can be imagined, this will prove to be an exhaustive undertaking. Finally, while it is important to recognize that use of LPS in research led to the discovery of the TLR4 pathway (56), as can be seen, declaring that the pathoadaptive response to human sepsis can be sufficiently modeled by a single LPS injection has its limitations. Yet this persistent line of inquiry has led to large and expensive clinical trials to filter endotoxin from the circulation in patients with sepsis, the results of which have been recently shown to have a negligible effect on survival (http://www.spectraldx.com/).
Collapse of the intestinal microbiome, emergence of a pathobiome and the immunopathology of critical illness- a framework to understand the sepsis continuum
Overview of the microbiome Broadly defined, the microbiome refers to all of the microbial consortia (both commensal and pathogenic bacteria, viruses, and fungi), their genes, their gene products (proteins, metabolites), their community structure (distribution, diversity, evenness) and the particulars of the environment in which they reside. As such, the microbiome is the microbial ecosystem of the body. In this manner, the scientific community speaks of an expanded and diverse human microbial ecosystem, moving beyond simple culture and antibiotic sensitivity. Rapid advances in DNA and RNA sequencing, mass spectrometry (MS/MS), and proteomics, have allowed for the measurement of multiple dynamic components of that ecosystem within a given sample (57). This revolution in microbiome sciences now considers microbial communities at the bottom of the ocean (58), on hospital surfaces and in homes (59) and at tissue and fluid samples sites (60) to be similar. Metagenomic sequencing and mass-spectrometry can now describe not only who is there, but also what they are doing, who are they communicating with, and how are they metabolically interacting with one another. Tools have become so powerful in their analytic capacity, they one can even predict the effect of the local environment (pH, redox state, phosphate, nitrogen, carbon, etc) on microbial community structure and function (61). The ability to measure at such high resolution detail has led investigators to define what might be considered a “health-promoting microbiome” versus a “disease-promoting microbiome” or pathobiome (57). When microbiome analyses of structure such as 16s rRNA, olygotyping (genus and isolate sequences) or when analyses of microbial function such as metabolite and proteomic measurements do not align to normal signatures, to older term dysbiosis has been used (62). Yet next generation sequencing and metabolomics are defining the degree to which microbial diversity, community structure and metabolite concentration is required for the host to maintain its resilience to physiologic and traumatic stress. This is becoming ever clearer in the lung and gut where their respective microbiomes interact with epithelial receptors that communicate via dendritic cells to elements of the systemic immune system and provide tonic and health promoting influences on overall host health maintenance (63). What remains unknown however is to what extent loss of the microbiome impairs immune responsiveness during critical illness and hence recovery from organ failure.
As mentioned previously, within hours following a sudden physiologic insult, the mammalian intestinal microbiome collapses in microbial density, membership composition, and overall community structure and function. Yet why the intestinal microbiota rapidly downregulate growth and metabolic function during physiologic stress is unknown. Perhaps the microbiota “sense” that an ill host cannot feed itself and therefore will undergo a period of inanition, which will necessarily limit their access to nutrients. If the host does survive, the “hibernating” microbiota will have properly invested in the host upon whom their survival may depend. If however the host dies, the intestinal microbiota can feed off the decomposing body and jump to a new host as predators feed off the dead carcass. In fact, compositional and functional changes of the intestinal microbiome feeding off a decomposing corpse is now well-described and displays highly characteristic changes (64–67). Yet a patient maintained on life support and exposed to multiple antibiotics represents an unusual scenario for the microbiota. As there is no evolutionary precedent for this circumstance, it is not surprising that the host immune system might respond in a pathoadaptive manner. Acquisition of hospital adapted pathogens may drive a pathoadaptive response during life support in which the pathogens are the driving force of the immunosuppression.
A major deficiency in the immunocentric view of sepsis is that it does not accommodate, either diagnostically or therapeutically, the growing body of evidence demonstrating the key role that the intestinal microbiome (microbiota that are programmed to induce a recovery-directed immune response) and pathobiome (healthcare adapted pathogen communities) play in recovery from critical illness. It is the position of this paper that the evolvability of the microbiome and pathobiome along the continuum of critical illness and its reverse causality at each time point and network interaction, imposes a level of irreconcilable implausibility for the immunocentric view. Microbes can shift their evolutionary trajectories within hours compared to host adaptation responses, which require days to weeks. An intestinal microbiome that rapidly diminishes in biomass, composition and function following an insult and that is not allowed to re-faunate because of the promiscuous and unavoidable use of antibiotics during the care of the critically ill, has major consequences on the immune system along various points of critically illness (68). Loss of tonic stimulation by the microbiota allows for niche acquisition of an emergent pathobiome within privileged sites, such as intestinal crypts. This pathobiome, replete with ancestral and newly acquired virulence and resistance genes, may become the driver of a pathoadaptive immune response (69). Given that emergent bacterial phenotypes are specific to each critically ill patient, how they reprogram and subvert immune elements in a context dependent manner is likely to be complex, difficult to detect, and highly individualized (67). If this is the case, therapeutic strategies derived from immune pathways informed by experiments from specific pathogen-free mice will not suffice. These models need to incorporate “accidental pathogens”, i.e. those that have not co-evolved with their hosts and who have long life-histories shaped by harsh environments. Due to these selective pressures, these short-sighted pathogens have all the raw goods to develop on-the-spot strategies to kill unfamiliar hosts either quickly, or slowing, depending on what circumstances call for. This is easily accomplished via interspecies and interkingdom quorum sensing, horizontal gene transfer, phage virus incorporation, and specialized antibiotic production to kill off competitors- all in a day’s work.
While a comprehensive review of all the mechanisms by which a microbe-microbe and host-microbe interactions affect host response systems is beyond the scope of the present review, a few points are worth discussing that often go unmentioned in discussions of sepsis. The first is that microbial virulence expression is a dynamic process that is highly dependent on the local microenvironmental context. Microbes that exist in resource-rich environment, rarely express virulence, however when resources are limited, invasion tactics are dynamically expressed in order to obtain nutrients from host cells (70–72). In order to be successful at invasion, microbes must first adhere to and invade tissues in an immunoelusive manner. Microbes achieve this by having evolved exquisitely sensitive information processing systems to “sense and respond” to local environmental cues such as pH, iron, phosphate, osmolality, bacterial population density (quorum sensing), host cell contact, etc (27, 72, 73). However, because virulence activation can be an asset one minute and a liability the next, information processing must be constant and fluid in response to the various physiologic contexts encountered in both space and time. During critical illness, as the microbiome collapses in both bacterial biomass and functional output, pathogen communities proliferate and develop stable strategies to co-exist by distancing themselves from immune clearance mechanisms (i.e biofilm production) or by directly disarming immune cells (74). During critical illness however, the presumption has always been that absent gross signs of inflammation or dysfunction in a particular organ where such pathobiomes might exist (i.e gut, lung), immune function and inflammation have little to do with these invisible microorganisms. Yet there is now compelling evidence that circulating immune cells routinely enter organs such as the lung and gut and can sample the colonizing microbial communities present on the mucosal surface and lumen, become educated and reprogrammed and then recirculate and home to distant sites when they exert a myriad of effects on various arms of immunity and inflammation (75–77). Much of this work appears to occur through neutrophils and T regulatory cells moving in and out of the gut and lung. Particularly implicated in this response are TH-17 cells (78). The microbiocentric view of sepsis might then suggest that the immune and inflammatory response to critical illness is directly shaped by how the inciting insult affects the resilience of the microbiome to recover to normal and by how it is able to resist a hostile takeover by a pathobiome. While the mechanistic details of this framework remain to be defined, it is important to note that luminal pathogens need not cause local mucosal inflammation or need not translocate to influence and redirect a pathoadaptive immune and inflammatory response to critical illness. As molecular tools and sequencing begin to allow for this hypothesis to be formally tested in humans along the entire continuum of the sepsis response, the terms “sterile sepsis” or “non-infected sepsis” will likely disappear.
No two pathogens are alike- niche acquisition, environmental selection and the promise of sequencing technology
Another aspect of confusion in the field of sepsis is the traditional view of infection among the critically ill as a monomicrobial disease. Tidy stories like the causality between Helicobacter pylori and peptic ulcer disease perpetuate the notion that a single pathogen can be identified to explain all infection-related diseases. However upon closer inspection, many patients harbor H. pylori, but do not express the disease phenotype and conversely many patients have peptic ulcer disease and remain culture negative for H. pylori. Such observations have led to the drafting of the molecular Koch’s postulates to explain microbial phenotype variation as a mechanism of infectious disease, not just the mere presence of the pathogen (79). We have previously provided a lengthy review of the molecular Koch’s postulates as they relate to sepsis and critical illness and have proposed that microbial phenotype and microbiome community structure can be a major driving force by which the intestinal microbiota contribute to the immunopathology of sepsis (80).
No two pathogen isolates are completely alike, even when grown from a single strain in pure culture where environmental conditions are held constant, spontaneous mutations occur (81). Add in to this mix interspecies and interkingdom telesensing, horizontal gene transfer, exposure to antibiotics, acquisition of phage viruses and selective pressure by predatory pathogens, and within hours, both microbiome and pathobiome membership, structure and phenotype take on emergent properties whose effect on the immune response will be different for each patient. In the case of the critically ill patient, targeting a single pathogen isolated from the blood, urine, or lung as the causative pathogen in the sepsis process belies the complexity within which that particular microbe ended up in that particular compartment. Invariably broad spectrum antibiotics are applied, the identified pathogen is eliminated along with many others, and the presumption of a monomicrobial cause of sepsis is falsely confirmed. There are indeed circumstances in which single microbes cause sepsis syndrome such as meniggoccocemia, MRSA infection from a defined source, P. aeruginosa pneumonia, and C difficile colitis, yet these are the exceptions not the rule in most critically ill patients. Yet what is lost in the framing of the pathogenesis of these infections is the mechanism by which these pathogens developed into healthcare- and host-adapted pathogens in the first place and how they disrupt a recovery-directed immune response in a critically ill host when the normal microbiota are eliminated by antibiotics.
Designing a path forward: changing models and terms
Among the various criticisms to explain our lack of progress in this field might be to recognize the fact that animal models used today are the same ones that have been used for decades. Mouse models need to be modified so they are more reflective of both human progress itself and the conditions in which critical illness results in life-threatening complications. This might include weeks of eating a western diet, exposure to antibiotics prior to the initiating insult, application of environmental (smoking, alcohol) pharmacological (opioids) and social isolation stress. These exposures would then need to be studied for their effects on the microbiome and host transcriptome. Independent effects of these stressors could be sorted out using germ free mouse conditions paired with specific knockouts. The mouse gut might also be “humanized” to reflect a pathobiome that is typical of a critically ill patient. For example, germ-free mice colonized by various patient pathobiomes could be co-housed with intention-to-treat mice where they might become naturally colonized by coprophagia (82). Conditioned mice could then be subjected to injury and infectious stressors such as burn injury, hemorrhagic shock, exposure to specific pathogens, etc. Mice would be properly resuscitated and treated with the appropriate antibiotics and surgery where indicated. Comparisons between survivors versus non-survivors within the treated group would yield the most important information. Spatially nested pathogen-host interactomes (region-specific intestinal tissues, lymph nodes, liver, spleen etc) could be analyzed using dual RNA-seq. Fecal transplants from sick versus healthy mice could then be transferred to germ-free mice and their independent effects on host immunity interrogated.
Neither multiple organ failure nor sepsis, in our opinion, should be used as a cause of death because they neither inform pathogenesis nor treatment strategies. It is often argued that there is a need for a term such as sepsis or SIRS in order to signal clinicians to the possibility of a rapidly evolving illness and to compare outcomes between treatments protocols, stratify patients in trials, etc. Yet in the context of the discussion herein presented, a universally acceptable definition of sepsis should, de facto, be unachievable given that it seeks to represent the net result of all possible inputs into a global response system within a single compartment (plasma). For this reason, the discriminative value of a sepsis scoring system that facilitates the management of patients, allows for the useful comparison between study patients and that informs disease pathogenesis and mechanism of treatment effects remains contemptuously debated and scientifically unfulfilled.
In order to develop a conceptual and organizational approach to the problem of destructive inflammation in response to a microbial stimulus, Carl Nathan has recently proposed the unifying concept of “nonresolving inflammation” (83). He argues that “the problem with inflammation is not how often it starts, but rather how often it fails to subside.” He posits that while nonresolving inflammation itself is not a cause of disease, it does contributes to disease pathogenesis. As previously mentioned, what is peculiar about sepsis as it is currently defined, is that it continues to be framed as either too much inflammation or not enough. Following trauma, burn injury or severe infection, immunity and inflammation need to flex up and flex down in a context dependent manner. Trying to define sepsis as either the systemic inflammatory response syndrome (SIRS) or persistent inflammation immunosuppression and catabolism syndrome (PICS) is an attempt to categorize subsets of patients and predict outcomes (84). However, these terms belie the very complexity and individualized responses of a given patient, their genes, dynamic gene expression, and the ongoing reciprocal interaction of the host immune/inflammatory system with its colonizing microbiota/pathobiota. The outputs of the various interactions cannot be categorized by describing symptoms or signs of clinical deterioration. As personalized medicine emerges to understand critical illness, the false generalizations of these attempts at categorization will be revealed.
A new term “non-resolving organ dysfunction syndrome (nRODs) might be used to replace all previous terms in this field. The term multiple organ failure suggests an unrecoverable injury to the organ. It is being used as a cause of death despite the etiology remaining unknown. Patients no longer die of a severe infection or a burn injury, they die of persistent inflammation, cachexia, and immunoparalysis, associated with, but not necessarily caused by, multi-drug resistant pathogens. Informed by this mischaracterization, therapy focuses on nitrogen loading, growth factors, immune enhancing and blocking agents. While refinements in these measure and others are needed, scoring systems and biomarkers that attempt to classify clinical presentations as “phenotypes” will fail as no two critically ill patients carry the same pathobiome, nor are exposed to the same physiologic perturbations, nor do they express the same host transcriptome at equal time points.
As the electronic medical record is now capable of capturing patient physiologic data and as personalized medicine generates unique identifiers in patients such as their microbiota, SNPs, proteomic signatures, etc, using the term nRODs may be preferable over any attempt to classify or score a critically ill patient. nRODs is agnostic to cause or solution, it only describes a state of organ dysfunction. The best treatment of nRODs will be informed by the physicians sense of recoverability from the initial insult, daily inputs from consultants such as neurologist and neurosurgeons, a sense of daily improvement in physiologic parameters such as white blood cell count, fever and cardiopulmonary function, the absence of intercurrent infections, and most importantly a sense of the patients and families wishes to continue care. Along this line of reasoning, the term nRODs may be sufficient. On the other hand the pathogenesis of nRODs will be informed by basic scientists working on each organ system using more relevant models beyond cecal ligation and puncture, endotoxin administration, hemorrhagic shock alone, and gut ischemia reperfusion injury in mice colonized by normal mouse microbiota who never received fluid resuscitation or antibiotics. Identifying mechanisms by which such products engage the host response gets at the root cause and mechanisms of nRODs. Understanding the various selective pressures that drive nRODs will require modeling and measurements that at the level of both host and pathogen transcriptome, at various time points and within various spatially nested sites. As the human brain cannot compute this amount of information, computational methods must be applied.
What is gained and what is lost using a terms such as nRODs? Clearly what is gained when systemic inflammation remains sustained in response to an infectious, physiologic or injurious insult, is the host’s attempt at damage control and containment. Yet what is lost with nRODs is the body’s ability to fine-tune inflammation commensurate with modern medicine’s ongoing attempt to maintain life in complete ignorance of the evolved adaptive response of host, microbiota and pathobiota. One such error of ignorance may be the routine elimination of the tonic stimulation provided by the normal microbiota to participate in the response to the ongoing insult. For example, it is known that there are more than 81 genes that are required to prevent the host from developing spontaneous inflammation, likely more (83). How these genes and their activating pathways are affected by critical care management and promiscuous antibiotic use is unknown. The body is accustomed to microbial products within a complex microbial community ecology constantly ligating and activating its pathogen recognition receptors in the lung, gut and elsewhere. While it is not yet understood how this process plays out in real time during critical illness, it still must somehow be managed. Given that there is no evolutionary precedent for survival from critical illness the way it is treated today, nRODs may simply be a matter of any combination of “loss of function mutations” in the host as a mechanism to manage “gain of function mutations” in colonizing pathogens. In the words of the great evolutionary biologist Leigh Van Valen who coined the term the Red Queen Effect (85), “organisms must constantly adapt, evolve, and proliferate not merely to gain reproductive advantage but also simply to survive pitted against ever-evolving opposing organisms in an ever-changing environment.” How this interaction plays out over the course of critical illness in a patient whose microbiome has ecologically collapsed and whose pathobiome is constantly evolving in response to antibiotics and life support measures remains to be elucidated.
It is time to recognize that we do not need the terms sepsis or multiple organ failure, SIRS or PICS to study why patients die of critically illness. In many ways these terms perpetuate a flawed paradigm of the actually clinical state that characterizes individual patients. The underlying disorder and its survivability at the time of ICU confinement, the patients’ response to treatment and the particulars of their newly acquired pathobiome and their lost microbiome should define their trajectory toward recovery or futility. Sepsis and its attendant serum biomarkers cannot fulfill this holistic and dynamic ecological view of critical illness, nor can multiple organ failure, nor any other term. The term nRODs is also inadequate, it defines neither recoverability nor futility. It strength is that it leaves behind the term sepsis and all of its baggage.
Conclusion
According to quantum physics, “no matter how much information we obtain or how powerful our computing abilities, the outcomes of physical processes cannot be predicted with certainty because they are not determined with certainty” (86). Two concepts in physics have been ignored in our attempt to understand the cause of mortality in the human response to severe injury and infection: emergent properties and reverse causality (82). That human and microbial cells are constantly exchanging information along the entire continuum of critically illness at every epithelial surface in the body, presents a daunting challenge to sort out the probabilistic from the deterministic. While it may seem impossible and economically implausible to account for all the time- and context dependent fluctuations herein proposed, scientists at the very edge of studying such fluctuations will tell us, it is not necessary. A good doctor does not need all of the information, just enough of it. Perhaps the first step for us practitioners is to part with our illusion that patients are actually dying of a definable disorder called “sepsis.”
Acknowledgments
Copyright form disclosure: Dr. Alverdy’s institution received funding from the National Institutes of Health (NIH). He received funding from the NIH and from Reshape Medical (royalties). Dr. Alverdy and Dr. Krezalek received support for article research from the NIH.
NIH grant support: 2R01GM062344-14
References
- 1.The Ascent of Man - BBC Four [Internet] BBC. [cited 2016 Oct 5] Available from: http://www.bbc.co.uk/programmes/b00wms4m.
- 2.Shankar-Hari M, Deutschman CS, Singer M. Do we need a new definition of sepsis? Intensive Care Med. 2015;41:909–911. doi: 10.1007/s00134-015-3680-x. [DOI] [PubMed] [Google Scholar]
- 3.Shankar-Hari M, Phillips GS, Levy ML, et al. Developing a New Definition and Assessing New Clinical Criteria for Septic Shock: For the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) JAMA. 2016;315:775–787. doi: 10.1001/jama.2016.0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vincent J-L, Rello J, Marshall J, et al. International study of the prevalence and outcomes of infection in intensive care units. JAMA. 2009;302:2323–2329. doi: 10.1001/jama.2009.1754. [DOI] [PubMed] [Google Scholar]
- 5.Beesley SJ, Lanspa MJ. Why we need a new definition of sepsis. Ann Transl Med. 2015;3:296. doi: 10.3978/j.issn.2305-5839.2015.11.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burnham JP, Lane MA, Kollef MH. Impact of Sepsis Classification and Multidrug-Resistance Status on Outcome Among Patients Treated With Appropriate Therapy. Crit Care Med. 2015;43:1580–1586. doi: 10.1097/CCM.0000000000001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lynn LA. The diagnosis of sepsis revisited - a challenge for young medical scientists in the 21st century. Patient Saf Surg. 2014;8:1. doi: 10.1186/1754-9493-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kempker JA, Martin GS. The Changing Epidemiology and Definitions of Sepsis. Clin Chest Med. 2016;37:165–179. doi: 10.1016/j.ccm.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Otto GP, Sossdorf M, Claus RA, et al. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care Lond Engl. 2011;15:R183. doi: 10.1186/cc10332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng S-C, Scicluna BP, Arts RJW, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol. 2016;17:406–413. doi: 10.1038/ni.3398. [DOI] [PubMed] [Google Scholar]
- 11.De Waele JJ, Rello J, Anzueto A, et al. Infections and use of antibiotics in patients admitted for severe acute pancreatitis: data from the EPIC II study. Surg Infect. 2014;15:394–398. doi: 10.1089/sur.2012.228. [DOI] [PubMed] [Google Scholar]
- 12.Seymour CW, Liu VX, Iwashyna TJ, et al. Assessment of Clinical Criteria for Sepsis: For the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) JAMA. 2016;315:762–774. doi: 10.1001/jama.2016.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zaborin A, Smith D, Garfield K, et al. Membership and behavior of ultra-low-diversity pathogen communities present in the gut of humans during prolonged critical illness. mBio. 2014;5:e01361–1314. doi: 10.1128/mBio.01361-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaborin A, Defazio JR, Kade M, et al. Phosphate-containing polyethylene glycol polymers prevent lethal sepsis by multidrug-resistant pathogens. Antimicrob Agents Chemother. 2014;58:966–977. doi: 10.1128/AAC.02183-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Savage RD, Fowler RA, Rishu AH, et al. The Effect of Inadequate Initial Empiric Antimicrobial Treatment on Mortality in Critically Ill Patients with Bloodstream Infections: A Multi-Centre Retrospective Cohort Study. PloS One. 2016;11:e0154944. doi: 10.1371/journal.pone.0154944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest. 2016;126:23–31. doi: 10.1172/JCI82224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen J, Vincent J-L, Adhikari NKJ, et al. Sepsis: a roadmap for future research. Lancet Infect Dis. 2015;15:581–614. doi: 10.1016/S1473-3099(15)70112-X. [DOI] [PubMed] [Google Scholar]
- 18.Muenzer JT, Davis CG, Chang K, et al. Characterization and modulation of the immunosuppressive phase of sepsis. Infect Immun. 2010;78:1582–1592. doi: 10.1128/IAI.01213-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Box GEP. Science and Statistics. J Am Stat Assoc. 1976;71:791–799. [Google Scholar]
- 20.Baker CC, Chaudry IH, Gaines HO, et al. Evaluation of factors affecting mortality rate after sepsis in a murine cecal ligation and puncture model. Surgery. 1983;94:331–335. [PubMed] [Google Scholar]
- 21.Remick DG, Bolgos GR, Siddiqui J, et al. Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock Augusta Ga. 2002;17:463–467. doi: 10.1097/00024382-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 22.Rittirsch D, Huber-Lang MS, Flierl MA, et al. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–36. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hecker A, Schneck E, Röhrig R, et al. The impact of early surgical intervention in free intestinal perforation: a time-to-intervention pilot study. World J Emerg Surg WJES. 2015;10:54. doi: 10.1186/s13017-015-0047-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baluna R, Rizo J, Gordon BE, et al. Evidence for a structural motif in toxins and interleukin-2 that may be responsible for binding to endothelial cells and initiating vascular leak syndrome. Proc Natl Acad Sci U S A. 1999;96:3957–3962. doi: 10.1073/pnas.96.7.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munford RS. Murine responses to endotoxin: another dirty little secret? J Infect Dis. 2010;201:175–177. doi: 10.1086/649558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kravchenko VV, Kaufmann GF, Mathison JC, et al. Modulation of gene expression via disruption of NF-kappaB signaling by a bacterial small molecule. Science. 2008;321:259–263. doi: 10.1126/science.1156499. [DOI] [PubMed] [Google Scholar]
- 27.Zaborina O, Lepine F, Xiao G, et al. Dynorphin activates quorum sensing quinolone signaling in Pseudomonas aeruginosa. PLoS Pathog. 2007;3:e35. doi: 10.1371/journal.ppat.0030035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fox AC, McConnell KW, Yoseph BP, et al. The endogenous bacteria alter gut epithelial apoptosis and decrease mortality following Pseudomonas aeruginosa pneumonia. Shock Augusta Ga. 2012;38:508–514. doi: 10.1097/SHK.0b013e31826e47e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prakash A, Sundar SV, Zhu Y-G, et al. Lung Ischemia-Reperfusion is a Sterile Inflammatory Process Influenced by Commensal Microbiota in Mice. Shock Augusta Ga. 2015;44:272–279. doi: 10.1097/SHK.0000000000000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuijt TJ, Lankelma JM, Scicluna BP, et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut. 2016;65:575–583. doi: 10.1136/gutjnl-2015-309728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martínez JL. Short-sighted evolution of bacterial opportunistic pathogens with an environmental origin. Front Microbiol. 2014;5:239. doi: 10.3389/fmicb.2014.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nesse RM, Stearns SC, Omenn GS. Medicine needs evolution. Science. 2006;311:1071. doi: 10.1126/science.1125956. [DOI] [PubMed] [Google Scholar]
- 33.Chawla LS, Fink M, Goldstein SL, et al. THE EPITHELIUM AS A TARGET IN SEPSIS. Shock Augusta Ga. 2016;45:249–258. doi: 10.1097/SHK.0000000000000518. [DOI] [PubMed] [Google Scholar]
- 34.Fink MP. Intestinal epithelial hyperpermeability: update on the pathogenesis of gut mucosal barrier dysfunction in critical illness. Curr Opin Crit Care. 2003;9:143–151. doi: 10.1097/00075198-200304000-00011. [DOI] [PubMed] [Google Scholar]
- 35.Oostdijk EAN, de Smet AMGA, Kesecioglu J, et al. The role of intestinal colonization with gram-negative bacteria as a source for intensive care unit-acquired bacteremia. Crit Care Med. 2011;39:961–966. doi: 10.1097/CCM.0b013e318208ee26. [DOI] [PubMed] [Google Scholar]
- 36.Besselink MG, van Santvoort HC, Renooij W, et al. Intestinal barrier dysfunction in a randomized trial of a specific probiotic composition in acute pancreatitis. Ann Surg. 2009;250:712–719. doi: 10.1097/SLA.0b013e3181bce5bd. [DOI] [PubMed] [Google Scholar]
- 37.Mirvis SE, Shanmuganathan K, Erb R. Diffuse small-bowel ischemia in hypotensive adults after blunt trauma (shock bowel): CT findings and clinical significance. AJR Am J Roentgenol. 1994;163:1375–1379. doi: 10.2214/ajr.163.6.7992732. [DOI] [PubMed] [Google Scholar]
- 38.Piton G, Belin N, Barrot L, et al. Enterocyte Damage: A Piece in the Puzzle of Post-Cardiac Arrest Syndrome. Shock Augusta Ga. 2015;44:438–444. doi: 10.1097/SHK.0000000000000440. [DOI] [PubMed] [Google Scholar]
- 39.Chalkias A, Scheetz MH, Gulati A, et al. Periarrest intestinal bacterial translocation and resuscitation outcome. J Crit Care. 2016;31:217–220. doi: 10.1016/j.jcrc.2015.09.015. [DOI] [PubMed] [Google Scholar]
- 40.Yoshiya K, Lapchak PH, Thai T-H, et al. Depletion of gut commensal bacteria attenuates intestinal ischemia/reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2011;301:G1020–1030. doi: 10.1152/ajpgi.00239.2011. [DOI] [PubMed] [Google Scholar]
- 41.Fink D, Romanowski K, Valuckaite V, et al. Pseudomonas aeruginosa potentiates the lethal effect of intestinal ischemia-reperfusion injury: the role of in vivo virulence activation. J Trauma. 2011;71:1575–1582. doi: 10.1097/TA.0b013e31821cb7e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coopersmith CM, Stromberg PE, Dunne WM, et al. Inhibition of intestinal epithelial apoptosis and survival in a murine model of pneumonia-induced sepsis. JAMA. 2002;287:1716–1721. doi: 10.1001/jama.287.13.1716. [DOI] [PubMed] [Google Scholar]
- 43.Inoue S, Unsinger J, Davis CG, et al. IL-15 prevents apoptosis, reverses innate and adaptive immune dysfunction, and improves survival in sepsis. J Immunol Baltim Md 1950. 2010;184:1401–1409. doi: 10.4049/jimmunol.0902307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klingensmith NJ, Coopersmith CM. The Gut as the Motor of Multiple Organ Dysfunction in Critical Illness. Crit Care Clin. 2016;32:203–212. doi: 10.1016/j.ccc.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mittal R, Coopersmith CM. Redefining the gut as the motor of critical illness. Trends Mol Med. 2014;20:214–223. doi: 10.1016/j.molmed.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spitz J, Hecht G, Taveras M, et al. The effect of dexamethasone administration on rat intestinal permeability: the role of bacterial adherence. Gastroenterology. 1994;106:35–41. doi: 10.1016/s0016-5085(94)94155-6. [DOI] [PubMed] [Google Scholar]
- 47.Spitz J, Yuhan R, Koutsouris A, et al. Enteropathogenic Escherichia coli adherence to intestinal epithelial monolayers diminishes barrier function. Am J Physiol. 1995;268:G374–379. doi: 10.1152/ajpgi.1995.268.2.G374. [DOI] [PubMed] [Google Scholar]
- 48.Yoshikawa K, Kurihara C, Furuhashi H, et al. Psychological stress exacerbates NSAID-induced small bowel injury by inducing changes in intestinal microbiota and permeability via glucocorticoid receptor signaling. J Gastroenterol. 2016 doi: 10.1007/s00535-016-1205-1. [DOI] [PubMed] [Google Scholar]
- 49.Schreiber F, Arasteh JM, Lawley TD. Pathogen Resistance Mediated by IL-22 Signaling at the Epithelial-Microbiota Interface. J Mol Biol. 2015;427:3676–3682. doi: 10.1016/j.jmb.2015.10.013. [DOI] [PubMed] [Google Scholar]
- 50.Hayakawa M, Asahara T, Henzan N, et al. Dramatic changes of the gut flora immediately after severe and sudden insults. Dig Dis Sci. 2011;56:2361–2365. doi: 10.1007/s10620-011-1649-3. [DOI] [PubMed] [Google Scholar]
- 51.Sorkine P, Szold O, Halpern P, et al. Gut decontamination reduces bowel ischemia-induced lung injury in rats. Chest. 1997;112:491–495. doi: 10.1378/chest.112.2.491. [DOI] [PubMed] [Google Scholar]
- 52.Rosman C, Wübbels GH, Manson WL, et al. Selective decontamination of the digestive tract prevents secondary infection of the abdominal cavity, and endotoxemia and mortality in sterile peritonitis in laboratory rats. Crit Care Med. 1992;20:1699–1704. doi: 10.1097/00003246-199212000-00017. [DOI] [PubMed] [Google Scholar]
- 53.Qin X, Sheth SU, Sharpe SM, et al. The mucus layer is critical in protecting against ischemia-reperfusion-mediated gut injury and in the restitution of gut barrier function. Shock Augusta Ga. 2011;35:275–281. doi: 10.1097/SHK.0b013e3181f6aaf1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caballero S, Carter R, Ke X, et al. Distinct but Spatially Overlapping Intestinal Niches for Vancomycin-Resistant Enterococcus faecium and Carbapenem-Resistant Klebsiella pneumoniae. PLoS Pathog. 2015;11:e1005132. doi: 10.1371/journal.ppat.1005132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nevado R, Forcén R, Layunta E, et al. Neomycin and bacitracin reduce the intestinal permeability in mice and increase the expression of some tight-junction proteins. Rev Esp Enfermedades Dig Organo Of Soc Esp Patol Dig. 2015;107:672–676. doi: 10.17235/reed.2015.3868/2015. [DOI] [PubMed] [Google Scholar]
- 56.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 57.Gilbert JA, Quinn RA, Debelius J, et al. Microbiome-wide association studies link dynamic microbial consortia to disease. Nature. 2016;535:94–103. doi: 10.1038/nature18850. [DOI] [PubMed] [Google Scholar]
- 58.Gilbert JA, Dupont CL. Microbial metagenomics: beyond the genome. Annu Rev Mar Sci. 2011;3:347–371. doi: 10.1146/annurev-marine-120709-142811. [DOI] [PubMed] [Google Scholar]
- 59.Lax S, Smith DP, Hampton-Marcell J, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science. 2014;345:1048–1052. doi: 10.1126/science.1254529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shogan BD, Belogortseva N, Luong PM, et al. Collagen degradation and MMP9 activation by Enterococcus faecalis contribute to intestinal anastomotic leak. Sci Transl Med. 2015;7:286ra68. doi: 10.1126/scitranslmed.3010658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nekrutenko A, Taylor J. Next-generation sequencing data interpretation: enhancing reproducibility and accessibility. Nat Rev Genet. 2012;13:667–672. doi: 10.1038/nrg3305. [DOI] [PubMed] [Google Scholar]
- 62.Wischmeyer PE, McDonald D, Knight R. Role of the microbiome, probiotics, and “dysbiosis therapy” in critical illness. Curr Opin Crit Care. 2016;22:347–353. doi: 10.1097/MCC.0000000000000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dickson RP. The microbiome and critical illness. Lancet Respir Med. 2016;4:59–72. doi: 10.1016/S2213-2600(15)00427-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Can I, Javan GT, Pozhitkov AE, et al. Distinctive thanatomicrobiome signatures found in the blood and internal organs of humans. J Microbiol Methods. 2014;106:1–7. doi: 10.1016/j.mimet.2014.07.026. [DOI] [PubMed] [Google Scholar]
- 65.Hyde ER, Haarmann DP, Lynne AM, et al. The living dead: bacterial community structure of a cadaver at the onset and end of the bloat stage of decomposition. PloS One. 2013;8:e77733. doi: 10.1371/journal.pone.0077733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Metcalf JL, Wegener Parfrey L, Gonzalez A, et al. A microbial clock provides an accurate estimate of the postmortem interval in a mouse model system. eLife. 2013;2:e01104. doi: 10.7554/eLife.01104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Seider K, Brunke S, Schild L, et al. The facultative intracellular pathogen Candida glabrata subverts macrophage cytokine production and phagolysosome maturation. J Immunol Baltim Md 1950. 2011;187:3072–3086. doi: 10.4049/jimmunol.1003730. [DOI] [PubMed] [Google Scholar]
- 68.Ojima M, Motooka D, Shimizu K, et al. Metagenomic Analysis Reveals Dynamic Changes of Whole Gut Microbiota in the Acute Phase of Intensive Care Unit Patients. Dig Dis Sci. 2016;61:1628–1634. doi: 10.1007/s10620-015-4011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Behnsen J, Jellbauer S, Wong CP, et al. The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity. 2014;40:262–273. doi: 10.1016/j.immuni.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zaborin A, Romanowski K, Gerdes S, et al. Red death in Caenorhabditis elegans caused by Pseudomonas aeruginosa PAO1. Proc Natl Acad Sci U S A. 2009;106:6327–6332. doi: 10.1073/pnas.0813199106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Romanowski K, Zaborin A, Valuckaite V, et al. Candida albicans isolates from the gut of critically ill patients respond to phosphate limitation by expressing filaments and a lethal phenotype. PloS One. 2012;7:e30119. doi: 10.1371/journal.pone.0030119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kohler JE, Zaborina O, Wu L, et al. Components of intestinal epithelial hypoxia activate the virulence circuitry of Pseudomonas. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1048–1054. doi: 10.1152/ajpgi.00241.2004. [DOI] [PubMed] [Google Scholar]
- 73.Patel NJ, Zaborina O, Wu L, et al. Recognition of intestinal epithelial HIF-1alpha activation by Pseudomonas aeruginosa. Am J Physiol Gastrointest Liver Physiol. 2007;292:G134–142. doi: 10.1152/ajpgi.00276.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koeppen K, Hampton TH, Jarek M, et al. A Novel Mechanism of Host-Pathogen Interaction through sRNA in Bacterial Outer Membrane Vesicles. PLoS Pathog. 2016;12:e1005672. doi: 10.1371/journal.ppat.1005672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brazil JC, Lee WY, Kolegraff KN, et al. Neutrophil migration across intestinal epithelium: evidence for a role of CD44 in regulating detachment of migrating cells from the luminal surface. J Immunol Baltim Md 1950. 2010;185:7026–7036. doi: 10.4049/jimmunol.1001293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sumagin R, Robin AZ, Nusrat A, et al. Transmigrated neutrophils in the intestinal lumen engage ICAM-1 to regulate the epithelial barrier and neutrophil recruitment. Mucosal Immunol. 2014;7:905–915. doi: 10.1038/mi.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Klesney-Tait J, Keck K, Li X, et al. Transepithelial migration of neutrophils into the lung requires TREM-1. J Clin Invest. 2013;123:138–149. doi: 10.1172/JCI64181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012;5:354–366. doi: 10.1038/mi.2012.24. [DOI] [PubMed] [Google Scholar]
- 79.Falkow S. Molecular Koch’s postulates applied to microbial pathogenicity. Rev Infect Dis. 1988;10(Suppl 2):S274–276. doi: 10.1093/cid/10.supplement_2.s274. [DOI] [PubMed] [Google Scholar]
- 80.Seal JB, Morowitz M, Zaborina O, et al. The molecular Koch’s postulates and surgical infection: a view forward. Surgery. 2010;147:757–765. doi: 10.1016/j.surg.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 81.Barrick JE, Yu DS, Yoon SH, et al. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature. 2009;461:1243–1247. doi: 10.1038/nature08480. [DOI] [PubMed] [Google Scholar]
- 82.Stappenbeck TS, Virgin HW. Accounting for reciprocal host-microbiome interactions in experimental science. Nature. 2016;534:191–199. doi: 10.1038/nature18285. [DOI] [PubMed] [Google Scholar]
- 83.Nathan C, Ding A. Nonresolving Inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 84.Gentile LF, Cuenca AG, Efron PA, et al. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg. 2012;72:1491–1501. doi: 10.1097/TA.0b013e318256e000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liow LH, Van Valen L, Stenseth NC. Red Queen: from populations to taxa and communities. Trends Ecol Evol. 2011;26:349–358. doi: 10.1016/j.tree.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 86.Hawking S, Mlodinow L. The Grand Design. Random House Publishing Group; 2010. [Google Scholar]