

Abstract
Background
With the advent of high throughput sequencing, the identification of genetic causes of cardiovascular disease (CVD) has become an integral part of medical diagnosis and management and at the forefront of personalized medicine in this field. The utility of whole exome sequencing (WES) for clinical diagnosis, risk stratification and management of inherited CVD has not been previously evaluated.
Methods and Results
We analyzed the results of WES in first two hundred adult patients with inherited CVD, who underwent genetic testing at the Yale Program for Cardiovascular Genetics. Genetic diagnosis was reached and reported with success rate of 26.5% (53 of 200 patients). This compares to 18% (36 of 200) that would have been diagnosed using commercially available genetic panels (p=0.04). WES was particularly useful for clinical diagnosis in patients with aborted sudden cardiac death (SCD), in whom the primary insult for the presence of both depressed cardiac function and prolonged QT had remained unknown. The analysis of the remaining cases using genome annotation and disease segregation led to discovery of novel candidate genes in another 14% of the cases.
Conclusions
WES is an exceptionally valuable screening tool for its capability to establish the clinical diagnosis of inherited cardiovascular diseases, particularly for poorly defined cases of SCD. By presenting novel candidate genes and their potential disease associations we also provide evidence for the utility of this genetic tool for identification of novel CVD genes. Creation and sharing of exome databases across centers of care should facilitate the discovery of unknown cardiovascular disease genes.
Keywords: sudden cardiac death, cardiomyopathy, arrhythmia (heart rhythm disorders), genetics
Introduction
Since the completion of the Human Genome Project in 2003, researchers have strived to develop fast and inexpensive methods for sequencing large-scale genetic data and from these efforts, next generation sequencing technology has emerged. An individual’s whole exome can now be sequenced at low cost in less than a week. Whole exome sequencing (WES) enables high throughput sequencing of the coding regions (>90%) of approximately 20,000 genes in a single analysis. Since it is approximated that more than 85% of mutations in single disease gene disorders reside within the exons/exon-intron boundaries1, WES is a high yield and cost effective alternative to whole genome sequencing for monogenic disorders. The use of WES has the potential to revolutionize the way we practice medicine by generating large-scale personalized genetic information.
Cardiovascular diseases (CVD) comprise the most common causes of death and disability in Western countries. Early twin studies have established the importance of genetic influences on most CVD2, 3. CVD display both single gene as well as complex inheritance patterns. Depending on the particular sub-category of CVD, many of the genes contributing to familial forms have been elucidated4, 5, while in other areas of CVD the underlying genes remain largely unknown.
CVD genetics is a rapidly expanding field and the need for practitioners to diagnose and treat individuals with familial forms of CVD is large. Although there have been several reports suggesting that genetic panels should be used as the first-line evaluation of CVD patients in the adult genetics clinic6, 7, there has been no report on use of WES in this context. Herein represents the first published report of 200 adult patients with familial CVD initially evaluated with proband-only clinical WES. The results will provide an estimate for the success rate of genetic diagnosis of the condition by WES in conjunction with clinical data and its utility in identifying novel candidate genes and providing a database for future discovery of novel disease genes.
Materials and Methods
Recruitment
Individuals referred to the Yale Program for Cardiovascular Genetics (YPCG) for genetic testing typically had undergone an extensive cardiac work-up and have a working diagnosis. This included, but was not limited to, electrocardiograms (EKG), echocardiograms, cardiac MRI (cMR), Holter monitoring, and electrophysiology studies. At the YPCG appointment, a board-certified genetic counselor and cardiovascular geneticist took family and medical histories and evaluated previous cardiac records. Genetic testing via WES was offered to patients, in compliance with the previously published guidelines for inherited CVD (i.e. Long QT syndrome (LQTS), hypertrophic cardiomyopathy (HCM), etc.) as a first line test8–12. Testing was performed at the Yale Center for Genome Analysis and interpreted by the DNA Diagnostic Laboratory (DNA Lab), a College of American Pathologists (CAP) and CLIA certified laboratory with input from YPCG. The Human Investigation Committee of the Yale University School of Medicine approved the study protocol. Consent was obtained from all subjects. The written consent includes release of information, including incidental findings to the patient and referring physicians and permission to extend the kindred.
Presenting Diagnosis
We made the best effort to categorize these complex patients into a single presenting diagnosis based on the indication for the referral. Sudden cardiac death (SCD) was defined as witnessed instantaneous circulatory arrest requiring resuscitation in the field for a previously stable subject without structural heart disease. This definition was used when referring physicians were unable to report a more specific diagnosis based on presentation and clinical data. In certain cases, the QRS morphologies, the intervals on EKG or the left ventricular ejection fraction (LVEF) were used for a working diagnosis, but these findings were often not perfectly consistent with the clinical presentation. Dilated non-ischemic cardiomyopathy (NICM) were grouped under DCM. If a clear diagnosis of a subtype of cardiomyopathy (dilated vs. hypertrophic) could not be reached, “nonspecific-NICM” was used.
Evaluation
The YPCG and the DNA Lab evaluate clinical WES in parallel. An algorithm (Figure 1) was created to detect mutations in genes known to cause inherited CVD conditions using a comprehensive internal list of all known published CVD genes. The list is regularly updated using newly published data. In 2012 the list had 88 genes, and currently it has 163 genes.
Figure 1.
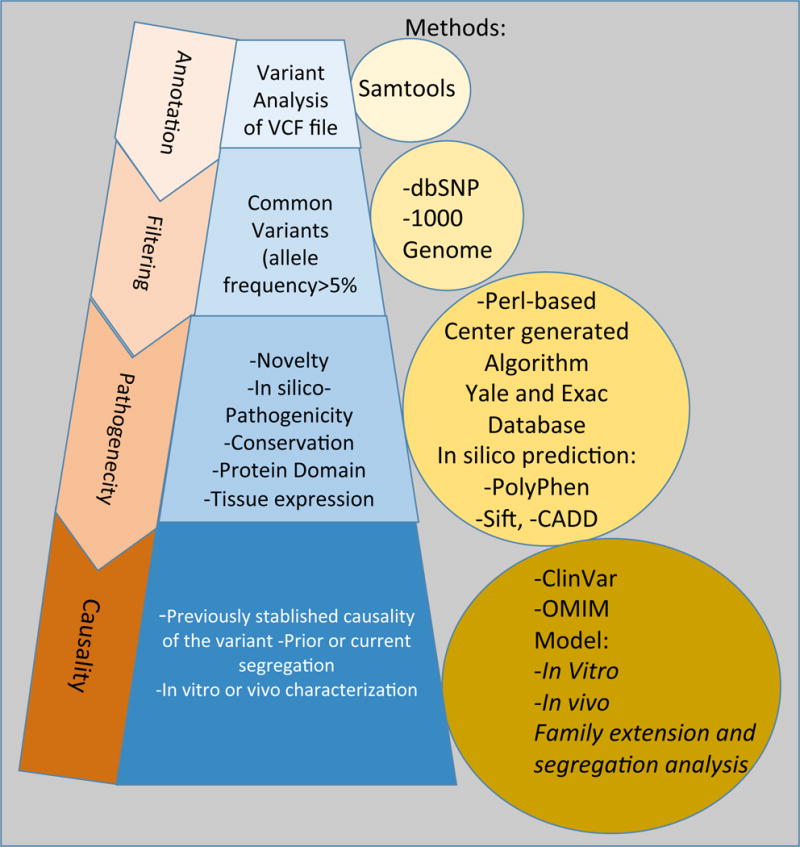
Pipeline to assess the pathogenicity of genetic variants identified in an Adult Cardiovascular Disease Genetics Clinic.
A genetic diagnosis was considered as established if a variant is classified as likely-pathogenic or pathogenic could be identified based on ACMG 2015 criteria14 (Table S1, S2). Accordingly, mutations were specified as variant of uncertain significance (VUS), if they were novel but potentially pathogenic, i.e. were mutations of established disease genes, whereby no segregation analysis or a functional study had been carried out to confirm their pathogenicity. In cases of a VUS result, every attempt was made to perform family segregation of the variants.
If a pathogenic mutation was not identified, the remainder of genetic data was examined using genomic annotation tools. Mutations were examined for novelty, conservation, the position of the encoded amino acid in relationship to critical domains, potential change of the 3D structure, the expression of the encoded protein in the heart and/or relevant tissues, and the protein function annotation. Deleterious mutations in annotated genes were prioritized based on 1) gene expression in the cardiovascular tissues and 2) characterization in vivo or in vitro with the focus on disease-relevant functions. The exome report created by YPGC was sent to the DNA Lab for further evaluation. Sanger sequencing was employed to confirm variant from WES until mid 2012. This practice was discontinued as most labs, including ours after multiple quality checks deemed it as unnecessary13. Sanger sequencing was carried out for segregation analysis. Copy number variation (CNV) could not be detected by the bioinformatics pipeline available at the time.
The DNA Lab reviewed the created reports and released official reports as specified by CAP, CLIA, and American College of Medical Genetics (ACMG) guidelines14, 15. Cases were reviewed individually in weekly meetings with an interdisciplinary team, established research scientist in the field of cardiovascular genetics, a board-certified cardiologist, internal medicine and cardiovascular medicine fellows, and a board-certified genetic counselor. In certain cases, in vitro or in vivo analyses were carried out for functional characterization of the candidate genes.
Sequencing
Genomic DNA was analyzed using WES as previously described.16 Roche/Nimble-Gen 2.1M Human Exome Array covers 34.0 Mb of genomic sequence and approximately 180,000 exons of 18,673 protein-coding genes. Briefly, DNA was fragmented and ligated to linkers followed by fractionation by agarose gel electrophoresis. Extracted DNA was PCR amplified and hybridized to the capture arrays. Bound genomic DNA was eluted, purified, and amplified by ligation-mediated PCR. The PCR products were purified and subjected to DNA sequencing on the Illumina platform.
Captured libraries were sequenced on the Illumina genome analyzer followed by Image analysis and base calling. Sequence reads were mapped to the reference genome (hg18/hg19) using the Maqprogram SAMtools. Resulting sequence data were processed using Maq software. SAMtools software was used to detect single nucleotide variants (SNVs) and insertion/deletion (indel) were subsequently filtered against reference genome as earlier described17. Filters were applied against public databases, initially with 1,000 Genomes, dbSNP, and the National Heart Blood and Lung Institute’s Exome Variant Server databases, and the Exome Aggregation Consortium’s ExAC Browser, after it became available on October 2014. Variants were annotated based on their effects on protein function and structure, in silico predication programs, PolyPhen-2 and SIFT, novelty, with a minor allele frequency threshold of <0.1, conservation, and tissue expression using a perl-based computer script. The results are reported in two different data sets–a dataset of all exome-wide variants and a list of variants within in the targeted genes designed specifically for the CVD of interest (see Fig. 1). Statistical comparison of CVD-causing pathogenic variant identified by WES versus those that would have been identified by commercially available panels (defined as standard genes represented by at least 2 of 3 commercial panels as of January 2016) was made using Pearson’s chi-squared test.
Quantitative Real-Time PCR (qRT-PCR)
Human right ventricle endomyocardial biopsy was obtained fluoroscopically. Total RNA was extracted using RNeasy Kit (QIAGEN, Valencia, CA), and the concentration was measured. 500ng of RNA was reversed transcribed using iScript cDNA synthesis kit (Bio-Rad). Real-time PCR was carried out using iQ SYBR Green Supermix kit (Bio-Rad). The relative mRNA expression level of CACNA1D was calculated using GAPDH as control. The presence of CACNA1D was further confirmed by electrophoresis using Real-time PCR products. The following primers were used: CACNA1D-F, 5′-gtgtcaggagtgcccagttt-3′; CACNA1D-R, 5′-ctgggtcctcttcagctacg-3′; GAPDH-F, 5′-gagtcaacggatttggtcgt-3′; GAPDH-R, 5′-ttgattttggagggatctcg-3′.
Results
Reportable Diagnosis
The average age was 46 ± 14 years, and 107 were men (53.5%) and 37 non-white (18.5%). The most common indications for referral and the success rate in making a reportable genetic diagnosis and/or identifying candidate genes are presented in Table 1.
Table 1.
Reasons for referral to the Yale Program for Cardiovascular Genetics Clinic and the genes with variants identified as VUS or as pathogenic (denoted in bold).
| Clinical Diagnosis | Patients N (% of total) | Pathogenic mutation identified N (%) | Likely Pathogenic mutation identified N (%) | VUS identified N (%) | Candidate gene identified N (%) | Genes |
|---|---|---|---|---|---|---|
| Connective Tissue Disease | 37(18.5) | 3(8.1) | 1(2.7) | 11(29.7) | 7(18.9) | COL1A1, COL5A1, COL5A2, ELN, FBN1, FBN2, FLNA, MYH11, MYLK, PTPN11, SKI, SMAD3, TGFBR2 |
| Sudden Cardiac Death | 35(17.5) | 8(22.8) | 3(8.5) | 9(26.4) | 6(17.6) | ACTN2, ANK2, AKAP9, CACNA1D, DPP6, DSG2, DSP, GYG1, KCNH2, LMNA, MYBPC3, MYH6, MYPN, NEXN, PNN, RBM20, RYR2, SCN5A, TGFB3, TNNI3, TTN |
| HCM | 28(14) | 8(28.5) | 5(17.5) | 9(32.1) | 2(7.1) | ACTN2, AKAP9, ABCC9, CALR3, JPH2, MYBPC3, MYH6, MYH7, PRKAG2, TCAP, TTN, TNNT2, TPM1, TRPM4 |
| DCM | 24(12) | 4(16.6) | 0(0) | 9(37.5) | 3(12.5) | BAG3, DSP, DSG2, HFE, LMNA, MYBPC3, MYH6, MYH7, PRDM16, PRKAG2, RBM20, SCN5A, TTN |
| FH/Lipodystrophy | 21 (10.5) | 6(28.5) | 2(9.5) | 1(4.7) | 5(23.8) | APOB, APOE, LDLR, LMNA, PLAT, PLIN1 |
| LQTS | 15(7.5) | 3(20) | 0(0) | 3(20) | 4(26.6) | AKAP9, ANK2, CAV3, CTNNA3, KCNQ1, RBM20, SCN5A, TTN |
| Atrial and/or ventricular arrhythmias | 10(5.0) | 0(0) | 0(0) | 4(40.0) | 0(0) | ABCC9, CACNB2, GPD1L, KCNE2, SYNE2 |
| BrS | 9(4.5) | 1(11.1) | 2(22.2) | 3(33.3) | 0(0) | CACNA1C, DSP, RYR2, SCN5A |
| Family History of SCD or LQTS | 9(4.5) | 0(0) | 1(11.1) | 5(55.5) | 1(11.1) | ANK2, CACNA1C, DES, DSG2, SCN4B, SCN5A, MYH6, TMEM43, TNNI3, VCL |
| Nonspecific NICM | 6(3) | 2(33.3) | 1(16.6) | 2(33.3) | 0(0) | AKAP9, LAMP2, LDB3, MIB1, MYH6, NEXN, PKP2, RYR2, SCN5A, TNNT2, TTN |
| Other | 6(3) | 0(0) | 2(33.3) | 0 (0) | 1(20) | DSP, EMD, NDUFV2, TNNT2 |
A minimum depth of 20 reads was achieved for 95% coverage of the reference genome. Overall, reportable genetic diagnosis was reached in 53 of 200 patients (26.5%) with an additional 56 patients (28%) reported as having a VUS. Twenty-nine candidate variants were identified (14.5%) in the remaining cases (Table 1, Table S1, S2). In comparison, 36 of our 200 cases (18%) would have been called with definitive diagnosisif we had used commercially available genetic panels, (p=0.04).
Connective tissue diseases, including Marfan syndrome, Ehlers Danlos syndrome (EDS), and familial thoracic aneurysm and dissection (TAA) was the most common reason for referral, comprising 18% of total patients (n=37). Genetic diagnosis was reached in 10.8%, with another 29.7% reported as having a VUS, and 18.9% resulting in a candidate variant identification. SCD was the second most common reason for referral, comprising 17.5% of total patients (n=35). Genetic diagnosis was reached in 31.4% of SCD patients, with another 26.7% reported as having VUS, and 17.6% resulting in candidate variant identification. 14% and 12% of patients had HCM and DCM, respectively (n=28 and n=24). Genetic diagnosis was reached in 46.4% of HCM cases, while in 32.1% of the cases a VUS was reported. In 7.1% of the HCM cases a novel candidate gene was identified and documented for future analyses. 16.6% of DCM patients had an established genetic diagnosis, 37.5% were reported as having a VUS, and in 12.5% a candidate variant was identified. For Lipodystrophy/Familial Hypercholesterolemia (FH) (n=21, 10.5%), genetic diagnosis was reached in 38% of cases, while in 4.7% of the cases a VUS was reported and in roughly 24% a candidate gene was identified.
The working diagnoses and identified gene variants in 200 patients presenting to the YPCG are listed in Table S1. Classification and supporting evidence based on ACMG 2015 criteria is presented for all gene variants. Candidate genes that are undergoing further analysis at YPCG are denoted by gray blocks.
Ambiguous Clinical Diagnosis Modified or Altered by the Genetic Diagnosis
A significant advantage of WES was establishing the clinical diagnosis for ambiguous cases, particularly in patients with SCD. We carefully characterized the thirty-five patients presenting with initial diagnosis of SCD of unspecified causes using WES results and clinical data. Most, if not all these patients had initially abnormal EKGs, often with a prolonged QT duration and variable degree of left ventricular systolic dysfunction. The working diagnosis by the referring physicians were arbitrarily assigned to either NICM or LQTS, with the final diagnosis awaiting the genetic results. Primary ventricular arrhythmia was the final diagnosis in 42.8% of cases, followed by LQTS (17.1%), DCM (14.2%), and HCM and Nonspecific-NICM (8.5% each). ARVC, CPVT, and RCM all comprised the final diagnosis in 9% of SCD cases. Genes represented on commercially available panels that can be used to evaluate cardiac arrhythmia and cardiomyopathy are represented in Table S3 and S4, respectively. It is apparent that the genetic diagnosis in most cases was only possible by using WES data as compared to individual disease-based panels. It is noteworthy that the medical management in many cases were altered based on genetic results by the referring physician.
Following, we present several cases to provide justification for use of WES to establish a final diagnosis.
Patient 6 (Table S1/S2/Figure 2A) was a 22-year old female with a history of X-linked periventricular heterotopia (PVH), hyperflexibility of the major joints and familial aneurysm of different vascular beds referred for targeted genetic testing for FLNA to confirm the diagnosis of PVH. Family history included a sister with congenital hip dislocation, easy bruising, joint hyperflexibility, carpal tunnel syndrome, a brother with scoliosis, joint hyperflexibility, and pectus excavatum; none had neurological findings of PVH. The index case’s mother had mitral valve prolapse, and scoliosis. Her maternal aunt had a pectus excavatum and pes planus. Her maternal grandmother had undergone an ascending TAA repair, often seen in PVH, an aneurysm of the renal artery, and no neurological symptoms. The maternal grandmother’s brother had died of an TAA rupture at age 52. The maternal great grandmother reportedly had hyperflexible joints.
Figure 2.
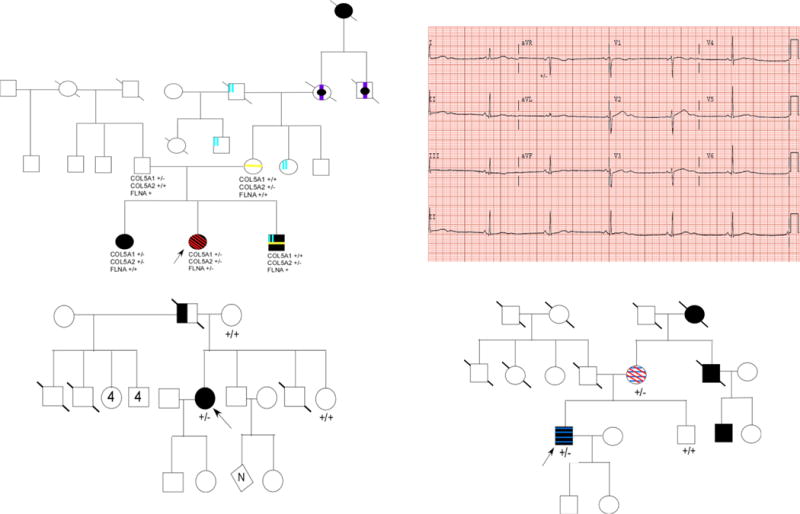
Pedigrees for discussed cases are shown. Circles represent females; squares represent males and symbols with a slash through them indicate deceased subjects. An arrow indicates the proband/person who had exome sequencing. (A) Pedigree of case 1 (patient 6). Individuals with hypermobility are indicated by black symbols; individuals not investigated for hypermobility are indicated by a black dot; individuals with aneurysm are indicated by a vertical purple line; individuals with scoliosis are indicated by a yellow vertical line; individuals with pectus are indicated with two blue vertical lines; individuals with periventricular heterotopia are indicated with red diagonal lines. Genotypes of COL5A1, COL5A2, and FLNA are shown below those individuals who underwent genetic testing. (B) Representative electrocardiogram (EKG) of case 4 (patient 152). Heart rate is 36 beat per minute and her corrected QTC was 630 msec. (C) Pedigree of case 4 (patient 152). Individuals with both torsade des pointes and congenital hearing loss are indicated by filled symbols; individuals reported to have permanent pacemakers are indicated by half-filled symbols. (D) Pedigree of case 5 (patient 97). Individuals with myocardial infarction and surgical intervention (i.e. CABG, PCI, etc.) are indicated with filled symbols; individuals with DVT are indicated by blue horizontal lines; individual with PE are indicated by red diagonal lines.
Given the complexity of the condition a WES of the index case was carried out, in place of targeted screening for a FLNA mutation and/or EDS genes. The analysis revealed a frameshift variant in FLNA (c. 4214delA), and missense variants in COL5A1 (c.5674G>A; p.D1771N), and COL5A2 (c.2379C>T; p.G702R) genes. Segregation analysis of the first-degree relatives revealed that the FLNA variant was de-novo, while her mother had the COL5A2 variant and patient’s father with no EDS had the COL5A1 variant. The index case’s sister had both COL5A1 and COL5A2 variants; her brother and her maternal grandmother had only the COL5A2. Thus, COL5A2 (p.G702R) segregated with the hyperflexible joints and with the TAA in the maternal side of the family as a separate entity from FLNA.
Patient 4 (Table S1/S2) was a 52-year-old female with status post an aborted ventricular fibrillation. An epicardial cardioverter defibrillation patch, a currently retreated device due to a high rate of patch crinkling18 was implanted in the late 1980s with 2 appropriate shocks identified by device interrogation. Family history was significant for her mother having heart failure. Later her defibrillator was upgraded to an ICD, while her patches remained in place. She then developed shortness of breath and was diagnosed with persistent atrial fibrillation, for which she underwent a radiofrequency ablation without resolution of her symptoms. An echocardiography examination revealed mildly reduced LVEF, severe diastolic dysfunction, mild pulmonary hypertension, and bi-atrial enlargement.
A WES was carried out to determine whether a forme fruste inherited RCM (in absence of lower extremity edema) versus the impact from epicardial defibrillation patches were responsible for her shortness of breath. Results revealed a TNNI3 mutation c.592C>G, p.L198V. In silico L198V creates a splice donor site with splice acceptors predicted in the 3′UTR of the gene. This amino acid substitution is found in the C-terminal of the gene, which contains a secondary actin-tropomyosin binding domain necessary for inhibiting cross-bridge cycling during diastole19. Mutations in this gene have been associated with RCM20, which was also consistent with echocardiographic findings of diastolic dysfunction and biatrial enlargement. Hence, this diagnosis was considered more plausible; the extremely risky removal of pads was abandoned. Unfortunately, later she developed ventricular arrhythmia and deceased after external defibrillation shocks failed to restore sinus rhythm.
Patient 59 (Table S1/S2) was a 20-year-old female with a family history of SCD and an aborted SCD, resuscitated by a defibrillator. She was found to have prolonged QTc, (later normalized) and a mildly depressed LVEF (40–45%). A follow up echocardiogram showed improvement of her LVEF to near normal (53%), hence, she was referred for genetic testing with a working diagnosis of aborted SCD and possibly LQTS. No genetic variants associated with LQTS but a variant in MYH6 c.3979 G>A, p.R1270C and a frameshift mutation in TTN c.45689delG, p.G8858fs were identified. Her younger sister, who had symptoms of CHF and mildly reduced LVEF by cMR but no SCD tested positive for both variants. The index case’s father had the MYH6 variant but no symptom or sign of heart failure on physical examination or by cardiac imaging. These findings led to the final diagnosis of TTN-related SCD and mild form of DCM in this patient.
Identification of Novel Candidate Genes
In cases where no pathogenic variant could be identified, the entire exome was examined for nonconservative and deleterious candidate variants in annotated genes expressed in the cardiovascular tissues. These variants were investigated for disease segregation and occasionally were characterized in an in vitro system. A list of candidate genes was created to serve as a reference for future exome analysis of cases with unknown disease genes. Once independent mutations are identified in a given gene, the data will be made publically available for independent replication before the causality is established. Creation and sharing of large exome databases across major genomic centers should facilitate establishing causal link between candidate genes and diseases of interest.
Patient 152 (Table S1/S2) was a 34-year old female with a history of congenital hearing loss, who presented with torsade des pointes (TdP), bradycardia with heart rates between 30–50 beats per minute, and a prolonged QT interval on EKG (Figure 2B). Patient’s father had been diagnosed with bradycardia and had undergone permanent pacemaker implantation and died at age 52 from a “sudden heart condition” (Figure 2C). A WES revealed a heterozygous nonsense variant in the CACNA1D gene in the proband. Mutations in this gene had been implicated in bradycardia but not LQT21–24. In addition, the encoded protein has been shown to be expressed only in mice atrium. To explain the ventricular arrhythmia in the subject, a human ventricular biopsy specimen was obtained from a human control, which revealed high expression levels of both mRNA and protein (Figure S1). These findings suggest a possible causal link between this mutation and LQTS. Since her unaffected mother and sister did not carry this variant, the variant is likely the cause of bradycardia in her father.
Patient 97 (Table S1) was a 44-year old male athlete, who presented with a second acute ST-elevation-MI (STEMI) in absence of major CAD risk factor. His first STEMI occurred while swimming. An angiography examination had revealed 100% right coronary artery (RCA) occlusion with a fresh thrombus, for which he underwent thrombectomy, angioplasty (PCI) and placement of a bare metal stent (BMS) and diffuse, critical stenosis of the left anterior descendent coronary artery, which was grafted. Perioperatively, he developed a popliteal deep venous thrombus (DVT). Two years after coronary artery bypass grafting (CABG), he developed chest pain after a long run. The coronary angiogram revealed again a fresh thrombus in the RCA at the site of the BMS, which was treated with a drug eluting stent and a second critical lesion in the left circumflex that was treated with PCI. His family history was significant for multiple DVTs in his mother in her twenties and a more recent massive pulmonary embolus (PE), CABG in a maternal uncle at the age 32, CAD stenting in a maternal cousin in his thirties, and acute myocardial infarction and death in his maternal grandmother at age 38 (Figure 2D). There was no relevant history on the paternal side of the family. A WES revealed no variants in dyslipidemia genes, but a novel nonconservative missense alteration in the PLAT gene, encoding tissue plasminogen activator (tPA). Segregation analysis supported its possible causality by identifying the mutation in his affected mother and its absence in the unaffected brother.
Incidental Pathological Findings
We identified actionable pathological variants in 11 out of 200 (5.5%) study subjects. This included pathogenic mutations in ATM, APC, BRCA2, BRIP1/FANCJ, HFE, LDLR, MSH6, and SCNN1B genes (Table S5). In these cases, patients and referring providers were informed of these findings.
Discussion
In this study we present our experience in the use of WES in 200 consecutive adult patients with familial CVD. The data provided is evidence for multiple advantages of WES. Utilizing WES, we have been successful at reaching a genetic diagnosis in a patient population with a spectrum of cardiovascular illnesses. Overall reportable genetic diagnosis (pathogenic or likely pathogenic variants) was reached in 26.5% of patients, with an additional 28% reported as having a VUS, and 14.5% resulting in (not reportable) candidate variant identification. We owe part of this success to the careful selection of those patients by cardiovascular medicine physicians that have pursued expertise in genetics, working alongside with the genetics team, consisted of a cardiovascular geneticist, a genetic counselor and cardiology fellows in training. Not infrequently, patients referred to the YPCG clinic had complex presentations and ambiguous diagnoses. We illustrate how genetic diagnosis aided in establishing a final clinical diagnosis in this group.
Genetic Diagnosis Assisted the Final Clinical Diagnosis
In our experience with clinical WES, one clear advantage has been in the establishment of clinical diagnosis. Often in SCD, primary diagnosis at the time of incident is unclear and difficult to determine whether it is a primary arrhythmogenic disorder versus cardiomyopathy. This makes the selection of phenotype-driven panels for genetic testing particularly challenging. In patients with preserved ejection fraction distinguishing the type of arrhythmia, i.e. LQTS vs. BrS, based on post resuscitation EKG is often impossible. Electrophysiological studies using pharmacological challenge have also limited use. The currently available pan-cardiovascular disease panels offered by different vendors have very different combinations of arrhythmia and cardiomyopathy genes (Tables S3 and S4) and none fully covers the entire list of known genes for these disorders. In comparison, WES is in a unique position in establishing a final clinical diagnosis when used in conjunction with the clinical data25.
In the current study, patient 6 with PVH and familial aneurysm and hyperflexibility of the major joints illustrates one such example. While a priori association between PVH and vascular traits26 were thought to be explained by a single gene mutation in the FLNA gene, a segregation analysis revealed that the classic EDS phenotypes, including TAA, segregated with a COL5A2 mutation in her family. The genetic diagnosis entirely changed the clinical management as the COL5A2 mutation carriers in the family were recommended to undergo echocardiographic screening for TAA.
Discovering The Pleotropic Effects of a Known Disease Gene
Another advantage of using WES has been the unraveling of novel traits for known disease genes. Ventricular arrhythmia was a novel trait associated with truncating TTN mutations in our patient population with an initially normal structural heart. As shown in the example of patient number 59, these subjects often had prolonged QTc, normal or near normal LVEF, and had been referred to us with a working diagnosis of LQTS. In many cases segregation analysis could help to establish causal link between TTN mutations and SCD. In such cases the correct diagnosis would not have been reached if a targeted sequencing had been performed using available arrhythmia panels (Table S3). In our WES patients, we reported six cases of truncating TTN mutations (patients number 59, 75, 84, 26, 114, 193 Table S1/S2) in patients with aborted SCD but no history of cardiomyopathy prior to their aborted SCD event. Our finding is consistent with prior reports of high incidence of ventricular arrhythmias in subjects with truncating TTN mutations25, 27.
Identification of Novel Candidate Genes
An advantage of WES has been the identification of novel candidate genes. One example is a nonsense mutation in CACNA1D gene in patient number 152 (Table S1/S2), who had documented TdP, prolonged QT interval, and congenital hearing loss. This gene has been associated with primary aldosteronism, seizures and neurological abnormalities (gain of function). Homozygous (and in one single case heterozygous) 3-bp insertion in the CACNA1D gene had been reported in 2 consanguineous Pakistani kindreds with sinoatrial node dysfunction and deafness28. CACNA1D -deficient mice are deaf due to the complete absence of L-type currents in cochlear inner hair cells and degeneration of outer and inner hair cells and have sinoatrial node dysfunction29. However, CACNA1D mutation has not been previously reported in LQTS and the gene is not reportedly not expressed in mice ventricular myocardium. Our investigation, however, showed that CACNA1D is expressed in control human ventricular myocardium and could account for the patient’s hearing loss and episode of TdP.
A second example is the patient number 97, who was a 44-year old triathlete in peak physical fitness with a history of coronary artery stent and CABG at 42 years of age and perioperative DVT, who presented with a second STEMI and recurrent thrombi in the native vessel or at the site of his first stent. A novel, likely pathogenic, nonconservative missense variant in the PLAT gene was identified, which segregated with DVT/PE in his family. Loss of function PLAT variants have been associated with decreased release of the thrombolytic protein tPA and thrombophilia and gain of function variants in this gene have been associated with increased tPA release and hyperfibrinolysis30–33. Common variants of PLAT gene have been associated with the risk of CVD34 and myocardial infarction35. It is particularly interesting that release of tPA is stimulated by exercise, presumably in response to a physiological need, and both of this patient’s STEMIs occurred during or directly following intense exercise. Based on the patient’s clinical history and genetic finding, indefinite continuation of dual antiplatelet therapy was opted. The causality of all these candidate genes can be established by independent replication or preferably by sharing of large exome databases between major genomic centers. Due to their highly speculative nature, most other identified candidate genes were not reported in this manuscript.
Incidental Pathological Findings
Exome sequencing can reveal variants in genes with actionable findings, as previously reported36, 37. Five and a half percent of patients referred to our clinic had actionable incidental findings (Table S5), many of whom had no personal or family history of cancer. These results were reported to patients as indicated by guidelines. With the identification of incidental findings, patients were referred to proper follow up for the condition identified. They were encouraged to discuss these results with their family physician and family members if applicable.
WES Compared with Available Panels
While we have shown that WES is an effective tool for adult cardiovascular genetic testing, the development of phenotype-based panels has grown. Of the 53 subjects (26.5%) identified with a CVD causing pathogenic variant by WES (Table S2), only 36 patients (18%) would have definitively been identified by commercially available panels (defined as standard genes represented by at least 2 of 3 commercial panels), a difference that is statistically significant (p=0.04). It is noteworthy that these numbers are based on panels current as of January 2016. However, the majority of WES were performed at a time when commercially available panels had considerably fewer gene sets with much less power to identify the causal variants. With many disease genes remaining unknown but discovered in an ongoing basis, the current panels will be soon deemed as incomplete.
Limitations
Our study has several limitations. Most importantly, the success of WES technique is greatly dependent on the current knowledge of disease genes. By the same token, it is the most appropriate screening test for low-yield cases for it provides a comprehensive database that can be revisited in future as the genetic literature grows or used for the discovery of novel genes. Another limitation is that we performed proband-only WES. Previous reports of the use of this technology in pediatric populations describe WES in duos and trios. This practice is often unfeasible in an adult CVD clinic, either because affected family members are deceased or geographically dispersed. For cases where a VUS was identified, every attempt was made to perform segregation analysis in the extended families (see Table S1). This reduces false positive or negative rates to a greater magnitude compared to genetic study of trios. Overall, the determination of pathogenicity in our center was made in accordance with the ACMG 2015 guidelines by using its major criteria. The major criteria for pathogenicity include presence of a radical or a de-novo mutation, and supportive in vitro or in vivo studies. A fourth limitation is the requirement for a multidisciplinary infrastructure for analyzing large data. Lastly, WES is not useful for the detection of chromosomal rearrangements, or mutations in noncoding regions. In addition, our analysis at the time did not include CNV. The applicability of CNV analysis in clinical cardiovascular genetics, however, remains undetermined38. Despite all our efforts, there are always potentials for false positive results. In this context, we like to state that a genetic diagnosis alone cannot establish the clinical diagnosis and should be strictly used in conjunction with the clinical data.
In conclusion, our study shows many advantages of WES for its capability to establish the correct clinical diagnosis, to identify novel traits for known disease genes, or novel candidate genes for CVD.
Supplementary Material
Clinical Perspective.
High throughput sequencing will soon revolutionize the way we practice medicine by generating large-scale, personalized, and relatively affordable genetic information. Cardiovascular disease (CVD) is the most common cause of death in developed countries and the need for practitioners to diagnose and treat individuals with familial forms of CVD is urgent. Although phenotype-based genetic panels are widely used as the first-line tool for genetic evaluation of CVD patients in the adult genetics clinic, there has been no report on the use of whole exome sequencing (WES) in this context. This work represents the first published report of patients with familial CVD evaluated with proband-only clinical WES. Using this diagnostic tool, 26.5% of subjects were identified with a disease causing pathogenic variant, compared to only 18% if commercially available genetic panels would have been used (p=0.04). We also demonstrate that WES is a valuable screening tool in establishing the clinical diagnosis of poorly defined cases of sudden cardiac death, as well as for the identification of novel CVD genes. Furthermore, we offer a blueprint for how CVD genetics clinics could offer precise and personalized services using WES, including the use of CVD specific algorithms, in silico prediction tools and publically available resource to help establish WES variant pathogenicity; the use of interdisciplinary team of physicians, researchers and genetics counselors for collective review of CVD cases and potential identification of causative mutations via WES; current success rate for the genetic diagnosis; and expected frequency and recommendations for communicating incidental findings.
Acknowledgments
Sources of Funding: This study was supported by the NIH grants 1R01HL12283 and 1R01HL122822 (to A.M.) and NIH training grant 2T32HL094301-06 (to S.S.)
Footnotes
Disclosures: None
References
- 1.Rabbani B, Tekin M, Mahdieh N. The promise of whole-exome sequencing in medical genetics. J Hum Genet. 2014;59:5–15. doi: 10.1038/jhg.2013.114. [DOI] [PubMed] [Google Scholar]
- 2.Mangino M, Spector T. Understanding coronary artery disease using twin studies. Heart. 2013;99:373–375. doi: 10.1136/heartjnl-2012-303001. [DOI] [PubMed] [Google Scholar]
- 3.Levine RS, Hennekens CH, Duncan RC, Robertson EG, Gourley JE, Cassady JC, et al. Blood pressure in infant twins: Birth to 6 months of age. Hypertension. 1980;2:I29–33. doi: 10.1161/01.hyp.2.4_pt_2.i29. [DOI] [PubMed] [Google Scholar]
- 4.Hannah-Shmouni F, Seidelmann SB, Sirrs S, Mani A, Jacoby D. The genetic challenges and opportunities in advanced heart failure. Can J Cardiol. 2015;31:1338–1350. doi: 10.1016/j.cjca.2015.07.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho CY. Hypertrophic cardiomyopathy. Heart Fail Clin. 2010;6:141–159. doi: 10.1016/j.hfc.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: Expanded panels offer limited additional sensitivity. Genet Med. 2015;17:880–888. doi: 10.1038/gim.2014.205. [DOI] [PubMed] [Google Scholar]
- 7.Li Z, Huang J, Zhao J, Chen C, Wang H, Ding H, et al. Rapid molecular genetic diagnosis of hypertrophic cardiomyopathy by semiconductor sequencing. J Transl Med. 2014;12:173. doi: 10.1186/1479-5876-12-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. Hrs/ehra expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: This document was developed as a partnership between the heart rhythm society (hrs) and the european heart rhythm association (ehra) Europace. 2011;13:1077–1109. doi: 10.1093/europace/eur245. [DOI] [PubMed] [Google Scholar]
- 9.Ganesh SK, Arnett DK, Assimes TL, Basson CT, Chakravarti A, Ellinor PT, et al. Genetics and genomics for the prevention and treatment of cardiovascular disease: Update: A scientific statement from the american heart association. Circulation. 2013;128:2813–2851. doi: 10.1161/01.cir.0000437913.98912.1d. [DOI] [PubMed] [Google Scholar]
- 10.Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA, et al. Genetic evaluation of cardiomyopathy–a heart failure society of america practice guideline. J Card Fail. 2009;15:83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 11.Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey DE, Jr, et al. 2010 accf/aha/aats/acr/asa/sca/scai/sir/sts/svm guidelines for the diagnosis and management of patients with thoracic aortic disease: Executive summary. A report of the american college of cardiology foundation/american heart association task force on practice guidelines, american association for thoracic surgery, american college of radiology, american stroke association, society of cardiovascular anesthesiologists, society for cardiovascular angiography and interventions, society of interventional radiology, society of thoracic surgeons, and society for vascular medicine. Catheter Cardiovasc Interv. 2010;76:E43–86. doi: 10.1002/ccd.22537. [DOI] [PubMed] [Google Scholar]
- 12.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strom SP, Lee H, Das K, Vilain E, Nelson SF, Grody WW, et al. Assessing the necessity of confirmatory testing for exome-sequencing results in a clinical molecular diagnostic laboratory. Genet Med. 2014;16:510–515. doi: 10.1038/gim.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. Acmg recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rehm HL, Bale SJ, Bayrak-Toydemir P, Berg JS, Brown KK, Deignan JL, et al. Acmg clinical laboratory standards for next-generation sequencing. Genet Med. 2013;15:733–747. doi: 10.1038/gim.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mani A, Meraji SM, Houshyar R, Radhakrishnan J, Mani A, Ahangar M, et al. Finding genetic contributions to sporadic disease: A recessive locus at 12q24 commonly contributes to patent ductus arteriosus. Proc Natl Acad Sci U S A. 2002;99:15054–15059. doi: 10.1073/pnas.192582999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ng PC, Levy S, Huang J, Stockwell TB, Walenz BP, Li K, et al. Genetic variation in an individual human exome. PLoS Genet. 2008;4:e1000160. doi: 10.1371/journal.pgen.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Molina JE, Benditt DG, Adler S. Crinkling of epicardial defibrillator patches. A common and serious problem. J Thorac Cardiovasc Surg. 1995;110:258–264. doi: 10.1016/S0022-5223(05)80032-7. [DOI] [PubMed] [Google Scholar]
- 19.Solaro RJ, Van Eyk J. Altered interactions among thin filament proteins modulate cardiac function. J Mol Cell Cardiol. 1996;28:217–230. doi: 10.1006/jmcc.1996.0021. [DOI] [PubMed] [Google Scholar]
- 20.Mogensen J, Kubo T, Duque M, Uribe W, Shaw A, Murphy R, et al. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin i mutations. J Clin Invest. 2003;111:209–216. doi: 10.1172/JCI16336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karnabi E, Qu Y, Mancarella S, Boutjdir M. Rescue and worsening of congenital heart block-associated electrocardiographic abnormalities in two transgenic mice. J Cardiovasc Electrophysiol. 2011;22:922–930. doi: 10.1111/j.1540-8167.2011.02032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matthes J, Yildirim L, Wietzorrek G, Reimer D, Striessnig J, Herzig S. Disturbed atrio-ventricular conduction and normal contractile function in isolated hearts from cav1.3-knockout mice. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:554–562. doi: 10.1007/s00210-004-0940-7. [DOI] [PubMed] [Google Scholar]
- 23.Qu Y, Baroudi G, Yue Y, Boutjdir M. Novel molecular mechanism involving alpha1d (cav1.3) l-type calcium channel in autoimmune-associated sinus bradycardia. Circulation. 2005;111:3034–3041. doi: 10.1161/CIRCULATIONAHA.104.517326. [DOI] [PubMed] [Google Scholar]
- 24.Rose RA, Sellan M, Simpson JA, Izaddoustdar F, Cifelli C, Panama BK, et al. Iron overload decreases cav1.3-dependent l-type ca2+ currents leading to bradycardia, altered electrical conduction, and atrial fibrillation. Circ Arrhythm Electrophysiol. 2011;4:733–742. doi: 10.1161/CIRCEP.110.960401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bagnall RD, Weintraub RG, Ingles J, Duflou J, Yeates L, Lam L, et al. A prospective study of sudden cardiac death among children and young adults. N Engl J Med. 2016;374:2441–2452. doi: 10.1056/NEJMoa1510687. [DOI] [PubMed] [Google Scholar]
- 26.Sheen VL, Jansen A, Chen MH, Parrini E, Morgan T, Ravenscroft R, et al. Filamin a mutations cause periventricular heterotopia with ehlers-danlos syndrome. Neurology. 2005;64:254–262. doi: 10.1212/01.WNL.0000149512.79621.DF. [DOI] [PubMed] [Google Scholar]
- 27.Campuzano O, Allegue C, Sarquella-Brugada G, Coll M, Mates J, Alcalde M, et al. The role of clinical, genetic and segregation evaluation in sudden infant death. Forensic Sci Int. 2014;242:9–15. doi: 10.1016/j.forsciint.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 28.Baig SM, Koschak A, Lieb A, Gebhart M, Dafinger C, Nurnberg G, et al. Loss of ca(v)1.3 (cacna1d) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci. 2011;14:77–84. doi: 10.1038/nn.2694. [DOI] [PubMed] [Google Scholar]
- 29.Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, et al. Congenital deafness and sinoatrial node dysfunction in mice lacking class d l-type ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 30.Petaja J, Rasi V, Vahtera E, Myllyla G. Familial clustering of defective release of t-pa. Br J Haematol. 1991;79:291–295. doi: 10.1111/j.1365-2141.1991.tb04535.x. [DOI] [PubMed] [Google Scholar]
- 31.Ladenvall P, Wall U, Jern S, Jern C. Identification of eight novel single-nucleotide polymorphisms at human tissue-type plasminogen activator (t-pa) locus: Association with vascular t-pa release in vivo. Thromb Haemost. 2000;84:150–155. [PubMed] [Google Scholar]
- 32.Sartori MT, Saggiorato G, Spiezia L, Varvarikis C, Carraro G, Patrassi GM, et al. Influence of the alu-repeat i/d polymorphism in t-pa gene intron 8 on the stimulated t-pa release after venous occlusion. Clin Appl Thromb Hemost. 2003;9:63–69. doi: 10.1177/107602960300900109. [DOI] [PubMed] [Google Scholar]
- 33.Stead NW, Bauer KA, Kinney TR, Lewis JG, Campbell EE, Shifman MA, et al. Venous thrombosis in a family with defective release of vascular plasminogen activator and elevated plasma factor viii/von willebrand’s factor. Am J Med. 1983;74:33–39. doi: 10.1016/0002-9343(83)91115-4. [DOI] [PubMed] [Google Scholar]
- 34.Kathiresan S, Yang Q, Larson MG, Camargo AL, Tofler GH, Hirschhorn JN, et al. Common genetic variation in five thrombosis genes and relations to plasma hemostatic protein level and cardiovascular disease risk. Arterioscler Thromb Vasc Biol. 2006;26:1405–1412. doi: 10.1161/01.ATV.0000222011.13026.25. [DOI] [PubMed] [Google Scholar]
- 35.Guella I, Duga S, Ardissino D, Merlini PA, Peyvandi F, Mannucci PM, et al. Common variants in the haemostatic gene pathway contribute to risk of early-onset myocardial infarction in the italian population. Thromb Haemost. 2011;106:655–664. doi: 10.1160/TH11-04-0247. [DOI] [PubMed] [Google Scholar]
- 36.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ceyhan-Birsoy O, Pugh TJ, Bowser MJ, Hynes E, Frisella AL, Mahanta LM, et al. Next generation sequencing-based copy number analysis reveals low prevalence of deletions and duplications in 46 genes associated with genetic cardiomyopathies. Mol Genet Genomic Med. 2016;4:143–151. doi: 10.1002/mgg3.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.