

Abstract
Thousands of human diseases could be treated by selectively controlling the expression of specific proteins in vivo. We demonstrate a new series of alkenyl amino alcohol (AAA) ionizable lipids that, when formulated into lipid nanoparticles (LNPs), are capable of delivering human mRNA with unprecedented levels of in vivo efficacy. This study highlights the importance of utilizing synthesis tools in tandem with biological inspiration to understand and improve nucleic acid delivery in vivo.
Keywords: RNA, drug delivery, nanoparticles, biomaterials, proteins
Graphical Abstract
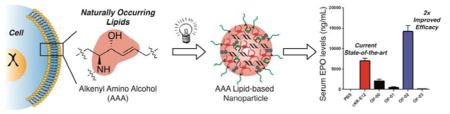
Nucleic acid therapies could be leveraged to treat thousands of genetic disorders, many of which are difficult or impossible to manage with present day therapeutic approaches. For example, the successful delivery of short interfering RNAs (siRNA) to cells in both rodents and non-human primates has been widely used for the treatment of hereditary diseases and cancer.[1] By contrast, the delivery of messenger RNA (mRNA) remains largely unexplored. Whereas siRNA sequences are employed to silence gene expression, mRNA therapeutics could be used to treat diseases caused by deficiencies in specific proteins.[2] This is because mRNA sequences can be translated into proteins once they are successfully transported into the cytoplasm of target cells. The implementation of mRNA therapeutics, therefore, could profoundly impact fields such as protein replacement therapy, vaccine development, and immune tolerization wherein the selective expression of proteins in vivo could treat disease.[3]
Before clinical translation can be realized, however, serious limitations with the in vivo delivery of mRNA must still be overcome. The high anionic charge density, size, and hydrophilicity of nucleic acids prevent meaningful levels of passive diffusion of mRNA across cell membranes.[4] To circumvent this barrier, our group and others have developed and implemented an array of lipid nanoparticles (LNPs) for the entrapment and subsequent delivery of nucleic acids in vivo.[1] Although these LNPs have been largely optimized for siRNA sequences, both Schlake’s group[5] and our research team[6] have recently employed LNPs derived from previously described components to deliver mRNA in vivo. Successful delivery was confirmed by quantifying serum protein levels, thereby establishing LNPs as viable delivery vehicles for mRNA.
Inspired by these results, we sought to design and synthesize novel LNP components capable of delivering mRNA with unprecedented levels of in vivo efficacy. In practice, LNPs are comprised of cholesterol (aids in stability),[7] a phospholipid (modifies bilayer structure),[1a, 8] a polyethylene glycol (PEG) derivative (decreases aggregation and non-specific uptake),[9] and an ionizable lipid (complexes negatively charged RNA and enhances endosomal escape).[10] Evidence within the siRNA delivery community has implicated the chemical structure and identity of the ionizable lipid as the most pivotal component for efficacy. Accordingly, several rationally designed[11] and combinatorial chemistry[10b, 12] methodologies have been explored to discover novel series of ionizable lipid materials capable of maximizing gene silencing at the lowest possible dose. This strategy both conserves precious therapeutic nucleic acid cargo and also serves to mitigate any possible issues with the toxicity of the LNPs themselves.
Interestingly, however, no reports detailing the creation of a new series of ionizable lipids for the expressed purpose of improving mRNA LNP delivery in vivo have yet been reported. We hypothesized that ionizable lipids based upon alkenyl amino alcohols (AAA), a functional group combination found in sphingosine and other bioactive molecules, could promote high levels of in vivo protein expression when formulated into mRNA LNPs (Figure 1).[13] We envisioned that we could furnish AAA ionizable lipids via a ring opening reaction between alkenyl epoxides (AE) with a polyamine core (Figure 2a).[12] It is important to note, however, that no AEs of suitable tail length are commercially available, nor are they trivial to synthesize on account of the difficulty of selectively oxidizing singular alkenes in the presence of electronically-similar carbon-carbon double bonds. As such, we report the first detailed procedures for AE synthesis and characterization beginning from biologically relevant fatty acid starting materials (Figure S1). AE-00 through AE-03 were then each reacted in turn with polyamine 1 to afford AAA ionizable lipids OF-00 through OF-03. While OF-00 through OF-03 represent the first four members of this series of materials, we hope that the chemical versatility of AE-00 through AE-03 will serve as inspiration for future generations of AAA ionizable lipids for nucleic acid delivery.
Figure 1.

Naturally occurring components of the cell membrane contain alkenyl amino alcohol (AAA) functionality serving as the inspiration for the development of AAA ionizable lipids. These lipids form the basis of lipid nanoparticles exhibiting highly efficient in vivo delivery of mRNA.
Figure 2.

a) Synthesis of OF-00 through OF-03, seminal members of the AAA class of ionizable lipids. b) Representative cryogenic transmission electron microscopy of OF-02 LNPs.
Lipids OF-00 through OF-03 were then formulated with cholesterol, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), C14-PEG-2000, and unmodified mRNA coding for human erythropoietin (EPO) into mRNA LNPs.[6] EPO was selected as a model protein to evaluate the relationship between ionizable lipid identity and mRNA LNP efficacy for two reasons: 1) the associated protein is secreted directly into the bloodstream allowing for robust protein quantification, and 2) EPO has potential therapeutic applications in such areas as anemia.[4, 14] Cryogenic transmission electron microscopy images of OF-02 LNPs detail a spherical morphology and a multilamellar structure (Figure 2b). Additional physical properties include a narrow polydispersity index (0.130) and an average particle diameter around 100 nm.
The nanoparticle diameters, polydispersity indices, and encapsulation efficiencies for each OF-00 through OF-03 LNP formulation can be found in Table S1. Ionizable lipid cKK-E12 was also formulated alongside these compounds to be used as a positive control in our study. cKK-E12 was chosen because it is capable of silencing Factor VII expression in mice at siRNA doses as low as 0.002 mg/kg, and as such it represents a benchmark ionizable lipid in the field of nucleic acid delivery.[12] Each resultant mRNA loaded LNP was then injected intravenously at a 0.75 mg/kg dose in C57BL/6 mice alongside phosphate buffered saline (PBS) as a negative control. At six hours, the serum EPO levels were quantified (Figure 3a). The PBS control imparted no significant EPO production in vivo, whereas positive control cKK-E12 LNPs promoted a serum EPO concentration of 7100 ± 700 ng/mL. Excitingly, OF-02 LNPs significantly outperformed benchmark lipid cKK-E12 LNPs, promoting an approximate two-fold increase in EPO concentration to 14200 ± 1500 ng/mL. Additionally, OF-02 outperformed two other benchmark ionizable lipids from the nucleic acid delivery field, namely 503013[15] and C12-200,[10b] whose respective LNP promoted in vivo EPO concentrations were 2800 ± 200 ng/mL and 7100 ± 500 ng/mL at an identical dose. To the best of our knowledge, therefore, OF-02 LNPs represent the most potent mRNA delivery vehicle reported to date in the scientific literature.
Figure 3.

a) In vivo expression of EPO following administration of AAA LNPs for delivery of mRNA. b) Batch-to-batch variability of OF-02 LNPs for EPO mRNA delivery in vivo. c) In vivo dose response curves for OF-02 and cKK-E12 LNPs. d) EPO expression following administration of OF-02 and cKK-E12 LNPs at 6 and 24 h. Data presented as mean + standard deviation (n = 3).
The OF-00, OF-01, and OF-03 LNPs also allow the deduction of structure/function relationships within this new series of AAA ionizable lipids. We note two general structure/function trends of interest. First, we note that only alkenes with a cis geometry promote in vivo efficacy – OF-00 and OF-01 exclusively differ in the cis/trans geometry of their alkenes, and only OF-00 produces meaningful EPO concentrations. Second, the optimal number and placement of two cis alkenes per tail matches those observed in optimized siRNA LNPs.[11, 16] While we are still discerning why these specific trends are observed, these empirical findings could potentially shape subsequent generations of AAA lipids. It is interesting to note, therefore, that only the linoleic acid derivative OF-02 promotes significantly higher levels of EPO expression than the positive control, although oleic acid derivative OF-00 also demonstrates modest activity promoting a serum EPO concentration of 2100 ± 500 ng/mL using the same dose.
With this information in hand, our attention then shifted from exploring the general properties of the new AAA series of ionizable lipids to further characterizing LNPs made from our lead material OF-02. The clinical translation of nucleic acid delivery vehicles is in part predicated on high reproducibility of the chemical constituents and formulation of LNPs. To test this, three independent batches of OF-02 were synthesized and then formulated into LNPs. The average serum concentration among all batches was found to be 13700 ± 1700 ng/mL and demonstrated minimal batch-to-batch variability (Figure 3b). Next, a dose response curve was collected at 0.75 mg/kg, 1.5 mg/kg, and 2.25 mg/kg total EPO mRNA dose for both OF-02 and cKK-E12 LNPs (Figure 3c). OF-02 LNPs outperformed their cKK-E12 counterparts roughly two-fold across all doses studied, reaching a maximum EPO concentration of 45400 ± 5300 ng/mL at the 2.25 mg/kg dose. It is also interesting to note that both sets of LNPs promote EPO production in a linear fashion with respect to dose. This trend implies that we have not yet reached a saturation point for the intracellular translation machinery, suggesting protein production is currently only limited by the dose of mRNA. Moreover, it is important to note that no animal mortality was observed at all doses studied, and that mice treated with both cKK-E12 and OF-02 LNPs displayed similar weight loss profiles at identical doses (Figure S2). OF-02 LNPs therefore represent a tunable handle for in vivo EPO production readily capable of exceeding normal human EPO levels (40 – 250 pg/mL) in our chosen mouse model.[17] Finally, OF-02 LNPs also outperformed their cKK-E12 counterparts at 24 hours, independent of dose (Figure 3d). The sharp decrease in EPO concentration as a function of time highlights one of the many exciting potential therapeutic advantages of mRNA delivery in vivo; in contrast to permanent gene replacement therapies, mRNA delivery offers transient, dose-response dependent protein expression in vivo, a property that could one day prove useful for a variety of genetic disorders.
Finally, we were interested to determine if the efficacy differences observed between ckk-E12 and OF-02 LNPs were due to variations in biodistribution. mRNA coding for luciferase was independently formulated with both ckk-E12 and OF-02 in the same fashion as for EPO delivery, and mouse organs were harvested 24 hours post injection. The tissues were subsequently imaged ex-vivo to measure the total luminescence per organ, demonstrating that mRNA from both ckk-E12 and OF-02 LNPs is predominantly translated in the liver with minimal translation in the spleen and negligible translation in other organs (Figure 4a,b). Quantification of this data also confirms nearly identical biodistribution profiles for the two formulations, suggesting that the increased efficacy of OF-02 LNPs is not due to a difference in tissue targeting (Figure 4c). Since more than 4000 human diseases are caused by liver genetic disorders such as hemophilias A and B, OF-02 LNPs represent a promising delivery vehicle for therapeutic mRNA delivery to the liver.[18]
Figure 4.

Representative luminescence biodistribution of a) cKK-E12 and b) OF-02 LNPs with luciferase mRNA ex-vivo and c) associated quantification. Data presented as mean + standard deviation (n = 3).
In summary, we have synthesized a new series of AAA ionizable lipid materials for mRNA LNP delivery. To the best of our knowledge, compounds OF-00 through OF-03 represent the first examples of AAA ionizable lipids in the scientific literature, and we hope that their alkene-epoxide precursors AE-00 through AE-03 can serve as versatile scaffolds for the synthesis of future generations of these ionizable lipids. OF-02 LNPs yielded a two-fold increase in EPO production in vivo as compared to benchmark LNPs in the literature across a broad linear dose-response window. This illustrates that OF-02 presents a tunable handle over in vivo protein expression, which is important in protein replacement therapies.[2] Batch-to-batch variability, dose response curves, and time course studies were coupled with biodistribution data, highlighting the exceptional potency with which these LNPs can deliver mRNA to the liver. Future work will study the potential of OF-02 LNPs for therapeutic applications and establish further groundwork necessary for translating this novel mRNA delivery vehicle to the clinic. In total, this study demonstrates efficient mRNA delivery with OF-02 as well as the importance of utilizing synthetic chemistry in tandem with biological inspirations to further improve and understand nucleic acid delivery in vivo.
Experimental Section
Animal Experiments
All animal studies were approved by the M.I.T. Institutional Animal Care and Use Committee and were consistent with local, state and federal regulations as applicable. LNPs were intravenously injected in female C57BL/6 mice (Charles River Labs, 18–22 grams) via the tail vein. After six or 24 hours, blood was collected via the tail vein and serum was isolated by centrifugation in serum separation tubes. Serum EPO levels were quantified with an ELISA assay (Human Erythropoietin Quantikine IVD ELISA Kit, R&D Systems, Minneapolis, MD). 24 hours after injection of Luc-mRNA LNPs, mice were injected intraperitoneally with 130 μL of D-luciferin (30 mg/mL in PBS). After fifteen minutes, mice were sacrificed and the organs were isolated (pancreas, spleen, liver, kidneys, lungs, heart, uterus and ovaries) and imaged with an IVIS imaging system (Perkin Elmer, Waltham, MA). Luminescence was quantified using LivingImage software (Perkin Elmer).
Supplementary Material
Acknowledgments
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship (Owen S. Fenton). This work was also supported in part by Shire Pharmaceuticals (Lexington, MA). E.A.A. is grateful for financial support through a Wellcome Trust-MIT post-doctoral fellowship. We also thank the Nanotechnology Materials Core Facility at the Koch Institute at MIT.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author. All additional experimental details (including chemical instrumentation and materials, LNP synthesis, LNP characterization, synthetic chemistry procedures, and percent weight gain data) can be found in full in the supplementary information of our report.
Contributor Information
Owen S. Fenton, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139. Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139
Kevin J. Kauffman, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139. Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139
Rebecca L. McClellan, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139
Dr. Eric A. Appel, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139
J. Robert Dorkin, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139. Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139.
Dr. Mark W. Tibbitt, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139
Dr. Michael W. Heartlein, Shire Pharmaceuticals, Lexington, MA 02421
Dr. Frank DeRosa, Shire Pharmaceuticals, Lexington, MA 02421
Prof. Robert Langer, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139. Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139. Institute for Medical Engineering and Science, Massachusetts Institute of Technology, Cambridge, MA 02139. Harvard-MIT Division of Health Science and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139
Prof. Daniel G. Anderson, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139. Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139. Institute for Medical Engineering and Science, Massachusetts Institute of Technology, Cambridge, MA 02139. Harvard-MIT Division of Health Science and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139
References
- 1.a) Kanasty R, Dorkin JR, Vegas A, Anderson D. Nat Mater. 2013;12:967–977. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]; b) Whitehead KA, Langer R, Anderson DG. Nat Rev Drug Discov. 2009;8:516–516. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sahin U, Kariko K, Tureci O. Nat Rev Drug Discov. 2014;13:759–780. doi: 10.1038/nrd4278. [DOI] [PubMed] [Google Scholar]
- 3.Leader B, Baca QJ, Golan DE. Nat Rev Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 4.Kormann MSD, Hasenpusch G, Aneja MK, Nica G, Flemmer AW, Herber-Jonat S, Huppmann M, Mays LE, Illenyi M, Schams A, Griese M, Bittmann I, Handgretinger R, Hartl D, Rosenecker J, Rudolph C. Nat Biotechnol. 2011;29:154–U196. doi: 10.1038/nbt.1733. [DOI] [PubMed] [Google Scholar]
- 5.Thess A, Grund S, Mui BL, Hope MJ, Baumhof P, Fotin-Mleczek M, Schlake T. Mol Ther. 2015;23:S55–S55. doi: 10.1038/mt.2015.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kauffman KJ, Dorkin JR, Yang JH, Heartlein MW, DeRosa F, Mir FF, Fenton OS, Anderson DG. Nano Lett. 2015:7300–7306. doi: 10.1021/acs.nanolett.5b02497. [DOI] [PubMed] [Google Scholar]
- 7.a) Lu JJ, Langer R, Chen JZ. Mol Pharmaceut. 2009;6:763–771. doi: 10.1021/mp900023v. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Allen TM, Cullis PR. Adv Drug Deliver Rev. 2013;65:36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 8.Zuhorn IS, Bakowsky U, Polushkin E, Visser WH, Stuart MCA, Engberts JBFN, Hoekstra D. Mol Ther. 2005;11:801–810. doi: 10.1016/j.ymthe.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 9.Mui BL, Tam YK, Jayaraman M, MAnsell S, Du XY, YYC Tam XY, Lin PJC, Chen S, Narayanannair JK, Rajeev KG, Manoharan M, Akinc A, Maier MA, Cullis P, Madden TD, Hope MJ. Mol Ther-Nucl Acids. 2013;2 doi: 10.1038/mtna.2013.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Sahay G, Querbes W, Alabi C, Eltoukhy A, Sarkar S, Zurenko C, Karagiannis E, Love K, Chen DL, Zoncu R, Buganim Y, Schroeder A, Langer R, Anderson DG. Nat Biotechnol. 2013;31:653–U119. doi: 10.1038/nbt.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Love KT, Mahon KP, Levins CG, Whitehead KA, Querbes W, Dorkin JR, Qin J, Cantley W, Qin LL, Racie T, Frank-Kamenetsky M, Yip KN, Alvarez R, Sah DWY, de Fougerolles A, Fitzgerald K, Koteliansky V, Akinc A, Langer R, Anderson DG. P Natl Acad Sci USA. 2010;107:9915–9915. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Semple SC, Akinc A, Chen JX, Sandhu AP, Mui BL, Cho CK, Sah DWY, Stebbing D, Crosley EJ, Yaworski E, Hafez IM, Dorkin JR, Qin J, Lam K, Rajeev KG, Wong KF, Jeffs LB, Nechev L, Eisenhardt ML, Jayaraman M, Kazem M, Maier MA, Srinivasulu M, Weinstein MJ, Chen QM, Alvarez R, Barros SA, De S, Klimuk SK, Borland T, Kosovrasti V, Cantley WL, Tam YK, Manoharan M, Ciufolini MA, Tracy MA, de Fougerolles A, MacLachlan I, Cullis PR, Madden TD, Hope MJ. Nat Biotechnol. 2010;28:172–U118. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- 12.Dong YZ, Love KT, Dorkin JR, Sirirungruang S, Zhang YL, Chen DL, Bogorad RL, Yin H, Chen Y, Vegas AJ, Alabi CA, Sahay G, Olejnik KT, Wang WH, Schroeder A, Lytton-Jean AKR, Siegwart DJ, Akinc A, Barnes C, Barros SA, Carioto M, Fitzgerald K, Hettinger J, Kumar V, Novobrantseva TI, Qin JN, Querbes W, Koteliansky V, Langer R, Anderson DG. P Natl Acad Sci USA. 2014;111:5753–5753. doi: 10.1073/pnas.1322937111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Bartke N, Hannun YA. J Lipid Res. 2009;50:S91–S96. doi: 10.1194/jlr.R800080-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hannun YA, Obeid LM. Nat Rev Mol Cell Bio. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 14.a) Kariko K, Muramatsu H, Keller JM, Weissman D. Mol Ther. 2012;20:948–953. doi: 10.1038/mt.2012.7. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu S, Ren JA, Hong ZW, Yan DS, Gu GS, Han G, Wang GF, Ren HJ, Chen J, Li JS. Nutr Clin Pract. 2013;28:120–127. doi: 10.1177/0884533612462744. [DOI] [PubMed] [Google Scholar]
- 15.Whitehead KA, Dorkin JR, Vegas AJ, Chang PH, Veiseh O, Matthews J, Fenton OS, Zhang YL, Olejnik KT, Yesilyurt V, Chen DL, Barros S, Klebanov B, Novobrantseva T, Langer R, Anderson DG. Nat Commun. 2014;5 doi: 10.1038/ncomms5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heyes J, Palmer L, Bremner K, MacLachlan I. J Control Release. 2005;107:276–287. doi: 10.1016/j.jconrel.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 17.Cazzola M, Mercuriali F, Brugnara C. Blood. 1997;89:4248–4267. [PubMed] [Google Scholar]
- 18.McClellan J, King MC. Cell. 2010;142:353–355. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.