

Abstract
Nuclear lamins are major architectural elements of the mammalian cell nucleus, and they have been implicated in the functional organization of the nuclear interior, possibly by providing structural support for nuclear compartments. Colocalization studies have suggested a structural role for lamins in the formation and maintenance of pre-mRNA splicing factor compartments. Here, we have directly tested this hypothesis by analysis of embryonic fibroblasts from knock-out mice lacking A- and C-type lamins. We show that the morphology and cellular properties of splicing factor compartments are independent of A- and C-type lamins. Genetic loss of lamins A/C has no effect on the cellular distribution of several pre-mRNA splicing factors and does not affect the compartment morphology as examined by light and electron microscopy. The association of splicing factors with the nuclear matrix fraction persists in the absence of lamins A/C. Live cell microscopy demonstrates that the intranuclear positional stability of splicing factor compartments is maintained and that the exchange dynamics of SF2/ASF between the compartments and the nucleoplasm is not affected by loss of lamin A/C. Our results demonstrate that formation and maintenance of intranuclear splicing factor compartments is independent of lamins A/C, and they argue against an essential structural role of lamins A/C in splicing factor compartment morphology.
INTRODUCTION
The mammalian cell nucleus contains numerous distinct compartments (Dundr and Misteli, 2001). One of the most prominent being the splicing factor compartments (SFCs), or speckles (Spector et al., 1983; Spector, 1993; Misteli, 2000). These nuclear domains are enriched in pre-mRNA splicing factors but also contain a multitude of other proteins (Mintz et al., 1999). By light microscopy, SFCs are irregularly shaped entities with a diameter of ∼1–2 μm. At the electron microscopic level, SFCs correspond to interchromatin granule clusters (IGCs) and perichromatin fibrils with IGCs representing the major part of the speckled staining pattern observed by fluorescence microscopy (Fakan et al., 1984; Spector, 1993; Fakan, 1994). Because IGCs contain little to no DNA and are devoid of newly synthesized RNA, it is unlikely that these structures correspond to major centers of active transcription (Monneron and Bernhard, 1969; Fakan, 1994). Splicing factor compartments may represent storage/assembly sites for splicing components because splicing factors are recruited from SFCs to sites of active transcription upon transcriptional activation (Jiménez-García and Spector, 1993; Huang and Spector, 1996; Misteli et al., 1997). In addition, movement of macromolecular complexes and unspliced RNAs from intron-containing genes toward SFCs has been described, pointing to a possible function of SFCs in posttranscriptional splicing (Johnson et al., 2000; Melc̆ák et al., 2000).
SFCs might be physically associated with structural elements of the nucleus because IGCs are resistant to nuclease, detergent and high salt extractions and are thus often considered part of the nuclear skeleton or nuclear matrix (Monneron and Bernhard, 1969; Spector et al., 1983). Furthermore, SFCs maintain their location within the cell nucleus over extended periods, possibly due to tethering to an intranuclear scaffold (Misteli et al., 1997; Kruhlak et al., 2000). What determines the morphological appearance of splicing factor compartments, how their intranuclear position is established, and how these intranuclear domains are maintained is unclear. A possible mechanistic explanation for their stability and structural integrity comes from the colocalization of SFCs with lamins A/C (Jagatheesan et al., 1999; Muralikrishna et al., 2001; Kumaran et al., 2002).
Lamins A/C, together with the B-type lamins, are the major components of the nuclear lamina (Fawcett 1966; Goldman et al., 2002). Lamins A and C arise by alternative splicing from the LMNA gene, whereas separate genes exist for B-type lamins (reviewed in Goldman et al., 2002). Lamins have traditionally been considered key components of the nuclear envelope to provide structural support for the nucleus and to serve as anchoring sites for chromatin (Glass and Gerace, 1990; Vlcek et al., 2001). More recently, lamins also have been found in the nuclear interior where they have been implicated in various processes, including transcription, DNA synthesis, and apoptosis (Goldman et al., 1992; Bridger et al., 1993; Moir et al., 1994; Hozák et al., 1995; Broers et al., 1999; Moir et al., 2000; Cohen et al., 2001; Goldman et al., 2002). One possible function for intranuclear lamins is to provide structural support for subnuclear compartments. In support, some antibodies show lamin A to colocalize with SFCs (Jagatheesan et al., 1999; Kumaran et al., 2002), although tagged lamin A has not been observed to accumulate in SFCs in intact, living cells (Moir et al., 1994; Broers et al., 1999). Furthermore, disruption of A- and B-type lamins results in redistribution of pre-mRNA splicing factors (Spann et al., 2002; Kumaran et al., 2002). In the present study, we set out to directly test the hypothesis that formation and maintenance of SFCs is dependent on lamins A/C by analyzing nuclei from mice lacking lamins A and C.
MATERIALS AND METHODS
Cell Lines and Transfection
Mouse embryonic fibroblast-positive and -negative cells (Sullivan et al., 1999) were used. Cells were maintained in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (vol/vol) (Hyclone Laboratories, Logan, UT), glutamine, and 1% penicillin/streptomycin (vol/vol) at 37°C in a humidified atmosphere containing 5% CO2. For fluorescence recovery after photobleaching (FRAP) experiments, attached cells were grown in live cell chambers (LabTekII; Nunc, Rochester, NY), transfected with SF2/ASF-GFP construct (Misteli et al., 1997), by using FuGENE 6 reagent and then incubated for 14 h.
In Vivo Microscopy and FRAP
For live cell microscopy, cells were grown to 95% confluence in Nalgene LabTekII chambers and observed using a Zeiss LSM 510 confocal microscope with the 488 nm line of an argon laser. The sample emission was detected at 545 nm. All experiments were performed at 37°C. Single confocal sections were acquired every 10 s. Experimental conditions for FRAP were described previously (Phair and Misteli, 2000). Five imaging scans were acquired, followed by a single bleach pulse of ∼590 ms by using a spot of ∼1 μm in diameter. Single-section images were then collected at ∼590 ms intervals. One hundred images were taken. FRAP recovery curves were generated from background-subtracted images. The total fluorescence was determined for each image and compared with the initial value to account for the amount of signal lost during the bleach pulse and during imaging (Phair and Misteli, 2000). All quantitative values represent averages from at least 12 cells from two independent experiments.
Immunocytochemistry, Light Microscopy, and Electron Microscopy
For immunocytochemical localizations, cells were grown on coverslips to 60% confluence, rinsed briefly in phosphate-buffered saline (PBS) (pH 7.4), and then fixed in 2% paraformaldehyde in PBS for 10 min and permeabilized in 0.5% Triton X-100. Before immunolabeling, cells were washed in PBS. Subsequently, the cells were incubated with mouse anti-SF2 (Zymed Laboratories, South San Francisco, CA), anti-U2-B″, or human anti-Sm antibody. All antibodies were diluted in 1% bovine serum albumin to block nonspecific binding and detected using Cy3- and fluorescein isothiocyanate-conjugated secondary antibodies (Jackson Immunoresearch Laboratories, West Grove, PA). Cells were counterstained with 4,6-diamino-2-phenylindole (DAPI) and embedded in Mowiol and viewed using an AX70 Provis (Olympus, Melville, NY) microscope equipped with a charge-coupled device camera (PXL with KAF 1400 chip; Photometrics, Tucson, AZ). IP lab software was used to collect digital images.
For electron microscopy observations, A/C +/+ and A/C -/- cells were fixed with 1% glutaraldehyde in 0.2 M PIPES, pH 6.95, and embedded in epon. Ultrathin sections were stained according to Bernhard (1969) for preferential visualization of IGC in ultrathin sections or with uranyl acetate. After staining, the sections were viewed using an EM 900 Zeiss electron microscope equipped with a KeenView charge-coupled device camera (Soft Imaging Systems, Münster, Germany).
Gel Electrophoresis and Immunoblotting
Cell lysates were resolved on 12% SDS-PAGE gels and transferred to nitrocellulose according to established protocols (Laemmli et al., 1970). Nitrocellulose membranes were washed with blocking buffer [∼5% gelatin (wt/vol), 0.05% Tween 20 in PBS] or with 5% Blotto (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C. Goat polyclonal IgG SC-6215 or SC-7293 (Santa Cruz Biotechnology) was used to detect lamins A/C. Nitrocellulose membranes were rinsed with blocking buffer several times, incubated with appropriate mouse anti-goat horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) for 1 h at room temperature. ECL reagents (Amersham Biosciences, Piscataway, NJ) were used for the immunological detection of proteins.
Nuclear Matrix Preparation
Lamin +/+ and -/- cells were grown on coverslips, washed with PBS at 4°C, and incubated for 10 min in ice-cold cytoskeleton buffer (10 mM NaCl, 300 mM sucrose, 10 mM PIPES, pH 6.8, 3 mM MgCl2, 0.5% Triton X-100, 1 mM phenylmethylsulfonyl fluoride [PMSF], 10 μM leupeptin, 10 μM pepstatin A, 15 μM E-64, 50 μM bestatin, 20 U/ml recombinant RNasin [Promega, Madison, WI] ribonuclease inhibitor). Then, cells were incubated in extraction buffer (250 mM ammonium sulfate, 300 mM sucrose, 10 mM PIPES, pH 6.8, 3 mM MgCl2, 0.5% Triton X-100, 1 mM PMSF, 10 μM leupeptin, 10 μM pepstatin A, 15 μM E-64, 50 μM bestatin, 20 U/ml recombinant RNasin [Promega] ribonuclease inhibitor) on ice for 5 min and digestion buffer (50 mM NaCl, 300 mM sucrose, 10 mM PIPES, pH 6.8, 3 mM MgCl2, 0.5% Triton X-100, 1 mM PMSF, 10 μM leupeptin, 10 μM pepstatin A, 15 μM E-64, 50 μM bestatin, 20 U/ml recombinant RNasin [Promega] ribonuclease inhibitor, 500 U/ml RQ1 RNase-free DNase [Promega]) at 37°C for 4 h. After each step, one coverslip was removed, and cells were fixed in 2% paraformaldehyde as described above.
RESULTS
SFCs Persist in the Absence of Lamins A/C
We sought to test whether lamins A/C are essential for the organization of SFCs by analysis of well characterized mouse embryonic fibroblasts (MEFs) from transgenic mice lacking lamins A/C (LMNA -/-) due to targeted disruption of the LMNA gene by insertion of Pgkneo (Sullivan et al., 1999). Immunoblot analysis and immunofluorescence microscopy using several specific antibodies against lamins A/C confirmed the previously reported absence of lamin A/C protein from the knock-out cells (Figure 1; our unpublished data). Absence of LMNA mRNA in LMNA -/- cells has been demonstrated previously (Sullivan et al., 1999).
Figure 1.
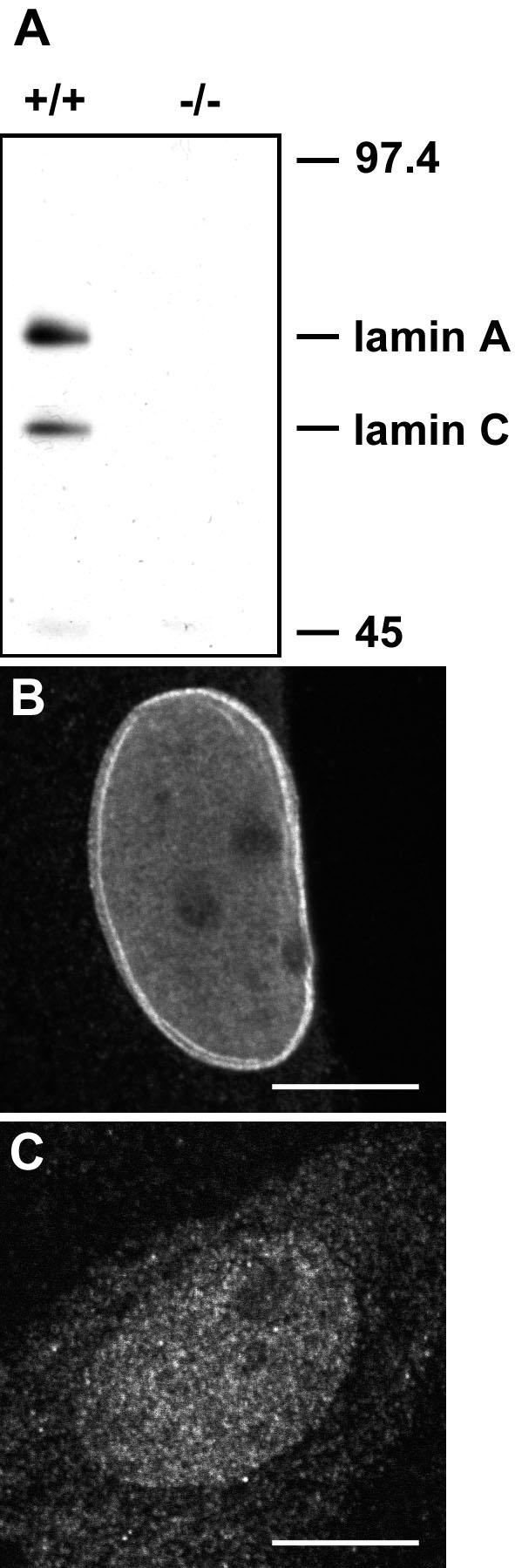
Absence of lamins A/C from LMNA -/- MEFs. Western blot (A) and (B and C) indirect immunofluorescence microscopy by using anti-lamin A/C polyclonal antibody. (B) In confocal sections, a bright rim staining at the nuclear periphery corresponding to the nuclear lamina can be recognized in LMNA +/+ cells. (C) The signal for lamin A/C is diffuse in LMNA -/- cells. The image in C is greatly overexposed. Bars, 10 μm.
To test whether loss of lamins A/C had any effect on SFC morphology, we localized the SFC resident SR protein splicing factor SF2/ASF, the snRNP protein U2-B″, and core snRNP splicing factors containing the Sm epitope in LMNA -/- cells or LMNA +/+ control cells (Krainer et al., 1990; Will and Lührmann, 1997). The characteristic speckled pattern observed in most mammalian cell lines and in LMNA +/+ cells was also found for all three epitopes in LMNA -/- cells (Figure 2, A–F). No significant differences in shape, size, or number of SFCs were observed in MEFs lacking lamins A and C compared with control cells (Figure 2, A–F). This observation demonstrates that A- and C-type lamins are not essential for the proper localization of members of three distinct classes of pre-mRNA splicing factors.
Figure 2.

Distribution and matrix extraction properties of splicing factors are not affected by loss of lamins A/C. (A–F) Indirect immunofluorescence detection of several splicing factors. (G–L) Sequential extraction of SF2/ASF splicing factor. Bar, 10 μm.
Despite the unchanged overall morphological appearance of SFCs, it was possible that the association properties of proteins with SFCs were affected by the absence of lamins A/C. To address this point, the behavior of splicing factors in a nuclear matrix extraction was assessed (Figure 2, G–L). Indirect immunofluorescence microscopy was used to monitor the effect of sequential treatment of LMNA -/- or LMNA +/+ control cells with Triton X-100, ammonium sulfate, and DNase on the localization of SF2/ASF (Figure 2, G–L). SF2/ASF in LMNA -/- cells was resistant to extraction to the same degree as in control cells, and the speckled organization of SF2/ASF was preserved in LMNA +/+ and LMNA -/- cells after each extraction step (Figure 2, G–L). Successful DNA digestion was confirmed by DAPI staining. Identical results were obtained for U2-B″ and Sm epitopes (our unpublished data). We concluded that lamins A/C are not essential for the accumulation of several types of pre-mRNA splicing factors in nuclear splicing factor compartments.
The Absence of Lamins A/C Does Not Influence the Mobility of SFCs or the Nucleoplasmic Exchange of SF2/ASF-GFP
Because lamins might be part of an internal nuclear scaffold, they might affect the dynamic properties of either whole SFCs or of the proteins accumulated within splicing factor compartments. To first test whether the positional stability of SFCs was lost in the absence of lamins A/C, we performed time-lapse fluorescence microscopy by using SF2/ASF fused to the green fluorescent protein as an in vivo marker for SFCs. GFP-SF2/ASF has previously been shown to be functionally active in alternative splice site selection in vivo and to colocalize with its endogenous counterpart (Misteli et al., 1997). GFP-SF2/ASF was transiently transfected into LMNA -/- cells and LMNA +/+ control cells, and the movement of entire SFCs was monitored over time periods from a few minutes up to 2 h using confocal microscopy (Figure 3, A and B). In agreement with previous results (Misteli et al., 1997; Kruhlak et al., 2000), SFCs changed their shape over time, and only a minor fraction changed their position during the observation period in LMNA +/+ cells (Figure 3A). Occasionally, fusion or fission events were observed. In LMNA -/- cells, identical SFC dynamics were observed (Figure 3B). Measurements of mean square changes over time confirmed quantitatively that there was no significant difference in SFC dynamics between the two cell lines, and identical results were observed in short-term and long-term observations (our unpublished data). These results suggest that loss of lamin A/C does not influence the positional stability of splicing factor compartments directly nor indirectly.
Figure 3.

The dynamics of SF2/ASF in the presence/absence of lamins A/C. Time-lapse confocal microscopy of SF2/ASF-GFP in LMNA +/+ (A) and LMNA -/- (B) cells reveal no significant differences in the overall dynamics of SFCs. Time points are indicated in seconds. The overlay shows the same cell at the beginning and the end of the observation period (60 s). (C–H) FRAP analysis of GFP-SF2/ASF dynamics. Images were acquired before bleaching and immediately after and during recovery. Selected images are shown. (C and E) Bleaching of an SFC. (D and F) Bleaching of a nucleoplasmic area. The position of the bleaching spot is indicated by an arrowhead. (G and H) Quantitative analysis of recovery kinetics in a SFC (G) or a nucleoplasmic region (H) did not show any significant difference between LMNA +/+ cells (black circles) and LMNA-/- cells (white circles). Values represent averages ± S. D. from at least 12 cells from two or more experiments. Bars, 10 μm.
Despite the overall stability of nuclear SFCs, it has been demonstrated that splicing factors, including SF2/ASF, only reside transiently in SFCs and are continuously and rapidly exchanged between SFCs and the nucleoplasm with a typical residence time in SFCs on the order of 30–60 s (Kruhlak et al., 2000; Phair and Misteli, 2000). We thus tested whether loss of lamins A/C plays a role in the exchange dynamics of SF2/ASF-GFP molecules with the nucleoplasm (Figure 3, C–H). To this end, we measured the apparent mobility of SF2/ASF-GFP in nuclear SFCs and in the nucleoplasm using FRAP (Figure 3, C–H; Phair and Misteli, 2000). Qualitative and quantitative analysis of FRAP experiments showed no significant difference in the recovery kinetics of GFP-SF2/ASF in lamin A/C -/- cells compared with lamin A/C +/+ cells, regardless of whether an SFC or an area in the nucleoplasm was bleached (Figure 3, C–H). We concluded that the presence of lamins A/C does not influence the dynamics of association and dissociation of SF2/ASF protein in living cells.
The Absence of Lamins A/C Does Not Result in Ultrastructural Changes of Nuclear SFCs
Although our results indicated that lamins A/C are not involved in the accumulation of splicing factors in SFCs, we could not exclude their participation in the structural organization of SFCs. To address this point, the ultrastructural appearance of IGCs in LMNA +/+ and LMNA -/- MEFs was analyzed by electron microscopy. Consistent with our light microscopy observations, IGCs were routinely observed in sections of both LMNA +/+ and LMNA -/- cells, and no differences in shape, number, or size of IGCs was observed between lamin A/C -/- and +/+ cell lines (Figure 4). Furthermore, no differences in the ultrastructural morphological appearance of IGCs from lamin A/C -/- and +/+ cells were detected (Figure 4). Similarly, other prominent nuclear structures such as nucleoli, perichromatin fibrils, and heterochromatin domains were unaffected by loss of lamins A/C (our unpublished data). These observations indicate that lamins A/C are not essential for the ultrastructural appearance of splicing factor compartments.
Figure 4.

Electron microscopy of cells lacking lamins A/C. (A and C) Control LMNA +/+ cells. (B and D) LMNA -/- cells. Samples were poststained using the EDTA-regressive method to visualize IGCs (dark granular areas). No differences in the morphology of stained IGCs in lamin A/C +/+ and lamin A/C -/- cells were observed. (A and B) Low magnification. (C and D) High magnification of the same cells. Nu, nucleolar area. Bars, 2 μm (A and B); 500 nm (C and D).
Lamin 2H10 Antibody Recognizes SFCs in LMNA -/- Cells
Much of the evidence to support a role for lamin A in formation of SFCs comes from extensive colocalization studies by using anti-lamin A antibody 2H10 (LA-2H10) (Jagatheesan et al., 1999; Muralikrishna et al., 2001; Kumaran et al., 2002). To ask whether this antibody recognized proteins other than lamins A and C, we performed immunofluorescence microscopy on LMNA -/- cells (Figure 5A). In those cells, we found a similar staining pattern with LA-2H10 as in LMNA +/+ MEFs. We observed normally appearing speckled SFC patterns in both cell types, and all SFCs detected by LA-2H10 colocalized with endogenous SF2/ASF (Figure 5A). Because the LMNA -/- cells do not contain lamin A or C, these results strongly suggest that the 2H10 antibody recognizes at least one additional protein. To address this issue, we probed whole cell extracts from either LMNA +/+ or -/- MEFs with the LA-2H10 antibody by Western blot (Figure 5B). In +/+ MEFs, in addition to a band corresponding to the expected molecular weight of lamin A, we find several addition bands (Figure 5B). These bands persist in the cells lacking lamins A and C (Figure 5B). These results suggest that this antibody recognizes epitopes in addition to lamin A/C. Our efforts to identify these epitopes have been unsuccessful.
Figure 5.

Antibody LA-2H10 in LMNA -/- MEFs. (A) Indirect immunofluorescence microscopy by using LA-2H10 monoclonal antibody. Both in LMNA +/+ and -/- MEFs, the antibody recognizes splicing factor compartments and colocalizes with SF2/ASF. (B) Western blot analysis of LMNA +/+ and -/- MEFs with control anti-lamin antibody (sc-6215; left) and LA-2H10 (right). LA-2H10 recognizes multiple bands both in control LMNA +/+ and in -/- MEFs.
DISCUSSION
We have here directly tested the hypothesis that lamins A/C are major structural components of pre-mRNA splicing factor compartments. Based on observation in MEFs from mice in which the gene encoding both lamins A and C has been knocked out, we suggest that lamins A and C are not essential for formation or maintenance of SFCs. Several lines of evidence support this conclusion. First, three resident proteins belonging to three functionally distinct classes of SFC residents localized normally in SFCs in the absence of lamins A/C. Second, in situ extraction and FRAP experiments demonstrate that the association and nucleoplasmic exchange of a pre-mRNA splicing factors in fixed and living cells were not affected by loss of lamins A/C. Third, the positional stability and general dynamic properties of SFCs was not altered in the absence of lamins A/C. Fourth, no changes in ultrastructure were present in IGCs in lamin A/C -/- cells. These results are unlikely due to the presence of remnant lamin A/C because by Western blot, Northern analysis, and immunofluorescence microscopy, no detectable levels of lamin A or C protein or mRNA can be detected in LMNA -/- cells (Sullivan et al., 1999). Furthermore, nuclei from LMNA -/- mice are more difficult to isolate and have distinct mechanical properties (Lammerding et al., 2004), suggesting architectural defects of nuclei in these cells presumably directly or indirect due to the loss of functional lamin A/C.
Although our experiments strongly suggest that lamins A/C are not essential for SFC morphology, they do not rule out a nonessential contribution of lamin A to speckle morphology. It remains to be determined what this role might be. Regardless of the putative function, the lamin A pool in SFCs is likely relatively small because no accumulation of lamin A or C tagged with green fluorescent protein (GFP) or epitope tags can be seen (Kruhlak et al., 2000; Broers et al., 1999). Our experiments do not address the role B-type lamins might play in SFC morphology. Simultaneous disruption of A- and B-type lamins has been shown to result in reorganization of SFCs (Spann et al., 2002), and it is thus possible that lamin B is the major contributor to SFC morphology. Our attempts to knock out lamin B in LMNA -/- cells invariably resulted in cell death, consistent with the notion that lamin B is an essential gene (Harborth et al., 2001). Although it is possible that B-type lamins might compensate for the loss of A-type lamins and rescue the loss of lamins A and C to maintain SFC morphology in LMNA -/- cells, we think this possibility is unlikely because lamin B is not upregulated in cells from LMNA -/- mice (Sullivan et al., 1999). Furthermore, loss of lamins A/C results in clear morphological alternations of the nuclear envelope, which are not rescued by B-type lamins (Sullivan et al., 1999). Regardless of whether lamin B rescues a potential structural role of lamin A and C in SFC morphology, the function of lamin A is clearly not essential or necessary.
It is not immediately obvious how the discrepancy between our observations and the body of work implicating lamin A in SFC morphology is explained. It is worth pointing out that much of the experimental data in support of a role for lamin A in SFC morphology is based on the use of antibody LA-2H10. Because the antibody in question fails to detect lamin A in its typical peripheral localization (Jagatheesan et al., 1999), it is possible that it recognizes other nuclear proteins as well, although the absence of staining could be rationalized by recognition of an epitope in lamin A that is specifically exposed in SFCs. Our observation of identical staining patterns in cells with and without lamin A/C and the detection, in our hands, of multiple bands both in LMNA +/+ and LMNA -/- cells, may suggest that the antibody cross-reacts with nonlamin epitopes. Regardless, our results rule out lamins A and C as prominent structural components of SFCs and further experiments are required to find potential structural components of this and other nuclear compartments. The identification of these molecules will be an important step toward understanding nuclear architecture.
Acknowledgments
We thank Dr. V. Parnaik for providing LA-2H10 antibody, Drs. Tatiana Karpova, Jim McNally, Zuzana Cvac̆ková, Alena Loužecká, Lucie Tomšíková and Simona Ryšavá for technical assistance. Light microscopy was performed at the National Cancer Institute Fluorescence Imaging Facility. We acknowledge Jackson Immunoresearch Laboratories for donation of reagents. This work was supported by grants IAA5039103, MSM111100003, AVOZ 5039906, 304/03/1121, 304/02/0342 and 304/04/0692 to I.R. and by a Traveling Fellowship from the Journal of Cell Science to J.V. T.M. is a Fellow of the Keith R. Porter Endowment for Cell Biology..
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E04–07–0645. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E04–07–0645.
References
- Bernhard, W. (1969). A new staining procedure for electron microscopical cytology. J. Ultrastruct. Res. 27, 250-265. [DOI] [PubMed] [Google Scholar]
- Bridger J.M., I.R. Kill, M. O.`Farrel, and C.J. Hutchinson. (1993). Internal lamin structures within G1 nuclei of dermal fibroblasts. J. Cell Sci. 104, 297-306. [DOI] [PubMed] [Google Scholar]
- Broers, J.L., Machiels, B.M., van Eys, G.J., Kuijpers, H.J., Manders, E.M., van Driel, R., and Ramaekers, F.C. (1999). Dynamics of the nuclear lamina as monitored by GFP-tagged A-type lamins. J. Cell Sci. 112, 3463-3475. [DOI] [PubMed] [Google Scholar]
- Cohen, M., Lee, K.K., Wilson, K.L., and Gruenbaum, Y. (2001). Transcriptional repression, apoptosis, human disease and the functional evolution of the nuclear lamina. Trends Biochem. Sci. 26, 41-47. [DOI] [PubMed] [Google Scholar]
- Dundr, M., and Misteli, T. (2001). Functional architecture in the cell nucleus. Biochem. J. 356, 297-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett, D.W. (1966). On the occurrence of a fibrous lamina on the inner aspect of the nuclear envelope in certain cells of vertebrates. Am. J. Anat. 119, 129-145. [DOI] [PubMed] [Google Scholar]
- Fakan, S., Leser, G., and Martin, T.E. (1984). Ultrastructural distribution of nuclear ribonucleoproteins as visualized by immunocytochemistry on thin sections. J. Cell Biol. 98, 358-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakan, S. (1994). Perichromatin fibrils are in situ forms of nascent transcripts. Trends Cell Biol. 4, 86-90. [DOI] [PubMed] [Google Scholar]
- Glass, J.R., and Gerace, L. (1990). Lamins A and C bind and assemble at the surface of mitotic chromosomes. J. Cell Biol. 111, 1047-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, A.E., Moir, R.D., Montag, L.M., Stewart, M., and Goldman, R.D. (1992). Pathway of incorporation of microinjected lamin A into the nuclear envelope. J. Cell Biol. 119, 725-735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, R.D., Gruenbaum, Y., Moir, R.D., Shumaker, D.K., and Spann, T.P. (2002). Nuclear lamins: building blocks of nuclear architecture. Genes Dev. 16, 533-547. [DOI] [PubMed] [Google Scholar]
- Harborth, J., Elbashir, S.M., Bechert, K., Tuschl, T., and Weber, K. (2001). Identification of essential genes in cultured mammalian cells using small interfering RNAs. J. Cell Sci. 114, 4557-4565. [DOI] [PubMed] [Google Scholar]
- Hozák, P., Sasseville, A.M., Raymond, Y., and Cook, P.R. (1995). Lamin proteins form an internal nucleoskeleton as well as a peripheral lamina in human cells. J. Cell Sci. 108, 635-644. [DOI] [PubMed] [Google Scholar]
- Huang, S., and Spector, D.L. (1996). Intron-dependent recruitment of pre-mRNA splicing factors to sites of transcription. J. Cell Biol. 133, 719-732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagatheesan, G., Thanumalayan, S., Muralikrishna, B., Rangaraj, N., Karande, A.A., and Parnaik, V.K. (1999). Colocalization of intranuclear lamin foci with RNA splicing factors. J. Cell Sci. 112, 4651-4661. [DOI] [PubMed] [Google Scholar]
- Jiménez-García, L.F., and Spector, D.L. (1993). In vivo evidence that transcription and splicing are coordinated by a recruiting mechanism. Cell 73, 47-59. [DOI] [PubMed] [Google Scholar]
- Johnson, C., Primorac, D., McKinstry, M., McNeil, J., Rowe, D., and Lawrence, J.B. (2000). Tracking COL1A1 RNA in osteogenesis imperfecta splice-defective transcripts initiate transport from the gene but are retained within the SC35 domain. J. Cell Biol. 150, 417-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krainer, A.R., Conway, G.C., and Kozak, D. (1990). Purification and characterization of pre-mRNA splicing factor SF2 from HeLa cells. Genes Dev. 4, 1158-1171. [DOI] [PubMed] [Google Scholar]
- Kruhlak, M.J., Lever, M.A., Fischle, W., Verdin, E., Bazett-Jones, D.P., and Hendzel, M.J. (2000). Reduced mobility of the alternate splicing factor (ASF) through the nucleoplasm and steady state speckle compartments. J. Cell Biol. 150, 41-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaran, R.I., Muralikrishna, B., and Parnaik, V.K. (2002). Lamin A/C speckles mediate spatial organization of splicing factor compartments and RNA polymerase II transcription. J. Cell Biol. 159, 783-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli, U.K., Molbert, E., Showe, M., and Kellenberger, E. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680-685. [DOI] [PubMed] [Google Scholar]
- Lammerding, J., Schulze, P.C., Takahashi, T., Kozlov, S., Sullivan, T., Kamm, R.D., Stewart, C.L., and Lee, R.T. (2004). Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Investig. 113, 370-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melčák, I., Cermanová, S., Jirsová, K., Koberna, K., Malínský, J., and Raška, I. (2000). Nuclear pre-mRNA compartmentalization: trafficking of released transcripts to splicing factor reservoirs. Mol. Biol. Cell 11, 497-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli, T., Cáceres, J.F., and Spector, D.L. (1997). The dynamics of a pre-mRNA splicing factor in living cells. Nature 387, 523-527. [DOI] [PubMed] [Google Scholar]
- Misteli, T. (2000). Cell biology of transcription and pre-mRNA splicing: nuclear architecture meets nuclear function. J. Cell Sci. 113, 1841-1849. [DOI] [PubMed] [Google Scholar]
- Mintz, P.J., Patterson, S.D., Neuwald, A.F., Spahr, C.S., and Spector, D.L. (1999). Purification and biochemical characterization of interchromatin granule clusters. EMBO J. 18, 4308-4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir, R.D., Montag-Lowy, M., and Goldman, R.D. (1994). Dynamic properties of nuclear lamins: lamin B is associated with sites of DNA replication. J. Cell Biol. 125, 1201-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir, R.D., Yoon, M., Khuon, S., and Goldman, R.D. (2000). Nuclear lamins A and B1: different pathways of assembly during nuclear envelope formation in living cells. J. Cell Biol. 151, 1155-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monneron, A., and Bernhard, W. (1969). Fine structural organization of the interphase nucleus in some mammalian cells. J. Ultrastruct. Res. 27, 266-288. [DOI] [PubMed] [Google Scholar]
- Muralikrishna, B., Dhawan, J., Rangaraj, N., and Parnaik, V.K. (2001). Distinct changes in intranuclear lamin A/C organization during myoblast differentiation. J. Cell Sci. 114, 4001-4011. [DOI] [PubMed] [Google Scholar]
- Phair, R.D., and Misteli, T. (2000). High mobility of proteins in the mammalian cell nucleus. Nature 404, 604-609. [DOI] [PubMed] [Google Scholar]
- Spann, T.P., Goldman, A.E., Wang, C., Huang, S., and Goldman, R.D. (2002). Alteration of nuclear lamin organization inhibits RNA polymerase II-dependent transcription. J. Cell Biol. 156, 603-608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector, D.L., Schrier, W.H., and Busch, H. (1983). Immunoelectron microscopic localization of snRNPs. Biol. Cell 49, 1-10. [DOI] [PubMed] [Google Scholar]
- Spector, D.L. (1993). Nuclear organization of pre-mRNA processing. Curr. Opin. Cell Biol. 5, 442-447. [DOI] [PubMed] [Google Scholar]
- Sullivan, T., Escalante-Alcalde, D., Bhatt, H., Anver, M., Bhat, N., Nagashima, K., Stewart, C.L., and Burke, B. (1999). Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 147, 913-920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlcek, S., Dechat, T., and Foisner, R. (2001). Nuclear envelope and nuclear matrix: interactions and dynamics. Cell Mol. Life Sci. 58, 1758-1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will, C.L., and Lührmann, R. (1997). snRNP structure and function. In: Eucaryotic mRNA Processing, ed. A.R. Krainer, New York, NY: Oxford University Press, 130-173.