

Abstract
Periodontal diseases are a major public health concern leading to tooth loss and also shown to be associated with several chronic systemic diseases. Smoking is a major risk factor for developing numerous systemic diseases, as well as periodontitis. While it is clear that smokers have a significantly enhanced risk for developing periodontitis leading to tooth loss, the population varies with regards to susceptibility to disease associated with smoking. This investigation focuses on identifying differences in four broad sets of variables consisting of: (a) host response molecules, (b) periodontal clinical parameters, (c) antibody measures for periodontal pathogens and oral commensal bacteria challenge, and (d) other variables of interest in a smoking population with (n = 171) and without periodontitis (n = 117). Subsequently, Bayesian network structured learning techniques (BNSL) techniques were used to investigate potential associations and cross-talk between the four broad sets of variables. BNSL revealed two broad communities with markedly different topology between the non-periodontitis and periodontitis smoking population. Confidence of the edges in the resulting network also showed marked variations within and between the periodontitis and non-periodontitis groups. The results presented validated known associations, as well as discovered new ones with minimal precedence that may warrant further investigation and novel hypothesis generation. Cross-talk between the clinical variables and antibody profiles of bacteria were especially pronounced in the case of periodontitis and mediated by the antibody response profile to P. gingivalis.
Keywords: Bioinformatics, Smoking, Periodontal disease, Host response, Inflammation
INTRODUCTION
Periodontal diseases are a major public health concern leading to tooth loss and associated chronic systemic diseases (1–7). Bacterial-induced periodontal diseases have been categorized into two general types related to connective tissue attachment loss and alveolar bone resorption, ie. gingivitis and periodontitis. Gingivitis is a reversible inflammation of the gum tissue (i.e., gingiva) caused by the presence of a biofilm that forms on the tooth surface and resolves rather quickly after the reinstitution of mechanical and chemical oral hygiene procedures. Most individuals will experience at least mild and transient gingivitis at some time in their life (8–10). Periodontitis characterized by persistent gingival inflammation, breakdown of the connective tissue (i.e., attachment apparatus surrounding teeth), and destruction of alveolar bone (11, 12) with epidemiologic data supporting the concept of differential susceptibility to periodontitis regarding onset, rate of progression, and severity across the population (13, 14).
Smoking is a major risk factor for developing numerous systemic diseases, as well as periodontitis (15–18). Smokers frequently present with lower levels of gingival bleeding than would be predicted based upon the level of tissue destruction of the periodontium (19) This is likely due to effects of the smoke derived xenobiotics on local vascular functions (19, 20), while tobacco smoke appears to amplify the inflammatory response to the microbial challenge (21–23). A report using NHANES III data determined that a population attributable risk (PAR) for current or former smoking was approximately 50% for exhibiting periodontitis (24). However, while it is clear that smokers have a significantly enhanced risk for developing periodontitis leading to tooth loss, the population varies with regards to susceptibility to disease associated with smoking (25, 26).
The oral ecology in an individual evolves over time with variations in quantity and quality of phyla, genera and species (27), as well as the genomic profile of the individual species (28–30). However, this evolution generally leads to an equilibrium between the microbiota and the individual’s oral environment, as a climax community. The resulting microbial communities or biofilms are complex ecosystems of bacteria that are somewhat unique to various ecological niches in the oral cavity (31). The microbiomes of the subgingival environment of periodontally healthy and periodontally diseased sites are quite distinct (27, 32). The accretion of tooth-associated bacterial biofilms elicits gingival inflammation as a result of bacterial virulence and metabolic factors affecting tissue vasculature. In sites colonized by more pathogenic biofilms, the inflammatory response results in destruction of connective tissue and alveolar bone, the classic features of periodontitis. Various extrinsic environmental factors can also affect the microbial composition in the oral cavity, as well as host response patterns. Recent studies have demonstrated clear effects of smoking on the composition of the microbiome at sites of periodontitis and peri-implantitis (33–36).
The accumulation of these biofilms also elicits a robust inflammatory response with the cellular infiltrate releasing a panoply of pro-inflammatory molecules that initiate clinical inflammatory measures of gingival redness and edema (14, 37, 38). Numerous studies of gingival crevicular fluid at inflamed sites have identified associated biomolecules, such as IL-1β, PGE2, IL-10, and acute phase response proteins, including plasminogen activator inhibitor-1 (PAI-1) and myeloperoxidase (MPO), which can contribute to local antibacterial responses (39). The current paradigm in periodontal disease is a local chronic infection with microbial dysbiosis (40) that triggers a persistent destructive inflammatory response, rather than direct toxic/noxious actions of the bacterial virulence factors (41). Beyond innate immune and inflammatory responses, substantial literature documents the production of specific local and systemic antibodies to oral bacteria (38). Bacterial species-specific antibody levels increase significantly with periodontitis and decrease following therapy (14, 38). Additionally, various studies have demonstrated alterations in the characteristics of the induced antibody in the presence of smoking (42–44). However, there is rather limited data regarding these adaptive immune responses in smokers presenting with a range of periodontal health and periodontal diseases.
The present study investigates changes in magnitude of four broad sets of variables consisting of: (a) host response molecules, (b) periodontal clinical parameters, (c) antibody measures of periodontal pathogens and oral commensal bacteria and (d) other variables of interest related to smoking population with (n = 171) and without periodontitis (n = 117). Subsequently, it investigates the potential associations and cross-talk between these variables using Bayesian network structured learning techniques (BNSL). BNSL is a probabilistic approach and widely used (45–47) to model direct as well as conditional dependencies between a given set of variables. In contrast to traditional statistical modeling, BNSL does not impose a pre-defined relationship between the given set of variables. It rather aims to discover these relationships from the data generated across the variables and abstract them as directed acyclic graphs (DAGs). The resulting DAGs can validate established associations as well as discover novel undocumented associations providing a more informed basis for hypothesis generation in addition to hypothesis testing. More importantly, a break down in associations across disease states is accompanied by marked changes in the topology or wiring pattern (48–50) of the DAGs as shown in this study. DAGs also serve as useful system-level abstractions of the concerted working of the variables that may not be readily apparent by investigating the variables in isolation. While BNSL has the potential to reveal causal relationships there are definite limitations to this approach. The DAG representation of the relationships between the variables in BNSL implicitly assumes absence of feedback between them. Such an assumption need not necessarily be true in general. Also, the number of potential structures in BNSL increases markedly with the number of variables discouraging an exhaustive search for all potential structures especially when the number of variables is large. PDAGs returned by BNSL essentially represents multiple structures that are probabilistically equivalent (equivalence class) as opposed to a single structure and may result in graphs comprising of directed as well as undirected edges. Thus BNSL can learn the associations between the variables only up to the equivalence class irrespective of the choice of the sample size. While directed edges may indicate potential causal relationships, undirected edges provide no such insights. However, it is important to note that causal associations can be determined ideally only through active interventions. Thus associations returned by BNSL need to be validated experimentally using appropriate interventions.
MATERIALS and METHODS
Patient Population and Clinical Parameters
The cohort included 30 periodontally healthy subjects (M:F – 7:23), 55 gingivitis patients (M:F – 14:41), and 184 periodontitis patients (M:F – 73:111) ages 21–65 years. The protocol for this study was approved by the University of Kentucky Institutional Review Board and all participants signed an appropriate consent form. A comprehensive oral and periodontal examination was completed to assess the periodontal health. Inclusion/exclusion criteria for participating in the study: must be smokers, able to complete a questionnaire and sign a consent form, have a minimum of 20 teeth, willing to have blood drawn, whole saliva collected, and have a full periodontal evaluation.
Three clinical parameters routinely used for periodontal evaluation: (a) mean probing pocket depth (PPD), (b) clinical attachment level (51), and (c) bleeding on probing (BOP) were obtained from all patients. Full-mouth mean pocket depth and attachment level measured in millimeters (mm), and bleeding on probing, measured by percentage of sites in the mouth that bleed were determined at 6 sites per tooth excluding third molars (52). The measurements were taken and recorded by a single examiner. Measures of BOP and PPD were used to categorize the patients recognizing that various patients fell into “gray” areas. These “gray” area patients were assigned to a category and simply contributed to the variation within the different groups. We used a mean PPD ≤2.5 mm for non-periodontitis (ie. Health and Gingivitis) and ≥2.5 mm for periodontitis. While all Gingivitis subjects were categorized accurately (>20% sites BOP; mean PPD ≤2.5 mm), a subset of healthy subjects (26) had mean PD ≥2.5 mm, but limited % BOP and no sites with >4 mm pocket depths. Two of the periodontitis subjects had mean PPD ≤2.5 mm, but these subjects also had high % BOP and numerous sites with >4 mm pocket depths.
Variables such as age (Age), pack years of smoking (Yrs), salivary cotinine (Cot) levels, and body mass index (BMI) that have been traditionally investigated in periodontitis and smoking population studies were also included in the analysis.
Serum Analyses
Serum from a venipuncture blood sample was originally obtained from a group of 301 smokers (age 21–65, 82 black, 219 white; 106 males, 195 females). After preliminary investigation and eliminating missing values the subjects were distributed to non-periodontitis (NP; n=117) and periodontitis (PD; n=171) groups. The serum samples were stored at −80°C until the assays were performed. An array of oral microorganisms were used in the assays, cultivated under standard conditions, and prepared for antigens as described previously (53). The bacteria included periodontal pathogens: Aggregatibacter actinomycetemcomitans (Aa) strain JP2, Porphyromonas gingivalis (Pg) ATCC 33277, Treponema denticola (Td) ATCC 35405, and a group of oral commensal bacteria that included Streptococcus sanguinis (Ss) ATCC 10556, Actinomyces naeslundii (An) ATCC 49340, Veillonella parvula (Vp) ATCC 10790, Capnocytophaga ochracea (Co) ATCC 33596. An ELISA was used to determine the level of IgG antibody to the bacteria (52). Purified human IgG was bound to the plate to produce a standard curve. Sample data was extrapolated from this curve, using a four parameter logistic curve fit (54).
Molecular markers of inflammatory responses in serum, including interleukin-1β (IL-1β), plasminogen activator inhibitor-1 (PAI-1), myeloperoxidase (MPO), and IL-10 were evaluated using Luminex beadlyte technology (EMD Millipore, Billerica, MA) or commercial high sensitivity PGE2 ELISA ELISA kits (Assay Design, Ann Arbor, MI) (42).
Salivary cotinine levels were measured in whole saliva using an ELISA (Salimetrics’ High Sensitivity Salivary Cotinine Quantitative enzyme immunoassay kit, Carlsbad, CA) as we have described previously (43).
Statistical Analyses
Parametric t-test was used to identify those variables whose magnitude changed significantly across non-periodontitis and periodontitis groups after controlling the false-discovery rate (55). Variables whose adjusted p-values were significant (p < 0.02) are reported. Subsequently, BNSL was used to model potential associations between 15 variables comprising critical proteins (IL-1β, PAI-1), clinical parameters (BOP, PD, CAL), antibody to periodontal pathogens (Aa, Pg, Td) and oral commensal bacteria (Co, An, Vp, Ss), and other variables (Age, Yrs, Cot) across periodontitis and non-periodontitis subjects in the smoking population. Several techniques, such as constraint-based and search-score approaches, have been traditionally proposed in the literature for BNSL (56–58). Recent studies have shown that hybrid techniques such as max-min hill climbing (MMHC) that use a combination of constraint-based and search-score frameworks in tandem to outperform either of these approaches (59). Constraint-based part of MMHC uses max-min parents-children approach to determine the skeleton of the underlying network. Subsequently, a Bayesian-greedy hill-climbing approach is used to determine the orientation of the edges in the skeleton. Partially directed acyclic graph (PDAG) representations that incorporate equivalence classes of the DAGs were subsequently generated (56, 57) in order to account for DAGs are probabilistically indistinguishable. Confidence of the edges (60, 61) in the PDAGs was determined from 1000 independently by resampling with replacement and edges with confidence less than 30% were deemed noisy, hence eliminated from further consideration. The remaining edges along with their confidences are reported as weighted graphs. Open-source implementation of the MMHC approach as a part of the bnlearn R package (62, 63) was used in the present study. Graphical layouts were generated using Gephi (64).
RESULTS
Adjusted p-values (p < 0.02) from a parametric t-test after controlling the false-discovery rate (55) revealed eight variables to exhibit a marked difference between the periodontitis (PD) and non-periodontitis (NP) groups (Table 1). As expected, the three clinical parameters (BOP, PPD, CAL) were significantly elevated in the periodontitis group as opposed to the NP group. As the groups were defined by clinical parameters, it was expected that these would be significantly different. Of the remaining variables, IL-1β, PAI-1, pack-years of smoking, and antibody to P. gingivalis and A. naeslundii were also significantly elevated in the periodontitis group. Since previous results had not evaluated variations in host responses across clinical presentation in smokers, alterations in targeted inflammatory mediators and antibody levels, particularly to P. gingivalis, were undefined previously, but not totally unexpected.
Table 1.
Average values and standard deviations of the 18 variables across between periodontitis and non-periodontitis smoking subjects.
| Variable | Non-Periodontitis | Periodontitis |
|---|---|---|
| Clinical/Demographic | ||
| Age (years) | 36.9±11.1 | 39.9±9.5 |
| BMI | 27.3±5.9 | 28.7±7.2 |
| BOP** (% >0) | 4.7±4.8 | 25.8±22 |
| PPD** (mouth mean mm) | 2.4±0.30 | 3.4±0.69 |
| CAL** (mouth mean mm) | 2.6±0.51 | 3.8±1 |
| Smoking | ||
| Pack/years** | 17.2±10 | 20.7±9.5 |
| Salivary cotinine (ng/mL) | 529.2±456.5 | 649.6±616.4 |
| Inflammatory Mediators | ||
| IL-1β** (pg/mL) | 39.1±56.7 | 79.6±99.8 |
| PAI-1** (ng/mL) | 110.7±139.3 | 160.4±157.5 |
| MPO (ng/mL) | 13056.5±7316.8 | 15238±9316.8 |
| PGE2 (ng/mL) | 531.5±818.6 | 472.9±709.8 |
| Serum Antibody (ng/mL) | ||
| A. actinomycetemcomitans | 15.3±12.9 | 18±13.6 |
| A. naeslundii** | 15.8±9.8 | 19.1±10.2 |
| C. ochracia | 10.2±5.6 | 11.7±7.3 |
| P. gingivalis** | 29±22 | 42.1±32 |
| S. sanguinis | 39.5±19.3 | 38.1±16.2 |
| T. denticola | 14.4±10.6 | 17.2±12.4 |
| V. parvula | 21.8±14.7 | 22±12.4 |
adjusted p-value < 0.02
Subsequently, Max-Min hill climbing approach was used to generate the Bayesian networks representing the interplay between 15 clinical and host response variables across the NP and PD smoking population. Confidence of the edges in the network was generated across 1000 independent bootstrap realizations. The five sets of variables are shown by distinct colors in Figs.1a–1b for clarity and the thickness of the edges is proportional to their confidence represented in these figures. Edges whose confidences were <30% were deemed noisy, hence not reported. The networks corresponding to NP and PD exhibit two broad communities revealing intricate associations and cross-talk between (clinical parameters, other variables, Community 1) and (antibacterial responses, Community 2). However, there were substantial differences in the wiring pattern between them. Confidence of the edges showed marked variations within and between the NP and PD groups. While the network corresponding to NP is disconnected (Fig. 1a) that of the periodontitis patients is weakly connected (i.e. it is possible to traverse between any two variables in the underlying undirected graph) (Fig. 1b).
Figure 1.
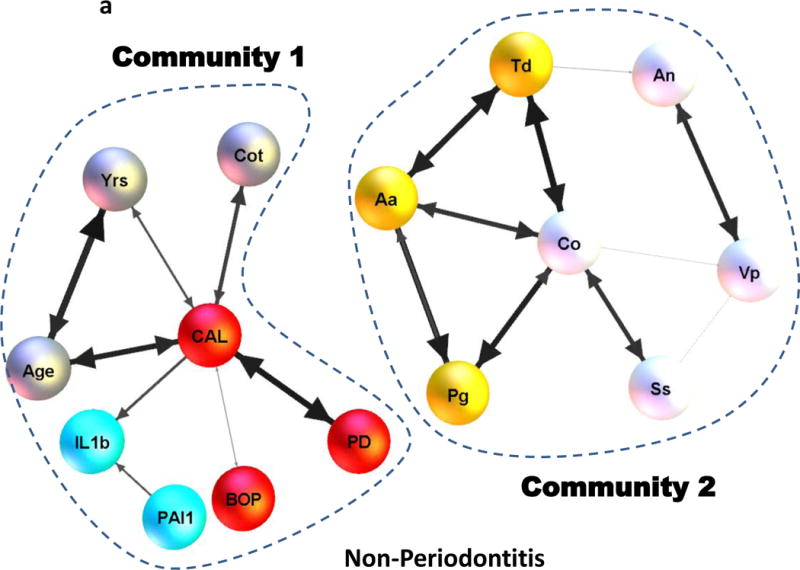
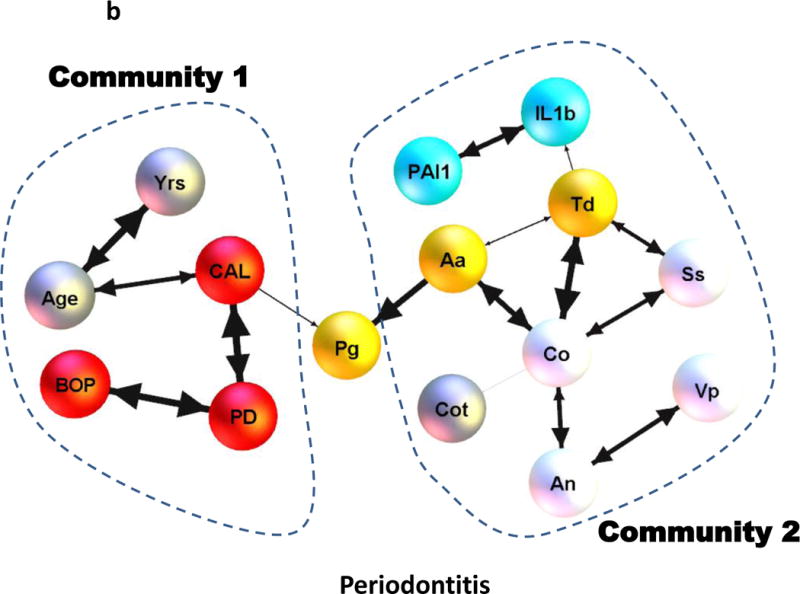
Bayesian network representing potential associations between four broad sets of variables consisting of: (i) host response molecules (IL1β, PAI-1), (ii) periodontal clinical parameters (PD, BOP, CAL), (iii) antibody measures of periodontal pathogens (Aa, Pg, Td) and oral commensal bacteria (Co, Vp, An, Ss) and other variables (Age, Yrs, Cot) of interest in the non-periodontitis and the periodontitis smoking population is shown in (a) and (b) respectively. The four broad sets of variables are shown by different colors for clarity with the dotted lines representing the two broad communities in (a) and (b). The thickness of the edges in (a) and (b) are directly proportional to the edge confidence estimated from (N = 1000) bootstrap realizations.
More importantly, in the case of periodontitis, antibody to P. gingivalis seems to act as a mediator establishing an association between otherwise unrelated variables (eg. CAL and Aa) and in turn establishing potential associations between the parameters of Community 1 and 2. Unlike the PD group, there were no apparent relationships between BOP and mean pocket depth (PPD) in the case of the NP group. Scatter plots of (%BOP vs. PPD) also validated these findings with the absence of a significant direct association in the NP group (r=−0.15) as opposed to the PD group (r=0.58, p<0.01) (Fig. 2a). Our analysis also revealed some of the associations to be preserved across the NP and periodontitis groups. This includes the correlation between antibody to the pathogen (Aa) and commensal bacterium (Co) being reflected by prominent edges in Figs. 1a–1b. Aa and Co also exhibited a significant direct association across NP (r=0.58, p<0.01) as well as the PD (r=0.49, p<0.01) group (Fig. 2a). Associations between (Co and Td) were significant across NP (r=0.67, p<0.01) and PD (r=0.61, p<0.01) groups. Associations between (Co and Pg) were also significant across NP (r=0.63, p<0.01) and PD (r=0.39, p<0.01) groups (Fig. 2b).
Figure 2.
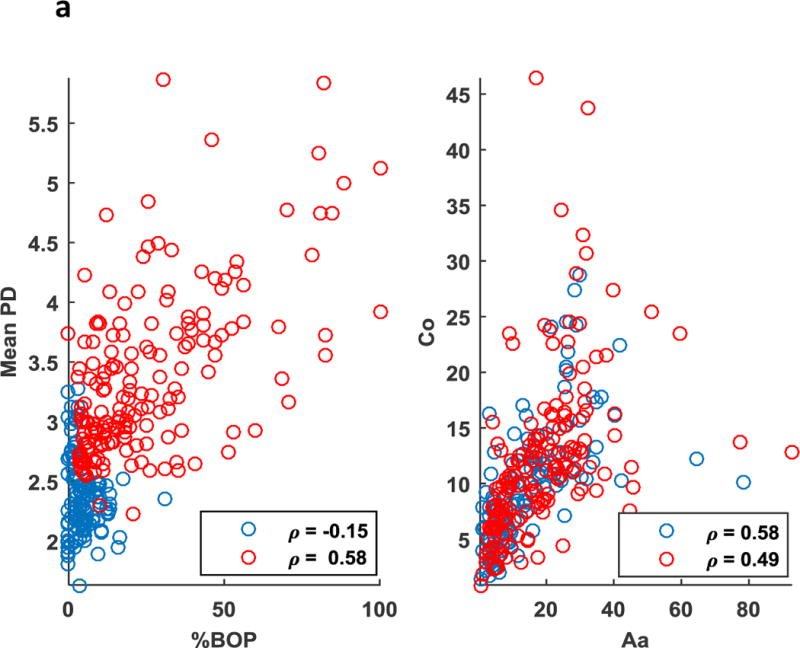
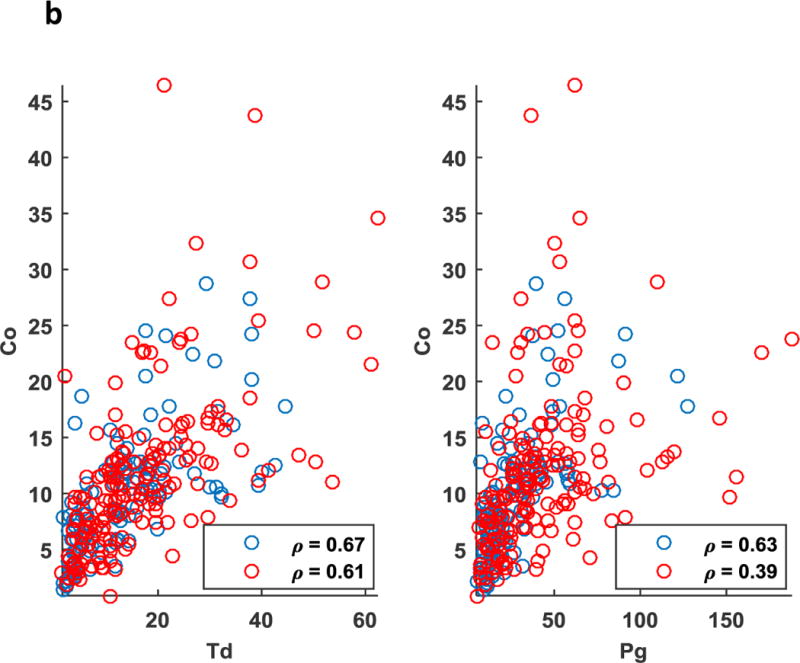
(a) Scatter plot representing direct association between clinical parameters (Mean BOP) and (Mean PPD) was significant in the periodontitis (red, p<0.01) as opposed to the non-periodontitis (blue) smoking population. Association between antibody to a periodontal pathogen (Aa) and a commensal bacteria (Co) was significant in periodontitis (red, p<0.01), as well as the non-periodontitis (blue, p<0.01) smoking population. (b) Scatter plot representing direct association between antibody to a periodontal pathogens (Td and Pg) and a commensal bacteria (Co) was significant in periodontitis (red, p<0.01), as well as the non-periodontitis (blue, p<0.01) smoking population.
Examining the associations of systemic inflammatory mediators to the clinical parameters in the population of smokers also provided an interesting profile. While numerous inflammatory biomarkers have been identified in serum and related to the extent and severity of periodontitis (23, 38, 65), the findings of this study in smokers provide a somewhat different outcome. Confidence of the edges corresponding to (IL-1β, PAI-1) was markedly higher in the periodontitis group as opposed to the non-periodontitis group, reflected by the discrepancy in the thickness of the edges in Fig. 1a–1b. This was also reflected by the marked decrease in the magnitude of the Pearson correlation between (IL–1β, PAI-1) from the PD group (r=0.34, p<0.01) compared to the NP group (r=0.20, p=0.03). Also, IL-1β was associated with the clinical parameter (mean CAL) in the case of NP (Fig. 1a), whereas it was connected to the host response variables through linkage with antibody to the pathogen (T. denticola) in the case of PD (Fig. 1b). Also, the markers (IL-1β, PAI-1) did not exhibit a direct association to clinical disease parameters or smoking measures in the PD group. These results suggest that smoking may disrupt control of local and systemic inflammatory responses that occur related to the health of the periodontium. Active smoking (i.e. salivary cotinine levels) exhibited a strong association with the clinical parameter (CAL, p<0.01) in the NP group (Fig. 1a), unlike that of the PD group. While obesity has been reported to be linked to expression of periodontitis in humans (66–70), our data from a population of individuals who were all smokers showed that BMI did not exhibit a significant association to any of the clinical parameters, inflammatory responses, or specific antibody in either PD or NP individuals, hence not included in Figs. 1a–1b. These observations may indicate that certain biologic effects of smoking could overwhelm any contribution that modifiers elicited by overweight/obesity might have in altering the presentation of this oral disease. MPO and PGE2 exhibited a significant association across NP (r=0.50, p<0.01) and the PD (r=0.37, p<0.01) groups solely based on the correlation coefficients and associated p-values. However, as can be seen with the scatter plots (Fig. 3) the relationship is highly noisy, suggesting that the correlations may primarily reflect outlier values of a few subjects in the groups and hence are not identified as important contributors to the community relationships identified in Fig. 1a–1b. Additional variables demonstrated minimal or no relationship to these community linkages and are also not presented in the figure.
Figure 3.
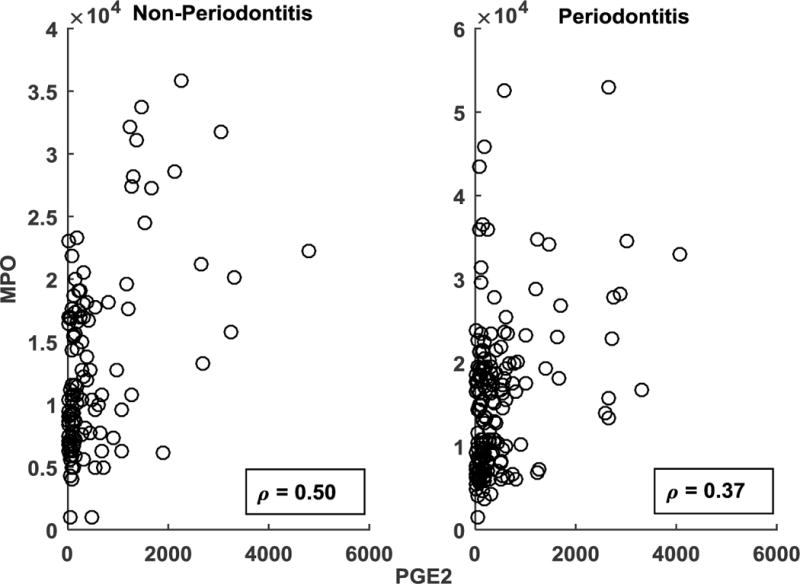
Scatter plot representing potential association between serum levels of MPO and PGE2 for the non-periodontitis and periodontitis smoking population. The correlation is rendered significant (p < 0.01) by a few outliers in each of the case.
DISCUSSION
Smoking is one of, if not the major cause, of preventable morbidity and mortality in the population with substantial negative effects on general and oral health (16–18, 71). Smoking has been clearly delineated as a major modifiable environmental risk factor in the extent/severity of periodontal disease through cross-sectional and longitudinal epidemiologic investigations (72). These data indicate that there is a 300–600% increase in periodontitis in smokers compared with non-smokers (73), and there clearly is a deleterious impact of the wide array of xenobiotics derived from smoking on the response to periodontal therapy (16, 74). Nevertheless, while a significantly enhanced proportion of smokers exhibit periodontitis compared to the non-smoking population, clinical evaluation of smoking and periodontitis demonstrate a proportion of the smoking population remain periodontally healthy or only develop gingivitis (75). Another facet of the broader literature was that a subset of smokers appeared periodontally healthy even with long term smoking (ie. pack years) and substantial continine levels. Thus, some features of either the microbial challenge or characteristics of the host response profile appear to afford these patients an enhanced capacity to retain tissue homeostasis in the oral cavity. Similar observations have been summarized in a recent report by Genco and Borgnakke (69).
We have previously demonstrated serum antibody levels to selected oral pathogens and commensal bacteria related to race/ethnicity, age, and periodontal disease extent in a population of smokers (43), as well as systemic inflammatory responses related to smoking level (42). This study extended these findings by examining both serum antibodies and inflammatory molecules related to smoking parameters, and periodontal condition to identify the impact of active smoking on the relationships among these various parameters. Serum levels of IL-1β were significantly elevated in the periodontitis patients. IL-1β has been identified to be a crucial molecule in chronic inflammatory responses, both locally and systemically (76). (77) (78). This pro-inflammatory mediator and bone resorbing cytokine does appear to contribute to the expression of periodontitis and has been linked to serum levels of C-reactive protein and altered IL-6 production appearing to play a role in periodontitis in type 2 diabetes mellitus (79). Our findings in smokers with periodontitis with the highest cotinine levels demonstrating elevated serum IL-1β provides additional data regarding the importance of this pro-inflammatory mediator in periodontitis.
A number of pro-inflammatory mediators are derived via the arachidonic acid pathway, including PGE2 (80), which has been implicated in the pathogenesis of gingival inflammation and periodontitis (81, 82). Additional biomarkers of inflammatory responses in the systemic circulation are components of the acute phase response, eg. PAI-1 (39). This array of molecules serves a range of functions with the goal of reestablishing systemic homeostasis following noxious challenge and have been related to periodontitis, periodontal therapy, and other chronic inflammatory diseases (83, 84) (85, 86) (87, 88) (83, 84). (89) (90), (91). Also, during the chronic inflammatory response reactive oxygen species (ROS) and free radicals (e.g., NO and NO2) are generated by neutrophils and macrophages, often through the action of myeloperoxidase (MPO) that is released into extracellular fluid at sites of inflammatory lesions (92, 93) and is affected by smoking (94) (95). Alterations in MPO have been identified to vary in periodontitis patients with/without treatment (86, 96, 97) (98) (86, 99).
Serum IgG antibody levels to a group of putative periodontal pathogens and oral commensal bacteria were also evaluated. As would be expected antibody to the periodontal pathogens were significantly elevated in the periodontitis smokers and significantly decreased in the periodontally healthy smokers. Substantial literature has demonstrated local and systemic adaptive immune responses to oral bacteria related to the presence, extent/severity, and therapeutic outcomes of periodontitis (38) (44) (51). We have found similar types of response differences in smokers related to the oral health of the individual; however, we extended the available literature by demonstrating a racial/ethnic contribution to these responses, as well as a difference in response profile to pathogenic compared to commensal oral bacteria (43). As importantly, the level of serum antibody to the oral bacteria has been shown to provide some reflection of the presence and burden of the particular microorganism within the oral cavity (100, 101). Thus, this measure is somewhat of a surrogate for the oral colonization, and could be suggested to reflect a challenge that the host sees as potentially deleterious and needs to be managed (100, 101). In this regard, the identification of antibody to A. actinomycetemcomitans interfacing with responses to P. gingivalis and T. denticola in the non-periodontitis smokers and also linked with clinical parameters in the periodontitis subset. Historically, periodontitis have been categorized as chronic adult and aggressive (102) and linked to microbiological characteristics with P. gingivalis and A. actinomycetemcomitans generally considered as representative of the major pathogens in these diseases (103) (38, 104). The majority of these studies intermixed both smokers and nonsmokers into their cohorts, and did often identify various clinical and biological differences related to smoking. However, our population comprised entirely of smokers presented a somewhat unique profile. In the non-periodontitis (NP) group antibody levels to P. gingivalis and A. actinomycetemcomitans were associated with antibody to an important commensal species, C. ochracea, albeit, the level of antibody to these bacteria were significantly lower than in the periodontitis group, as was expected (38, 43). Interestingly, in the periodontitis patients antibody to C. ochracea, generally reflecting microbial challenge was associated with the group of commensal bacteria and provided a strong link to responses to the oral pathogens, ie. Aa, Pg, Td. This host response identifying these members of the microbial ecology was also uniquely related to the clinical and smoking community variables via responses to P. gingivalis. Thus, in the periodontitis patients, where P. gingivalis represents a hallmark bacteria and has been proposed as a keystone pathogen, the relationships in this smoking population reflected the capacity of P. gingivalis to interface clinical disease with a range of host responses.
These differences in responses were also obvious in demonstrating significant correlations of serum PGE2, IL-1β, and PAI-1 with various smoking parameters. In contrast, the serum antibodies lacked this type of relationship. We subsequently examined the direct relationship between the individual serum inflammatory mediators and the adaptive immune responses in subsets of patients stratified by salivary cotinine levels. The results demonstrated that in the subsets with lower cotinine levels, antibodies to a number of the commensal bacteria were significantly negatively correlated with serum PGE2 and MPO levels. In contrast, in the high cotinine group, antibody levels to the pathogens, and particularly P. gingivalis were significantly positively correlated with some of the inflammatory mediators (42). Thus, while antibodies were not directly correlated with cotinine and smoking levels, there appeared to be some interface between the systemic inflammatory and adaptive responses related to the interactions of smoking and periodontal disease.
The present study investigated variations in the magnitude of expression of four broad categories of variables between periodontitis and non-periodontitis smoking population. Subsequently, potential cross-talk and associations between these variables were investigated using a BNSL. In contrast to traditional statistical modeling BNSL does not impose any constraint on the associations between the variables rather discovers direct as well as indirect associations from the given data. This aspect of BNSL is especially useful in hypothesis generation. BNSL accommodates all the variables simultaneously and has the ability to provide system-level insights and potential cross-talk between them, thus, BNSL is markedly different from traditional reductionist approaches that investigate only subsets of variables in isolation. More importantly, BNSL has the ability to validate established associations, such as P. gingivalis responses in periodontitis and the relationship of age and amount of smoking to clinical measure of periodontal disease. Additionally, this approach can help to ascertain novel associations with minimal precedence, such as the relationship of select inflammatory mediators with clinical presentation on non-periodontitis smokers, inflammatory mediators linked more strongly to the microbial challenge in periodontitis, the centrality of the response to C. ochracea with response profiles to both commensals and pathogens, and the apparent ability of P. gingivalis to provide a bridge in linking bacterial responses to clinical parameters of disease in the periodontitis subset of smokers. More importantly, the results revealed marked variation in associations, their confidence as well as the overall network topology between the periodontitis and non-periodontitis groups. Whether there are molecules in the xenobiotic mix of smoking products that trigger unique host response profiles, or this relationship reflects a general noxious challenge to host tissues and cells that results in some specificity for eliciting the inflammatory and adaptive responses remains to be determined. Thus, while periodontitis is considered to reflect a dysregulated host response to subgingival biofilms, these data suggest that smoking has the capacity to disrupt these responses to an even greater degree, thus increasing the risk for disease initiation/progression.
Acknowledgments
This work was supported by a USPHS grant from the NIH/NCRR P20 RR020145 and the services of the Center for Clinical and Translational Sciences at the University of Kentucky (NCRR and the National Center for Advancing Translational Sciences, NIH UL1 RR033173).
References
- 1.Seymour GJ, Ford PJ, Cullinan MP, Leishman S, Yamazaki K. Relationship between periodontal infections and systemic disease. Clinical microbiology and infection: the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2007;13(Suppl 4):3–10. doi: 10.1111/j.1469-0691.2007.01798.x. [DOI] [PubMed] [Google Scholar]
- 2.Belstrom D, Damgaard C, Nielsen CH, Holmstrup P. Does a causal relation between cardiovascular disease and periodontitis exist? Microbes and infection/Institut Pasteur. 2012;14:411–418. doi: 10.1016/j.micinf.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 3.Jimenez M, Krall EA, Garcia RI, Vokonas PS, Dietrich T. Periodontitis and incidence of cerebrovascular disease in men. Ann Neurol. 2009;66:505–512. doi: 10.1002/ana.21742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kinane D, Bouchard P, Group EoEWoP Periodontal diseases and health: Consensus Report of the Sixth European Workshop on Periodontology. Journal of clinical periodontology. 2008;35:333–337. doi: 10.1111/j.1600-051X.2008.01278.x. [DOI] [PubMed] [Google Scholar]
- 5.Preshaw PM, Alba AL, Herrera D, et al. Periodontitis and diabetes: a two-way relationship. Diabetologia. 2012;55:21–31. doi: 10.1007/s00125-011-2342-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bobetsis YA, Barros SP, Offenbacher S. Exploring the relationship between periodontal disease and pregnancy complications. J Am Dent Assoc. 2006;137(Suppl 2):7S–13S. doi: 10.14219/jada.archive.2006.0403. [DOI] [PubMed] [Google Scholar]
- 7.Noble JM, Borrell LN, Papapanou PN, Elkind MS, Scarmeas N, Wright CB. Periodontitis is associated with cognitive impairment among older adults: analysis of NHANES-III. J Neurol Neurosurg Psychiatry. 2009;80:1206–1211. doi: 10.1136/jnnp.2009.174029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loe H, Theilade E, Jensen SB. Experimental Gingivitis in Man. The Journal of periodontology. 1965;36:177–187. doi: 10.1902/jop.1965.36.3.177. [DOI] [PubMed] [Google Scholar]
- 9.Eke PI, Thornton-Evans G, Dye B, Genco R. Advances in surveillance of periodontitis: the Centers for Disease Control and prevention periodontal disease surveillance project. The Journal of periodontology. 2012;83:1337–1342. doi: 10.1902/jop.2012.110676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eke PI, Dye BA, Wei L, Thornton-Evans GO, Genco RJ, Cdc Periodontal Disease Surveillance workgroup: James Beck GDRP Prevalence of periodontitis in adults in the United States: 2009 and 2010. Journal of dental research. 2012;91:914–920. doi: 10.1177/0022034512457373. [DOI] [PubMed] [Google Scholar]
- 11.Zarco MF, Vess TJ, Ginsburg GS. The oral microbiome in health and disease and the potential impact on personalized dental medicine. Oral diseases. 2012;18:109–120. doi: 10.1111/j.1601-0825.2011.01851.x. [DOI] [PubMed] [Google Scholar]
- 12.Teles FR, Teles RP, Uzel NG, et al. Early microbial succession in redeveloping dental biofilms in periodontal health and disease. Journal of periodontal research. 2012;47:95–104. doi: 10.1111/j.1600-0765.2011.01409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cobb CM. Microbes, inflammation, scaling and root planing, and the periodontal condition. J Dent Hyg. 2008;82(Suppl 3):4–9. [PubMed] [Google Scholar]
- 14.Kinane DF, Bartold PM. Clinical relevance of the host responses of periodontitis. Periodontol 2000. 2007;43:278–293. doi: 10.1111/j.1600-0757.2006.00169.x. [DOI] [PubMed] [Google Scholar]
- 15.Van Dyke TE, Sheilesh D. Risk factors for periodontitis. J Int Acad Periodontol. 2005;7:3–7. [PMC free article] [PubMed] [Google Scholar]
- 16.Chambrone L, Preshaw PM, Rosa EF, et al. Effects of smoking cessation on the outcomes of non-surgical periodontal therapy: a systematic review and individual patient data meta-analysis. Journal of clinical periodontology. 2013;40:607–615. doi: 10.1111/jcpe.12106. [DOI] [PubMed] [Google Scholar]
- 17.Thomson WM, Sheiham A, Spencer AJ. Sociobehavioral aspects of periodontal disease. Periodontol 2000. 2012;60:54–63. doi: 10.1111/j.1600-0757.2011.00405.x. [DOI] [PubMed] [Google Scholar]
- 18.Filoche SK, Cornford E, Gaudie W, Wong M, Heasman P, Thomson WM. Smoking, chronic periodontitis and smoking cessation support: reviewing the role of dental professionals. The New Zealand dental journal. 2010;106:74–77. [PubMed] [Google Scholar]
- 19.Dietrich T, Bernimoulin JP, Glynn RJ. The effect of cigarette smoking on gingival bleeding. The Journal of periodontology. 2004;75:16–22. doi: 10.1902/jop.2004.75.1.16. [DOI] [PubMed] [Google Scholar]
- 20.Dietrich T, Hoffmann K. A comprehensive index for the modeling of smoking history in periodontal research. Journal of dental research. 2004;83:859–863. doi: 10.1177/154405910408301107. [DOI] [PubMed] [Google Scholar]
- 21.Barros SP, Offenbacher S. Modifiable risk factors in periodontal disease: Epigenetic regulation of gene expression in the inflammatory response. Periodontol 2000. 2014;64:95–110. doi: 10.1111/prd.12000. [DOI] [PubMed] [Google Scholar]
- 22.Winkler AR, Nocka KN, Williams CM. Smoke exposure of human macrophages reduces HDAC3 activity, resulting in enhanced inflammatory cytokine production. Pulmonary pharmacology & therapeutics. 2012;25:286–292. doi: 10.1016/j.pupt.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 23.Johannsen A, Susin C, Gustafsson A. Smoking and inflammation: evidence for a synergistic role in chronic disease. Periodontol 2000. 2014;64:111–126. doi: 10.1111/j.1600-0757.2012.00456.x. [DOI] [PubMed] [Google Scholar]
- 24.Tomar SL, Asma S. Smoking-attributable periodontitis in the United States: findings from NHANES III. National Health and Nutrition Examination Survey. The Journal of periodontology. 2000;71:743–751. doi: 10.1902/jop.2000.71.5.743. [DOI] [PubMed] [Google Scholar]
- 25.Palmer RM, Wilson RF, Hasan AS, Scott DA. Mechanisms of action of environmental factors–tobacco smoking. Journal of clinical periodontology. 2005;32(Suppl 6):180–195. doi: 10.1111/j.1600-051X.2005.00786.x. [DOI] [PubMed] [Google Scholar]
- 26.Yeh HL, Kuo LT, Sung FC, Chiang CW, Yeh CC. GSTM1, GSTT1, GSTP1, and GSTA1 genetic variants are not associated with coronary artery disease in Taiwan. Gene. 2013 doi: 10.1016/j.gene.2013.02.052. [DOI] [PubMed] [Google Scholar]
- 27.Paster BJ, Olsen I, Aas JA, Dewhirst FE. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol 2000. 2006;42:80–87. doi: 10.1111/j.1600-0757.2006.00174.x. [DOI] [PubMed] [Google Scholar]
- 28.van Winkelhoff AJ, Rijnsburger MC, van der Velden U. Clonal stability of Porphyromonas gingivalis in untreated periodontitis. Journal of clinical periodontology. 2008;35:674–679. doi: 10.1111/j.1600-051X.2008.01285.x. [DOI] [PubMed] [Google Scholar]
- 29.Dogan B, Kipalev AS, Okte E, Sultan N, Asikainen SE. Consistent intrafamilial transmission of Actinobacillus actinomycetemcomitans despite clonal diversity. The Journal of periodontology. 2008;79:307–315. doi: 10.1902/jop.2008.070270. [DOI] [PubMed] [Google Scholar]
- 30.Yoshino T, Laine ML, van Winkelhoff AJ, Dahlen G. Genotype variation and capsular serotypes of Porphyromonas gingivalis from chronic periodontitis and periodontal abscesses. FEMS Microbiol Lett. 2007;270:75–81. doi: 10.1111/j.1574-6968.2007.00651.x. [DOI] [PubMed] [Google Scholar]
- 31.Periasamy S, Kolenbrander PE. Mutualistic biofilm communities develop with Porphyromonas gingivalis and initial, early, and late colonizers of enamel. J Bacteriol. 2009;191:6804–6811. doi: 10.1128/JB.01006-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar PS, Leys EJ, Bryk JM, Martinez FJ, Moeschberger ML, Griffen AL. Changes in periodontal health status are associated with bacterial community shifts as assessed by quantitative 16S cloning and sequencing. J Clin Microbiol. 2006;44:3665–3673. doi: 10.1128/JCM.00317-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wade WG. The oral microbiome in health and disease. Pharmacological research. 2013;69:137–143. doi: 10.1016/j.phrs.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 34.Shchipkova AY, Nagaraja HN, Kumar PS. Subgingival microbial profiles of smokers with periodontitis. Journal of dental research. 2010;89:1247–1253. doi: 10.1177/0022034510377203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsigarida AA, Dabdoub SM, Nagaraja HN, Kumar PS. The Influence of Smoking on the Peri-Implant Microbiome. Journal of dental research. 2015;94:1202–1217. doi: 10.1177/0022034515590581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Camelo-Castillo AJ, Mira A, Pico A, et al. Subgingival microbiota in health compared to periodontitis and the influence of smoking. Frontiers in microbiology. 2015;6:119. doi: 10.3389/fmicb.2015.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benakanakere M, Kinane DF. Innate cellular responses to the periodontal biofilm. Frontiers of oral biology. 2012;15:41–55. doi: 10.1159/000329670. [DOI] [PubMed] [Google Scholar]
- 38.Ebersole JL, Dawson DR, 3rd, Morford LA, Peyyala R, Miller CS, Gonzalez OA. Periodontal disease immunology: ‘double indemnity’ in protecting the host. Periodontol 2000. 2013;62:163–202. doi: 10.1111/prd.12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ebersole JL, Cappelli D. Acute-phase reactants in infections and inflammatory diseases. Periodontol 2000. 2000;23:19–49. doi: 10.1034/j.1600-0757.2000.2230103.x. [DOI] [PubMed] [Google Scholar]
- 40.Hajishengallis G. Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends in immunology. 2014;35:3–11. doi: 10.1016/j.it.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garlet GP. Destructive and protective roles of cytokines in periodontitis: a re-appraisal from host defense and tissue destruction viewpoints. Journal of dental research. 2010;89:1349–1363. doi: 10.1177/0022034510376402. [DOI] [PubMed] [Google Scholar]
- 42.Ebersole JL, Steffen MJ, Thomas MV, Al-Sabbagh M. Smoking-related cotinine levels and host responses in chronic periodontitis. Journal of periodontal research. 2014;49:642–651. doi: 10.1111/jre.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayman L, Steffen MJ, Stevens J, et al. Smoking and periodontal disease: discrimination of antibody responses to pathogenic and commensal oral bacteria. Clinical and experimental immunology. 2011;164:118–126. doi: 10.1111/j.1365-2249.2010.04314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Apatzidou DA, Riggio MP, Kinane DF. Impact of smoking on the clinical, microbiological and immunological parameters of adult patients with periodontitis. Journal of clinical periodontology. 2005;32:973–983. doi: 10.1111/j.1600-051X.2005.00788.x. [DOI] [PubMed] [Google Scholar]
- 45.Jansen R, Yu H, Greenbaum D, et al. A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science. 2003;302:449–453. doi: 10.1126/science.1087361. [DOI] [PubMed] [Google Scholar]
- 46.Sachs K, Perez O, Pe’er D, Lauffenburger DA, Nolan GP. Causal protein-signaling networks derived from multiparameter single-cell data. Science. 2005;308:523–529. doi: 10.1126/science.1105809. [DOI] [PubMed] [Google Scholar]
- 47.Friedman N. Inferring cellular networks using probabilistic graphical models. Science. 2004;303:799–805. doi: 10.1126/science.1094068. [DOI] [PubMed] [Google Scholar]
- 48.Taylor IW, Linding R, Warde-Farley D, et al. Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nature biotechnology. 2009;27:199–204. doi: 10.1038/nbt.1522. [DOI] [PubMed] [Google Scholar]
- 49.Greene CS, Krishnan A, Wong AK, et al. Understanding multicellular function and disease with human tissue-specific networks. Nature genetics. 2015;47:569–576. doi: 10.1038/ng.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lage K, Hansen NT, Karlberg EO, et al. A large-scale analysis of tissue-specific pathology and gene expression of human disease genes and complexes. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:20870–20875. doi: 10.1073/pnas.0810772105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tangada SD, Califano JV, Nakashima K, et al. The effect of smoking on serum IgG2 reactive with Actinobacillus actinomycetemcomitans in early-onset periodontitis patients. The Journal of periodontology. 1997;68:842–850. doi: 10.1902/jop.1997.68.9.842. [DOI] [PubMed] [Google Scholar]
- 52.Ebersole JL, Holt SC, Hansard R, Novak MJ. Microbiologic and immunologic characteristics of periodontal disease in Hispanic americans with type 2 diabetes. The Journal of periodontology. 2008;79:637–646. doi: 10.1902/jop.2008.070455. [DOI] [PubMed] [Google Scholar]
- 53.Ebersole JL. Humoral immune responses in gingival crevice fluid: local and systemic implications. Periodontol 2000. 2003;31:135–166. doi: 10.1034/j.1600-0757.2003.03109.x. [DOI] [PubMed] [Google Scholar]
- 54.Cappelli D, Steffen MJ, Holt SC, Ebersole JL. Periodontitis in pregnancy: clinical and serum antibody observations from a baboon model of ligature-induced disease. The Journal of periodontology. 2009;80:1154–1165. doi: 10.1902/jop.2009.080199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Annals of Statistics. 2001;29:1165–1188. [Google Scholar]
- 56.Pearl J. Causality: Models, reasoning and inference. Cambridge University Press; 2009. [Google Scholar]
- 57.Koller D, Friedman N. Probabilistic graphical models: Principles and techniques. MIT Press; 2009. [Google Scholar]
- 58.Glymour C, Cooper GF. Computation, causation, and discovery. AAAI Press; 1999. [Google Scholar]
- 59.Tsamardinos I, Brown LE, Aliferis CF. The max-min hill-climbing Bayesian Network structure learning algorithm. Machine Learning. 2006;65:31–78. [Google Scholar]
- 60.Friedman N, Goldszmidt M, Wyner A. Data analysis with Bayesian Networks: A bootstrap approach UAI ’99: Procceding of the 15th Annual Conference on Uncertainty in Artificial Intelligence. Morgan Kaufmann; 1999. pp. 196–201. [Google Scholar]
- 61.Scutari M, Nagarajan R. Identifying significant edges in graphical models of molecular networks. Artificial intelligence in medicine. 2013;57:207–217. doi: 10.1016/j.artmed.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scutari M. Learning Bayesian Networks with bnlearn R package. Journal of Statistical Software. 2010;35:1–22. [Google Scholar]
- 63.Nagarajan R, Scutari M, Lèbre S. Bayesian networks in R. Springer. 2013;122:125–127. [Google Scholar]
- 64.Bastian M, Heymann S, Jacomy M. Gephi an open source software for exploring and manipulating networks International AAAI Conferecne on Weblogs and Social Media. 2009 [Google Scholar]
- 65.Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nature reviews Immunology. 2015;15:30–44. doi: 10.1038/nri3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li Y, Lu Z, Zhang X, et al. Metabolic syndrome exacerbates inflammation and bone loss in periodontitis. Journal of dental research. 2015;94:362–370. doi: 10.1177/0022034514561658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reynolds MA. Modifiable risk factors in periodontitis: at the intersection of aging and disease. Periodontol 2000. 2014;64:7–19. doi: 10.1111/prd.12047. [DOI] [PubMed] [Google Scholar]
- 68.Linden GJ, Lyons A, Scannapieco FA. Periodontal systemic associations: review of the evidence. The Journal of periodontology. 2013;84:S8–S19. doi: 10.1902/jop.2013.1340010. [DOI] [PubMed] [Google Scholar]
- 69.Genco RJ, Borgnakke WS. Risk factors for periodontal disease. Periodontol 2000. 2013;62:59–94. doi: 10.1111/j.1600-0757.2012.00457.x. [DOI] [PubMed] [Google Scholar]
- 70.Reeves AF, Rees JM, Schiff M, Hujoel P. Total body weight and waist circumference associated with chronic periodontitis among adolescents in the United States. Arch Pediatr Adolesc Med. 2006;160:894–899. doi: 10.1001/archpedi.160.9.894. [DOI] [PubMed] [Google Scholar]
- 71.Huttunen R, Heikkinen T, Syrjanen J. Smoking and the outcome of infection. Journal of internal medicine. 2011;269:258–269. doi: 10.1111/j.1365-2796.2010.02332.x. [DOI] [PubMed] [Google Scholar]
- 72.Machtei EE, Dunford R, Hausmann E, et al. Longitudinal study of prognostic factors in established periodontitis patients. Journal of clinical periodontology. 1997;24:102–109. doi: 10.1111/j.1600-051x.1997.tb00474.x. [DOI] [PubMed] [Google Scholar]
- 73.Grossi SG, Skrepcinski FB, DeCaro T, Zambon JJ, Cummins D, Genco RJ. Response to periodontal therapy in diabetics and smokers. The Journal of periodontology. 1996;67:1094–1102. doi: 10.1902/jop.1996.67.10s.1094. [DOI] [PubMed] [Google Scholar]
- 74.Fiorini T, Musskopf ML, Oppermann RV, Susin C. Is There a Positive Effect of Smoking Cessation on Periodontal Health? A Systematic Review. The Journal of periodontology. 2013 doi: 10.1902/jop.2013.130047. [DOI] [PubMed] [Google Scholar]
- 75.Scott DA, Singer DL. Suppression of overt gingival inflammation in tobacco smokers - clinical and mechanistic considerations. Int J Dent Hyg. 2004;2:104–110. doi: 10.1111/j.1601-5037.2004.00079.x. [DOI] [PubMed] [Google Scholar]
- 76.Graves DT, Oates T, Garlet GP. Review of osteoimmunology and the host response in endodontic and periodontal lesions. Journal of oral microbiology. 2011:3. doi: 10.3402/jom.v3i0.5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kornman KS. Interleukin 1 genetics, inflammatory mechanisms, and nutrigenetic opportunities to modulate diseases of aging. Am J Clin Nutr. 2006;83:475S–483S. doi: 10.1093/ajcn/83.2.475S. [DOI] [PubMed] [Google Scholar]
- 78.Diehl SR, Kuo F, Hart TC. Interleukin 1 genetic tests provide no support for reduction of preventive dental care. J Am Dent Assoc. 2015;146:164–173 e164. doi: 10.1016/j.adaj.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 79.Javed F, Al-Askar M, Al-Hezaimi K. Cytokine profile in the gingival crevicular fluid of periodontitis patients with and without type 2 diabetes: a literature review. The Journal of periodontology. 2012;83:156–161. doi: 10.1902/jop.2011.110207. [DOI] [PubMed] [Google Scholar]
- 80.Tsai CC, Hong YC, Chen CC, Wu YM. Measurement of prostaglandin E2 and leukotriene B4 in the gingival crevicular fluid. J Dent. 1998;26:97–103. doi: 10.1016/s0300-5712(96)00084-x. [DOI] [PubMed] [Google Scholar]
- 81.Salvi GE, Yalda B, Collins JG, et al. Inflammatory mediator response as a potential risk marker for periodontal diseases in insulin-dependent diabetes mellitus patients. The Journal of periodontology. 1997;68:127–135. doi: 10.1902/jop.1997.68.2.127. [DOI] [PubMed] [Google Scholar]
- 82.Lamster IB, Novak MJ. Host mediators in gingival crevicular fluid: implications for the pathogenesis of periodontal disease. Crit Rev Oral Biol Med. 1992;3:31–60. doi: 10.1177/10454411920030010501. [DOI] [PubMed] [Google Scholar]
- 83.Buduneli N, Buduneli E, Kardesler L, Lappin D, Kinane DF. Plasminogen activator system in smokers and non-smokers with and without periodontal disease. Journal of clinical periodontology. 2005;32:417–424. doi: 10.1111/j.1600-051X.2005.00694.x. [DOI] [PubMed] [Google Scholar]
- 84.Kinnby B. The plasminogen activating system in periodontal health and disease. Biol Chem. 2002;383:85–92. doi: 10.1515/BC.2002.008. [DOI] [PubMed] [Google Scholar]
- 85.Taylor B, Tofler G, Morel-Kopp MC, et al. The effect of initial treatment of periodontitis on systemic markers of inflammation and cardiovascular risk: a randomized controlled trial. European journal of oral sciences. 2010;118:350–356. doi: 10.1111/j.1600-0722.2010.00748.x. [DOI] [PubMed] [Google Scholar]
- 86.Behle JH, Sedaghatfar MH, Demmer RT, et al. Heterogeneity of systemic inflammatory responses to periodontal therapy. Journal of clinical periodontology. 2009;36:287–294. doi: 10.1111/j.1600-051X.2009.01382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Strongin AY. Proteolytic and non-proteolytic roles of membrane type-1 matrix metalloproteinase in malignancy. Biochimica et biophysica acta. 2010;1803:133–141. doi: 10.1016/j.bbamcr.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. Journal of cellular physiology. 2012;227:493–507. doi: 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jankun J, Al-Senaidy A, Skrzypczak-Jankun E. Can inactivators of plasminogen activator inhibitor alleviate the burden of obesity and diabetes? (Review) International journal of molecular medicine. 2012;29:3–11. doi: 10.3892/ijmm.2011.810. [DOI] [PubMed] [Google Scholar]
- 90.Ma Z, Paek D, Oh CK. Plasminogen activator inhibitor-1 and asthma: role in the pathogenesis and molecular regulation. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology. 2009;39:1136–1144. doi: 10.1111/j.1365-2222.2009.03272.x. [DOI] [PubMed] [Google Scholar]
- 91.Paulus P, Jennewein C, Zacharowski K. Biomarkers of endothelial dysfunction: can they help us deciphering systemic inflammation and sepsis? Biomarkers: biochemical indicators of exposure, response, and susceptibility to chemicals. 2011;16(Suppl 1):S11–21. doi: 10.3109/1354750X.2011.587893. [DOI] [PubMed] [Google Scholar]
- 92.Klebanoff SJ. Myeloperoxidase. Proceedings of the Association of American Physicians. 1999;111:383–389. doi: 10.1111/paa.1999.111.5.383. [DOI] [PubMed] [Google Scholar]
- 93.Lau D, Baldus S. Myeloperoxidase and its contributory role in inflammatory vascular disease. Pharmacology & therapeutics. 2006;111:16–26. doi: 10.1016/j.pharmthera.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 94.Rudolph TK, Rudolph V, Baldus S. Contribution of myeloperoxidase to smoking-dependent vascular inflammation. Proceedings of the American Thoracic Society. 2008;5:820–823. doi: 10.1513/pats.200807-063TH. [DOI] [PubMed] [Google Scholar]
- 95.Buduneli N, Kardesler L, Isik H, et al. Effects of smoking and gingival inflammation on salivary antioxidant capacity. Journal of clinical periodontology. 2006;33:159–164. doi: 10.1111/j.1600-051X.2006.00892.x. [DOI] [PubMed] [Google Scholar]
- 96.Hirschfeld J, Dommisch H, Skora P, et al. Neutrophil extracellular trap formation in supragingival biofilms. International journal of medical microbiology: IJMM. 2015;305:453–463. doi: 10.1016/j.ijmm.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 97.Cao CF, Smith QT. Crevicular fluid myeloperoxidase at healthy, gingivitis and periodontitis sites. Journal of clinical periodontology. 1989;16:17–20. doi: 10.1111/j.1600-051x.1989.tb01606.x. [DOI] [PubMed] [Google Scholar]
- 98.Correa JD, Madeira MF, Resende RG, et al. Association between polymorphisms in interleukin-17A and -17F genes and chronic periodontal disease. Mediators of inflammation. 2012;2012:846052. doi: 10.1155/2012/846052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Buchmann R, Hasilik A, Van Dyke TE, Lange DE. Amplified crevicular leukocyte activity in aggressive periodontal disease. Journal of dental research. 2002;81:716–721. doi: 10.1177/154405910208101012. [DOI] [PubMed] [Google Scholar]
- 100.Ebersole JL, Taubman MA. The protective nature of host responses in periodontal diseases. Periodontol 2000. 1994;5:112–141. doi: 10.1111/j.1600-0757.1994.tb00021.x. [DOI] [PubMed] [Google Scholar]
- 101.Ebersole JL, Taubman MA, Smith DJ, Frey DE, Haffajee AD, Socransky SS. Human serum antibody responses to oral microorganisms. IV. Correlation with homologous infection. Oral Microbiol Immunol. 1987;2:53–59. doi: 10.1111/j.1399-302x.1987.tb00290.x. [DOI] [PubMed] [Google Scholar]
- 102.Armitage GC. Learned and unlearned concepts in periodontal diagnostics: a 50-year perspective. Periodontol 2000. 2013;62:20–36. doi: 10.1111/prd.12006. [DOI] [PubMed] [Google Scholar]
- 103.Lamont RJ, Hajishengallis G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends in molecular medicine. 2015;21:172–183. doi: 10.1016/j.molmed.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kinane DF, Mooney J, Ebersole JL. Humoral immune response to Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in periodontal disease. Periodontol 2000. 1999;20:289–340. doi: 10.1111/j.1600-0757.1999.tb00164.x. [DOI] [PubMed] [Google Scholar]
- 105.Zerzan JT, Morden NE, Soumerai S, et al. Trends and geographic variation of opiate medication use in state Medicaid fee-for-service programs, 1996 to 2002. Medical care. 2006;44:1005–1010. doi: 10.1097/01.mlr.0000228025.04535.25. [DOI] [PubMed] [Google Scholar]