

Abstract
Understanding the genetic basis of reproductive isolation is a long-standing goal of speciation research. In recently diverged populations, genealogical discordance may reveal genes and genomic regions that contribute to the speciation process. Previous work has shown that conspecific colonies of Acropora that spawn in different seasons (spring and autumn) are associated with highly diverged lineages of the phylogenetic marker PaxC. Here, we used 10 034 single-nucleotide polymorphisms to generate a genome-wide phylogeny and compared it with gene genealogies from the PaxC intron and the mtDNA Control Region in 20 species of Acropora, including three species with spring- and autumn-spawning cohorts. The PaxC phylogeny separated conspecific autumn and spring spawners into different genetic clusters in all three species; however, this pattern was not supported in two of the three species at the genome level, suggesting a selective connection between PaxC and reproductive timing in Acropora corals. This genome-wide phylogeny provides an improved foundation for resolving phylogenetic relationships in Acropora and, combined with PaxC, provides a fascinating platform for future research into regions of the genome that influence reproductive isolation and speciation in corals.
Keywords: speciation, temporal reproductive isolation, phylogeny, spawning, PaxC, control region
1. Introduction
Molecular phylogenies are archival road maps of biodiversity, providing the fundamental framework for interpreting evolutionary history and adaptation.
Accurately inferring evolutionary relationships, however, is made complicated by discordant trees from different markers, as a result of gene duplication, incomplete lineage sorting, selective sweeps and introgression/hybridization [1–3]. Conversely, genealogical incongruence can also provide important insight into the regions of the genome that contribute to adaptation, reproductive isolation and speciation [4–6]. In the initial stages of speciation, regions of the genome that generate reproductive isolation may diverge quickly, as they experience reduced effective recombination compared with other regions [7–9]. Thus, in recently diverged populations where reproductive isolation is incomplete, genealogical discordance may reveal genes and genomic regions that contribute to speciation.
An important trait affecting reproductive isolation in scleractinian corals is the timing of reproduction [10–14]. Timing of reproduction in broadcast spawners is particularly important because gametes are viable for only a few hours, so individuals that spawn more than a few hours apart are unlikely to cross-fertilize [15]. In Western Australia, there are two coral spawning seasons (spring and autumn), and in some species there are two seasonal reproductive cohorts in the population [16–18].
Acropora is the most speciose genus of hard corals, with 134 species [19] organized into 19 species groups based on skeletal similarities [20]. To date, molecular phylogenetic studies of Acropora have focused on the mitochondrial DNA Control Region (CR) and the nuclear PaxC intron [11,21–26] (hereafter referred to as PaxC), which have produced significantly different phylogenies [11,22,27]. Incongruence between these regions has been attributed primarily to introgressive hybridization [11,22,23,27], but recent evidence suggests that PaxC is under selection and is associated with differences in spawning time, owing to two highly diverged PaxC lineages (ΦST = 0.98) that are correlated with different seasonal spawning cohorts in A. samoensis and A. tenuis [17,28]. Resolution of this issue is important because PaxC is the most commonly used phylogenetic marker in coral studies, but a selective connection may render PaxC unreliable as a phylogenetic marker. Next-generation sequencing technologies provide affordable, genome-wide resolution to resolve phylogenetic discordance, and improve phylogenetic resolution (e.g. [30–33]). Here, we construct the first genome-wide phylogeny for the scleractinian coral genus Acropora using a genotyping-by-sequencing (GBS) approach, and compare it with molecular phylogenies from PaxC and the mtDNA Control Region. Tests of congruence among these phylogenies provide a clearer understanding of the evolution of PaxC, and an improved foundation for resolving phylogenetic relationships and patterns of speciation in corals.
2. Material and methods
(a). Sample collection
Specimens of 20 species of Acropora were collected from a wide latitudinal range in Western Australia (figure 1) and stored in 100% ethanol. Three individuals were sampled for each of 17 species from 10 species groups (electronic supplementary material, table S1; except A. stoddarti and A. gemmifera with n = 2), for which accompanying reproductive data were not collected. In three species, reproductive data were collected in the field by examining the size and colour of oocytes in broken branches and classifying the colonies as autumn or spring spawners (following protocols in [17,34]). These included eight colonies of A. millepora (four autumn spawners from Ningaloo Reef and four spring spawners from Ashmore Reef; two of each were included in the GBS dataset), and 16 colonies (eight spring spawners and eight autumn spawners) each of A. samoensis (from [17]) and A. tenuis (from [28]). Voucher specimens were retained for most samples and are housed at the Western Australian Museum (electronic supplementary material, table S1).
Figure 1.
Geographical locations in Western Australia from where the 20 Acropora species were collected (boxes) for this study.
(b). Sequencing
DNA used for Sanger sequencing was extracted from branch tips using DNeasy extraction kits for animal tissue (Qiagen, USA). Partial sequences of the mtDNA CR and the PaxC 46/47 intron were amplified using the primers and protocols described in [17]. We attempted to include three replicates from each species, but some samples could not be amplified across both genes (see the electronic supplementary material, table S1). DNA fragments were sequenced in both directions at BGI Hong Kong, edited manually in Sequencher v. 4.5 (Gene Codes Corp., Ann Arbor, MI, USA), and aligned using ClustalW and Muscle in MEGA6 [35]. Heterozygotes were identified in the PaxC sequences, and IUPAC nucleotide ambiguity codes were assigned to heterozygous bases.
Genome-wide single-nucleotide polymorphism (SNP) data were generated at Diversity Arrays Technology (DArT). DArTseq is GBS technology which represents a combination of DArT complexity reduction methods and next-generation sequencing platforms, and is similar to the widely applied RADseq methodology [36]. Four methods of complexity reduction were tested, and the PstI-HpaII method was selected. Genomic DNA was processed in digestion/ligation reactions principally as per [33], but replacing a single PstI-compatible adaptor with two different adaptors corresponding to two different restriction enzyme overhangs. Sequencing was carried out on a single lane of an Illumina Hiseq2500 and processed using proprietary DArT analytical pipelines (see the electronic supplementary material for further detail on DArTseq methods). Sequences were blasted against a Symbiodinium reference genome to ensure that only sequences belonging to the coral host and not the symbiont were included in the dataset; however, no symbionts were detected among the markers anyway due to the strength of DArT PL's filtering software (which also filters viral and/or bacterial sequences in SNP marker selection; see the electronic supplementary material for further detail).
(c). Data analysis
Phylogenetic relationships were estimated for PaxC and the CR using a Bayesian statistical framework implemented in MrBayes v. 3.1.2 [37], and maximum-likelihood (ML) analyses in PhyML v. 3.0 [38] (detail in the electronic supplementary material). Both genes contained numerous indels, which were coded as single base changes. p-distances between autumn- and spring-spawning cohorts were calculated in MEGA6 [35]. For the SNP analyses, the SNPs were extracted from the sequences (read length approx. 100 bp) and concatenated into supermatrices, using IUPAC codes for heterozygous loci. ML analyses were conducted in RAxML [39] using the GTR + gamma model of sequence evolution and support for each node was assessed with 100 bootstrap replicates. Because the concatenation of variable SNPs artificially inflates branch lengths [40], we used the acquisition bias correction implemented in RAxML to generate the final topology. In addition to the tree-based methods, we also conducted admixture analysis in ADMIXTURE 1.3 [41], and a principal components analysis (PCA) in PLINK [42] for both the entire dataset and on subsets of the data comprising sympatric spring and autumn A. samoensis colonies (n = 16) and allopatric A. tenuis colonies (n = 16).
To test statistical congruence among the different datasets, we used the congruence among distance matrices (CADM) test [43] in the APE package in R [44] using Kimura2 evolutionary distance, and compared the CR, PaxC and the three SNP trees with differing genotype call rates (100%, 90% and 70%). Post-hoc tests were used to identify which datasets were congruent with one another. Contrary to most other tests, the null hypothesis of the CADM test is complete incongruence of the datasets [43].
3. Results
(a). Sequencing
In total, 85 individuals were sequenced for PaxC and 84 were sequenced for CR. After the indels were coded as single base changes and sequences were aligned and trimmed, segments of the CR and PaxC sequences consisted of 1036 bp and 357 bp, respectively, from 20 species of Acropora. Eighty-six individuals were genotyped using DArTseq methodologies, and after filtering away poor-quality sequences (see methods in the electronic supplementary material), a total of 44 356 loci remained. We filtered DArT loci to include loci that were present at a minimum depth of 8× and were present in greater than 70% of samples (hereafter referred to as minimum genotype call rate), resulting in 10 034 (23%) loci remaining for phylogenetic analyses. The resolution of the inferred tree topology substantially increased as the SNP data matrix increased in size; using genotype call rates of 100% (413 loci) and greater than or equal to 90% (3085 loci) produced topologies with much lower bootstrap support in the internal branches than when using a call rate of greater than or equal to 70% (10 034 loci; electronic supplementary material, figure S1). As a result, the data matrix with a genotype call rate of greater than or equal to 70% (i.e. up to 30% missing data per locus) was used in downstream analyses. Within each species, approximately 18% of loci were polymorphic, and the average frequency of homozygotes for the reference allele was 0.72 (±0.4; electronic supplementary material, table S2).
(b). Tree congruence: Control Region, PaxC and single-nucleotide polymorphisms
The CADM global test rejected incongruence (p < 0.001), indicating that at least one pair of distance matrices were not completely incongruent, although the overall degree of congruence was not high (Kendall's W statistic = 0.43). Posthoc Mantel tests comparing the degree of similarity between the evolutionary distance matrices showed CR and PaxC were correlated, and the SNP datasets were correlated with each other, but there were no correlations either between the PaxC and SNP datasets or between the CR and SNP datasets (table 1). When compared visually, the PaxC tree was more similar to the final SNP tree (figure 2) than to the CR tree (three discrepancies between PaxC and SNP versus nine discrepancies between PaxC and CR; electronic supplementary material, table S3), therefore the statistical correlation between PaxC and CR, but not between PaxC and the SNP distance matrices, serves to illustrate the significant difference in phylogenetic congruence between single gene trees and genome-wide phylogenies.
Table 1.
Results of Mantel tests conducted in CADM showing p-values on the top diagonal and congruence values on the bottom diagonal. Significant values are shown in italics.
| PaxC | CR | SNP 100 | SNP 90 | SNP 70 | |
|---|---|---|---|---|---|
| PaxC | — | 0.0010 | 0.8801 | 0.8761 | 0.9071 |
| CR | 0.782 | — | 0.9730 | 0.9570 | 0.6883 |
| SNP 100 | −0.060 | −0.101 | — | 0.0010 | 0.0010 |
| SNP 90 | −0.060 | −0.085 | 0.946 | — | 0.0010 |
| SNP 70 | −0.067 | −0.029 | 0.727 | 0.812 | — |
Figure 2.

Comparison of phylogenetic trees in the genus Acropora using (a) 10 034 SNPs, (b) mtDNA CR and (c) PaxC, with collapsed nodes to illustrate major patterns (see the electronic supplementary material for uncollapsed SNP trees). Branch support values are ML bootstrap values and Bayesian posterior probabilities. Major Acropora clades are indicated by Roman numerals; spring and autumn spawners are shown in blue and red; symbols after species indicate polyphyletic lineages. Trees are mid-point rooted.
The most striking aspect of phylogenetic discordance in this study was that the PaxC tree placed the spring and autumn spawners of all three species (A. millepora, A. samoensis and A. tenuis) into different and well-supported clusters within the major clades (figure 2); however, this pattern was evident in only A. samoensis in the SNP tree and not in A. millepora or A. tenuis (figure 2). The spring and autumn spawners were not separated in any species in the CR tree, which had low phylogenetic signal, as evidenced by the short branch lengths. The differences in PaxC sequences between spring and autumn spawners in A. millepora, A. samoensis and A. tenuis were characterized by multiple fixed differences and phylogenetically informative indels (figure 3). The p-distance between the autumn and spring spawners was highest in A. samoensis (p = 0.017; figure 3a) and lowest in A. tenuis (p = 0.011; figure 3c).
Figure 3.
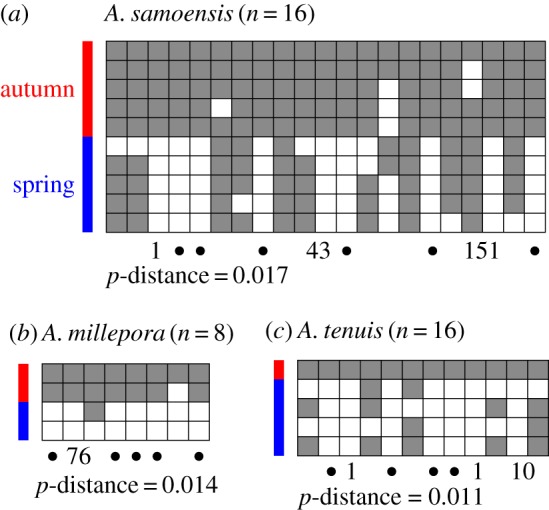
Comparisons between haplotypes of PaxC in autumn and spring spawners in (a) A. samoensis, (b) A. millepora and (c) A. tenuis. Each box represents a nucleotide base; the top row represents the most common haplotype in each species (dark shading), and light shading illustrates a different nucleotide. Only unique haplotypes within each reproductive cohort are shown. Fixed differences between the spring and autumn spawners are shown by black circles; the numbers indicate the presence/absence of an indel and detail the length (number of base pairs) in fixed indels. p = mean p-distance between autumn and spring spawners in each species. (Online version in colour.)
(c). Phylogenetic relationships
All methods of analysis recovered three of the four phylogenetically discrete Acropora clades from [11] (the monotypic fourth clade was not recovered because A. latistella was not included in this study), although clade III in PaxC and clade IV in CR were weakly supported by bootstrap values (figure 2). High posterior probabilities extended to finer relationships in the final SNP tree, offering greater resolution of evolutionary relationships than the CR or PaxC (figure 2). Many species in the SNP tree were polyphyletic, with colonies split within clades (A. subulata, A. pulchra, A. stoddarti, A. gemmifera, A. muricata, A. tenuis, A. selago, A. florida, A. samoensis and A. divaricata; figure 2), and one species that was monophyletic in the SNP tree was polyphyletic in the CR tree (A. spicifera; figure 2). Three species were polyphyletic with colonies split between clades III and IV in all of the CR, PaxC and the SNP trees (A. digitifera, A. aspera and A. lutkeni; figure 2).
The SNP phylogeny supported some of the traditional morphologically based species groups, and not others. For example, the morphologically based selago group constituted a phylogenetic group (with the exception of A. loisettae, which was in a different clade; electronic supplementary material, table S1 and figure S1c), and similarly the humilis group constituted a phylogenetic group, which was split into two clades (electronic supplementary material, table S1 and figure S1c). Conversely, the aspera group did not form any kind of phylogenetic group (electronic supplementary material, table S1 and figure S1c).
Phylogeographic patterns varied between species. Some species appeared to show geographical differences between conspecific colonies (e.g. A. divaricata, A, stoddarti, A. digitifera; electronic supplementary material, table S1), but in others that were collected from widespread locations such as the Abrolhos (S28°) or Montebellos (S21°) and the Kimberley (S13°–15°) there was little genetic difference between conspecifics (e.g. A. spicifera, A. humilis, A. cytherea, A. intermedia; electronic supplementary material, table S1). Furthermore, in some species, conspecific colonies from the same location were more genetically different to one another than conspecifics from other locations (e.g. A. aspera, A. subulata, A. lutkeni, A. donei; electronic supplementary material, table S1).
The PCA based on the SNP dataset split the genus into three clusters that corresponded to the three phylogenetic clades (figure 4a). These groups were also identified by the admixture analysis confirming that the three phylogenetic clades correspond to clearly defined genetic groups, with only two individuals having more than 25% admixture (A. selago2 and A. loisettae3; electronic supplementary material, figure S2). When the A. samoensis and A. tenuis SNP datasets were analysed alone, the results showed clear clustering between sympatric spring and autumn spawners in A. samoensis (figure 4b), but not a clear split between the autumn and spring spawners in A. tenuis (figure 4c).
Figure 4.
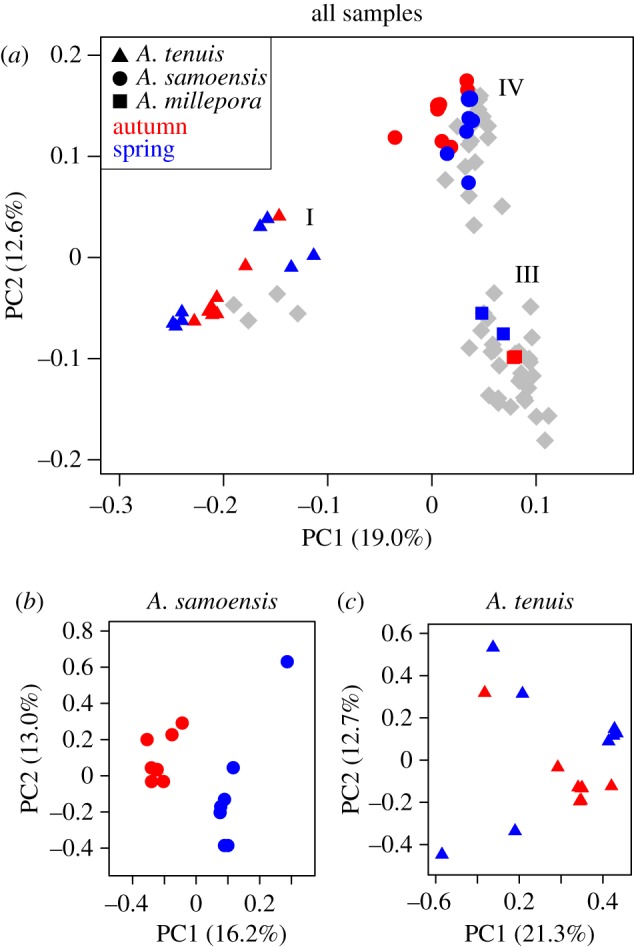
Plots of the first two axes from principal components analysis (PCA) for (a) all Acropora species examined, (b) A. samoensis seasonal cohorts and (c) A. tenuis seasonal cohorts. Autumn and spring spawners are shown in red and blue, respectively.
4. Discussion
This study is the first to use genome-wide patterns of differentiation to explore phylogenetic relationships in reef-building corals. We combined traditional phylogenetic reconstruction using single-gene approaches, with high-throughput sequencing technologies to reconstruct phylogenies and explore patterns of divergence in the widely distributed and speciose coral genus Acropora. Our results provide insight into the influence of reproductive timing on the evolutionary patterns in this group, and a springboard for future research into the genes that influence reproductive isolation in Acropora.
There are three possible explanations for incongruence between the phylogenies presented in this study. First, PaxC could be a rapidly evolving gene that presents a highly resolved phylogeny, with which other loci will eventually become phylogenetically concordant. However, there is less resolution at branch tips in the PaxC tree than the SNP tree, indicating that the PaxC phylogeny is not more highly resolved, making this explanation unlikely. A second possible explanation is that PaxC is under selection associated with coral spawning season [17]. The PaxC gene in anthozoans is closely related to Pax6 in higher-order animals, which is involved in developing eyes [45–48]. While anthozoans do not have eyes, they are sensitive to light, and they use light cues to control spawning on a range of time scales [49–52]; hence PaxC might function as a type of photoreceptor that cues spawning [28]. A third possible explanation is that PaxC is simply a hitchhiker linked to other aspects of reproductive isolation, as quantitative trait loci for different traits under divergent selection can co-localize on the genetic linkage map [53]. Further investigation is required to separate these two possibilities, and will provide a fascinating avenue for future research into the regions of the genome that influence reproductive isolation and speciation in corals. Regardless of the cause, this result is significant because a selective connection to the timing of reproduction in Acropora indicates that PaxC should be used with caution as a phylogenetic marker in corals, as it can produce spurious relationships.
Irrespective of whether it is functional or a hitchhiker, our analysis suggests that PaxC is located in a genomic region that contributes to reproductive isolation in Acropora corals. Identifying genes and genomic regions that confer isolation is a major goal of speciation research, providing insight into ecological settings, evolutionary forces and molecular mechanisms that drive the divergence of populations [54,55]. Genes that confer reproductive isolation may very well be leading indicators of evolutionary relationships and define the branches of what will ultimately become the species tree. The level of genetic differentiation between PaxC sequences associated with autumn- and spring-spawning cohorts was higher in A. samoensis than in A. tenuis and A. millepora (figure 3), and at the genome level reproductive populations were more distinct in A. samoensis than in A. tenuis (figure 4). This is likely to be a combination of recent polymorphism and incomplete reproductive barriers in A. tenuis (and some phylogeographic structure; see [28]). For example, some individuals of A. tenuis in far northwestern Australia have been observed spawning twice a year, in both autumn and spring, providing a conduit for gene flow between the reproductive groups in this species [18]. In addition, greater divergence in PaxC in A. samoensis may reflect a longer period of temporal isolation between autumn and spring spawners in this old species; fossils date A. samoensis to 9.4–9.8 Myr old [56], and fossils of A. slovenica (also in the A. humilis group and very similar to A. samoensis) to the Oligocene (approximately 28–34 Myr old) [57].
The PCA identified three major genetic clusters that corresponded to the three major phylogenetic clades, indicating these are distinct evolutionary lineages. The genome-wide phylogeny offered greater resolution of evolutionary relationships for more recently diverged taxa than the CR or PaxC, and this resolution was sufficient to reveal numerous polyphyletic species. In some polyphyletic species, conspecifics were in different positions in the SNP and CR trees (e.g. A. donei, A. muricata, A. spicifera and A. pulchra), suggesting mitochondrial introgression between species, consistent with other studies [11,27,58–60]. Conversely, in three polyphyletic species in particular (A. aspera, A. digitifera and A. lutkeni), conspecifics occurred in the same position in all phylogenetic analyses, they were well supported in all trees, and patterns were consistent between the mitochondrial and nuclear genes, indicating that these are indeed phylogenetic lineages that represent morphologically cryptic species. The degree of differentiation between the phylogenetic lineages in these three species is substantial, as the lineages are split across two clades; moreover, the differentiation is not associated with geography as might be expected in A. aspera or in A. lutkeni (electronic supplementary material, table S1), and may possibly be related to habitat (e.g. [61]). Advances in molecular techniques in the past two decades have revealed a plethora of cryptic species that are widespread across scleractinian coral genera [61–65], and their continued identification is important for estimates of species richness, endemism, range distributions and, ultimately, the conservation of this ecologically important group of corals.
Supplementary Material
Acknowledgements
We thank Madeline Chase for assistance with the RAxML analysis, S. Palumbi, A. Chen and three anonymous reviewers for very helpful advice on the manuscript, and J. Underwood for helpful discussion on methodology and analysis. Z.T.R. was supported by Woodside Energy and the Woodside Collection (Kimberley) project.
Data accessibility
All data from this article are publicly available on the Dryad Digital Repository, including raw DArT-seq data and aligned DNA sequences for PaxC and CR (http://dx.doi.org/10.5061/dryad.831g1 [66]). Phylogenetic trees supporting this article are included in the electronic supplementary material.
Authors' contributions
N.L.R. and L.T. conceived the study, designed the study, participated in field collection, carried out the molecular laboratory work, conducted the data analysis and drafted the manuscript; S.S. conducted statistical analyses and drafted the manuscript; Z.T.R. collected samples, identified and curated specimens, and drafted the manuscript; W.J.K. designed the study, conducted statistical analyses and drafted the manuscript; M.S.J. conceived the study, designed the study, conducted data analysis and drafted the manuscript. All authors gave final approval for publication.
Competing interests
We have no competing interests.
Funding
This work was supported by the Australian Coral Reef Society though a postgraduate research award to N.L.R. and L.T.
References
- 1.Maddison WP. 1997. Gene trees in species trees. Syst. Biol. 46, 523–536. ( 10.1093/sysbio/46.3.523) [DOI] [Google Scholar]
- 2.Hahn MW, Nakhleh L. 2015. Irrational exuberance for resolved species trees. Evolution 70, 7–17. ( 10.1111/evo.12832) [DOI] [PubMed] [Google Scholar]
- 3.Mallet J, Besansky N, Hahn MW. 2015. How reticulated are species? Bioessays 38, 140–149. ( 10.1002/bies.201500149) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fontaine MC, et al. 2015. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science 347, 1258524 ( 10.1126/science.1258524) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stankowski S, Streisfield MA. 2015. Introgressive hybridization facilitates adaptive divergence in a recent radiation of monkeyflowers. Proc. R. Soc. B 282, 20151666 ( 10.1098/rspb.2015.1666) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamichhaney S, et al. 2015. Evolution of Darwin's finches and their beaks revealed by genome sequencing. Nature 518, 371–375. ( 10.1038/nature14181) [DOI] [PubMed] [Google Scholar]
- 7.Wu C. 2001. The genic view of the process of speciation. J. Evol. Biol. 14, 851–865. ( 10.1046/j.1420-9101.2001.00335.x) [DOI] [Google Scholar]
- 8.Dopman EB, Perez L, Bogdanowicz SM, Harrison RG. 2005. Consequences of reproductive barriers for genealogical discordance in the European corn borer. Proc. Natl Acad. Sci. USA 102, 14 706–147 011. ( 10.1073/pnas.0502054102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Via S. 2009. Natural selection in action during speciation. Proc. Natl Acad. Sci. USA 106, 9939–9946. ( 10.1073/pnas.0901397106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knowlton N, Mate JL, Guzman HM, Rowan R, Jara J. 1997. Direct evidence for reproductive isolation among the three species of the Montastrea annularis complex in Central America (Panama and Honduras). Mar. Biol. 127, 705–711. ( 10.1007/s002270050061) [DOI] [Google Scholar]
- 11.van Oppen MJH, McDonald BJ, Willis BL, Miller DJ. 2001. The evolutionary history of the coral genus Acropora (Scleractinia, Cnidaria) based on mitochondrial and a nuclear marker: reticulation, incomplete lineage sorting or morphological convergence? Mol. Biol. Evol. 18, 1315–1329. ( 10.1093/oxfordjournals.molbev.a003916) [DOI] [PubMed] [Google Scholar]
- 12.Fukami H, Omori M, Shimoike K, Hayashibara T, Hatta M. 2003. Ecological and genetic aspects of reproductive isolation by different spawning times in Acropora corals. Mar. Biol. 142, 679–684. ( 10.1007/s00227-002-1001-8) [DOI] [Google Scholar]
- 13.Wolstenholme JK. 2004. Temporal reproductive isolation and gametic compatibility are evolutionary mechanisms in the Acropora humilis species group (Cnidaria; Scleractinia). Mar. Biol. 144, 567–582. ( 10.1007/s00227-003-1209-2) [DOI] [Google Scholar]
- 14.Nakajima Y, Nishikawa A, Iguchi A, Sakai K. 2012. The population genetic approach delineates the species boundary of reproductively isolated corymbose acroporid corals. Mol. Phylogen. Evol. 63, 527–531. ( 10.1016/j.ympev.2012.01.006) [DOI] [PubMed] [Google Scholar]
- 15.Levitan DR, Fogarty ND, Jara J, Lotterhos KE, Knowlton N. 2011. Genetic, spatial, and temporal components of precise spawning synchrony in reef building corals of the Montastraea annularis species complex. Evolution 65, 1254–1270. ( 10.1111/j.1558-5646.2011.01235.x) [DOI] [PubMed] [Google Scholar]
- 16.Rosser NL, Gilmour JP. 2008. New insights into patterns of coral spawning on Western Australian reefs. Coral Reefs 27, 345–349. ( 10.1007/s00338-007-0335-6) [DOI] [Google Scholar]
- 17.Rosser NL. 2015. Asynchronous spawning in sympatric populations of a hard coral reveals cryptic species and ancient genetic lineages. Mol. Ecol. 24, 5006–5019. ( 10.1111/mec.13372) [DOI] [PubMed] [Google Scholar]
- 18.Gilmour JP, Underwood JN, Howells EJ, Gates E, Heyward AJ. 2016. Biannual spawning and temporal reproductive isolation in Acropora corals. PLoS ONE 11, e0150916 ( 10.1371/journal.pone.0150916) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wallace CC, Done BJ, Muir PR. 2012. Revision and catalogue of worldwide staghorn corals of Acropora and Isopora (Scleractinia: Acroporidae) in the Museum of Tropical Queensland. Mem. Queensland Museum Nat. 57, 1–255. [Google Scholar]
- 20.Wallace CC. 1999. Staghorn corals of the world a revision of the genus Acropora. Melbourne, Australia: CSIRO Publishing. [Google Scholar]
- 21.van Oppen MJH, Willis BL, van Vugt HWJA, Miller DJ. 2000. Examination of species boundaries in the Acropora cervicornis group (Scleractinia, Cnidaria) using nuclear DNA sequence analyses. Mol. Ecol. 9, 1363–1373. ( 10.1046/j.1365-294x.2000.01010.x) [DOI] [PubMed] [Google Scholar]
- 22.Marquez LM, van Oppen MJH, Willis BL, Reyes A, Miller DJ. 2002. The highly cross-fertile coral species, Acropora hyacinthus and Acropora cytherea, constitute statistically distinguishable lineages. Mol. Ecol. 11, 1339–1349. ( 10.1046/j.1365-294X.2002.01526.x) [DOI] [PubMed] [Google Scholar]
- 23.van Oppen MJH, Koolmees EM, Veron JEN. 2004. Patterns of evolution in the scleractinian coral genus Montipora (Acroporidae). Mar. Biol. 144, 9–18. ( 10.1007/s00227-003-1188-3) [DOI] [Google Scholar]
- 24.Vollmer SV, Palumbi SR. 2007. Restricted gene flow in the Caribbean staghorn coral Acropora cervicornis: implications for the recovery of endangered reefs. J. Hered. 98, 40–50. ( 10.1093/jhered/esl057) [DOI] [PubMed] [Google Scholar]
- 25.Richards ZT, Miller DJ, Wallace CC. 2013. Molecular phylogenetics of geographically restricted Acropora species: implications for threatened species conservation. Mol. Phylogen. Evol. 69, 837–851. ( 10.1016/j.ympev.2013.06.020) [DOI] [PubMed] [Google Scholar]
- 26.Ohki S, Kowalski RK, Kitanobo S, Morita M. 2015. Changes in spawning time led to the speciation of the broadcast spawning corals Acropora digitifera and the cryptic species Acropora sp. 1 with similar gamete recognition systems. Coral Reefs 34, 1189–1198. ( 10.1007/s00338-015-1337-4). [DOI] [Google Scholar]
- 27.Richards ZT, van Oppen MJH, Wallace CC, Willis BL, Miller DJ. 2008. Some rare Indo-Pacific coral species are probable hybrids. PLoS ONE 3, e3240 ( 10.1371/journal.pone.0003240) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosser NL. 2016. Demographic history and asynchronous spawning shape genetic differentiation among populations of the hard coral Acropora tenuis in Western Australia. Mol. Phylogen. Evol. 98, 89–96. ( 10.1016/j.ympev.2016.02.004) [DOI] [PubMed] [Google Scholar]
- 29.Nadeau NJ, Jiggins CD. 2010. A golden age for evolutionary genetics? Genomic studies of adaptation in natural populations. Trends Genet. 26, 484–492. ( 10.1016/j.tig.2010.08.004) [DOI] [PubMed] [Google Scholar]
- 30.Rubin BER, Ree RH, Moreau CS. 2012. Inferring phylogenies from RAD sequence data. PLoS ONE 7, e33394 ( 10.1371/journal.pone.0033394) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCormack JE, Hird SM, Zellmer AJ, Carstens BC, Brumfield RT. 2013. Applications of next-generation sequencing to phylogeography and phylogenetics. Mol. Phylogen. Evol. 66, 526–538. ( 10.1016/j.ympev.2011.12.007) [DOI] [PubMed] [Google Scholar]
- 32.Escudero M, Eaton DAR, Hahn M, Hipp AL. 2014. Genotyping-by-sequencing as a tool to infer phylogeny and ancestral hybridization: a case study in Carex (Cyperaceae). Mol. Phylogenet. Evol. 79, 359–367. ( 10.1016/j.ympev.2014.06.026) [DOI] [PubMed] [Google Scholar]
- 33.Rivers DM, Darwell CT, Althoff DM. 2016. Phylogenetic analysis of RAD-seq data: examining the influence of gene genealogy conflict on analysis of concatenated data. Cladistics 32, 672–681. ( 10.1111/cla.12149) [DOI] [PubMed] [Google Scholar]
- 34.Rosser NL. 2013. Biannual coral spawning decreases at higher latitudes on Western Australian reefs. Coral Reefs 32, 455–460. ( 10.1007/s00338-012-0986-9) [DOI] [Google Scholar]
- 35.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. ( 10.1093/molbev/mst197) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davey JL, Blaxter MW. 2010. RADseq: Next-generation population genetics. Brief. Funct. Genomics 9, 416–423. ( 10.1093/bfgp/elq031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. ( 10.1093/bioinformatics/btg180) [DOI] [PubMed] [Google Scholar]
- 38.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. ( 10.1093/sysbio/syq010) [DOI] [PubMed] [Google Scholar]
- 39.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. ( 10.1093/bioinformatics/btu033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leache AD, Banbury BL, Felsenstein J, Nieto-Montes de Oca A, Stamatakis A. 2015. Short tree, long tree, right tree, wrong tree: new acquisition bias corrections for inferring SNP phylogenies. Syst. Biol. 64, 1032–1047. ( 10.1093/sysbio/syv053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alexander DH, Novembre J, Lange K. 2009. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. ( 10.1101/gr.094052.109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Purcell S, et al. 2007. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 81, 559–575. ( 10.1086/519795) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campbell V, Legendre P, Lapointe F. 2011. The performance of the Congruence Among Distance Matrices (CADM) test in phylogenetic analysis. BMC Evol. Biol. 11, 407 ( 10.1186/1471-2148-11-64) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paradis E, Claude J, Strimmer K. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. ( 10.1093/bioinformatics/btg412) [DOI] [PubMed] [Google Scholar]
- 45.Callaerts P, Halder G, Gehring W. 1997. PAX-6 in development and evolution. Annu. Rev. Neurosci. 20, 483–532. ( 10.1146/annurev.neuro.20.1.483) [DOI] [PubMed] [Google Scholar]
- 46.Miller DJ, Hayward DC, Reece-Hoyes JS, Scholten I, Catmull J, Gehring WJ, Callaerts P, Larsen JE, Ball EE. 2000. Pax gene diversity in the basal cnidarian Acropora millepora (Cnidaria, Anthozoa): implications for the evolution of the Pax gene family. Proc. Natl Acad. Sci. USA 97, 4475–4480. ( 10.1073/pnas.97.9.4475) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Jong D. 2005. Characterisation of homologs of nervous system patterning genes in the staghorn coral, Acropora millepora (Cnidaria; Anthozoa; Scleractinia). PhD thesis, James Cook University, Australia.
- 48.Matus DQ, Pang K, Daly M, Martindale MQ. 2007. Expression of Pax gene family members in the anthozoan cnidarian, Nematostella vectensis. Evol. Dev. 9, 25–38. ( 10.1111/j.1525-142X.2006.00135.x) [DOI] [PubMed] [Google Scholar]
- 49.van Woesik R, Lacharmoise F, Koksal S. 2006. Annual cycles of solar insolation predict spawning times of Carribbean corals. Ecol. Lett. 9, 390–398. ( 10.1111/j.1461-0248.2006.00886.x) [DOI] [PubMed] [Google Scholar]
- 50.Brady AK, Hilton JD, Vize PD. 2009. Coral spawn timing is a direct response to solar light cycles and is not an entrained circadian response. Coral Reefs 28, 677–680. ( 10.1007/s00338-009-0498-4) [DOI] [Google Scholar]
- 51.Sweeney AM, Boch CA, Johnsen S, Morse DE. 2011. Twilight spectral dynamics and the coral reef invertebrate spawning response. J. Exp. Biol. 214, 770–777. ( 10.1242/jeb.043406) [DOI] [PubMed] [Google Scholar]
- 52.Reitzel AM, Tarrant AM, Levy O. 2013. Circadian clocks in the Cnidaria: environmental entrainment, molecular regulation and organismal outputs. Intergr. Comp. Biol. 53, 118–130. ( 10.1093/icb/ict024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hawthorne DJ, Via S. 2001. Genetic linkage facilitates ecological specialization and reproductive isolation in pea aphids. Nature 412, 904–907. ( 10.1038/35091062) [DOI] [PubMed] [Google Scholar]
- 54.Orr HA, Masly JP, Presgraves DC. 2004. Speciation genes. Curr. Opin. Genet. Dev. 14, 675–679. ( 10.1016/j.gde.2004.08.009) [DOI] [PubMed] [Google Scholar]
- 55.Rieseberg LH, Blackman BK. 2010. Speciation genes in plants. Ann Bot. 106, 439–455. ( 10.1093/aob/mcq126) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santodomingo N. 2014. Miocene reef-coral diversity of Indonesia: unlocking the murky origins of the Coral Triangle. PhD thesis, Utrecht University, The Netherlands.
- 57.Wallace CC, Bosellini FR. 2014. Acropora (Scleractinia) from the Oligocene and Miocene of Europe: species longevity, origination and turnover following the Eocene–Oligocene transition. J. Syst. Paleontol. 12, 447–469. ( 10.1080/14772019.2014.930525). [DOI] [Google Scholar]
- 58.Veron JEN. 1995. Corals in space and time: the biogeography and evolution of the Scleractinia. Ithaca, NY: Cornell University Press. [Google Scholar]
- 59.Willis BL, van Oppen MJH, Miller DJ, Vollmer SV, Ayre DJ. 2007. The role of hybridization in the evolution of reef corals. Annu. Rev. Ecol. Evol. Syst. 37, 489–517. ( 10.1146/annurev.ecolsys.37.091305.110136) [DOI] [Google Scholar]
- 60.Kenyon JC. 1997. Models of reticulate evolution in the coral genus Acropora based on chromosome numbers: parallels with plants. Evolution 51, 756–767. ( 10.2307/2411152) [DOI] [PubMed] [Google Scholar]
- 61.Warner PA, Van Oppen MJH, Willis BL. 2015. Unexpected cryptic species diversity in the widespread coral Seriatopora hystrix masks spatial-genetic patterns of connectivity. Mol. Ecol. 24, 2993–3008. ( 10.1111/mec.13225) [DOI] [PubMed] [Google Scholar]
- 62.Flot J-F, Blanchot J, Charpy L, Cruaud C, Licuanan WF, Nakano Y, Payri C, Tillier S et al. 2011. Incongruence between morphotypes and genetically delimited species in the coral genus Stylophora: phenotypic plasticity, morphological convergence, morphological stasis or interspecific hybridization? BMC Ecol. 11, 22 ( 10.1186/1472-6785-11-22) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ladner JT, Palumbi SR. 2012. Extensive sympatry, cryptic diversity and introgression throughout the geographic distribution of two coral species complexes. Mol. Ecol. 21, 2224–2238. ( 10.1111/j.1365-294X.2012.05528.x) [DOI] [PubMed] [Google Scholar]
- 64.Schmidt-Roach S, Lundgren P, Miller KJ, Gerlach G, Noreen AME, Andreakis N. 2013. Assessing hidden species diversity in the coral Pocillopora damicornis from Eastern Australia. Coral Reefs 32, 161–172. ( 10.1007/s00338-012-0959-z) [DOI] [Google Scholar]
- 65.Richards ZT, Berry O, van Oppen MJH. 2016. Cryptic genetic divergence within threatened species of Acropora coral from the Indian and Pacific Oceans. Conserv. Genet. 17, 577–591. ( 10.1007/s10592-015-0807-0) [DOI] [Google Scholar]
- 66.Rosser NL, Thomas L, Stankowski S, Richards ZT, Kennington WJ, Johnson MS. 2017. Data from: Phylogenomics provides new insight into evolutionary relationships and genealogical discordance in the reef-building coral genus Acropora. Dryad Digital Repository. ( 10.5061/dryad.831g1) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data from this article are publicly available on the Dryad Digital Repository, including raw DArT-seq data and aligned DNA sequences for PaxC and CR (http://dx.doi.org/10.5061/dryad.831g1 [66]). Phylogenetic trees supporting this article are included in the electronic supplementary material.