

Abstract
N-acetyl serotonin (NAS) as a melatonin precursor has neuroprotective actions. Nonetheless, it is not clarified how NAS protects neuronal cells against oxidative stress. Recently, we have reported that N-palmitoyl serotonins possessed properties of antioxidants and neuroprotection. Based on those, we hypothesized that NAS, a N-acyl serotonin, may have similar actions in oxidative stress-induced neuronal cells, and examined the effects of NAS based on in vitro and in vivo tests. NAS dose-dependently inhibited oxidative stress-induced cell death in HT-22 cells. Moreover, NAS suppressed glutamate-induced apoptosis by suppressing expression of AIF, Bax, calpain, cytochrome c and cleaved caspase-3, whereas it enhanced expression of Bcl-2. Additionally, NAS improved phosphorylation of tropomyosin-related kinase receptor B (TrkB) and cAMP response element-binding protein (CREB) as well as expression of brain-derived neurotrophic factor (BDNF), whereas the inclusion of each inhibitor of JNK, p38 or Akt neutralized the neuroprotective effect of NAS, but not that of ERK. Meanwhile, NAS dose-dependently reduced the level of reactive oxygen species, and enhanced the level of glutathione in glutamate-treated HT-22 cells. Moreover, NAS significantly increased expression of heme oxygenase-1, NAD(P)H quinine oxidoreductase-1 and glutamate-cysteine ligase catalytic subunit as well as nuclear translocation of NF-E2-related factor-2. Separately, NAS at 30 mg/kg suppressed scopolamine-induced memory impairment and cell death in CA1 and CA3 regions in mice. In conclusion, NAS shows actions of antioxidant and anti-apoptosis by activating TrkB/CREB/BDNF pathway and expression of antioxidant enzymes in oxidative stress-induced neurotoxicity. Therefore, such effects of NAS may provide the information for the application of NAS against neurodegenerative diseases.
Abbreviations: AIF, Apoptosis-inducing factor; Akt, Protein kinase B; Bax, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma 2; BDNF, Brain-derived neurotrophic factor; calpain, Ca2+-dependent, non-lysosomal cysteine protease; CREB, cAMP response element-binding protein; DCFDA, 2, 7-Dichlorofluorescein diacetate; ERK, Extracellular signal-regulated kinase 1/2; GCLC, Glutamate-cysteine ligase catalytic subunit; GSH, Glutathione; HO-1, Heme oxygenase-1; JNK, c-Jun N-terminal kinase 1/2; MAPK, Mitogen-activated protein kinase; NAS, N-acetyl serotonin; NQO-1, NAD(P)H quinine oxidoreductase-1; Nrf2, NF-E2-related factor-2; ROS, Reactive oxygen species; TrkB, Tropomyosin-related kinase receptor B
Keywords: Antioxidant enzymes, Apoptosis, BDNF, Memory impairment, N-acetyl serotonin, Oxidative stress
Graphical abstract
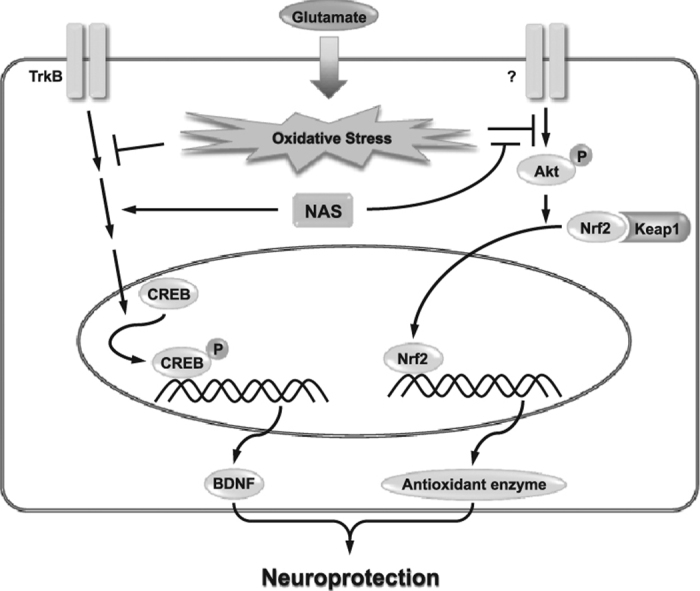
Highlights
-
•
NAS protects apoptosis induced by oxidative stress in neuronal cells.
-
•
NAS exerts an antioxidant property in neuronal cells.
-
•
NAS improves activation of BDNF/TrkB/CREB pathway in neuronal cells.
-
•
NAS enhances activation of Akt/Nrf2/Antioxidant enzyme pathway in neuronal cells.
-
•
NAS recovers memory and neuronal cells in scopolamine-treated mice.
1. Introduction
Alzheimer's disease, one of the neurodegenerative diseases, is induced by accumulation of amyloid β-peptides in neuronal cells [1]. In early stage, patients of Alzheimer's disease have memory impairment clinically [1]. Besides, the accumulation of amyloid β-peptides in neuronal cells is known as generating elevation of reactive oxygen species (ROS) [1]. Consequently, oxidative stress leads to cell death in neuronal cells [2]. Therefore, the regulation of ROS generation is very important to prevent or treat Alzheimer's disease. Meanwhile, glutamate (PubChem CID: 611) treatment of neuronal cells is known to cause generation of ROS through receptor-mediated excitotoxicity [3] or non-receptor-mediated cytotoxicity [2]. Therefore, the glutamate treatment for in intro assay is commonly used for screening antioxidant activity of a novel drug candidate in neuronal cells. Scopolamine (PubChem CID: 5184) is known as an antagonist of muscarinic acetylcholine receptor [4], and causing memory impairment in rodents [5], [6] and humans [7]. In addition, scopolamine induces oxidative stress in brain [8], and reduces formation of brain-derived neurotrophic factor (BDNF) in neuronal cells [9]. Therefore, administration of scopolamine for in vivo experiment is generally used as a model of memory impairment for Alzheimer's disease.
N-acetyl serotonin (NAS; PubChem CID: 903) as an endogenous N-acyl serotonin is better known as a precursor of melatonin [10], is produced from serotonin by serotonin N-acetyltransferase (EC 2.3.1.87) [11], and is converted to melatonin by hydroxyindole-O-methyltransferase (EC 2.1.1.4) in pineal gland [12] and retina cells [13]. NAS is known to possess stronger antioxidant activity than melatonin [14]. Recently, there have been reports that NAS can protect organ-resident cells against ischemic injury in intestine [15] and brain [16]. Besides, NAS leads to activation of tropomyosin-related kinase receptor B (TrkB) in neuronal cells [17]. Moreover, NAS is included in a library of 1040 FDA-approved compounds collected by the National Institute of Neurological Disorders and Stroke [16]. Nonetheless, it is still not clarified how NAS protects neuronal cells against oxidative stress.
Recently, we found that N-acyl serotonins possessed properties of excellent antioxidants and neuroprotection in glutamate-treated neuronal cells [2] and scopolamine-treated mice [9]. Although N-palmitoyl serotonin at submicromolar range possessed exceptional neuroprotective properties, it was not tested clinically. Therefore, it was hypothesized that NAS might have similar properties of N-palmitoyl serotonin. In this study, we investigated antioxidant activity and neuroprotective action of NAS in glutamate-treated HT-22 cells, a murine hippocampus-derived cell line [18], and scopolamine-induced memory impairment in mice. Finally, we propose novel molecular mechanisms for neuroprotective properties of NAS using immunoblotting analysis, and hematoxylin and eosin (H&E) staining assay. Herein, we report how NAS has neuroprotective properties in oxidative stress-induced apoptosis and scopolamine-induced memory impairment. Therefore, such effects of NAS may provide further information for the application of NAS as a therapeutic or preventive agent for neurodegenerative diseases.
2. Material and methods
2.1. Materials
DMEM, 1× PBS and 1× TBS were purchased from Welgene, Inc. (Gyeongsan, Gyeongbuk, Korea). FBS, 0.25% trypsin-EDTA, penicillin-streptomycin and 2,7-dichlorofluorescein diacetate (DCFDA) were obtained from Invitrogen (Carlsbad, CA, USA). NAS, scopolamine, K252a (a TrkB inhibitor; PubChem CID: 9912412), PD98059 (an extracellular signal-regulated kinase 1/2 (ERK) inhibitor; PubChem CID: 4713), SP600125 (a c-Jun N-terminal kinase 1/2 (JNK) inhibitor; PubChem CID: 8515), SB203580 (a p38 inhibitor; PubChem CID: 176155) and MK-2206 (an protein kinase B (Akt) inhibitor; PubChem CID: 24964624) were purchased from Cayman Chemical Company, Inc. (Ann Arbor, MI, USA). EZ-Cytox cell viability assay kit was obtained from DAEILLAB SERVICE Co. (Seoul, Korea). Specific antibodies against apoptosis-inducing factor (AIF), Bcl-2-associated X protein (Bax), B-cell lymphoma 2 (Bcl-2), Ca2+-dependent, non-lysosomal cysteine protease (calpain), caspase-3, cleaved caspase-3, TrkB, phospho-TrkB, phospho-cAMP response element-binding protein (CREB), heme oxygenase-1 (HO-1), glutamate-cysteine ligase catalytic subunit (GCLC), NF-E2-related factor-2 (Nrf2) and β-actin as well as a horseradish peroxidase-conjugated IgG secondary antibody were purchased from Cell signaling Technology, Inc. (Danvers, MA, USA). Specific antibodies against NAD(P)H quinine oxidoreductase-1 (NQO-1) and cytochrome c were obtained from Abcam, Inc. (Cambridge, UK). A specific antibody against BDNF was purchased from Santa cruz biotechnology, Inc. (Dallas, TX, USA). MuseTM Annexin V & Dead Cell Assay Kit and MuseTM Mitopotential Assay Kit were obtained from Merck Millipore, Inc. (Darmstadt, Germany). All other chemicals used in this study were analytical grade.
2.2. Animals
ICR mice (5–6 weeks, 25–30 g), known as Swiss CD-1 mice [19], were procured from Dehan Biolink Co. (Eumseong, Korea), and housed in cages (3 mice per cage) under specific pathogen-free condition (21–24 °C; 40–60% relative humidity) with a 12-h light/dark cycle, and free access to standard rodent food (Sangyang Co., Osen, Korea) and water. All behavioral experiments were carried out in a room adjacent to the same ambient conditions, conducted according to the Guide for Care and Use of Laboratory Animals of the National Research Council (NRC, 1996), and approved by the Committee of Animal Care and Experiment of Chungnam National University, Korea (CNU-00698).
2.3. Behavior tests
2.3.1. Morris water maze test
Morris water maze test was performed according to the method reported previously [9]. One hour before the trial, mice were administrated with NAS (1 or 30 mg/kg, p.o.). After 3 days, the mice were intra peritoneally administrated with scopolamine (2 mg/kg) following the treatment with NAS. The dose of NAS was determined in consideration of the effective concentration of its in vitro data.
2.3.2. Passive avoidance test
The passive avoidance test was performed following the method reported previously [9]. One hour before the acquisition trial, mice were orally administrated with NAS prior to the administration with scopolamine.
2.4. Hematoxylin and eosin staining assay
The H&E staining assay was performed following the previous method [9]. Mice were anesthetized, and perfused with ice-cold PBS and then ice-cold 2% paraformaldehyde solution in PBS (pH 7.4). Isolated brains were stored in 2% paraformaldehyde solution for one week, and then embedded in paraffin. Serial coronal sections (4 μm thickness) including dorsal hippocampus were cutted off with a rotary microtome (Leica RM 2165, Leica Microsystems Ltd., Wetzlar, Germany). Paraffin-embedded sections of brains were deparaffinized and rehydrated. After H&E staining, the histopathological changes were assessed under a light microscope (400-fold magnification).
2.5. Cell culture
HT-22 cells were maintained in DMEM medium with 10% FBS, 100 units/ml of penicillin and 100 μg/ml of streptomycin at 37 °C in a humidified atmosphere of 5% CO2. Passages 3–10 of the HT-22 cells were used in this study. All the experiments include a vehicle control group containing 0.1% DMSO.
2.6. Cell viability assay
Cell viability was evaluated following the previous method [2]. HT-22 cells were preincubated with or without NAS (50–500 μM) 30 min prior to glutamate treatment. After 12 h, cell viability was assessed using EZ-Cytox cell viability assay kit according to the manufacturer's instruction. The absorbance at 450 nm was measured by a microplate reader (DU650, Beckman Coulter, Brea, CA, USA).
2.7. Flow cytometry analysis
Apoptotic bodies and mitochondrial membrane potential were analyzed following the previous method [20]. HT-22 cells were preincubated with or without NAS before glutamate challenge. After 12 h, all the cells were harvested, and then washed with 1× PBS twice. Mitochondrial membrane potential and apoptosis were measured using MuseTM Mitopotential Assay Kit and MuseTM Annexin V & Dead Cell Assay Kit, respectively according to the manufacturer's instruction. Stained cells were analyzed by a flow cytometer (MuseTM Cell Analyzer, Merck Millipore, Inc.) with Muse 1.1.2 analysis software.
2.8. Measurement of Intracellular reactive oxygen species level
The intracellular ROS level was measured following the previous method [2]. Glutamate-treated HT-22 cells were stained with 10 μM DCFDA in Hank's balanced salt solution for 30 min in the darkness, and then the fluorescence was measured by a microplate reader (Beckman Coulter DTX 880 Multimode Detector, Brea, CA, USA) at an excitation wavelength of 485 nm and an emission wavelength of 525 nm.
2.9. Measurement of Intracellular glutathione
The intracellular glutathione (GSH) levels were measured following the previous method [2]. Supernatants separated from cell homogenates were analyzed for GSH level using a Quanticrom Glutathione Assay Kit obtained from BioAssay Systems (Hayward, CA, USA) according to the manufacturer's protocol.
2.10. Extraction of Nuclear and Cytosolic protein
Nuclear and cytosolic proteins were performed following a process reported previously [2] with a Nuclear Extraction Kit from Cayman Chemical Company, Inc. (Ann Arbor, MI, USA). Nuclear and cytosolic proteins were fractionated according to the instructions of the manufacturer. Briefly, cells were harvested and centrifuged (300×g, 5 min) at 4 °C. The cell pellets were mixed with hypotonic buffer containing phosphatase and protease inhibitors. After 10 min incubation on ice, 10% Nonidet P-40 Assay Reagent was added to the cells. Nuclei were recovered by centrifugation (14,000×g, 30 sec), and the supernatant was kept as cytoplasmic extract at −80 °C until use. The nuclei were extracted with Nuclear Extraction Buffer for 30 min on ice. Insoluble material was removed by centrifugation (14,000×g, 10 min). Finally, the supernatant was used as a nuclear extract.
2.11. Immunoblotting analysis
Immunoblotting analysis was evaluated following the previous method [2]. PVDF membranes including blotted proteins were visualized by WEST OneTM western blot detection system (iNtRON Biotechnology, Inc, Gyeonggi-do, Korea). The level of target proteins was compared to that of a loading control (β-actin or non-phosphorylated proteins), and the results were expressed as a ratio of density of each protein identified by a protein standard size marker (BIOFACT, Daejeon, Korea). The relative density of the protein expression was quantitated by Matrox Inspector software (version 2.1 for Windows; Matrox Electronic Systems Ltd., Dorval, Quebec, Canada).
2.12. Statistical Analysis
The experimental results were listed as means±SD for in vitro studies or SEM for in vivo studies. One-way ANOVA was used for multiple comparisons (GraphPad Prism version 5.03 for Windows, San Diego, CA, USA). If there was a significant variation between treated groups, the Dunnett's or Turkey's tests were applied. Differences at the *P<0.05 and **P<0.01 levels were considered statistically significant.
3. Results
3.1. Protective effect of N-acetyl serotonin on oxidative stress-induced apoptosis in HT-22 cells
NAS is known to have properties of antioxidant and neuroprotection [14], [16]. In this regard, we investigated effect of NAS on glutamate or hydrogen peroxide-induced cell death in HT-22 cells. When HT-22 cells were preincubated with NAS (50–500 μM) before glutamate challenge (5 mM), NAS dose-dependently inhibited glutamate-induced cell death in HT-22 cells, and NAS at 250 μM almost restored cell viability (Fig. 1a). Likewise, NAS protected neuronal cells from hydrogen peroxide-induced cytotoxicity (Fig. 1b). Moreover, NAS not only reduced apoptotic bodies (Fig. 2a), but also recovered mitochondrial membrane potential in glutamate-treated HT-22 cells (Fig. 2b). Based on these results, it is suggested that NAS possesses neuroprotective action against oxidative stress-mediated apoptosis in neuronal cells.
Fig. 1.

Inhibitory effect of N-acetyl serotonin on oxidative stress-induced cytotoxicity in HT-22 cells. HT-22 cells were seeded on a 96 well-plate, and then incubated for 24 h. The above cells were preincubated with or without NAS (50–500 μM) for 30 min before glutamate challenge (5 mM) or hydrogen peroxide (500 μM). After 12 h, cell viability was measured as described in materials and methods. Data are the mean±SD values of triple determinations. *P<0.05 and **P<0.01 versus Glutamate-treated group. A, glutamate; B, hydrogen peroxide.
Fig. 2.

Inhibitory effects of N-acetyl serotonin on glutamate-induced apoptosis and depolarization of mitochondrial membrane in HT-22 cells. HT-22 cells were seeded on a 60 mm dish, and then incubated for 24 h. The above cells were preincubated with or without NAS before glutamate challenge. After 12 h, apoptosis or mitochondrial membrane potential were analyzed by flow cytometry as described in materials and methods. A, apoptosis; B, mitochondrial potential.
3.2. Regulatory effects of N-acetyl serotonin on expression of pro-apoptotic factors and activation of TrkB/CREB/BDNF pathway
Since we found that NAS exerted anti-apoptotic action, we further investigated effect of NAS on expression of apoptosis-related factors and activation of TrkB/CREB/BDNF pathway in glutamate-treated HT-22 cells. As shown in Fig. 3a, NAS dose-dependently reduced expression of pro-apoptotic factors such as Bax, calpain, cytochome c, cleaved caspase-3 and AIF while it restored the level of Bcl-2, an anti-apoptotic factor. Especially, NAS at 500 μM completely blocked all the pro-apoptotic factors, and recovered Bcl-2 level. In addition, NAS recovered phosphorylation of TrkB and CREB as well as expression of BDNF (Fig. 3b). Interestingly, the compound at 500 μM also restored the levels of p-TrkB or p-CREB to control group. Overall, it is suggested that NAS protects neuronal cells against oxidative stress-induced apoptosis by regulating both expression of apoptosis-related factors and activation of TrkB/CREB/BDNF pathway.
Fig. 3.

Effects of N-acetyl serotonin on expression of anti- or pro-apoptotic factors and activation of TrkB, CREB or BDNF. HT-22 cells were incubated with glutamate in the presence or absence of NAS for 12 h. The expression of Bcl-2, Bax, AIF, calpain, cytochrome c (Cyto C), cleaved caspase-3 (CleaCas-3), phospho-TrkB, TrkB, phospho-CREB, BDNF or β-actin was determined as described in materials and methods. Similar results were obtained in three independent experiments. *P<0.05 and **P<0.01 versus Glutamate-treated group. A, Bcl-2, Bax, AIF, calpain, cytochrome c and cleaved caspase-3; B, p-TrkB, TrkB, p-CREB and BDNF.
3.3. Inhibitory effect of N-acetyl serotonin on the generation of reactive oxygen species induced by glutamate
After we found that NAS regulated the expression of apoptosis-related factors and the activation of TrkB/CREB/BDNF pathway, we turned to the effect of NAS on the generation of ROS in glutamate-treated HT-22 cells because NAS is known as possessing antioxidant activity [14], [16]. As shown in Fig. 4, NAS dramatically reduced ROS level at ≥50 μM (Fig. 4a), and significantly recovered GSH level at ≥100 μM (Fig. 4b) in glutamate-treated HT-22 cells. Concerning such effect of NAS, we further investigated the effect of NAS on expression of antioxidant enzymes such as HO-1, NQO-1 and GCLC. As shown in Fig. 4c, NAS dose-dependently enhanced the expression of the enzymes at 12 h. Meanwhile, in early stage (2 h), it enhanced nuclear translocation of Nrf2 while decreasing the Nrf2 level in cytosol (Fig. 4d). It is suggested that NAS can promote the expression of antioxidant enzymes by activating nuclear translocation of Nrf2. Consequently, NAS possesses strong antioxidant activity.
Fig. 4.

Effect of N-acetyl serotonin on ROS generation, glutathione deprivation, antioxidant enzymes and Nrf2 translocation. Experimental proceedings were described in Fig. 1 or Fig. 3. The levels of intracellular ROS and GSH were measured as described in materials and methods. Data are the mean±SD values of triple determinations. Separately, the expression of HO-1, NQO-1, GCLC, Nrf2, Lamin B or β-actin was determined as described in materials and methods. Similar results were obtained in three independent experiments. *P<0.05 and **P<0.01 versus Glutamate-treated group. A, ROS; B, GSH; C, HO-1, NQO-1 and GCLC; D, nuclear and cytosolic Nrf2.
3.4. Effect of N-acetyl serotonin on activation of intermediate proteins
Since we observed that NAS regulated expression of antioxidant enzymes by translocating Nrf2 into nucleus, we were interested in the intermediate protein related to nuclear translocation of Nrf2. Thus, we further examined effect of NAS on activation of intermediate proteins such as ERK, JNK, p38 or Akt, because the activation of the proteins is known to be related to Nrf2 translocation in neuronal cells [2]. When HT-22 cells were preincubated with NAS in combination with each inhibitor of ERK (PD98059), JNK (SP600125), p38 (SB203580) or Akt (MK-2206) before glutamate treatment, SP600125, SB203580 or MK-2206 significantly neutralized neuroprotective effect of NAS, but not PD98059 (Fig. 5a). Especially, MK-2206 among those inhibitors remarkably reversed the action of NAS. Moreover, it significantly reduced nuclear translocation of Nrf2 induced by NAS, and increased the levels of cytosolic Nrf2 (Fig. 5b). Collectively, these results suggest that the activation of Akt is closely associated with nuclear translocation of Nrf2 in neuronal cells. Therefore, NAS can regulate expression of antioxidant enzymes mainly through activation of Akt/Nrf2 pathway.
Fig. 5.

Effects of inhibitors of intermediate proteins on cell viability and nuclear translocation of Nrf2. HT-22 cells were preincubated with or without N-acetyl serotonin in combination with each inhibitor of intermediate proteins for 30 min before glutamate challenge. After 12 h, cell viability was measured as described in materials and methods. Data are the mean±SD values of triple determinations. Separately, HT-22 cells were preincubated with or without N-acetyl serotonin and MK-2206 (5 μM) for 30 min before glutamate challenge. After 2 h, the expression of Nrf2 or Lamin B was determined as described in materials and methods. Similar results were obtained in three independent experiments. *P<0.05 and **P<0.01, or #P<0.05 and ##P<0.01 versus Glutamate-treated group, or glutamate with NAS-treated group, respectively. A, cell viability; B, nuclear and cytosolic Nrf2.
3.5. Protective effect of N-acetyl serotonin on memory and neuronal cells in scopolamine-treated mice
Finally, after we demonstrated that NAS possessed neuroprotective properties by activating BDNF/TrkB/CREB pathway, and promoting Akt/Nrf2/antioxidant enzymes pathway in in vitro tests, we evaluated the action of NAS in scopolamine-induced memory impairment model, an animal model of Alzheimer's disease [5], [6]. When mice were orally administrated with NAS (1 or 30 mg/kg) 30 min prior to scopolamine challenge, NAS at 30 mg/kg not only reduced the escape latency time (Fig. 6a), but also improved the number of platform (Fig. 6b) as well as the latency time (Fig. 6c) in scopolamine-induced memory impairment in mice. Moreover, NAS at the same dose enhanced the number of neuronal cells in CA1 (Fig. 7a) and CA3 regions (Fig. 7b). Taken together, NAS exerts neuroprotective action by protecting neuronal cells in the animal model of Alzheimer's disease.
Fig. 6.

Protective effect of N-acetyl serotonin on memory in scopolamine-treated mice. Mice were orally administrated with NAS (1 or 30 mg/kg) before scopolamine treatment (2 mg/kg, intraperitoneally). After 1 h, the animals were tested for passive avoidance (Acquisition trial) or Morris water maze. Data are the mean±SEM values of sextuple determinations. **P<0.01 versus Scopolamine-treated group. A, Escape latency time; B, number of platform area crossing; C, Latency time.
Fig. 7.

Protective effect of N-acetyl serotonin on neuronal cells in hippocampal CA1 and CA3 regions of scopolamine-treated mice. Experimental proceedings were described in Fig. 6. Neuronal cells staining in tissue sections were determined as described in materials and methods. Similar results were obtained in three independent experiments. **P<0.01 versus Scopolamine-treated group. A, CA1 region; B, CA3 region.
4. Discussion
NAS, which is known to be a melatonin precursor [10] is an endogeneous N-acyl serotonin, and possesses some bioactive effects such as antioxidant activity [14], anti-ischemia [15], [16] and TrkB activation [17]. Nonetheless, the effect of NAS on antioxidant enzymes in neuronal cells is unknown. Recently, we reported that N-acyl serotonins exerted neuroprotection against oxidative stress-induced cytotoxicity by activating antioxidant enzymes [2]. Above all, N-palmitoyl serotonin was known to possess strong antioxidant activity. However, its clinical application is limited because it has been not tested clinically yet. Instead, we turned our interest to the action of NAS, because NAS is one of N-acyl serotonin, and one of the 1040 FDA-approved compounds [16]. In this respect, NAS may be used for clinical study for neurodegenerative diseases.
Glutamate cytotoxicity in nerve cells is associated with ROS generation through receptor-mediated excitotoxicity or non-receptor-mediated oxidative toxicity [2], [3], and is involved in neuronal apoptosis including caspase-dependent and independent signaling cascades [21], [22]. In this respect, it was supposed that the neuroprotective action of NAS might be related to regulation of pro-apoptotic factors induced by glutamate in neuronal cells. In support of this, NAS not only reduced formation of apoptotic cells, but also restored mitochondrial membrane potential by regulating pro-apoptotic factors such as Bax, calpain, cytochrome c, cleaved caspase-3 and AIF as well as anti-apoptotic factor such as Bcl-2 in glutamate-treated neuronal cells. All these results support the above hypothesis.
Concerning neuroprotective action of NAS, one possible mechanism may be related to TrkB/CREB/BDNF pathway. In support of this, NAS is known to induce TrkB activation [17] and BDNF formation [23]. In addition, BDNF is known to activate Trk receptors, and to protect neuronal cells from oxidative stress-induced cell death [24], [25], [26]. Moreover, we found that N-palmitoyl serotonin promoted activation of TrkB and formation of BDNF in glutamate-treated neuronal cells (Unpublished data) as well as in scopolamine-treated mice [9]. Actually, in this study, NAS also led to the activation of TrkB/CREB/BDNF pathway in glutamate-treated HT-22 cells, as well as the improvement of memory and the increase in the number of neuronal cells in scopolamine-treated mice. Based on these, it is suggested that an important mechanism for neuroprotection of NAS may be due to the activation of TrkB/CREB/BDNF pathway in neuronal cells.
Another possible mechanism for neuroprotection of NAS may be associated with the regulation of antioxidant enzymes. In support of this, NAS is known to possess antioxidant activity in in vitro and in vivo tests [14], [16]. Consistent with this, we reported that N-acyl serotonins reduced ROS generation, and enhanced expression of antioxidant enzymes in neuronal cells and the brain tissue of mice [2], [9]. Such effect of N-acyl serotonins is concerned with nuclear translocation of Nrf2 [2]. In the present study, NAS not only inhibited ROS generation, but also enhanced expression of antioxidant enzymes through activation of Akt/Nrf2 pathway in neuronal cells. Therefore, it is suggested that NAS may activate expression of antioxidant enzymes mainly through activation of Akt/Nrf2 pathway. Such effect of NAS is another important mechanism for its neuroprotective actions, although the expression of antioxidant enzymes by NAS is not clarified in the relation with TrkB signaling pathway. Although NAS shows a neuroprotective action similar to that of N-palmitoyl serotonin, the effective concentration of NAS is much higher than that of N-palmitoyl serotonin. Thus, there may be a great difference in the fate of NAS. In this respect, the further study on the metabolism or pharmacokinetics of N-acyl serotonins may be necessary to reveal the efficacy of NAS and other N-acyl serotonins in in vivo system.
5. Conclusions
The present study demonstrates that NAS exerts neuroprotective actions by regulating activation of both TrkB/CREB/BDNF pathway and Akt/Nrf2/antioxidant enzymes pathway. These findings may reveal a new feature for the mechanism of NAS in preventing neuronal cells against oxidative stress-induced cytotoxicity. Such effects may provide further information regarding the application of NAS with BDNF-promoting and antioxidant enzyme-promoting actions as a neuroprotective agent. Further, other animal models of neurodegenerative diseases may be required to confirm the neuroprotective actions of NAS with BDNF-promoting and antioxidant enzyme-promoting actions for potential clinical purpose.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (2014R1A2A2A04005236).
References
- 1.Maltsev A.V., Bystryak S., Galzitskaya O.V. The role of beta-amyloid peptide in neurodegenerative diseases. Ageing Res. Rev. 2011;10(4):440–452. doi: 10.1016/j.arr.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Jin M.C., Yoo J.M., Sok D.E., Kim M.R. Neuroprotective effect of N-acyl 5-hydroxytryptamines on glutamate-induced cytotoxicity in HT-22 cells. Neurochem. Res. 2014;39(12):2440–2451. doi: 10.1007/s11064-014-1448-2. [DOI] [PubMed] [Google Scholar]
- 3.Newell D.W., Barth A., Papermaster V., Malouf A.T. Glutamate and non-glutamate receptor mediated toxicity caused by oxygen and glucose deprivation in organotypic hippocampal cultures. J. Neurosci.: Off. J. Soc. Neurosci. 1995;15(11):7702–7711. doi: 10.1523/JNEUROSCI.15-11-07702.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klinkenberg I., Blokland A. The validity of scopolamine as a pharmacological model for cognitive impairment: a review of animal behavioral studies. Neurosci. Biobehav. Rev. 2010;34(8):1307–1350. doi: 10.1016/j.neubiorev.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Wiener N.I., Messer J. Scopolamine-induced impairment of long-term retention in rats. Behav. Biol. 1973;9(2):227–234. doi: 10.1016/s0091-6773(73)80157-9. [DOI] [PubMed] [Google Scholar]
- 6.Dennis S.G. Temporal aspects of scopolamine-induced one-way memory dissociation in mice. J. Comp. Physiol. Psychol. 1974;86(6):1052–1058. doi: 10.1037/h0037650. [DOI] [PubMed] [Google Scholar]
- 7.Petersen R.C. Scopolamine induced learning failures in man. Psychopharmacology. 1977;52(3):283–289. doi: 10.1007/BF00426713. [DOI] [PubMed] [Google Scholar]
- 8.Giridharan V.V., Thandavarayan R.A., Konishi T. Amelioration of scopolamine induced cognitive dysfunction and oxidative stress by Inonotus obliquus - a medicinal mushroom. Food Funct. 2011;2(6):320–327. doi: 10.1039/c1fo10037h. [DOI] [PubMed] [Google Scholar]
- 9.Min A.Y., Doo C.N., Son E.J., Sung N.Y., Lee K.J., Sok D.E., Kim M.R. N-palmitoyl serotonin alleviates scopolamine-induced memory impairment via regulation of cholinergic and antioxidant systems, and expression of BDNF and p-CREB in mice. Chem. Biol. Interact. 2015;242:153–162. doi: 10.1016/j.cbi.2015.09.016. [DOI] [PubMed] [Google Scholar]
- 10.Sandrock A.W., Jr., Leblanc G.G., Wong D.L., Ciaranello R.D. Regulation of rat pineal hydroxyindole-O-methyltransferase: evidence of S-adenosylmethionine-mediated glucocorticoid control. J. Neurochem. 1980;35(3):536–543. doi: 10.1111/j.1471-4159.1980.tb03688.x. [DOI] [PubMed] [Google Scholar]
- 11.Voisin P., Namboodiri M.A., Klein D.C. Arylamine N-acetyltransferase and arylalkylamine N-acetyltransferase in the mammalian pineal gland. J. Biol. Chem. 1984;259(17):10913–10918. [PubMed] [Google Scholar]
- 12.Jackson R.L., Lovenberg W. Isolation and characterization of multiple forms of hydroxyindole-O-methyltransferase. J. Biol. Chem. 1971;246(13):4280–4285. [PubMed] [Google Scholar]
- 13.Pevet P., Balemans M.G., Legerstee W.C., Vivien-Roels B. Circadian rhythmicity of the activity of hydroxyindole-O-methyl transferase (HIOMT) in the formation of melatonin and 5-methoxytryptophol in the pineal, retina, and harderian gland of the golden hamster. J. Neural Transm. 1980;49(4):229–245. doi: 10.1007/BF01252128. [DOI] [PubMed] [Google Scholar]
- 14.Wolfler A., Abuja P.M., Schauenstein K., Liebmann P.M. N-acetylserotonin is a better extra- and intracellular antioxidant than melatonin. FEBS Lett. 1999;449(2–3):206–210. doi: 10.1016/s0014-5793(99)00435-4. [DOI] [PubMed] [Google Scholar]
- 15.Ben Shahar Y., Sukhotnik I., Bitterman N., Pollak Y., Bejar J., Chepurov D., Coran A., Bitterman A. Effect of N-Acetylserotonin on Intestinal Recovery Following Intestinal Ischemia-Reperfusion Injury in a Rat. Eur. J. Pediatric Surg. 2016;26(1):47–53. doi: 10.1055/s-0035-1559886. [DOI] [PubMed] [Google Scholar]
- 16.Zhou H., Wang J., Jiang J., Stavrovskaya I.G., Li M., Li W., Wu Q., Zhang X., Luo C., Zhou S., Sirianni A.C., Sarkar S., Kristal B.S., Friedlander R.M., Wang X. N-acetyl-serotonin offers neuroprotection through inhibiting mitochondrial death pathways and autophagic activation in experimental models of ischemic injury. J. Neurosci.: Off. J. Soc. Neurosci. 2014;34(8):2967–2978. doi: 10.1523/JNEUROSCI.1948-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jang S.W., Liu X., Pradoldej S., Tosini G., Chang Q., Iuvone P.M., Ye K. N-acetylserotonin activates TrkB receptor in a circadian rhythm. Proc. Natl. Acad. Sci. USA. 2010;107(8):3876–3881. doi: 10.1073/pnas.0912531107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maher P., Davis J.B. The role of monoamine metabolism in oxidative glutamate toxicity. J. Neurosci.: Off. J. Soc. Neurosci. 1996;16(20):6394–6401. doi: 10.1523/JNEUROSCI.16-20-06394.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chapin R.E., George J.D., Lamb J.Ct. Reproductive toxicity of tricresyl phosphate in a continuous breeding protocol in Swiss (CD-1) mice. Fundam. Appl. Toxicol.: Off. J. Soc. Toxicol. 1988;10(2):344–354. doi: 10.1016/0272-0590(88)90320-x. [DOI] [PubMed] [Google Scholar]
- 20.Khan A., Gillis K., Clor J., Tyagarajan K. Simplified evaluation of apoptosis using the Muse cell analyzer. Post. Biochem. 2012;58(4):492–496. [PubMed] [Google Scholar]
- 21.Xu X., Chua C.C., Kong J., Kostrzewa R.M., Kumaraguru U., Hamdy R.C., Chua B.H. Necrostatin-1 protects against glutamate-induced glutathione depletion and caspase-independent cell death in HT-22 cells. J. Neurochem. 2007;103(5):2004–2014. doi: 10.1111/j.1471-4159.2007.04884.x. [DOI] [PubMed] [Google Scholar]
- 22.Antipova T.A., Sapozhnikova D.S., Stepanichev M.Y., Onufriev M.V., Gulyaeva N.V., Seredenin S.B. Effects of selective anxiolytic afobazole on active caspase-3. Bull. Exp. Biol. Med. 2010;149(2):201–203. doi: 10.1007/s10517-010-0907-2. [DOI] [PubMed] [Google Scholar]
- 23.Yoo D.Y., Nam S.M., Kim W., Lee C.H., Won M.H., Hwang I.K., Yoon Y.S. N-acetylserotonin increases cell proliferation and differentiating neuroblasts with tertiary dendrites through upregulation of brain-derived neurotrophic factor in the mouse dentate gyrus. J. Vet. Med. Sci. 2011;73(11):1411–1416. doi: 10.1292/jvms.11-0123. [DOI] [PubMed] [Google Scholar]
- 24.Pringle A.K., Sundstrom L.E., Wilde G.J., Williams L.R., Iannotti F. Brain-derived neurotrophic factor, but not neurotrophin-3, prevents ischaemia-induced neuronal cell death in organotypic rat hippocampal slice cultures. Neurosci. Lett. 1996;211(3):203–206. doi: 10.1016/0304-3940(96)12745-2. [DOI] [PubMed] [Google Scholar]
- 25.Almeida R.D., Manadas B.J., Melo C.V., Gomes J.R., Mendes C.S., Graos M.M., Carvalho R.F., Carvalho A.P., Duarte C.B. Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 2005;12(10):1329–1343. doi: 10.1038/sj.cdd.4401662. [DOI] [PubMed] [Google Scholar]
- 26.Rossler O.G., Giehl K.M., Thiel G. Neuroprotection of immortalized hippocampal neurones by brain-derived neurotrophic factor and Raf-1 protein kinase: role of extracellular signal-regulated protein kinase and phosphatidylinositol 3-kinase. J. Neurochem. 2004;88(5):1240–1252. doi: 10.1046/j.1471-4159.2003.02255.x. [DOI] [PubMed] [Google Scholar]