

Abstract
Hermansky-Pudlak syndrome (HPS) is a rare autosomal recessive disorder characterized by oculocutaneous albinism and a lack of dense granules in platelets. HPS types 1 and 4 are associated with a granulomatous enterocolitis that is phenotypically indistinguishable from Crohn’s disease. We present two cases of HPS-associated Crohn’s disease phenotype in which the patients were refractory to standard medical management. The pathophysiology of HPS is mediated by single-gene defects that alter endosome trafficking, and we hypothesize that this mechanism leads to the observed association with a CD phenotype.
Introduction
Hermansky-Pudlak syndrome (HPS) is a genetic disorder of lysosome-related organelles characterized by oculocutaneous albinism and platelet dysfunction. HPS is associated with granulomatous enterocolitis that pathologically and phenotypically resembles Crohn’s disease (CD).1 The reason for this association is unknown, although the specific genetic defects that cause HPS have been characterized.
Case Report
Case 1: A 27-year-old Puerto Rican man with HPS and CD phenotype presented with abdominal pain and hematochezia. He was diagnosed with CD-like granulomatous enterocolitis at age 11. He was reportedly treated with infliximab and azathioprine with good response. After 6 years of treatment, he was lost to follow-up. He was not on any medications between ages 17 and 27. He had occasional abdominal pain and hematochezia that would self-resolve. Two months prior to presentation he developed constant severe abdominal pain, hematochezia, and weight loss.
On physical exam, he demonstrated oculocutaneous albinism, a diffusely tender and distended abdomen, and painful anal fissures. Lab results showed hemoglobin 7.2 g/dL, C-reactive protein (CRP) 87 mg/L, and albumin 2.2 g/dL. Sigmoidoscopy showed continuous ulceration and stenosis of the rectosigmoid. Subsequent magnetic resonance enterography showed multifocal colorectal stenoses and transverse colon dilation to 11 cm.
He was given intravenous steroids, which did not resolve his abdominal pain or hematochezia. He underwent laparoscopic loop ileostomy due to colonic distention and steroid refractory disease. He tolerated the operation well, with no significant blood loss. His abdominal pain resolved, and he was discharged with subsequent initiation of infliximab and azathioprine.
Eight weeks later, he presented with massive rectal hemorrhage and underwent an emergent total proctocolectomy. His colon specimen showed hemorrhagic mucosa and diffuse ulceration (Figure 1). Histologic evaluation showed non-caseating granulomas (Figure 2).
Figure 1.
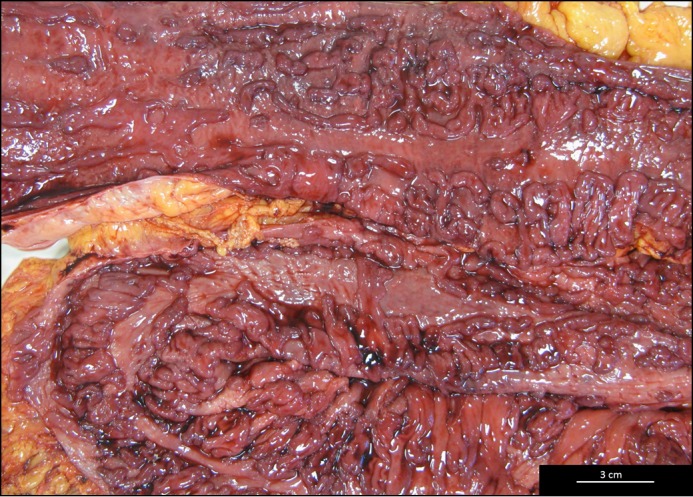
Gross appearance of colon after emergent colectomy showing circumferential ulceration of distal 90 cm.
Figure 2.

Colon biopsy histology showing a cluster of non-caseating granulomata (hematoxylin and eosin x40).
Case 2: A 25-year-old Puerto Rican male with HPS and CD phenotype with perianal disease presented with rectal pain, hematochezia, and urgency. He had been maintained on infliximab for 5 years, but then he developed breakthrough symptoms despite dose escalation. He eventually developed urgency and rectal bleeding that did not respond to infliximab therapy.
On physical exam he demonstrated oculocutaneous albinism, nontender abdomen, and painful anal fissures. Lab results showed hemoglobin 12.1 g/dL, CRP 38 mg/L, and albumin 4.3 g/dL. His infliximab level was 18 μg/mL (≥0.4 μg/mL indicates detection of infliximab), and his anti-infliximab antibodies level was 463 ng/mL (≥22 ng/mL indicates detection of antibodies to infliximab). Sigmoidoscopy showed severe, continuous inflammation of the rectosigmoid (Figure 3). Pathology demonstrated cryptitis with crypt abscesses, dense inflammation with loss of normal architecture, ulcerations, and melanosis coli (Figure 4).
Figure 3.
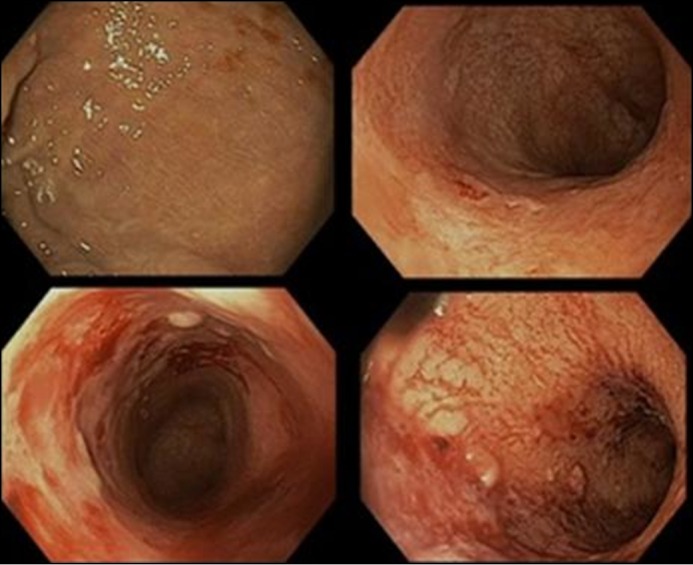
Severe ulcerations with spontaneous bleeding of distal 30 cm of colon.
Figure 4.

Rectosigmoid biopsy histology showing melanosis coli representing lipofuscin deposition (hematoxylin and eosin x10).
He was changed to adalimumab and azathioprine but did not have any response. Adalimumab was discontinued, and he started tacrolimus. He had clinical response with normalization of his CRP to <3 mg/L but developed rising creatinine. He continued azathioprine and started vedolizumab. On follow-up at 12 weeks, he had improved urgency and occasional blood in his stool, but his CRP was elevated to 19 mg/L.
Discussion
Originally described in 1959 by Drs. Hermansky and Pudlak, HPS is now known to be a rare autosomal recessive disease of lysosome-related organelles.2 There are several subtypes, but all share oculocutaneous albinism and platelet dysfunction.1 Different subtypes have associated conditions and differ in the affected gene.3 HPS type 1 is the most common subtype and is associated with Puerto Rican heritage due to a founder mutation in this population. HPS types 1 and 4 are associated with granulomatous enterocolitis that is phenotypically similar to CD.1,4,5 A case series of 4 patients reported that 3 of 4 patients had perianal disease, all were treated with infliximab, and 2 of 4 patients had complete response to infliximab at 6 weeks.6 Similar to the patient in Case 1, two of their patients underwent colectomy, one of which was due to severe acute rectal hemorrhage. In a case series of 8 HPS patients with colitis seen on colonoscopy, 1 patient showed perianal disease and 2 patients were treated with infliximab.1 The patients apparently responded well, but the duration of follow-up on infliximab was not reported. Both of our patients were previously on infliximab, but the patient in Case 2 lost response while the patient in Case 1 did not respond on re-exposure to infliximab.
It is not clear from the known pathophysiology of HPS whether conventional therapy for CD is as effective in HPS-associated enterocolitis as in CD. Therapeutic agents that target tumor necrosis factor α (TNFα) and cell adhesion molecules are not likely targeting the specific defect and may be overwhelmed by proinflammatory conditions. This may explain the difficulty of achieving and maintaining response to conventional therapies in HPS-associated enterocolitis. In Case 2, the treating gastroenterologist had significant experience using calcineurin inhibitors to induce remission as an alternative, steroid-sparing strategy given the patient’s poor response to an anti-TNFα strategy. Because of these poor responses to treatment, some clinicians argue for aggressive surgical management of HPS-associated CD-like colitis and perianal disease.7
Prior case reports have focused on the presence of ceroid lipofuscin as a possible explanation for HPS-associated CD phenotype, but the precise mechanism remains unknown. We propose an alternative hypothesis based on recent advances in the understanding of the pathophysiology of both HPS and CD. HPS type 1 is a result of a mutation in Hps1, which encodes the HPS1 protein. Similarly, HPS type 4 is caused by a mutation in Hps4, which encodes the HPS4 protein. HPS1 and HPS4 form a protein complex called biogenesis of lysosome-related organelles complex 3 (BLOC-3).8,9 BLOC-3 mediates endosome trafficking through effects on Rab GTP-ase proteins, in particular Rab32 and Rab38.10 BLOC-3-mediated dysfunction of Rab32 and Rab38 underlies the phenotype of albinism and absent platelet dense granules in HPS.10,11
An absence of platelet dense bodies leads to fewer procoagulant small molecules expressed from platelets, but it is not clear whether the absence of these small molecules from platelets directly affects disease manifestations on the cellular level. Impaired coagulation likely increases disease severity and complicates colonic hemorrhages as seen in Case 1 and in prior case reports. In addition to its effect on phenotype, Rab32 is also thought to impart an antibacterial effect to gut epithelial lysosomes. In fact, Salmonella typhi secretes a protein that proteolytically targets Rab32, allowing it to survive within host macrophages.12 Additional studies show a role for Rab32 in the cellular reaction to other intracellular pathogens, including Listeria monocytogenes and Mycobacterium leprae.13
Impaired endosomal trafficking is also implicated in the pathogenesis of CD. Rab13 is an endosomal trafficking protein that is responsible for tight junction maintenance in gut epithelium and is abnormally localized in CD.14 Additionally, the ATG16L1 gene is involved in autophagy and phagocytosis, and this has a functional effect on inflammation. CD patients with the risk allele ATG16L1-T300A had evidence of altered mucosal bacteria composition in inflamed tissue compared to the protective allele ATG16L1-T300.15 There is currently no evidence to suggest that BLOC-3 interacts directly with Rab13 or ATG16L1; however, they underscore the importance of endosome trafficking in the pathophysiology of Crohn’s disease. We speculate that the BLOC-3-mediated dysfunction of the Rab proteins propagates the mucosal inflammation indistinguishable from CD in HPS types 1 and 4.
We presented two cases of HPS associated with CD phenotype whose clinical courses and pathologic findings demonstrate the typical characteristics of these diseases. The involvement of endosome trafficking in both HPS and CD warrants further investigation. Defining this connection could help explain the pathogenesis of both diseases and elucidate possible therapeutic targets that may lead to novel drug therapies with distinct mechanism of action.
Disclosures
Author contributions: MA Sofia prepared the manuscript and figures, and is the article guarantor. A. Sakuraba and DT Rubin prepared the manuscript.
Financial disclosure: None to report.
Informed consent was obtained for this case report.
Previous presentation: This case was presented at the 2015 ACG Annual Meeting; October 19–21, 2015; Honolulu, Hawaii.
References
- 1.Hussain N, Quezado M, Huizing M, et al. . Intestinal disease in Hermansky-Pudlak syndrome: Occurrence of colitis and relation to genotype. Clin Gastroenterol Hepatol. 2006; 4(1):73–80. [DOI] [PubMed] [Google Scholar]
- 2.Hermansky F, Pudlak P. Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow: Report of two cases with histochemical studies. Blood. 1959; 14(2):162–9. [PubMed] [Google Scholar]
- 3.Salvaggio HL, Graeber KE, Clarke LE, Schlosser BJ, Orlow SJ, Clarke JT. Mucocutaneous granulomatous disease in a patient with Hermansky-Pudlak syndrome. JAMA Dermatol. 2014; 150(10):1083–7. [DOI] [PubMed] [Google Scholar]
- 4.Mora AJ, Wolfsohn DM. The management of gastrointestinal disease in Hermansky-Pudlak syndrome. J Clin Gastroenterol. 2011; 45(8):700–2. [DOI] [PubMed] [Google Scholar]
- 5.Thrash B, Patel M, Shah KR, Boland CR, Menter A. Cutaneous manifestations of gastrointestinal disease: Part II. J Am Acad Dermatol. 2013; 68(2):211.e1–211.e33. [DOI] [PubMed] [Google Scholar]
- 6.Grucela AL, Patel P, Goldstein E, Palmon R, Sachar DB, Steinhagen RM. Granulomatous enterocolitis associated with Hermansky-Pudlak syndrome. Am J Gastroenterol. 2006; 101(9):2090–5. [DOI] [PubMed] [Google Scholar]
- 7.Hazzan D, Seward S, Stock H, et al. . Crohn's-like colitis, enterocolitis and perianal disease in Hermansky-Pudlak syndrome. Colorectal Dis. 2006; 8:539–43. [DOI] [PubMed] [Google Scholar]
- 8.Martina JA, Moriyama K, Bonifacino JS. BLOC-3, a protein complex containing the Hermansky-Pudlak syndrome gene products HPS1 and HPS4. J Biol Chem. 2003; 278(31):29376–84. [DOI] [PubMed] [Google Scholar]
- 9.Nazarian R, Falcón-Pérez JM, Dell'Angelica EC. Biogenesis of lysosome-related organelles complex 3 (BLOC-3): A complex containing the Hermansky-Pudlak syndrome (HPS) proteins HPS1 and HPS4. Proc Natl Acad Sci U S A. 2003; 100(15):8770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerondopoulos A, Langemeyer L, Liang JR, Linford A, Barr FA. BLOC-3 mutated in Hermansky-Pudlak syndrome is a Rab32/38 guanine nucleotide exchange factor. Curr Biol. 2012; 22(22):2135–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ninkovic I, White JG, Rangel-Filho A, Datta YH. The role of Rab38 in platelet dense granule defects. J Thromb Haemost. 2008; 6(12):2143–51. [DOI] [PubMed] [Google Scholar]
- 12.Spano S, Galan JE. A Rab32-dependent pathway contributes to Salmonella typhi host restriction. Science. 2012; 338(6109):960–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solano-Collado V, Rofe A, Spanò S. Rab32 restriction of intracellular bacterial pathogens [published online Sep 20, 2016]. Small GTPases. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohira M, Oshitani N, Hosomi S, et al. . Dislocation of Rab13 and vasodilator-stimulated phosphoprotein in inactive colon epithelium in patients with Crohn's disease. Int J Mol Med. 2009; 24(6):829–35. [DOI] [PubMed] [Google Scholar]
- 15.Sadaghian Sadabad M, Regeling A, de Goffau MC, et al. . The ATG16L1-T300A allele impairs clearance of pathosymbionts in the inflamed ileal mucosa of Crohn's disease patients. Gut. 2015; 64(10):1546–52. [DOI] [PubMed] [Google Scholar]