

Abstract
Proteomes of even well characterized organisms still contain a high percentage of proteins with unknown or uncertain molecular and/or biological function. A significant fraction of those proteins is predicted to have catalytic properties. Here we aimed at identifying the function of the Saccharomyces cerevisiae Ydr109c protein and its human homolog FGGY, both of which belong to the broadly conserved FGGY family of carbohydrate kinases. Functionally identified members of this family phosphorylate 3- to 7-carbon sugars or sugar derivatives, but the endogenous substrate of S. cerevisiae Ydr109c and human FGGY has remained unknown. Untargeted metabolomics analysis of an S. cerevisiae deletion mutant of YDR109C revealed ribulose as one of the metabolites with the most significantly changed intracellular concentration as compared with a wild-type strain. In human HEK293 cells, ribulose could only be detected when ribitol was added to the cultivation medium, and under this condition, FGGY silencing led to ribulose accumulation. Biochemical characterization of the recombinant purified Ydr109c and FGGY proteins showed a clear substrate preference of both kinases for d-ribulose over a range of other sugars and sugar derivatives tested, including l-ribulose. Detailed sequence and structural analyses of Ydr109c and FGGY as well as homologs thereof furthermore allowed the definition of a 5-residue d-ribulokinase signature motif (TCSLV). The physiological role of the herein identified eukaryotic d-ribulokinase remains unclear, but we speculate that S. cerevisiae Ydr109c and human FGGY could act as metabolite repair enzymes, serving to re-phosphorylate free d-ribulose generated by promiscuous phosphatases from d-ribulose 5-phosphate. In human cells, FGGY can additionally participate in ribitol metabolism.
Keywords: carbohydrate metabolism, enzyme kinetics, mass spectrometry (MS), metabolomics, pentose phosphate pathway (PPP), protein motif, Saccharomyces cerevisiae, structural model, FGGY carbohydrate kinase family, ribulose
Introduction
A major challenge in the post-genomic era is that a large fraction of protein-coding genes remain functionally unknown or poorly characterized in all sequenced genomes (1, 2). Even in a well characterized organism such as Saccharomyces cerevisiae, the number of protein-coding genes with no known biological function, based on database searches in UniProt, amounts to ∼30%, which corresponds to about 2000 proteins. In this study, we investigated the function of two proteins of unknown function, the S. cerevisiae Ydr109c protein (Q04585) and its human homolog FGGY (Q96C11). Both proteins contain the highly conserved FGGY_N and FGGY_C Pfam domains. The members of the FGGY family of carbohydrate kinases, of which more than 8000 sequences are known according to the Pfam database, are widespread across the various kingdoms of life and show a high functional diversification (3). They phosphorylate C3 to C7 sugars or sugar derivatives, and a divergent subfamily of the FGGY protein family is involved in quorum sensing by phosphorylating the signaling molecule autoinducer-2 (AI-2, a furanosyl borate diester) (3). There are seven Swiss-Prot-reviewed FGGY domain-containing proteins encoded by the human genome as follows: sedoheptulokinase (Q9UHJ6); xylulose kinase (O75191); glycerol kinase (P32189); glycerol kinase 2 (Q14410); putative glycerol kinase 3 (Q14409); putative glycerol kinase 5 (Q6ZS86); and FGGY carbohydrate kinase domain-containing protein (Q96C11; designated hereafter as “FGGY”). The S. cerevisiae genome encodes four Swiss-Prot-reviewed FGGY domain-containing proteins: xylulose kinase (P42826); glycerol kinase (P32190); Mpa43 (P53583); and Ydr109c (Q04585). The motivation behind this study was the existence of a functionally uncharacterized carbohydrate kinase (Ydr109c) in S. cerevisiae with a homologous protein in humans (FGGY), which has been linked to S-ALS2 and bipolar disorder.
The first study reporting an FGGY association to S-ALS was by Dunckley et al. (4). Performing a genome-wide association study comparing healthy controls and S-ALS patients of European Caucasian descent living in the United States, the authors reported 10 statistically significant single nucleotide polymorphisms associated with S-ALS. The most significant gene associated with S-ALS was FGGY (FLJ10986). Assessment of FGGY expression in the same study using Western blotting indicated the presence of FGGY protein in cerebrospinal fluid, spinal cord, small intestine, lung, kidney, liver, and fetal brain. Association of the FGGY gene with S-ALS could, however, not be confirmed in subsequent studies using different cohorts (5–9). The contradictory results on the involvement of FGGY in S-ALS were suggested to be due to the variable causes and complexity of the disease itself (10). An exome sequencing study, which was carried out in a family with three female patients affected by bipolar disorder and one unaffected male sibling, identified heterozygous, very rare, and likely protein-damaging variants in eight brain-expressed genes, including FGGY (11). These variants were shared by the three affected siblings but were not present in the unaffected sibling and in more than 200 controls. Replication and functional studies would, however, be required to confirm disease association and test causality, respectively, of the identified variants. Although these observations suggest a possible link of the FGGY gene with neurodegenerative or psychiatric disorders, the overall evidence supporting this link thus remains limited at this stage.
In recent years, metabolomics has emerged as a new tool for discovery of enzyme function. Untargeted metabolomics has enabled us to analyze metabolites in biological samples in a much more comprehensive way and is a powerful technique for hypothesis generation. A remaining challenge of this methodology is metabolite identification; the data obtained via untargeted metabolomics contains thousands of metabolite features, with relatively few being identified in the end (12). In contrast to untargeted metabolomics, targeted metabolomics serves to identify and/or quantify a more or less limited set of preselected metabolites. In the field of enzymology, metabolomics or metabolite profiling techniques may be exploited to identify endogenous enzyme substrates. Ewald et al. (13) studied the effect of single enzymatic gene deletions in central carbon metabolism and of environmental changes on the metabolome of S. cerevisiae. 30–40% of the enzymatic gene deletions tested led to a very local metabolic response in proximity of the enzyme deficiency (often accumulation of the substrate of the deleted enzyme) (13). The observations suggest that this approach is a viable strategy for enzyme function identifications through comparative metabolomics analyses of wild-type cells and cells deficient in metabolic enzymes of unknown function. A notable advantage of this type of approach over in vitro substrate screens with purified enzymes is the higher likelihood of identifying the true physiological or endogenous substrate(s) of the deleted enzyme under investigation (14). Two recent examples of enzyme identifications in connection with the pentose phosphate pathway and using LC-MS-based metabolite profiling in samples derived from enzyme-deficient organisms are yeast sedoheptulose 1,7-bisphosphatase (SHB17) (15) and mammalian sedoheptulokinase (SHPK) (16).
LC-MS-based metabolite profiling can involve full scan or tandem MS methods. In the full scan MS methods, only the m/z of parent ions and/or adducts of the parent ions are utilized along with the retention time characteristic of each molecule to identify detected metabolites. Tandem MS methods increase the potential for metabolite identification by allowing the generation of m/z fingerprints (MS2 spectra), obtained by fragmenting the parent ions, that can then be matched with those of metabolite standards. We used a combination of untargeted full scan MS, ddMS2, and targeted methods to search for the endogenous substrate of the S. cerevisiae Ydr109c protein using ydr109cΔ knock-out strains. We found that ribulose was one of the most significantly changed metabolites, accumulating in the ydr109cΔ mutants as compared with the wild-type control strains. d-Ribulose was subsequently shown to be the preferred substrate of the yeast Ydr109c kinase as well as for its human homolog FGGY in vitro. In contrast to yeast cells, ribulose formation in human HEK293 cells could only be detected when ribitol was supplemented to the cultivation medium. Under this condition, FGGY knockdown led to ribulose accumulation in the HEK293 cells. Taken together, our results establish the molecular identity of d-ribulokinase in yeast and humans. Furthermore, combined sequence and structural analyses allowed us to identify a conserved signature motif that enables the prediction of d-ribulokinase activity with high confidence for FGGY protein family members.
Results
YDR109C Gene Deletion Leads to Ribulose Accumulation in Different S. cerevisiae Strains
The YDR109C gene is currently annotated as an uncharacterized open reading frame in the Saccharomyces Genome Database (SGD), which means, according to the SGD glossary, that “there are no specific experimental data demonstrating that a gene product is produced in S. cerevisiae.” In addition, no molecular or biological function has been assigned to this gene yet. Therefore, we started by investigating YDR109C expression in our prototrophic WT strain by quantitative RT-PCR. The YDR109C transcript was readily detected in exponentially growing wild-type cells (Fig. 1), with an average measured cycle threshold (Ct) value of 26.3 ± 0.3 (mean ± S.D.; n = 3) as compared with an average measured Ct value of 23.8 ± 0.5 (mean ± S.D.; n = 3) for the mannosyltransferase ALG9, which is commonly used as a reference gene for quantitative RT-PCR studies in S. cerevisiae (17). The YDR109C transcript was not detectable in our ydr109cΔ prototrophic knock-out strain (Fig. 1). These results show that YDR109C is transcribed in S. cerevisiae and indicate that our knock-out strain is a good model to explore the function of this gene. The Ydr109c protein was also detected and quantified (119 molecules/cell) in a proteomics study (18).
FIGURE 1.

Expression levels of the YDR109C gene in the prototrophic yeast strains used in this study. Total RNA was extracted from exponentially growing cells of the WT and ydr109cΔ strains as well as the ydr109cΔ strain transformed with the p41Hyg 1-F::YDR109C plasmid (rescue) or the corresponding empty plasmid. The expression fold change of the YDR109C gene in the indicated strains relative to the WT strain was calculated using the 2−ΔΔCt method. The expression level of the YDR109C gene in each sample was either normalized to reference gene ACT1 or ALG9. Means and standard deviations of three biological replicates are shown.
The Ydr109c protein sequence contains the widely conserved Pfam FGGY_N and FGGY_C domains, suggesting that it functions, as other members of the FGGY protein superfamily, as a kinase acting on sugars or sugar derivatives. To identify endogenous substrate candidates of this putative sugar kinase, we analyzed the polar metabolites extracted from our prototrophic WT and ydr109cΔ strains using LC-HRMS. Two complementary methods, ZIC-HILIC coupled to ddMS2 and reverse phase chromatography coupled to full scan MS with polarity switching, were used. Ribulose was found to be the metabolite with the highest fold change (more than 30-fold increase in KO versus WT), among the ones confirmed to be produced endogenously by the co-cultivation method described below, using the ZIC-HILIC-ddMS2 method in negative ionization mode (Fig. 2A and supplemental Table S1). We repeated the same analysis in metabolite extracts derived from a ydr109cΔ deletion strain in the auxotrophic background BY4741 and the corresponding WT strain. As for the prototrophic strains, ribulose was identified as the most significantly changed metabolite, accumulating in the auxotrophic ydr109cΔ mutant, among the metabolites detected in negative mode (23-fold increase in KO versus WT; supplemental Table S2). Identification of the accumulating compound as ribulose was based on accurate mass (m/z 149.0445), co-elution with a d-ribulose standard (Fig. 2A), and MS2 fragmentation pattern (Fig. 2B). Polar metabolites were separated better using ZIC-HILIC; the bulk of yeast polar metabolites eluted very early with our reverse phase chromatography method and was therefore not used for further experiments in this study. Using the ZIC-HILIC-based method, we were able to separate ribulose from other pentoses (Fig. 2C). We were, however, not able to separate the d- and l-forms of ribulose. Although we could detect a number of other sugars and sugar derivatives, including glucose, arabinose, mannitol, ribitol, maltose, xylose, galactose, and 2-deoxyribose (identification based on accurate mass and co-elution with standards) in the analyzed yeast metabolite extracts, only ribitol showed significantly different levels (>2-fold higher in the prototrophic and auxotrophic KO strains than in the corresponding WT strains; supplemental Tables S1–S4) upon YDR109C deletion in addition to ribulose. Taken together, these analyses highlighted ribulose as a strong endogenous substrate candidate for the putative Ydr109c kinase.
FIGURE 2.

Ribulose accumulates in an S. cerevisiae ydr109cΔ strain. A, extracted ion chromatograms (m/z 149.0445) obtained after ZIC-HILIC-MS analysis of ydr109cΔ and wild-type metabolite extracts as well as a d-ribulose analytical standard. B, head-to-tail comparison of the MS2 fragmentation pattern of the parent ion m/z 149.0445 detected in the ydr109cΔ extract at retention time of 4.77 min and the d-ribulose standard. C, extracted ion chromatograms (m/z 149.0445) of pentose and pentulose analytical standards analyzed by ZIC-HILIC-MS. The d- and l-forms of the sugars were not separable using ZIC-HILIC chromatography; all other isomers were separable at peak maxima. D, overlay of extracted ion chromatograms (m/z 149.0445 in red and m/z 154.0612 in black) obtained after ZIC-HILIC-MS analysis of a ydr109cΔ metabolite extract generated from cells cultivated in the presence of d-[U-13C]glucose as the sole carbon source and supplemented with non-labeled d-ribulose, d-ribose, or d-xylulose standards.
Free ribulose has not been described so far as an endogenous metabolite in S. cerevisiae, and there is also no entry for ribulose in the YMDB (19). Our detection of ribulose accumulation in yeast strains grown on controlled minimal medium containing d-glucose as the sole carbon source suggested that yeast cells can form free ribulose from d-glucose. We wanted to consolidate this observation via stable isotope labeling (SIL) experiments in which we replaced the non-labeled glucose with d-[U-13C]glucose in an otherwise identical cultivation medium. In these experiments, we observed a +5 m/z shift for the pentose peak (monoisotopic mass of 154.0612) accumulating in the ydr109cΔ mutant and perfectly co-eluting with a supplemented d-[12C]ribulose standard (Fig. 2D). As can also be seen in Fig. 2D, supplemented non-labeled d-xylulose and d-ribose standards, which elute in close proximity to the d-ribulose standard (see Fig. 2C), eluted slightly later than the labeled pentose accumulating in the ydr109cΔ mutant. These results consolidate the identity of the compound building up after deletion of the YDR109C gene as ribulose and show that S. cerevisiae can produce this compound from d-glucose. Using a 13C internal standard isotope dilution MS method and biovolume measurement by Coulter counter, we estimated an intracellular ribulose concentration of 0.054 ± 0.010 and 2.2 ± 0.3 mm (means ± S.D.; n = 6) for the prototrophic WT and ydr109cΔ strains, respectively.
Effect of YDR109C Deletion on Metabolite Levels Other than Ribulose and Ribitol
The ZIC-HILIC-ddMS2 data obtained with the prototrophic strains were further analyzed to investigate whether the levels of additional metabolites were significantly affected in response to YDR109C gene deletion. Principal component analysis (PCA) of the mTIC-normalized negative and positive mode data produced clusters separating WT and ydr109cΔ samples in a PC1 versus PC2 plot in which all the replicates were within the 95% CI of their group centroids (Fig. 3, A and B). Because PCA is an unsupervised visualization method, which is not guaranteed to preserve well the distances between the original untransformed data points, the partial least squares-discriminant analysis (PLS-DA) supervised method was additionally used to investigate the separability between the sample groups and to find features important in differentiating the WT from the ydr109cΔ strain. The data showed clear separation of the WT and ydr109cΔ replicate groups using PLS-DA as well, with again all the replicates lying within the 95% CI of their respective group (Fig. 3, C and D). The supplemental Tables S1–S4 contain a column with variable importance in projection scores for all the listed m/z features, reflecting their importance for PLS-DA separation of the WT and ydr109cΔ samples.
FIGURE 3.

Multivariate statistics analyses of the metabolite profiles obtained for the wild-type and ydr109cΔ prototrophic stains using ZIC-HILIC-MS. In the score plots shown, the green and red oval shapes represent the 95% confidence intervals for the wild-type (WT; green + symbols) and ydr109cΔ (KO; red Δ symbols) replicates, respectively. A, PCA score plot showing principal component 1 (PC1) versus PC2 for the negative mode mTIC normalized data. B, PCA score plot for the positive mode mTIC normalized data. C, PLS-DA score plot showing component 1 versus component 2 for the negative mode mTIC normalized data. Leave-one-out cross-validation statistics were R2 = 0.99 and Q2 = 0.98 for component 1, and R2 = 1.00 and Q2 = 0.99 for component 2. D, PLS-DA score plot for the positive mode mTIC normalized data. Leave-one-out cross-validation statistics were R2 = 0.98 and Q2 = 0.96 for component 1 and R2 = 0.99 and Q2 = 0.96 for component 2. mTIC, metabolic total ion chromatogram.
Fold changes between the prototrophic WT and ydr109cΔ strains for each metabolite feature detected by ZIC-HILIC-ddMS2 and associated p values were calculated using Welch's t test for unequal variances. Unexpectedly, the levels of as many as 92 and 213 non-redundant metabolite features having m/z matches in the KEGG database were found to be changed at least 2-fold and with a p value lower than 0.05 between the two strains in the negative and positive ionization modes, respectively. These numbers dropped to 26 and 69 non-redundant metabolite features, respectively, when only features with additional m/z matches in the YMDB (19) were retained (supplemental Tables S1 and S3). Interestingly, several intermediates of the arginine synthesis pathway (N-acetylglutamate, ornithine, N-acetylornithine, and N-acetylglutamate semialdehyde; supplemental Table S3) as well as several intermediates or derivatives of the kynurenine pathway for tryptophan catabolism (tryptophan itself, formylkynurenine, kynurenine, 3-hydroxykynurenine, 3-hydroxyanthranilate, kynurenic acid, and xanthurenic acid) ranged among the most significantly changed metabolites, and accordingly, some of those metabolites also had the highest scores in the PLS-DA. Given that those two pathways do not share any obvious connection with ribulose metabolism, we wanted to test whether similar changes could also be found upon YDR109C deletion in a different genetic background. We therefore also analyzed the ZIC-HILIC-ddMS2 data obtained for the auxotrophic WT and ydr109cΔ strains using multivariate and univariate statistics. In strong contrast to the prototrophic strains, the auxotrophic WT and ydr109cΔ strains showed much more similar metabolite profiles, and corresponding samples did not form two separate clusters after PCA of the non-targeted metabolomics data obtained in negative or positive ionization mode (data not shown). Data analysis using the Welch's t test yielded nevertheless 8 and 21 significantly changed metabolite features (2 and 11 metabolite features when retaining only features with matches in both the KEGG and YMDB databases; supplemental Tables S2 and S4) in the auxotrophic ydr109cΔ strain compared with the WT strain in the negative and positive ionization mode, respectively. Comparing those changes to the ones observed for the prototrophic strains, only two metabolites differed significantly between WT and KO in both genetic backgrounds in the negative mode (ribulose and ribitol), and nine metabolites were changed significantly in the KO versus the WT strain in both genetic backgrounds in the positive mode (all metabolites listed in supplemental Table S4, except for 4-aminobutanoate and methionine sulfoxide). As described above, this showed that the ribulose and ribitol accumulations observed upon YDR109C deletion are robust changes likely to be specifically linked to this gene, whereas the metabolite changes observed in the arginine synthesis and tryptophan degradation pathways upon YDR109C deletion in the prototrophic strain are background-specific.
To further test which of the KO versus WT metabolite changes in the prototrophic background were specifically caused by YDR109C deficiency, we generated a rescue strain (KOres or ydr109cΔ rescue) expressing YDR109C under the control of the endogenous promoter from a low copy number plasmid conferring resistance to hygromycin B (p41Hyg 1-F) in the ydr109cΔ background. Using quantitative RT-PCR, we measured two times higher YDR109C transcript levels in the rescue strain than in the corresponding wild-type strain (Fig. 1), validating the rescue strategy at the gene expression level. The YDR109C transcript was not detectable in a ydr109cΔ strain transformed with an empty plasmid (KOcnt or ydr109cΔ empty plasmid; Fig. 1). Polar metabolites extracted from the KOcnt and KOres strains were analyzed using ZIC-HILIC-ddMS2 in positive and negative ionization mode. Although ribulose levels were consistently lower in the rescue strain than in the empty vector control strain (KOcnt/KOres ratio of 1.7 with a p value of 0.0000005 calculated using the unequal variances Welch's t test; n = 6), the rescue efficiency was only very partial at the metabolite level given the more than 30-fold higher levels of ribulose measured in the non-transformed ydr109cΔ strain compared with the prototrophic wild-type control strain (supplemental Table S1). Except for N-acetylglutamate, N-acetylglutamate 5-semialdehyde, ribitol, and deoxyribose, other metabolite changes observed in the prototrophic ydr109cΔ strain compared with the wild type were either not found or were found to vary in opposite direction in the KOcnt versus KOres strain comparison (data not shown). Such results typically would suggest that those supplementary changes in the metabolite profiles of the original strains were due to the presence of secondary mutations present in the ydr109cΔ mutant but not in the wild-type control strain. However, given the incomplete rescue of the ribulose metabolic phenotype, it seems that the rescue plasmid used led, for reasons that remain unclear, to the formation of a transcript that does not allow reconstitution of wild-type protein and/or wild-type enzyme activity levels, potentially explaining why other metabolic changes were not rescued.
We next adapted a recently published SIL workflow (20) for improved untargeted metabolomics data analysis to our experimental model. Our prototrophic wild-type and ydr109cΔ strains were cultivated in parallel in controlled minimal medium supplemented either with non-labeled d-glucose or with d-[U-13C]glucose as the sole carbon source (both the non-labeled and the fully labeled glucose were added at a final concentration of 2% (w/v)). Polar metabolites were extracted from the four cultivations, and the 13C-labeled extracts derived from both the wild type and the ydr109cΔ cells were pooled. This labeled pooled sample was added as an internal standard into the individual non-labeled metabolite extracts of the wild-type and ydr109cΔ strains, and the supplemented samples were analyzed by ZIC-HILIC-ddMS2 in positive or negative ionization mode. Data analysis was performed using an extended version of the MetExtract software (20, 21). The data filtering based on SIL-specific isotopic patterns and subsequent grouping of 12C and 13C feature pairs greatly enriches the processed mass spectrometry dataset in small molecules that are produced intracellularly and assists with metabolite identification by the number of carbon atoms that can be deduced for each metabolite from the difference in mass between the 13C and 12C ions. Using this strategy, we again confirmed that ribulose (as well as ribitol) is produced endogenously by S. cerevisiae cells from d-glucose, but we also could extend this conclusion to all the other detected metabolite features that had a matching U-13C counterpart, and we could use this information to improve metabolite identification in our untargeted metabolomics dataset, with a focus on the metabolites that were found up- or down-regulated in the ydr109cΔ strain (supplemental Tables S1 and S3). This added an additional level of confidence to identifications for metabolites that were not represented in our in-house metabolite library (and conversely also allowed questioning of metabolite identifications based on accurate mass matches in the KEGG and YMDB databases; see, for example, the most significantly changed metabolite, detected in the positive mode, identified as N-acetyl-l-glutamate-5-semialdehyde by the KEGG and YMDB accurate mass match, but for which the co-cultivation method revealed a carbon number that does not concur with this identification). Notably, the identities of some of the arginine synthesis pathway intermediates (N-acetylglutamate and ornithine) as well as of the kynurenine pathway intermediates or derivatives (tryptophan, formylkynurenine, kynurenine, 3-hydroxykynurenine, 3-hydroxyanthranilate, kynurenic acid, and xanthurenic acid) that were found to significantly change between the prototrophic WT and ydr109cΔ strains were in this way further consolidated (supplemental Tables S1 and S3).
Deficiency of Another FGGY Protein Family Member Encoded by the S. cerevisiae Genome, Mpa43, Does Not Lead to Pentose Accumulation
BLAST searches revealed that the S. cerevisiae genome encodes a protein (Mpa43) that is highly similar to the Ydr109c protein. Mpa43 is a smaller protein (542 amino acids) than Ydr109c (715 amino acids), and the N- and C-terminal sequences of the two proteins do not share sequence similarity. However, about 70% of the Ydr109c protein sequence (from amino acids 41–552) aligns well with Mpa43, showing 28% sequence identity. Mpa43 also contains the conserved FGGY_N and FGGY_C domains. While nothing is known on the subcellular localization of Ydr109c, the Mpa43 protein was detected in highly purified mitochondria in high throughput studies (22, 23). This is in disagreement with scores obtained with the TargetP program (24), which predicted Mpa43 to be neither mitochondrial nor targeted to the secretory pathway, although for Ydr109c it computed the highest score for a mitochondrial localization (with, however, a low reliability). Given the protein sequence similarities between Ydr109c and Mpa43, we also analyzed metabolite extracts derived from a prototrophic strain knocked out for the MPA43 gene. In strong contrast to the findings described for the ydr109cΔ strain, the metabolite profile of the mpa43Δ strain showed only few significant metabolite level changes compared with the wild-type strain, and the mpa43Δ and wild-type metabolite profiles were not clearly separable using PCA (data not shown). Importantly, we could not detect an increase in free ribulose (or any other pentose) or ribitol levels in the mpa43Δ strain, suggesting that Ydr109c and Mpa43 are not isozymes.
FGGY Silencing in Human Embryonic Kidney Cells Leads to Increased Ribulose Levels under Certain Conditions
The human genome contains a homolog of the yeast YDR109C gene, designated FGGY (40% identity at the amino acid sequence level). As for the yeast protein, the molecular and biological roles of the human FGGY protein remain largely unknown. Based on our results in the yeast model, we searched for ribulose or other pentoses in metabolite extracts derived from HEK293 cells and from the hepatocyte cell line PH5CH8 using a targeted ZIC-HILIC-MS method. Extracted ion chromatograms (m/z = 149.0445) did not reveal the presence of ribulose in either the HEK293 or PH5CH8 cells when cultivated in DMEM supplemented with 5 mm d-glucose (in addition to the 25 mm glucose already contained in the DMEM formulation). The analyses were repeated in a HEK293 cell line stably expressing an FGGY-specific small hairpin RNA (shRNA) and in PH5CH8 cells transfected with FGGY-specific small interfering RNAs (siRNAs). Despite a knockdown efficiency of about 60% in both cell types at the mRNA level (Fig. 4A and data not shown), ribulose could not be detected in any of the conditions tested.
FIGURE 4.

Ribulose accumulates in FGGY knockdown human cells exposed to ribitol. HEK293 FGGY knockdown (KD) and control cells were cultivated in DMEM supplemented with 10 mm ribitol. Metabolites and total RNA were extracted 41 h after addition of ribitol, and pentoses were measured using a targeted ZIC-HILIC-MS method. A, FGGY knockdown efficiency was evaluated using quantitative RT-PCR. The relative expression level of FGGY was estimated using the 2−ΔΔCt method. The expression level of the FGGY gene in each sample was either normalized to reference gene ACTB or GAPDH. Means and standard deviations of 12 (FGGY KD cells) and 11 (control cells) biological replicates are shown. B, box-and-whisker plot representing the area of the ribulose peak (m/z 149.0445, [M − H]− ion in the negative mode). Statistical analysis by Welch's t test confirmed that the mean ribulose peak area was significantly increased in the FGGY KD cells compared with the control cells (p value = 0.0005; six biological replicates for each cell line). KD, knockdown.
Given that ribulose was shown not to be taken up by human fibroblasts (25), we supplemented the basal cell culture medium with the potential ribulose precursor ribitol, both at a concentration of 5 or 10 mm, for the PH5CH8 and HEK293 cells. In addition, we tested supplementation with 5 mm d-arabinose for the HEK293 cells. d-Arabinose and ribitol can be metabolized in bacteria via pathways that include d-ribulose as an intermediate (26, 27). Although we detected intracellular d-arabinose in the HEK293 cells cultivated in the presence of this pentose, we did not detect any intracellular ribulose 48 h after the addition of d-arabinose, without or with FGGY silencing. In contrast, in HEK293 cells cultivated in the presence of ribitol, we detected ribulose, and the latter accumulated to higher levels in cells knocked down for FGGY (Fig. 4B). When non-labeled ribitol was replaced by [U-13C]ribitol in these experiments, a +5 m/z shift was observed for the peak co-eluting with a ribulose standard (data not shown), confirming the identity of the peak as ribulose and showing that the ribulose measured derived from the supplemented ribitol under the cultivation conditions used. PH5CH8 cells cultivated in the presence of ribitol did not produce measurable amounts of ribulose, even when FGGY was knocked down (data not shown). Taken together, these results indicate that free ribulose is not produced in detectable amounts by the cell lines tested here under standard cultivation conditions, but that in certain cell types ribitol can be oxidized to ribulose. The increased ribulose levels measured in HEK293 FGGY knockdown cells cultivated in the presence of ribitol also confirm that FGGY can use ribulose as a substrate in a living cell.
Recombinant Yeast Ydr109c and Human FGGY Specifically Convert d-Ribulose to d-Ribulose 5-Phosphate in the Presence of ATP
Our findings in yeast and mammalian cells suggested that ribulose is an endogenous substrate of the yeast Ydr109c and human FGGY proteins. Additionally being members of the FGGY family of sugar kinases, this strongly indicated that Ydr109c and FGGY are ribulokinases. To confirm this hypothesis and find out whether the enzymes act on d-ribulose or l-ribulose (not separated by our liquid chromatography method preceding the MS analysis), we expressed recombinant N-terminally His-tagged Ydr109c and FGGY in a bacterial system and purified the proteins by Ni2+ affinity chromatography for subsequent enzyme activity assays.
The purified His-Ydr109c protein generated a band at the expected size (83 kDa) as shown by SDS-PAGE analysis and Western blotting using an anti-His antibody (Fig. 5, A and B). Using the PK/LDH coupled spectrophotometric assay, we tested the putative kinase activity of recombinant Ydr109c on 19 different sugars or sugar derivatives (9 pentoses, 4 sugar alcohols, d-glucose, d-gluconate, d-glycerol, d-ribulose-5-P, d-ribose-1-P, and d-ribose-5-P) at a concentration of 1 mm (Table 1). Enzymatic activity was only detected in the presence of d-ribulose. With this substrate, the enzyme showed Michaelis-Menten kinetics, and we determined a Km of 217 ± 15 μm and a Vmax of 22 ± 2 μmol·min−1·mg protein−1 (means ± S.D., n = 3). The recombinant His-Ydr109c protein was very unstable and lost activity upon freezing and thawing or during prolonged purification procedures. Therefore, enzyme activity assays with recombinant His-Ydr109c were carried out within 24 h after affinity purification, without prior desalting, keeping the protein at 4 °C throughout the purification procedure and until the measurements.
FIGURE 5.

SDS-PAGE and Western blotting analyses of recombinant His-Ydr109c and His-FGGY proteins. The indicated purified His-Ydr109c (A and B) and His-FGGY (C and D) fractions were analyzed by SDS-PAGE followed by Coomassie Blue staining and Western blotting using an antibody directed against the N-terminal polyhistidine tag fused to each of the recombinant proteins. Fractions eluted from an Ni2+ affinity column were analyzed either before (His-Ydr109c) or after desalting (His-FGGY). The expected molecular mass of the His-Ydr109c and His-FGGY proteins is 82.8 and 63.7 kDa, respectively. MW, molecular mass.
TABLE 1.
Substrate specificity of the carbohydrate kinase activity of Ydr109c and FGGY
The kinase activities of the recombinant purified His-Ydr109c and His-FGGY proteins were measured spectrophotometrically using the PK/LDH assay with 19 different sugars or sugar derivatives (all at a concentration of 1 mm). Enzymatic activities were corrected by control assays run in the absence of carbohydrate substrate. The results shown are mean relative activities ± S.D. resulting from three replicative measurements. ND, not detected; S.D., standard deviation.
| Substrate | FGGY activity | Ydr109c activity |
|---|---|---|
| % | % | |
| d-Ribulose | 100 ± 2 | 100 ± 3 |
| l-Ribulose | 8 ± 1 | ND |
| Ribitol | 21 ± 1 | ND |
| d-Xylulose | 1 ± 0.3 | ND |
| l-Xylulose | 1 ± 0.3 | ND |
| d-Glucose | ND | ND |
| Arabitol | ND | ND |
| Erythritol | ND | ND |
| l-Arabinose | ND | ND |
| d-Arabinose | ND | ND |
| d-Ribose | 1 ± 0.3 | ND |
| Glycerol | ND | ND |
| d-Ribulose 5-phosphate | ND | ND |
| Gluconate | ND | ND |
| 2-Deoxy-d-ribose | ND | ND |
| d-Lyxose | ND | ND |
| d-Ribose 5-phosphate | ND | ND |
| d-Mannitol | ND | ND |
| d-Ribose 1-phosphate | ND | ND |
The human recombinant His-FGGY protein was, unlike its yeast homolog, very stable. SDS-PAGE and Western blotting analyses of the fractions collected after Ni2+ affinity purification and desalting showed a major band at about 50 kDa, i.e. below the expected mass of 64 kDa for the His-FGGY protein (Fig. 5, C and D). LC-MS/MS analysis after trypsin digestion of the purified protein preparation confirmed, however, the sequence identity of the protein, the detected peptides showing the best match to the Q96C11-1 (UniProt) sequence, with 76% sequence coverage (data not shown). The reason for protein migration at a lower apparent molecular mass during SDS-PAGE remains unknown. To determine the substrate specificity of the enzyme, we screened the same 19 compounds that we used for Ydr109c substrate specificity testing, at two different concentrations (100 μm and 1 mm). The recombinant human FGGY protein clearly showed the highest activity with d-ribulose, but it also phosphorylated ribitol and l-ribulose at lower rates (Table 1). We determined a Km of 97 ± 25 μm and a Vmax of 5.6 ± 0.4 μmol·min−1·mg protein−1 (means ± SDs, n = 3) for the d-ribulokinase activity and a Km of 1468 ± 541 μm and a Vmax of 2.4 ± 0.3 μmol·min−1·mg protein−1 (means ± S.D., n = 4) for the ribitol kinase activity. Human FGGY is thus 35-fold more efficient as a d-ribulokinase than as a ribitol kinase. Purified human His-FGGY protein was found to be enzymatically active even after 1 year of storage at −80 °C.
The identity of the pentose phosphate product formed by human recombinant FGGY when incubated with its preferred substrate d-ribulose was confirmed by analyzing the enzymatic reaction mixture by LC-MS/MS. A major compound contained in this mixture displayed a detected mass corresponding to the theoretical mass of ribulose-5-P, co-eluted with a d-ribulose 5-phosphate analytical standard, and showed the same MS2 fragmentation pattern as this analytical standard (Fig. 6, A and B). After incubation of recombinant FGGY with ribitol or l-ribulose (both at a concentration of 1 mm), we also detected the masses of the expected ribitol 5-phosphate (theoretical m/z 231.0264; detected m/z 231.0273) or l-ribulose 5-phosphate (theoretical m/z 229.0108; detected m/z 229.0116) products in the reaction mixtures (data not shown); further support (co-elution and MS2 spectrum match with standards) for these compound identities could, however, not be gathered as appropriate analytical standards were not commercially available.
FIGURE 6.

Confirmation of the product of the reaction catalyzed by human FGGY as ribulose 5-phosphate. A, a reaction mixture resulting from the PK/LDH coupled d-ribulokinase assay was analyzed using ZIC-HILIC-ddMS2. The extracted ion chromatogram (m/z 229.0108, negative ionization mode) shows a peak at retention time 7.92 min that co-elutes with the external analytical standard d-ribulose 5-phosphate. B, head-to-tail comparison of the MS2 fragmentation pattern of the parent ion m/z 229.0108 eluting at 7.92 min during analysis of the enzyme assay mixture and of the co-eluting ion from the external d-ribulose 5-phosphate standard.
Yeast Ydr109c and Human FGGY Are Homologs of a Proteobacterial d-Ribulokinase Involved in Ribitol Metabolism
A specific d-ribulokinase was first reported in Klebsiella aerogenes by Neuberger et al. (28), and the gene encoding this enzyme (rbtK) was cloned in 1998 from Klebsiella pneumoniae (27). Certain enteric bacteria, including many Klebsiella strains, but only a few Escherichia coli strains (e.g. E. coli C, but not E. coli K12 and B), can use the pentitols d-arabitol and ribitol as sole carbon sources. Although both pentitols are catabolized via oxidation followed by phosphorylation, specific transporters, repressors, and enzymes encoded by two different operons govern the metabolism of each of these pentitols. The K. pneumoniae ribitol operon includes a ribitol transporter, a ribitol dehydrogenase, the d-ribulokinase, and a repressor that is induced by d-ribulose (27). The existence of this ribitol operon greatly helped us to identify “true” d-ribulokinases in other bacterial species, and we found d-ribulokinase genes clustering with ribitol dehydrogenase genes in other γ-proteobacteria, α-proteobacteria, and the β-proteobacterium Burkholderia sp. Ch1-1 using the SEED viewer browser (29). The K. pneumoniae d-ribulokinase sequence (UniProt A6TBJ4) was also used to identify d-ribulokinase ortholog candidates in eukaryotic species using blastp searches. This analysis showed that d-ribulokinase is conserved in many animal species, fungi, and plants with close to 40% amino acid sequence identity or more to the bacterial sequence. Among those ortholog candidates, S. cerevisiae Ydr109c shared 39% amino acid sequence identity (E-value of 6e-114), and human FGGY (RefSeq isoform b or UniProt Q96C11-1) shared 42% amino acid sequence identity (E-value of 7e-143) with the bacterial d-ribulokinase protein. The S. cerevisiae protein Mpa43 also aligned with the latter but showed lower amino acid sequence conservation (25% identity; E-value of 2e-31).
Fig. 7 shows a multiple sequence alignment (MSA) of d-ribulokinase candidate sequences selected across different kingdoms of life (γ-, α-, and β-proteobacteria, yeast, Drosophila, zebrafish, Arabidopsis, mice, and humans). It also includes the yeast Mpa43 protein sequence. Despite the considerable evolutionary distance between most of the chosen species, highly conserved sequence motifs can be found over the entire length of the d-ribulokinase sequence. Strikingly, the yeast Ydr109c protein shows, however, an approximate 20-amino acid N-terminal sequence extension and an approximate 100-amino acid sequence insertion toward the C-terminal extremity that are found in none of the other d-ribulokinase protein sequences analyzed. Although the N-terminal sequence may be involved in targeting the protein to a specific subcellular compartment, we cannot currently speculate on the role of the C-terminal insertion. Using the strain sequence alignment function in SGD, we found that the 20-amino acid N-terminal extension and the 100-amino acid C-terminal insertion are highly conserved within the S. cerevisiae species. Using the fungal sequence alignment function in SGD, it appears that Ydr109c homologs in other budding yeasts such as Saccharomyces paradoxus, Saccharomyces bayanus, Saccharomyces uvarum, and Saccharomyces mikatae also include an N-terminal extension and C-terminal insertion that is absent from bacterial, animal, and plant d-ribulokinase proteins (in many of the budding yeast species analyzed, the Ydr109c homologous proteins have a size of more than 700 amino acids as opposed to the smaller protein size of around 550 amino acids displayed by bacterial, animal, and plant d-ribulokinase proteins); these additional sequences show, however, as opposed to the rest of the Ydr109c protein sequence, no significant sequence similarity between the different yeast species analyzed. These observations suggest that the additional sequences found in the S. cerevisiae Ydr109c protein are “real” and do not result from erroneous gene structure annotation or genome sequencing errors. This was further consolidated by the fact that our own sequencing of the Ydr109c ORF that we PCR-amplified from S. cerevisiae genomic DNA yielded a result that was in perfect agreement with the sequence contained in the SGD database.
FIGURE 7.

Multiple sequence alignment of d-ribulokinase proteins across different kingdoms of life. Conserved amino acids are highlighted in different shades of blue according to the degree of conservation (the darker the shading the higher the conservation). The top 20 specificity determining positions (SDPs) are highlighted in red. The number above the SDPs indicates the ranking based on the scores calculated by the GroupSim+ConsWin software (number 1 corresponds to the SDP with the highest score). Amino acids predicted to interact with the pentose substrate based on structural homology models of the yeast Ydr109c and human FGGY proteins are highlighted in yellow. The UniProt identifiers of the protein sequences shown are A6TBJ4 (K. pneumoniae), B6XGJ3 (Providencia alcalifaciens), M9RKB9 (Octadecabacter arcticus), B9K586 (Agrobacterium vitis), I2IVR0 (Burkholderia sp. Ch1-1), P53583 (Mpa43, S. cerevisiae), Q04585 (Ydr109c, S. cerevisiae), Q96C11 (H. sapiens), A2AJL3 (Mus musculus), Q6NUW9 (Danio rerio), Q9VZJ8 (Drosophila melanogaster), and F4JQ90 (Arabidopsis thaliana). Note that the alignment includes the yeast Mpa43 protein, which, unlike the other proteins shown, most likely does not act as a d-ribulokinase. Accordingly, the d-ribulokinase signature motif defined in this study (TCSLV sequence highlighted by the box frame) is not strictly conserved in the MPA43 protein.
Zhang et al. (3) performed a detailed bioinformatics analysis of proteins belonging to the FGGY carbohydrate kinase family to better understand the evolutionary mechanisms underlying functional diversification in this family. They assembled a confidently annotated reference set (CARS) of 446 FGGY proteins with high quality functional annotations based not only on sequence homology but also on experimental evidence (if available) and on genomic as well as pathway context. The CARS protein set comprised only three eukaryotic FGGY family members (1 glycerol kinase and 2 xylulose kinases), all the other proteins were of bacterial origin. The CARS proteins were used for phylogenetic analyses and to predict amino acid residue positions that are important for the recognition of a specific substrate within isofunctional groups of proteins (also referred to as specificity-determining positions or SDPs). Here, we extended the phylogenetic analysis of this CARS protein set by adding the protein sequences that we confidently identified as d-ribulokinases and used for the MSA shown in Fig. 7, including the S. cerevisiae Ydr109c and human FGGY proteins. As expected, the resulting phylogenetic tree displayed a very similar topology to the one constructed by Zhang et al. (3), with most of the proteins forming tight clusters according to their enzymatic function (e.g. glycerol kinase cluster, xylulokinase cluster) and suggesting a divergent evolution model (supplemental Fig. S1). We also found a more complex branching for the l-ribulokinase (AraB) group whose members split into several subgroups interspersed with the gluconokinase and d-ribulokinase groups. All the d-ribulokinase sequences that we included additionally in the phylogenetic analysis cluster with the d-ribulokinase group that evolved from one of the l-ribulokinase subgroups (supplemental Fig. S1). This suggests that the prokaryotic and eukaryotic d-ribulokinases evolved from the same common ancestor by divergent evolution.
We further extended our d-ribulokinase sequence analyses by applying the GroupSim+ConsWin method (30) for the identification of sequence positions determining the sugar substrate specificity (SDPs) of d-ribulokinase proteins. The GroupSim method predicts SDPs based on sequence information only. ConsWin is a heuristic that can be combined with the GroupSim method to improve SDP predictions by taking into account sequence conservation of neighboring amino acids. SDP predictions were made by applying GroupSim+ConsWin to the MSA built from our extended CARS dataset (enriched in d-ribulokinase sequences), where sequences were grouped according to the high quality functional annotations (for isofunctional groups spanning multiple subgroups only the subgroup with the highest number of protein sequences was kept; see under “Experimental Procedures” for more details). The top 20 SDPs identified for the yeast and human d-ribulokinases (Ydr109c and FGGY, respectively) are highlighted in red in Fig. 7. The majority of these SDPs correspond to residues that are highly conserved between prokaryotic and eukaryotic d-ribulokinases (Fig. 7). It should be noted that among the 12 SDPs with strictly conserved residues in all of the d-ribulokinase sequences shown in Fig. 7, three positions contain different residues in the yeast Mpa43 protein (Ser to Val, Cys to Gly, and Ala to Ser substitutions), which, together with the absence of ribulose accumulation in the yeast mpa43Δ deletion strain, further supports that this protein most likely does not function as a d-ribulokinase. As described below, these predictions, as well as structural predictions provided the basis for the identification of a sequence motif that appears to be specific for d-ribulokinases.
Structural Homology Modeling of Yeast and Human d-Ribulokinases and Definition of a d-Ribulokinase Signature Motif
Three-dimensional structural homology models of the yeast Ydr109c and human FGGY proteins were generated using the crystal structure of a Yersinia pseudotuberculosis FGGY carbohydrate kinase (PDB code 3L0Q chain A and 3GGA chain A for FGGY and Ydr109c, respectively) as a template (Fig. 8). Of the 20 proteins with an FGGY_N domain for which three-dimensional structures have been deposited in PDB, this Y. pseudotuberculosis protein Q665C6 has the highest sequence similarity to the yeast Ydr109c and human FGGY proteins (39 and 51% amino acid sequence identity, respectively). A d-xylulose molecule is contained in the putative active site of the PDB 3L0Q crystal structure, and the protein is annotated as a xylulose kinase in the PDB. Sequence analysis clearly suggests, however, that this protein is in fact a d-ribulokinase, given the high sequence similarity with the yeast Ydr109c and human FGGY proteins and the presence of the TCSLV motif, identified here as a d-ribulokinase signature motif as described below.
FIGURE 8.

Structural homology models of yeast Ydr109c and human FGGY proteins. A, homology models of the Ydr109c protein (A) and the FGGY protein (B) created using template structures from PDB entries 3GG4 chain A and 3L0Q chain A, respectively, of Y. pseudotuberculosis d-ribulokinase. Residues corresponding to the top 20 specificity determining positions predicted in this study are highlighted in red. C, homology model of the FGGY protein in complex with the ligand d-xylulose (light green color). The FGGY_C and FGGY_N domains are highlighted in dark blue and pink, respectively.
We used the available ligand information from the 3L0Q structure to position d-xylulose within our human FGGY structural models (Fig. 8C) and to identify the localization of the active site as well as amino acid residues important for substrate binding and/or catalysis. For yeast Ydr109c, a molecular docking approach was used to determine both ligand position and orientation, as the original structure of 3GGA chain A was not bound to any ligand molecule. As for other FGGY carbohydrate kinases, a catalytic cleft is formed at the interface between the two conserved actin-like ATPase domains (Pfam domains FGGY_N and FGGY_C). It has been shown for other members of the family that the sugar substrate binds deeply within the catalytic pocket and interacts mainly with residues of the FGGY_N domain whereas the ATP co-substrate binds more toward the opening of the cleft interacting with both the FGGY_N and FGGY_C domains (31). Accordingly, the xylulose ligand in our d-ribulokinase structural models contacts only residues of the FGGY_N domain (Fig. 8C for the human protein; not shown for the yeast protein). Using the human FGGY homology model, we found that the residues Thr-86, Ile-259, Asp-260, Ala-261, and His-262 can take part in van der Waals interactions and that Cys-87, Asp-260, and Glu-328 have the potential to form hydrogen bonds with the substrate (residues highlighted in yellow in Fig. 7). Based on the Ydr109c homology model, the residues Cys-117, Ile-301, and Asp-302 are potentially involved in van der Waals interactions and Thr-116, Cys-117, Lys-224, Asp-302, Tyr-304, Glu-378, and Arg-449 potentially form hydrogen bonds with the substrate (also highlighted in yellow in Fig. 7). It can be seen in Fig. 7 that all the residues predicted to be important for sugar substrate binding using the structural models coincide or are found in close proximity to identified SDPs. Mapping the top 20 SDPs onto the human and yeast homology models, it can also be seen in Fig. 8, A and B, that most of them, although sometimes distant from each other in sequence, are located in or near the catalytic cleft in the structural models.
Two highly conserved motifs in the MSA shown in Fig. 7 stand out by featuring residues important for substrate specificity or binding as predicted by both the SDP or structural modeling approaches: the TCSLV motif (starting with Thr-86 in FGGY and Thr-116 in Ydr109c) and the IDA(H/Y) motif (starting with Ile-259 in FGGY and Ile-301 in Ydr109c). Using the master MSA used for construction of the phylogenetic tree, we found that the TCSLV motif is strictly conserved in the entire d-ribulokinase cluster of sequences but is not found in other isofunctional groups of the FGGY protein family, whereas the IDA(H/Y) motif was also retrieved in a few l-ribulokinase sequences. The TCSLV motif was further validated as a d-ribulokinase signature motif using a second master MSA based on more than 600 reviewed FGGY family members retrieved from UniProt (see “Experimental Procedures”). Interestingly, the Val residue of this signature motif is replaced by an Ala residue in the Mpa43 protein. This second master MSA also allowed us to confirm the existence of multiple distinctive subgroups within the l-ribulokinase functional group and to identify additional conserved sequence motifs distinguishing the l-ribulokinase subgroups in a homologous position to the TCSLV motif in the d-ribulokinase group (namely TGTST, TSST, TGSSP, TGSTP, MMHGY, and TACTM). Zhang et al. (3) also made SDP predictions and, based on structural information available in PDB, selected five (generally non-consecutive) SDPs as “signature residues” for all the subgroups in their FGGY CARS dataset. Of the five SDPs selected for the d-ribulokinase subgroup (Thr, Cys, Glu, Ala, and Tyr), three (Thr, Glu, and Ala) coincide with SDPs predicted using a different method in this study (see under “Experimental Procedures”), and two (Thr and Cys) are comprised in the d-ribulokinase signature motif defined and validated here using protein datasets containing a higher number of evolutionary more distant d-ribulokinase sequences than in the study by Zhang et al. (3).
Finally, using the described d-ribulokinase and l-ribulokinase sequence motifs, we analyzed the phylogenetic spread of these kinases based on FGGY_N domain sequences retrieved for 34 different phyla from the Pfam database, as described under “Experimental Procedures.” Strikingly, as can be seen in Fig. 9, d-ribulokinase is much more widely conserved in eukaryotes (including animals, plants, and fungi), whereas l-ribulokinases are more broadly distributed in prokaryotes. In prokaryotes, d-ribulokinase is found only in proteobacterial lineages as well as in Verrucomicrobia and Armatimonadetes. No ribulokinase homologs (neither d- nor l-) were found in Archaea.
FIGURE 9.
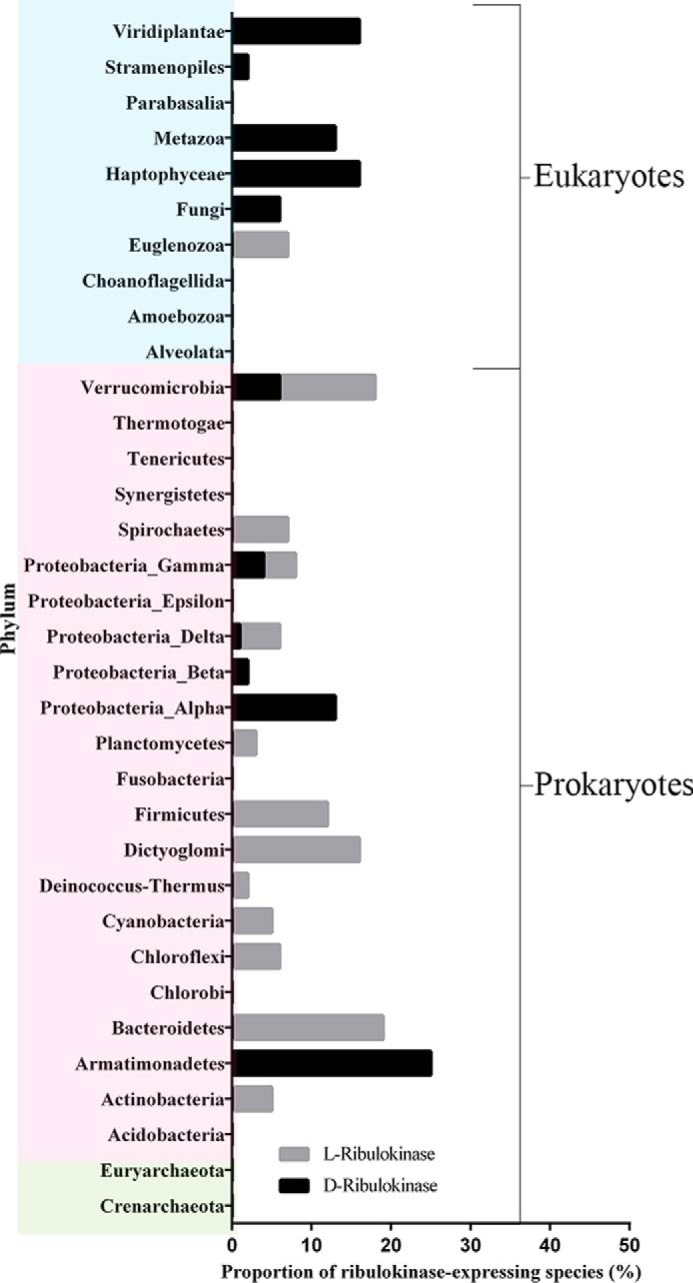
Phyletic spread of d-ribulokinase and l-ribulokinases. The taxonomic distribution of d- and l-ribulokinase proteins was determined using the FGGY_N domain sequences present in the Pfam database at the time of analysis and the d-ribulokinase and l-ribulokinase motifs defined in this study (TCSLV and TGTST, TSST, TGSSP, TGSTP, MMHGY, and TACTM, respectively). Eukaryotic, bacterial, and archaeal phyla are represented on blue, pink, and green backgrounds, respectively.
Discussion
Identification of YDR109C and FGGY as Genes Encoding a Specific Eukaryotic d-Ribulokinase
In this study, we aimed at identifying the molecular and biological roles of two homologous proteins of unknown function (Ydr109c in yeast and FGGY in humans), both of which are members of the FGGY protein family of carbohydrate kinases. To reach this objective, we started by comparing the polar metabolite profiles of yeast cells deleted in the YDR109C gene and of wild-type control cells using non-targeted LC-HRMS or LC-MS/MS. One of the most prominent and robust changes, observed in both a prototrophic and an auxotrophic genetic background and partially rescued when restoring YDR109C expression in the deletion strain, was the accumulation, in the ydr109cΔ strain, of a pentose identified as ribulose by comparing elution time, accurate mass, and MS2 fragmentation pattern with those of a ribulose standard. Ribulose did not accumulate in a yeast strain deleted for a closely related protein encoded by the S. cerevisiae genome (Mpa43), suggesting that Ydr109c is the only kinase responsible for free ribulose conversion in this organism. Labeling experiments in which d-[U-13C]glucose was used as the sole carbon source confirmed that yeast cells can produce free ribulose from d-glucose and that this pentose accumulates upon YDR109C deletion. In contrast, we could not detect free ribulose in the two human cell lines tested in this study (HEK293 and PH5CH8), even after silencing of the YDR109C homologous gene FGGY. In the HEK293 cells, but not in the PH5CH8 cells, ribulose became detectable upon supplementation of the cultivation medium with ribitol. Under those conditions, FGGY knockdown led to increased ribulose levels in the HEK293 cells. Finally, in vitro enzymatic assays with recombinant purified Ydr109c and FGGY proteins confirmed that both act as ATP-dependent sugar kinases and showed that d-ribulose is by far the best substrate for those enzymes. Taken together, these findings demonstrate that Ydr109c and FGGY phosphorylate d-ribulose in budding yeast and human cells, respectively, and associate the eukaryotic d-ribulokinase activity, which had been reported to exist in guinea pig liver before (32, 33), with a protein sequence. Although numerous other metabolites were significantly changed in the ydr109cΔ prototrophic strain compared with the corresponding wild-type strain in addition to ribulose, the facts that we did not observe a majority of those other changes in a different genetic background and that they were not restored in a prototrophic rescue strain do not allow us to firmly associate those changes with the YDR109C gene based on our current results.
d-Ribulokinase Is the Major Ribulokinase in Eukaryotes, while l-Ribulokinase Is More Widely Distributed in Prokaryotes
Prior to this study, a specific d-ribulokinase involved in ribitol metabolism had been cloned from K. pneumoniae (27). The d-ribulokinases from Aerobacter aerogenes (34) and K. aerogenes (28) have been extensively purified and characterized. The enzyme was shown to be active as a homodimer, and Km values for d-ribulose of 85 and 400 μm were reported for the Aerobacter and Klebsiella enzymes, respectively. A Vmax of 71 μmol·min−1·mg protein−1 was found for the Klebsiella d-ribulokinase, and this enzyme showed low side activities with ribitol (Km of 220 mm, Vmax of 12 μmol·min−1·mg protein−1) and d-arabitol (Km of 140 mm, Vmax of 6.6 μmol·min−1·mg protein−1) (28). The kinetic properties for d-ribulose are similar to the ones determined in this study for the S. cerevisiae and human d-ribulokinases (Km of 217 μm and Vmax of 22 μmol·min−1·mg protein−1 for Ydr109c; Km of 97 μm and Vmax of 5.6 μmol·min−1·mg protein−1 for FGGY). For the human enzyme, we also found lower but detectable activities with ribitol and l-ribulose; d-arabitol was not tested as it could not be obtained commercially at the time of this study. The highly similar enzymatic properties shared by those enzymes from evolutionary divergent organisms, in addition to the high sequence identity, support that they are orthologous proteins.
The molecular identification of the eukaryotic d-ribulokinase allowed us to incorporate eukaryotic d-ribulokinase sequences into phylogenetic analyses. In agreement with an extensive previous study on the evolution of functional specificities of prokaryotic members of the FGGY carbohydrate kinase family (3), we found that eukaryotic d-ribulokinases, just as the bacterial d-ribulokinases, have evolved from an l-ribulokinase (AraB) FGGY subgroup. Based on our sequence and structural analyses, we also defined and validated a d-ribulokinase signature motif (TCSLV), which can now be used with high confidence to functionally identify FGGY protein sequences as d-ribulokinase. Using this motif, we found that d-ribulokinase is conserved in only a few bacterial lineages, although it is widespread in eukaryotes (see Fig. 9). Notable exceptions of eukaryotic species that do not encode a d-ribulokinase are Schizosaccharomyces pombe, Caenorhabditis elegans, and trypanosomatid species. By contrast, l-ribulokinase, which is required for l-arabinose metabolism in bacteria, is much more broadly conserved in prokaryotes than d-ribulokinase, but it is not found in eukaryotic species, except for trypanosomatid species such as Leptomonas, Trypanosoma, and Leishmania (Fig. 9); the genomes of S. pombe and C. elegans do not encode either l- or d-ribulokinase. In summary, while l-ribulokinase is more broadly distributed in prokaryotes, d-ribulokinase is more broadly conserved in eukaryotes. Bacterial species that encode d-ribulokinase can also encode l-ribulokinase (e.g. K. pneumoniae), but many bacterial species encode only l-ribulokinase. Similarly, the trypanosomatid species that encode l-ribulokinase do not encode d-ribulokinase; unlike for many bacteria, however, the eukaryotic genomes that contain a d-ribulokinase gene do not contain an l-ribulokinase gene. So, whereas bacterial genomes can contain two types of ribulokinases, eukaryotic genomes generally encode either the d- or the l-ribulokinase form (Fig. 9).
Although this study on eukaryotic proteins and previous studies on bacterial proteins (28, 34) have shown that d-ribulokinase is highly specific for d-ribulose, l-ribulokinase is much more promiscuous in terms of sugar substrate specificity. E. coli l-ribulokinase (AraB) for example has been reported to use d-ribulose with a catalytic efficiency that is only 2–3-fold lower than its best substrate l-ribulose (35). In addition, AraB showed significant activity with l-xylulose, l-arabitol, and ribitol and also acted on d-xylulose (35). This enzyme could therefore also be designated 2-ketopentokinase. The substrate promiscuity of l-ribulokinase as well as the significant sequence similarity between l-ribulokinase and d-ribulokinase are certainly the reasons for many misannotations of d-ribulokinases as l-ribulokinases and vice versa in gene and protein databases. Human FGGY, for example, displays more than 40% sequence identity with the K. pneumoniae d-ribulokinase, but it also shares 25% sequence identity with the E. coli l-ribulokinase. Our enzymatic characterizations have clearly established the yeast and human ribulokinases as d-ribulokinases. This study, and more particularly the d-ribulokinase sequence signature that we defined, should therefore help to correct the numerous database misannotations of FGGY protein family members and more specifically of the ribulokinases. Although the numerous bacterial species that only encode an l-ribulokinase may not be able to grow on ribitol due to the lack of an inducible ribitol utilization pathway, including the specific d-ribulokinase (see below), the substrate promiscuity of their l-ribulokinase may ensure that free d-ribulose can be metabolized to a certain degree. Conversely, eukaryotic species, which for the most part only encode the more specific d-ribulokinase, should have a good metabolic capacity for d-ribulose, but more poorly, if at all, metabolize ribitol or l-ribulose.
Known and Putative Physiological Roles of d-Ribulokinase in Bacteria and Eukaryotes
Fig. 10 shows an overview of known and hypothetical reactions and pathways leading to the formation and metabolization of d-ribulose in various organisms. In K. pneumoniae and other enterobacteria that can use ribitol as the sole carbon source (36), the d-ribulokinase gene is located in a ribitol utilization operon that contains in addition a ribitol transporter, a ribitol dehydrogenase, and a repressor; d-ribulose is an inducer of this operon (27). In addition, Elsinghorst and Mortlock (26) reported in 1988 that in E. coli B a d-ribulokinase gene is contained in the l-fucose regulon. The latter encodes the enzymes required for l-fucose utilization but can also be induced by d-arabinose, which can then be metabolized to d-ribulose and d-ribulose-5-P via l-fucose isomerase and d-ribulokinase, respectively (26). In the bacterial species that encode d-ribulokinase, this enzyme thus allows to direct carbon from ribitol or d-arabinose to the pentose phosphate pathway via phosphorylation of the d-ribulose intermediate that is formed in the respective sugar utilization pathways. We found that d-ribulokinase sequences contained in either the ribitol or some enterobacterial l-fucose operons share high sequence similarity with yeast Ydr109c and human FGGY and contain the d-ribulokinase signature motif defined in this study.
FIGURE 10.

Known and putative routes involved in d-ribulose metabolism. Overview of reactions and pathways leading to the production or utilization of d-ribulose. Some of the pathways shown are only known to occur in certain microorganisms, as detailed in the main text. Dotted arrows represent hypothetical reactions for which no corresponding gene has been identified yet in any organism. The d-ribulokinase enzyme, for which the eukaryotic gene has been identified in this study, is highlighted.
The only previous reports on a eukaryotic d-ribulokinase activity, in our knowledge, go back to the early 1960s, when Kameyama and Shimazono (32, 33) detected such an activity in guinea pig liver. A corresponding gene has not been cloned since. In these early studies, the authors had become interested in the metabolism of d-ribulose in mammals after having found that d-ribulose can be formed from d-gluconate in guinea pig liver extracts (37). We did not find subsequent studies on this putative pathway, but d-gluconate to d-ribulose conversion may be initiated by a side activity of l-gulonate 3-dehydrogenase (encoded by the CRYL1 gene), an enzyme involved in the pentose pathway for d-glucuronate catabolism in mammals (38, 39). The subsequent step in the pentose pathway is the decarboxylation of 3-dehydro-l-gulonate to l-xylulose (40); a similar reaction could convert 3-dehydro-d-gluconate to d-ribulose. The gene encoding 3-dehydro-l-gulonate decarboxylase has not yet been identified. The mechanism of formation of d-gluconate in mammalian cells, if it occurs at all, also remains unclear. The existence of a mammalian pathway for d-ribulose formation from d-gluconate therefore remains highly speculative.
Using LC-MS-based metabolite profiling and isotopic labeling, we could measure d-ribulose formation from externally supplemented ribitol in human HEK293 cells but not from glucose. By contrast, in S. cerevisiae, we were able to detect formation of d-ribulose from glucose, although the conversion of ribitol to d-ribulose was not observed in this organism. Our results suggest that, whereas yeast cells constantly produce detectable amounts of free d-ribulose under standard cultivation conditions from glucose, this may not be the case for mammalian cells. However, our observations in the HEK293 cells suggest that ribitol can serve as a precursor for d-ribulose at least in certain mammalian cell types; d-ribulose was not detected in PH5CH8 cells (this study) or human fibroblasts (25) cultivated in the presence of ribitol, and no ribitol dehydrogenase activity could be detected in human erythrocyte lysates (25). Ribitol dehydrogenase activity has, however, been measured during early studies with partially purified enzyme preparations from rat liver (41) and from guinea pig liver mitochondria (42). Comparing gene expression profiles of HEK293 and PH5CH8 cells could be a promising strategy to identify the putative dehydrogenase responsible for the oxidation of ribitol to d-ribulose in certain mammalian cell types. The prototrophic yeast strain used in this study did not grow in rich medium (yeast extract and peptone) supplemented with 100–200 mm ribitol instead of 100–200 mm glucose. Moreover, when adding 5 mm [U-13C]ribitol to minimal medium containing 2% glucose, we could not detect any 13C-labeled ribulose in cellular extracts prepared from the yeast cultivations, neither for the wild-type nor for the ydr109cΔ strains. This suggests that, unlike for certain mammalian cell types and in agreement with previous observations (43), ribitol is not metabolized by S. cerevisiae cells.
Pathogenic (e.g. Candida albicans) and osmotolerant (e.g. Zygosaccharomyces rouxii) yeast species are known to produce high amounts of d-arabitol (44, 45). Based on studies using [2-14C]glucose, it was shown that C. albicans produces d-arabitol by dephosphorylating d-ribulose-5-P and then reducing d-ribulose by an NAD-dependent d-arabitol 2-dehydrogenase (d-ribulose reductase; Ard1) (46). The latter enzyme was also purified and characterized from Candida tropicalis (47). Many of the d-arabitol-producing yeast species are also able to utilize this pentitol as the sole carbon source (45). The d-arabitol utilization pathway most likely involves oxidation of arabitol to d-ribulose by d-arabitol 2-dehydrogenase and phosphorylation of d-ribulose to the pentose phosphate pathway intermediate d-ribulose-5-P by the homolog of the d-ribulokinase protein identified in this study. Ydr109c is indeed well conserved in Candida as well as in osmotolerant Zygosaccharomyces species. S. cerevisiae does not produce d-arabitol (44), and it is unlikely that d-ribulokinase participates in d-arabitol utilization in this species.
Why then, one may ask, has d-ribulokinase been conserved in species such as baker's yeast and higher eukaryotes, including humans? The most plausible endogenous precursor, in these species, for the d-ribulokinase substrate is certainly d-ribulose-5-P. Therefore, we propose that a possible physiological role of d-ribulokinase in species or cell types that do not produce significant amounts of free d-ribulose from pentitols or other pentose precursors may be to preserve the d-ribulose-5-P pool or to prevent potentially toxic accumulations of free d-ribulose by “re-phosphorylating” d-ribulose formed by nonspecific phosphatase activities from d-ribulose-5-P. As such, d-ribulokinase could be added to the growing list of so-called metabolite repair enzymes, i.e. enzymes that function to remove useless or sometimes toxic metabolites formed via side activities of metabolic enzymes (48, 49). In both S. cerevisiae and higher eukaryotes, low molecular weight phosphatases of the haloacid dehydrogenase protein superfamily may contribute to free ribulose formation from d-ribulose-5-P. Some of these phosphatases are quite promiscuous, and in S. cerevisiae, the poorly characterized Ykr070w and Ynl010w haloacid dehalogenase phosphatases have recently been shown to hydrolyze d-ribulose-5-P in addition to a range of other phosphomonoesters tested (50). In earlier studies, a partially purified acid phosphatase preparation, but not alkaline phosphatase preparation, from Z. rouxii was shown to display ribulose-5-P phosphatase activity (51). The existence of such phosphatase activities in mammals is supported by the presence of free ribulose in the urine of humans and fasted rats (52) and of elevated pentitol levels measured in patients with inborn errors in the pentose phosphate pathway. In humans, ribitol is usually present at very low levels in extracellular fluids (less than 6 μm in plasma and cerebrospinal fluid (53)), but this pentitol as well as d-arabitol accumulate in patients with ribose-5-phosphate isomerase (53) or transaldolase (54) deficiencies. In a patient with ribose-5-phosphate isomerase deficiency, millimolar levels of ribitol and d-arabitol were measured in cerebrospinal fluid (53). In these disorders, the amounts of free pentoses formed via hydrolysis of phosphopentose precursors thus seem to exceed the capacity of ribokinase and the herein identified d-ribulokinase, leading to the reduction of excess pentoses and their accumulation as pentitols. FGGY silencing did not lead to detectable ribulose levels in the human cell lines used in this study when cultivated without ribitol supplementation. This may be explained by the only partial knockdown achieved by the shRNA method used and/or low ribulose-5-P phosphatase activity in those cell lines.
Alternatively, the simultaneous presence of d-ribulose-5-P phosphatase and d-ribulokinase activities in a eukaryotic cell could in principle contribute to fine-tuning the regulation of the pentose phosphate pathway flux by substrate cycling (55) between ribulose-5-P and ribulose. As opposed to the metabolite repair hypothesis, the participation of d-ribulokinase in metabolic flux regulation through substrate cycling would, however, call for the existence of a specific and (for example allosterically) regulated ribulose-5-P phosphatase activity rather than a nonspecific and non-regulated production of free ribulose by promiscuous phosphatases.
While this work was ongoing, a new enzymatic activity producing CDP-ribitol was identified in mammals by three independent groups (56–58). This activity is carried by the ISPD protein, which acts as a CDP-ribitol pyrophosphorylase using ribitol-5-P and CTP to form CDP-ribitol. Furthermore, CDP-ribitol was shown to be used by the transferases FKTN and FKRP to incorporate ribitol-5-P from CDP-ribitol into a phosphorylated O-mannosyl glycan (CoreM3) of the α-dystroglycan glycoprotein (57, 58). Abnormal glycosylation of α-dystroglycan, a receptor for matrix and synaptic proteins, can lead to congenital syndromes that are characterized by muscle, brain, and/or eye disorders (59, 60). Mutations in ISPD, FKTN, and FKRP had been known to cause dystroglycanopathies, but until these recent molecular identifications, their role in disease development had remained unknown. The metabolic pathway leading to the formation of ribitol-5-P, the substrate of ISPD, remains unknown. Some of the bacterial homologs of ISPD are fused to a reductase that converts d-ribulose-5-P to ribitol-5-P (61). Such an activity would allow to produce CDP-ribitol from the pentose phosphate pathway intermediate d-ribulose-5-P without the need of a sugar kinase. In the mammalian system, the results obtained by Gerin et al. (58) indicate that, at physiological levels of ribitol, the pathway leading to ribitol-5-P formation involves a sorbinil-sensitive aldose reductase and may indeed be independent of the FGGY protein studied here. However, at supraphysiological ribitol levels, the authors could show that FGGY silencing clearly leads to decreased CDP-ribitol formation in HEK293 cells. Under such conditions, FGGY is thus involved in CDP-ribitol formation, either by directly phosphorylating ribitol or by phosphorylating d-ribulose formed from ribitol. Our results show indeed that HEK293 cells can oxidize ribitol to d-ribulose and that the latter is a 35-fold better substrate for FGGY than ribitol in terms of catalytic efficiency. The observation that sorbinil does not inhibit CDP-ribitol formation in HEK293 cells cultivated in the presence of externally added ribitol favors, however, the hypothesis of a direct phosphorylation of ribitol by FGGY under these conditions. Interestingly, ribitol supplementation in cultivation medium or drinking water led to increased CDP-ribitol levels in mammalian cells and mice, respectively, and ribitol supplementation partially restored α-dystroglycan glycosylation in fibroblasts from patients with ISPD mutations (58). These observations suggest that in patients with mutations in ISPD, but also FKTN and FKRP, dietary ribitol supplementation could exert therapeutic effects via a pathway that depends on the ribitol and/or d-ribulose kinase activity of FGGY.
Experimental Procedures
Chemicals
Unless otherwise indicated, chemicals were from Sigma. All solvents used were HiPerSolv CHROMANORM LC-MS grade from VWR Scientific. Most of the analytical standards were either from Sigma, Carbosynth, or Roche Applied Science and when possible were of greater than 90% purity; LC-MS grade chemicals were used when available. Cell culture media and supplements as well as trypsin were purchased from Life Technologies, Inc. The yeast minimal medium Yeast Nitrogen Base (YNB) with ammonium sulfate and peptone were from MP Biomedicals. Hygromycin B was purchased from Cayman Chemical Co.
Microbial Strains, Cell Lines, and Plasmids
To reduce metabolic and physiological biases introduced by auxotrophic markers, prototrophic S. cerevisiae strains were used for the majority of yeast experiments shown in this study. To analyze the impact of two specific gene deletions on the metabolome, strains of the prototrophic deletion collection (MATa can1Δ::STE2pr-SpHIS5 his3Δ1 lyp1Δ0 ho−), created as described previously (62), in which the YDR109C and MPA43 genes were replaced by the KanMX cassette were used. Those ydr109cΔ::KanMX and mpa43Δ::KanMX knock-out strains are designated as ydr109cΔ and mpa43Δ strains throughout this article. As a wild-type control, we used an isogenic strain from the same deletion collection in which the non-functional HO allele was replaced by the KanMX cassette, except for co-cultivation experiments where an FY4 MATa prototrophic wild-type strain was used. For some experiments, auxotrophic BY4741 strains (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) without or with deletion of the YDR109C gene by replacement with the KanMX cassette were used (Euroscarf).
The BL21(DE3)pLysS E. coli cells used for recombinant protein expression were from Life Technologies, Inc. The stable HEK293 FGGY knockdown cell line and the corresponding control cell line were provided by Dr. Guido Bommer (58). The PH5CH8 cell line was provided by Dr. Nobuyuki Kato.
The Gateway plasmid pDONR221, used to create Entry clones with attL sites upstream and downstream of the gene of interest, was from Invitrogen. The Gateway plasmid pDEST527 was a gift from Dominic Esposito (Addgene plasmid 11518). The empty Gateway plasmid p41Hyg 1-F GW was a gift from Leonid Kruglyak and Sebastian Treusch (Addgene plasmid 58547) (63).
Construction of Plasmids for Recombinant Protein Expression and Rescue Experiments
To express the yeast YDR109C gene in a bacterial system, the coding sequence was PCR-amplified from prototrophic wild-type S. cerevisiae FY4 (MATa) genomic DNA using the YDR109cFwd and YDR109cRev primers (supplemental Table S5) and high fidelity Phusion DNA polymerase (Thermo Fisher Scientific). The primers were designed to contain attB sites, and the Gateway Cloning strategy (Thermo Fisher Scientific) was followed according to the manufacturer's instructions to clone the purified attB-flanked PCR product into the pDONR221 Entry vector, and to subsequently subclone the insert into the pDEST527 Destination vector. This resulted in the pDEST527-YDR109C expression plasmid allowing for IPTG-inducible production of N-terminally His6-tagged fusion protein in E. coli.
Similarly, to produce recombinant human FGGY protein, the coding sequence of the human FGGY gene was amplified from the cDNA IMAGE clone ID 4871664 (Source Bioscience) using the FGGYfwd and FGGYrev primers (supplemental Table S5). The PCR product was cloned into the pDONR221 vector and subcloned into the pDEST527 vector, as described above, to obtain the pDEST527-FGGY expression vector.
A plasmid (p41Hyg 1-F GW-YDR109C) to rescue the yeast metabolic phenotype after YDR109C deletion was prepared by Gateway insertion of the YDR109C coding sequence as well as the 840 nucleotides upstream of the ATG start codon and 300 nucleotides downstream of the stop codon into the low copy number CEN vector p41Hyg 1-F GW (63). The YDR109C gene sequence was PCR-amplified from prototrophic wild-type S. cerevisiae FY4 (MATa) genomic DNA using the YDR109cFwdres and YDR109cRevres primers (supplemental Table S5). All the recombinant vectors described above were confirmed by Sanger sequencing (Eurofins Scientific) to contain the expected insert sequences in the correct orientation.
Yeast Recombinant Protein Expression and Purification
Chemically competent One Shot BL21(DE3)pLysS E. coli cells were transformed by heat shock with the pDEST527-YDR109C plasmid according to the manufacturer's instructions. Positive transformants were selected for on LB-agar plates containing 100 μg/ml ampicillin. A single positive transformant colony was cultured in LB containing 100 μg/ml ampicillin and 2% (w/v) d-glucose at 37 °C until the A600 reached 0.7. Recombinant His-Ydr109c expression was induced with the addition of 500 μm IPTG, and cultures were grown for another 20 h at 37 °C. Cells were harvested by centrifugation at 4500 × g for 15 min at 4 °C, and the cell pellet was stored at −80 °C until further processing.
For protein extraction, cells were resuspended in a lysis buffer (50 μl of buffer per 1 ml of original bacterial culture) containing 20 mm HEPES, pH 7.4, 1 mm DTT, 0.5 mm PMSF, and 1× cOmplete EDTA-free protease inhibitor cocktail (Roche Applied Science) and sonicated on ice (Branson Digital sonifier 250; 5 pulses of 0.5 s at an amplitude of 50% separated by 1-min breaks to minimize sample heat up). The insoluble fraction was removed by centrifugation at 15,000 × g for 40 min at 4 °C, and the supernatant was collected for a one-step purification and enzyme activity assays on the same day, given important activity losses observed in preliminary experiments upon additional purification steps and/or freeze-thaw cycles.
The His-Ydr109c protein was purified using a 1-ml HisTrap HP column (GE Healthcare) on an ÄKTA protein purifier (GE Healthcare), keeping the temperature at 4 °C throughout the procedure. Before purification, the imidazole concentration in the protein extract was adjusted to 10 mm, and the preparation was filtered on a surfactant-free cellulose acetate membrane (1.2 μm pore size, Sartorius). After equilibration of the HisTrap column with 20 ml of buffer A (25 mm Tris-HCl, 300 mm NaCl, pH 8) containing 10 mm imidazole, the protein filtrate was applied onto the column at a flow rate of 1 ml/min. Non-specifically bound proteins were removed by washing the column with 20 ml of 18.7 mm imidazole in buffer A. The His-tagged protein was eluted by applying a 20-min linear imidazole gradient (18.7–300 mm) in buffer A, during which 1-ml fractions were collected and kept on ice. Pure fractions containing His-tagged protein at the expected molecular mass were identified by SDS-PAGE and Western blotting analysis using an anti-His antibody (mouse-derived, 1:1500 dilution in PBS with 0.1% Tween 20 (v/v); GE Healthcare) and a fluorescent secondary antibody (IRDye 680RD, goat-derived, 1:10,000 dilution in Odyssey blocking buffer; Westburg, Leusden, Netherlands).
Human Recombinant Protein Expression and Purification
The human recombinant N-terminal His6-tagged FGGY protein was produced and purified similarly as the His-Ydr109c protein, with slight modifications in the protocol. Protein expression was induced at an A600 of 0.4 by addition of 200 μm IPTG, and E. coli cells were harvested 18 h later. For the purification on the HisTrap HP column, a more shallow 20-min imidazole gradient (18.7–184 mm) was used. Unlike His-Ydr109c, the His-FGGY protein was very stable. An additional desalting step was performed for the latter protein on a 5-ml Hi-Trap desalting column (GE Healthcare). 1 ml of active fraction eluted from the HisTrap HP affinity column was loaded, and a buffer at pH 7.5 containing 20 mm Tris-HCl and 25 mm NaCl was applied at a flow rate of 2.5 ml/min. The desalted purified His-FGGY protein fractions were stored at −80 °C.
Sugar Kinase Activity Assay
The kinase activities of the recombinant Ydr109c and FGGY proteins were determined using the PK/LDH assay system allowing the coupling of ADP formation by the kinase to NADH oxidation by LDH. The rate of NADH consumption, equivalent to the rate of sugar phosphorylation, was determined spectrophotometrically in a plate reader (Infinite M200 PRO, TECAN) or spectrophotometer (SPECORD 210 Plus, Analytik Jena) by monitoring the absorbance at 340 nm at 30 °C in a reaction mixture (total volume of 200 μl or 1 ml for the plate reader and spectrophotometer, respectively) containing 25 mm HEPES, pH 7.1, 5 mm ATP-MgCl2, 0.16 mm NADH, 1 mm phosphoenolpyruvate, 8 units/ml pyruvate kinase, 8 units/ml l-LDH, and the indicated concentrations of the sugars and sugar derivatives tested as substrates. The enzymatic reaction was launched by addition of recombinant Ydr109c or FGGY protein, at a final concentration of 4.3 and 1.9 μg/ml, respectively.
Yeast Cultivation, Measurement of Cell Volume, and Metabolite Extraction
The S. cerevisiae WT and ydr109cΔ (or mpa43Δ) KO prototrophic strains were streaked onto YPD agar plates containing 200 μg/ml geneticin (G418). Single colonies were inoculated into 5 ml of YPD (2% glucose) for overnight precultivation at 30 °C. Precultures were diluted 100 times in 5 ml of YNB supplemented with 2% glucose and incubated at 30 °C with shaking at 200 rpm for 10 h. The final cultivars were obtained by inoculating 25 ml of YNB with 2% glucose, in a 250-ml flask, at an initial A600 of 0.01. A similar cultivation method was used for the auxotrophic strains except that the YNB medium was supplemented with 80 mg/liter uracil, 80 mg/liter histidine, 80 mg/liter methionine, and 240 mg/liter leucine. Cell number and cell volume were determined in aliquots taken from the yeast cultivations using a Multisizer 3 Coulter Counter equipped with a 30-μm measurement capillary (Beckman Coulter) after diluting the samples 1:400 in ISOTON II solution (Prophac).
Metabolites were extracted using a protocol adapted from Ref. 64, when the A600 of the final cultivars was between 3.0 and 3.4. Two-ml aliquots of yeast cultivar were quenched by adding 8 ml of 60% (v/v) methanol at −40 °C. After a 2-min incubation at −40 °C, the cells were separated from the quenching solution by centrifuging at 4400 × g at −10 °C for 5 min. Metabolites were extracted from the pellet by addition of 2 ml of chloroform (−20 °C), 1 ml of methanol (−80 °C), and 0.8 ml of 10 mm ammonium acetate at pH 7.1 (4 °C), followed by 45 min of vortexing at −20 °C. The chloroform and aqueous phases were separated by centrifugation at 4400 × g at −10 °C for 10 min. 1 ml of the upper aqueous phase containing polar metabolites was dried under vacuum at −4 °C overnight, and the dried samples were stored at −80 °C. Before analysis, the samples were resuspended in 80% (v/v) acetonitrile or Milli-Q water for ZIC-HILIC-MS/MS or reverse phase-MS analysis, respectively, and filtered on regenerated cellulose membranes with a 0.2-μm pore size (Phenomenex).
Yeast RNA Extraction and Quantitative RT-PCR
To isolate yeast RNA, samples were obtained from cultivations produced in the same way as for metabolite extraction. Cells were harvested by centrifugation (5 min at 4500 × g at 4 °C) from 20 ml of cultures that had reached an A600 of 3.0–3.4 and resuspended in 800 μl of TriPure isolation reagent (Sigma). The samples were incubated at room temperature for 10 min and then transferred to 2-ml Precellys homogenizer tubes (Peqlab) along with 300 mg of acid-washed glass beads (425–600 mm diameter, Sigma). The samples were homogenized in the Precellys for 30 s at 6000 rpm and 5–10 °C. 200 μl of chloroform were added to the cell lysates, and samples were shaken for 15 min at 1400 rpm and at room temperature, followed by static incubation for 5 min. Samples were centrifuged for 15 min at 12,000 × g and at 4 °C. The aqueous supernatants were added to 500 μl of 4 °C isopropyl alcohol, followed by gentle mixing by inverting tubes and incubated at room temperature for 10 min. The samples were then centrifuged for 15 min at 12,000 × g at 4 °C, and the RNA pellet was washed with 1 ml of 70% ethanol. After an additional 5-min centrifugation at 12,000 × g at 4 °C, the RNA pellets were dried, resuspended in 30 μl of RNase-free water, and incubated at 55 °C for 10 min. Residual genomic DNA was removed from RNA samples by rigorous DNase treatment (TURBO DNase treatment kit, Life Technologies, Inc.) following the manufacturer's protocol. Complementary DNA was synthesized from ∼0.5 μg of DNase-treated RNA using the RevertAid H Minus First Strand cDNA synthesis kit and oligo(dT)18 primers (Thermo Fisher Scientific), following the manufacturer's instructions. The resulting cDNA samples were diluted 20 times in water, and 2 μl of these dilutions were used in 20 μl of qPCRs to determine the expression levels of the YDR109C gene. The qPCR primer sequences are given in supplemental Table S5; the ACT1 and ALG9 genes were used as housekeeping reference genes. qPCRs were run on a Lightcycler 480 instrument (Roche Applied Science) using the SYBR Green Supermix reagent (Bio-Rad) and 0.25 μm gene-specific forward and reverse primers. The qPCR cycle settings were 95 °C for 5 min, then 45 cycles at 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s with acquisition of fluorescence information followed by a melting curve.
Yeast Stable Isotope Labeling Experiments and Production of a U-13C-Labeled Internal Standard for Ribulose Quantification
A co-cultivation protocol for improved untargeted LC-HRMS-based metabolomics analyses was adapted from Ref. 20. Three replicate cultures of each of the MATa FY4 WT and the ydr109cΔ S. cerevisiae prototrophic strains were inoculated from single colonies on YPD plates into 5 ml of YNB supplemented with 2% (w/v) non-labeled glucose and incubated overnight at 30 °C with shaking. Aliquots of the overnight pre-cultures were diluted 100-fold into 5 ml of YNB containing non-labeled glucose or [U-13C]glucose as the sole carbon source at a concentration of 2% and incubated for 9 h at 30 °C with shaking. Main cultures were launched by inoculating 25 ml of YNB containing 2% non-labeled glucose or [U-13C]glucose at a starting A600 of 0.01. For each strain, three non-labeled culture replicates and one labeled culture replicate were prepared. Polar metabolites were extracted using the method described above from each of the culture replicates. The 13C-labeled WT and ydr109cΔ extracts were mixed in a 1:1 ratio. This pooled labeled extract was then added as an internal standard into the individual non-labeled extracts in a 1:1 ratio for subsequent ZIC-HILIC-ddMS2 measurements and data analysis by an extended version of the MetExtract software (20, 21). The pooled 13C-labeled extract was also used for estimation of the intracellular concentration of d-ribulose in selected WT and KO metabolite extracts by adding this internal standard into the samples and into non-labeled d-ribulose standard solutions in a 1:1 ratio. Supplemented samples (1 ml) were dried under vacuum at −4 °C overnight and stored at −80 °C until further use. Before ZIC-HILIC-ddMS2 analysis, dried samples were reconstituted as described above.
In addition, the 13C-labeled ydr109cΔ extract prepared as described above was used to further confirm the identity of the pentose peak accumulating in the knock-out strain by mixing the labeled extract in a 1:1 ratio with non-labeled d-ribulose, d-ribose, or d-xylulose standards (all at an initial concentration of 100 μm) directly followed by ZIC-HILIC-ddMS2 analysis.
Cell Culture and Knockdown of FGGY in Human HEK293 and PH5CH8 Cells
HEK293 cells stably transduced with shRNA expression constructs and PH5CH8 cells were cultured in DMEM (Life Technologies, Inc.) containing 4.5 g/liter glucose, 1 mm pyruvate, and 10% fetal bovine serum. The HEK293 cultivation medium was additionally supplemented with 1% penicillin/streptomycin (Life Technologies, Inc.), 1 μg/ml puromycin (InvivoGen), and 2 mm GlutaMax (Life Technologies, Inc.). The cell lines were incubated at 37 °C in an atmosphere of 5% CO2 in air.
The stable HEK293 cell line with doxycycline-inducible expression of an FGGY-specific shRNA and a corresponding control cell line were created using a lentiviral transduction strategy based on the pTRIPZ plasmid and selection using puromycin as described previously (58). In this study, we used the FGGY knockdown cell line obtained with the pIG271 plasmid, which was constructed by inserting a PCR-amplified oligonucleotide pair into the empty pTRIPZ plasmid, starting from the following sequences: hFGGY1_s 5′-TGC TGT TGA CAG TGA GCG ACA TCG AGC AGT CAG TCA AGT TTA GTG AAG CCA CAG ATG TA-3′ and hFGGY1_as 5′-TCC GAG GCA GTA GGC ACC ATC GAG CAG TCA GTC AAG TTT ACA TCT GTG GCT TCA CTA-3′ (58). About 100,000 cells (in 1 ml of medium) were initially seeded per well into 12-well plates (Nunc). Expression of shRNA was induced by supplementing media with 1 μg/ml doxycycline after 16 h of incubation, and various ribulose precursors were added to the media at the same time. Metabolites and RNA were extracted from different replicate wells 41 h later.
In the PH5CH8 cell line, transient knockdown of FGGY was achieved by transfection of a gene-specific siRNA pool (ON-TARGETplus SMARTpool from Dharmacon) using Lipofectamine 2000 (Life Technologies, Inc). In parallel, control cells were transfected in the same way with non-targeting siRNAs (ON-TARGETplus non-targeting pool from Dharmacon). About 100,000 cells (in 2 ml of medium) were seeded per well into 6-well plates (Nunc). Cells were transfected the following day using 30 nm siRNA, and 6 μl of transfection reagent in the absence of antibiotics in the Opti-MEM culture medium according to the manufacturer's instructions. The culture medium was replaced by fresh DMEM containing various ribulose precursor candidates at the indicated concentrations about 8 h after addition of the RNA-Lipofectamine 2000 complexes. After 24 h, a second round of siRNA transfection followed by medium exchange was performed as described above. Metabolites and RNA were extracted from different replicate wells 48 h later.
Metabolite and RNA Extraction from Human Cells and Quantitative RT-PCR
To extract metabolites from human cells cultivated in 6-well plates, the media were removed, and the cells were washed with 1 ml of 0.9% (w/v) NaCl. To the washed cells, 400 μl of −20 °C methanol and 400 μl of cold milliQ water were added; cells were detached using a cell scraper, keeping the plates on ice; and the cell lysate was collected into 2-ml tubes on ice. To the cell lysate, 400 μl of −20 °C chloroform were added, and the mixture was vortexed for 20 min at 1400 rpm at 4 °C, followed by a 5-min centrifugation at 15,000 × g at 4 °C. 300 μl of the upper aqueous phase, containing polar metabolites, were dried under vacuum at −4 °C, and the dried samples were stored at −80 °C until metabolite analysis. For cells cultivated in 12-well plates, the volume of the reagents added for metabolite extraction was halved.
To extract RNA from the HEK293 and PH5CH8 cells, media were removed, and the cells were resuspended in 1 ml of TriPure Isolation Reagent (Sigma). Total RNA was isolated from the TriPure cell extracts after phase separation using chloroform according to the manufacturer's instructions. The RNA concentration was determined by measuring the absorbance at 260 nm, and the samples were used directly for cDNA synthesis or stored at −80 °C.
To measure FGGY transcript levels in HEK293 and PH5CH8 knockdown cells, cDNA synthesis and qPCRs were performed as described above for measuring gene expression in yeast cells, except that 1.8 μg of RNA was engaged in the cDNA synthesis reactions and reverse transcription reactions were diluted 10-fold before addition to the qPCR mixture. The sequences of the qPCR primers used for the FGGY gene as well as for the ACTB and GAPDH reference genes are given in supplemental Table S5.
LC-MS and LC-MS/MS Methods
Metabolites were analyzed using a Dionex UltiMate 3000 LC system coupled to a Q Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific). Nitrogen was supplied by a Genius 1022 high purity generator (Peak Scientific Instruments, Ltd.). Chromatographic separation was performed using either a SeQuant ZIC-HILIC column (150 × 2.1 mm, 3.5 μm, 100 Å; Merck) fitted with a SeQuant ZIC-HILIC guard column (14 × 1 mm, 5 μm, 200 Å; Merck) or an Acquity UPLC HSS T3 column (150 × 2.1 mm, 1.8 μm, 100 Å; Waters) fitted with a VanGuard Acquity UPLC HSS T3 guard column (5 × 2.1 mm, 1.8 μm, 100 Å; Waters). The LC method for separation of metabolites using the ZIC-HILIC column was adapted from Ref. 65 with the solvent flow modified to a constant flow rate of 150 μl/min and column re-equilibration time increased to 10 min. Briefly, using buffers A (0.1% formic acid in water) and B (0.08% formic acid in acetonitrile), the solvent gradient was 80% B to 20% B from 0 to 30 min, 20% B to 5% B from 30 to 31 min, an isocratic step at 5% B from 31 to 39 min to wash the column, 5% B to 80% B from 39 to 40 min, and another isocratic step at 80% B from 40 to 50 min to re-equilibrate the column. The column oven was set to 20 °C. For the LC-MS method involving reverse phase chromatography on the Acquity HSS T3 column, the same buffers A and B were used. The column oven and solvent flow rate were set to 40 °C and 350 μl/min, respectively. For each run, the column was equilibrated with 1% buffer B from 0 to 1 min, followed by a gradient of 1% B to 90% B from 1 to 30 min, an isocratic step at 90% B from 30 to 32 min, a decrease from 90% B to 1% B from 32 to 33 min, and another isocratic step at 1% B from 33 to 35 min to re-equilibrate the column.
For non-targeted metabolomics, mass spectrometry detection was done either in full scan mode with negative and positive electrospray ionization switching or using a ddMS2 method in either negative or positive ionization mode. For the ZIC-HILIC-ddMS2 method, the following settings were used for the heated electrospray ionization (HESI-2) in both the positive and negative modes: sheath gas flow rate of 40; auxiliary gas flow rate of 10; sweep gas flow rate of 2; capillary temperature of 250 °C; S-lens RF level of 50; and auxiliary gas heater temperature of 300 °C. The spray current was set to 2.5 kV in negative mode and 3.5 kV in positive mode. The HESI-2 probe settings used with the reverse phase full scan method in negative ionization mode were the same as for the ZIC-HILIC-ddMS2 method, whereas for the reverse phase full scan method in positive ionization mode the settings were the following: sheath gas, auxiliary gas, and sweep gas flow rates set to 49, 12, and 2, respectively; spray voltage of 3.5 kV; capillary temperature of 259 °C; S-lens RF level of 50; and auxiliary gas heater temperature of 300 °C. Full scan MS with positive/negative switching was done with a mass resolution of 70,000, an automatic gain control set to 3 × 106, a maximal injection time of 200 ms, and an m/z scan range of 70–1050. For ddMS2 experiments, a survey full scan with these same parameters was run, and MS2 data were recorded for the top 7 m/z features with the highest abundance in the survey scan. The parameters for MS2 recording were as follows: a mass resolution of 17,500; an automatic gain control set to 105; a maximal injection time of 50 ms; a parent ion isolation width of 2 m/z; a normalized collision energy of 40% to fragment the parent ion; a trigger function underfill ratio of 1% with isotope exclusion for MS2 trigger, and a dynamic exclusion of 10 s to minimize redundant MS2 acquisitions.
A targeted ZIC-HILIC-MS method with selected ion monitoring was used to measure ribulose in human cell extracts. In this method, the quadrupole scanning range was restricted to m/z 149–150. All other parameters were the same as for the ZIC-HILIC-ddMS2 method.
MS File Conversions and Data Analysis
Files with .raw format were converted to the mzXML format using the “MSconvert” package of the “ProteoWizard” software, using “R.” For data acquired with the ddMS2 method, MS1 scan events were separated from MS2 scan events. The MZmine-2.20 software (66) was used for untargeted metabolite profiling data analysis. All parameter settings used for the various modules of MZmine 2.20 are given in the supplemental Experimental Procedures. For the co-cultivation experiments, additional data analyses were performed using the MetExtract software (21). For targeted analyses, chromatographic peak areas of interest were integrated with the Qual browser utility in Xcalibur (Thermo Scientific).
Statistical Analysis
The statistical analyses of peak height data tables obtained from the MZmine 2.20 analysis were performed with the MetaboAnalyst 3.0 online tool (67). All the peaks having any match in KEGG, adduct search match, and/or an in-house metabolite library match (within 10 ppm difference in m/z) were retained, and the heights of all the individual peaks retained for each sample were normalized to the sum of all the retained peak heights for this sample (also referred to as metabolic TIC or mTIC normalized data). Univariate Welch's t test, multivariate PCA, and multivariate PLS-DA were performed on the mTIC normalized data. The PLS-DA model was evaluated by using leave-one-out cross-validation. Higher values of the R2 statistic indicated that the PLS-DA model was well fitted. Higher values of the Q2 statistic indicated that the model was not overfitted.
Multiple Sequence Alignments, Phylogenetic Analysis, and Prediction of Specificity Determining Positions
Multiple sequence alignments were generated with the MUSCLE software tool (3.8 online version) (68) using default settings, except if otherwise indicated. To build a phylogenetic tree for FGGY protein family members, we used the CARS of 446 FGGY protein sequences assembled by Zhang et al. (3), which we extended by a set of 11 additional sequences that we confidently identified as d-ribulokinases based on our own experimental data, sequence conservation, and/or genome context (UniProt entries A6TBJ4, B6XGJ3, M9RKB9, B9K586, I2IVR0, Q04585, Q96C11, A2AJL3, Q6NUW9, Q9VZJ8, and F4JQ90). The CARS reference set of sequences from Ref. 3 was built upon 31 FGGY kinase sequences (28 from bacteria and 3 from eukaryotes) with experimentally assigned molecular and biological functions. It was then expanded to 446 kinase sequences by propagation to bacterial homologs with at least 30% sequence identity to one of the 31 starting sequences as well as a conserved genomic and pathway context supporting their functional assignments (3). Within the CARS dataset, 25 clusters with more than 30% sequence identity can be distinguished covering a total of nine different substrate specificities, i.e. functions. Some isofunctional groups (e.g. glycerol kinases and l-ribulose kinases) thus span multiple sequence similarity clusters. The d-ribulokinase cluster of the original CARS protein set contained seven sequences (UniProt ID O52716 and SEED_PEGids fig|272943.3.peg.3604, fig|204722.1.peg.2375, fig|216596.1.peg.7085, fig|224911.1.peg.3226, fig|288000.5.peg.1100, and fig|224914.1.peg.3039). The MSA generated based on the extended CARS sequence set (enriched in d-ribulokinase sequences) was used to perform a phylogenetic analysis with the FastTree 2.1.9 program (69) using a maximum-likelihood model (Jones-Taylor-Thornton, 1992 + CAT) (70) and default settings. As in Ref. 3, the endonuclease subunit of exinuclease ABC (UvrC) was used as an outgroup to determine the root of the phylogenetic tree.
Another MSA was generated to identify SDPs in the d-ribulokinase family of protein sequences. For this MSA, only the sequences contained in the largest cluster for each isofunctional group within the CARS protein set (Clust_IDs 48, 137, 11, 25, 124, 22, 85, 252, and 13 from Ref. 3) as well as the 11 additional d-ribulokinase sequences that we confidently annotated added to the d-ribulokinase cluster (Clust_ID 25) were used (total of 337 sequences). SDP prediction was performed using the GroupSim+ConsWin method (30). The MSA prepared with MUSCLE was re-formatted manually to match the input format used by the GroupSim+ConsWin software (grouping of the sequences according to their function and for each sequence the addition of the group name as suffix to the protein name).
Based on (a) an MSA generated from d-ribulokinase sequences from evolutionary distant organisms, (b) the SDPs predicted for the d-ribulokinase family of proteins, and (c) structural information obtained from d-ribulokinase homology models generated in this study, we defined a motif (TCSLV) that specifically characterizes the d-ribulokinase group of the FGGY protein superfamily. This motif was validated using the MSA generated as described above for the phylogenetic analyses and using an additional MSA generated with the Clustal Omega tool (default settings) (71) from about 634 reviewed FGGY family members retrieved from UniProt. The latter protein sequence set contained 10 isofunctional groups, including the 9 groups represented in the CARS protein set (kinases acting on d-glycerol, l-ribulose, d-ribulose, l-rhamnulose, d-gluconate, l-fuculose, d-xylulose, l-xylulose, and erythritol), and in addition a group of sedoheptulokinases.
Generation and Analysis of Structural Homology Models
For the FGGY (Homo sapiens) and Ydr109c (S. cerevisiae) proteins, no dedicated crystal structures were publicly available at the time of writing this work. Therefore, homology models for structural analysis were created using crystal structures for an ortholog from Y. pseudotuberculosis (UniProt Q665C6; sequence identity 51% to FGGY and 39% to Ydr109c; for the modeling, PDB structures 3L0Q, chain A, and 3GG4, chain A, were used). These structures were favored as templates over an l-ribulokinase crystal structure from Bacillus halodurans (PDB code 3QDK), because this l-ribulokinase has a lower sequence identity to both human FGGY (27%) and yeast Ydr109c (24%) and a less well resolved crystal structure (resolution of 2.31 Å as compared with 1.61 Å for 3L0Q and 2 Å for 3GG4).
To derive structural models from the chosen template protein, multiple candidate homology models were first generated, and the best model for each protein was then determined using the quality assessment score of the software VERIFY3D (72). Specifically, the candidate models were obtained using I-TASSER (73), Swiss-Model (74), RaptorX (75), and ModBase (76) (default parameters were used for all software tools). For both the FGGY and Ydr109c proteins, the homology model derived from ModBase provided the highest VERIFY3D score and was chosen for further analyses.
Structural visualizations of the selected homology models, including the estimation of hydrogen bond and van der Waals interactions in the binding pocket, were generated with the software Chimera (77). Because the ligand d-xylulose was included in the template crystal structure for FGGY (PDB code 3L0Q), but not in the template providing the best scored structural model for Ydr109c (PDB code 3GG4), molecular docking simulations with this ligand were performed in the Ydr109c structure using the software LeadIT/FlexX (version 2.1.5) (78). The top 30 docking poses were then determined, and binding affinity estimates were calculated for each pose by applying the LeadIT/HYDE tool (79). More details on the used docking pipeline can be found in Ref. 80. Finally, structural visualizations for the docking pose with the lowest estimated binding affinity were created using Chimera (77).
Taxonomic Distribution of d-Ribulokinase and l-Ribulokinases
To analyze the taxonomic distribution of d-ribulokinase and l-ribulokinases, we downloaded the FGGY_N domain sequences from the Pfam database for all the phyla possessing at least three such sequences (Archaea: Crenarchaeota and Euryarchaeota; Bacteria: Acidobacteria, Actinobacteria, Armatimonadetes, Bacteroidetes, Chlorobi, Chloroflexi, Cyanobacteria, Deinococcus-Thermus, Dictyoglomi, Firmicutes, Fusobacteria, Planctomycetes, α-Proteobacteria, β-Proteobacteria, γ-Proteobacteria, δ-Proteobacteria, ϵ-Proteobacteria, Spirochaetes, Synergistetes, Tenericutes, Thermotogae, Verrucomicrobia; Eukaryota: Alveolata, Amoebozoa, Choanoflagellida, Euglenozoa, Fungi, Haptophyceae, Metazoa, Parabasalia, Stramenopiles, and Viridiplantae). The d-ribulokinase and l-ribulokinase motifs defined in this study (TCSLV and TGTST, TSST, TGSSP, TGSTP, MMHGY, and TACTM, respectively) were used to estimate, for each phylum, the proportion of d- and l-ribulokinases compared with all the proteins containing an FGGY_N domain in this phylum.
Author Contributions
C. S. and C. L. L. conceived the study, designed the experiments, and mainly wrote the manuscript. C. L. L. supervised the study. C. S. performed the experiments. C. S. and C. L. L. analyzed and interpreted most of the data. E. G. generated the structural homology models and analyzed the models. All authors contributed to manuscript writing and manuscript proofreading. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
pDEST527 was a gift from Dominic Esposito (Addgene plasmid 11518), and p41Hyg 1-F GW was a gift from Leonid Kruglyak and Sebastian Treusch (Addgene plasmid 58547). We thank Amy A. Caudy (Donnelly Centre for Cellular and Biomolecular Research, University of Toronto, Toronto, Canada) for providing the yeast prototrophic strains, Guido Bommer (de Duve Institute and Université Catholique de Louvain, Brussels, Belgium) for providing the FGGY knockdown and control HEK293 cell lines, and Nobuyuki Kato (Okayama University, Japan) for providing the PH5CH8 cell line. We thank Rainer Schuhmacher and Christoph Bueschl (University of Natural Resources and Life Sciences, Vienna, Austria) for analyzing the co-cultivation stable isotope labeling data. We thank Céline Leclercq and Jenny Renaut (Luxembourg Institute of Science and Technology) for LC-MS/MS analysis of the purified FGGY protein. We also thank Wolfram Weckwerth and Lena Fragner (University of Vienna, Vienna, Austria) and Christian Jäger (Luxembourg Centre for Systems Biomedicine) for valuable help and guidance with preliminary GC-MS experiments. Finally, we thank Paul Jung (Luxembourg Centre for Systems Biomedicine) for critical reading of the manuscript and Daniel Kay (Luxembourg Centre for Systems Biomedicine) for proofreading certain sections of this manuscript.
This work was supported by the Fonds National de la Recherche Luxembourg AFR Ph.D. Grant 6885527 and a Fondation du Pélican de Mie et Pierre Hippert-Faber Scholarship (to C. S.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables S1–S5 and Fig. S1.
- S-ALS
- sporadic amyotrophic lateral sclerosis
- CARS
- confidently annotated reference set
- CI
- confidence interval
- FKRP
- fukutin-related protein
- FKTN
- fukutin
- HRMS
- high resolution mass spectrometry
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- ISPD
- isoprenoid synthase domain-containing protein
- KEGG
- Kyoto Encyclopedia of Genes and Genomes
- LC
- liquid chromatography
- MS2
- mass spectrometry-mass spectrometry
- mTIC
- metabolic total ion chromatogram
- PCA
- principal component analysis
- PK/LDH
- pyruvate kinase/lactate dehydrogenase
- PLS-DA
- partial least squares discriminant analysis
- SDP
- specificity determining positions
- SGD
- Saccharomyces Genome Database
- SIL
- stable isotope labeling
- UPLC
- ultra performance liquid chromatography
- YMDB
- yeast metabolome database
- ZIC-HILIC-ddMS2
- zwitterionic-hydrophilic interaction liquid chromatography-data-dependent mass spectrometry mass spectrometry
- qPCR
- quantitative PCR
- MSA
- multiple sequence alignment
- PDB
- Protein Data Bank.
References
- 1. Hanson A. D., Pribat A., Waller J. C., and de Crécy-Lagard V. (2010) ‘Unknown’ proteins and ‘orphan’ enzymes: the missing half of the engineering parts list–and how to find it. Biochem. J. 425, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Niehaus T. D., Thamm A. M., de Crécy-Lagard V., and Hanson A. D. (2015) Proteins of unknown biochemical function: a persistent problem and a roadmap to help overcome it. Plant Physiol. 169, 1436–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang Y., Zagnitko O., Rodionova I., Osterman A., and Godzik A. (2011) The FGGY carbohydrate kinase family: insights into the evolution of functional specificities. PLoS Comput. Biol. 7, e1002318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dunckley T., Huentelman M. J., Craig D. W., Pearson J. V., Szelinger S., Joshipura K., Halperin R. F., Stamper C., Jensen K. R., Letizia D., Hesterlee S. E., Pestronk A., Levine T., Bertorini T., Graves M. C., et al. (2007) Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N. Engl. J. Med. 357, 775–788 [DOI] [PubMed] [Google Scholar]
- 5. Chen Y., Zeng Y., Huang R., Yang Y., Chen K., Song W., Zhao B., Li J., Yuan L., and Shang H. F. (2012) No association of five candidate genetic variants with amyotrophic lateral sclerosis in a Chinese population. Neurobiol. Aging 33, 2721. [DOI] [PubMed] [Google Scholar]
- 6. Chiò A., Schymick J. C., Restagno G., Scholz S. W., Lombardo F., Lai S. L., Mora G., Fung H. C., Britton A., Arepalli S., Gibbs J. R., Nalls M., Berger S., Kwee L. C., Oddone E. Z., et al. (2009) A two-stage genome-wide association study of sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 18, 1524–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Daoud H., Valdmanis P. N., Dion P. A., and Rouleau G. A. (2010) Analysis of DPP6 and FGGY as candidate genes for amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 11, 389–391 [DOI] [PubMed] [Google Scholar]
- 8. Fernandez-Santiago R., Sharma M., Berg D., Illig T., Anneser J., Meyer T., Ludolph A., and Gasser T. (2011) No evidence of association of FLJ10986 and ITPR2 with ALS in a large German cohort. Neurobiol. Aging 32, 551. [DOI] [PubMed] [Google Scholar]
- 9. Van Es M. A., Van Vught P. W., Veldink J. H., Andersen P. M., Birve A., Lemmens R., Cronin S., Van Der Kooi A. J., De Visser M., Schelhaas H. J., Hardiman O., Ragoussis I., Lambrechts D., Robberecht W., Wokke J. H., et al. (2009) Analysis of FGGY as a risk factor for sporadic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 10, 441–447 [DOI] [PubMed] [Google Scholar]
- 10. Cai B., Tang L., Zhang N., and Fan D. (2014) Single-nucleotide polymorphism rs6690993 in FGGY is not associated with amyotrophic lateral sclerosis in a large Chinese cohort. Neurobiol. Aging 35, 1512. [DOI] [PubMed] [Google Scholar]
- 11. Kerner B., Rao A. R., Christensen B., Dandekar S., Yourshaw M., and Nelson S. F. (2013) Rare genomic variants link bipolar disorder with anxiety disorders to CREB-regulated intracellular signaling pathways. Front. Psychiatry 4, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Patti G. J., Yanes O., and Siuzdak G. (2012) Innovation: metabolomics: the apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 13, 263–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ewald J. C., Matt T., and Zamboni N. (2013) The integrated response of primary metabolites to gene deletions and the environment. Mol. Biosyst. 9, 440–446 [DOI] [PubMed] [Google Scholar]
- 14. Saghatelian A., Trauger S. A., Want E. J., Hawkins E. G., Siuzdak G., and Cravatt B. F. (2004) Assignment of endogenous substrates to enzymes by global metabolite profiling. Biochemistry 43, 14332–14339 [DOI] [PubMed] [Google Scholar]
- 15. Clasquin M. F., Melamud E., Singer A., Gooding J. R., Xu X., Dong A., Cui H., Campagna S. R., Savchenko A., Yakunin A. F., Rabinowitz J. D., and Caudy A. A. (2011) Riboneogenesis in yeast. Cell 145, 969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wamelink M. M., Struys E. A., Jansen E. E., Levtchenko E. N., Zijlstra F. S., Engelke U., Blom H. J., Jakobs C., and Wevers R. A. (2008) Sedoheptulokinase deficiency due to a 57-kb deletion in cystinosis patients causes urinary accumulation of sedoheptulose: elucidation of the CARKL gene. Hum. Mutat. 29, 532–536 [DOI] [PubMed] [Google Scholar]
- 17. Teste M.-A., Duquenne M., François J. M., and Parrou J.-L. (2009) Validation of reference genes for quantitative expression analysis by real-time RT-PCR in Saccharomyces cerevisiae. BMC Mol. Biol. 10:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kulak N. A., Pichler G., Paron I., Nagaraj N., and Mann M. (2014) Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11, 319–324 [DOI] [PubMed] [Google Scholar]
- 19. Jewison T., Knox C., Neveu V., Djoumbou Y., Guo A. C., Lee J., Liu P., Mandal R., Krishnamurthy R., Sinelnikov I., Wilson M., and Wishart D. S. (2012) YMDB: the yeast metabolome database. Nucleic Acids Res. 40, D815–D820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bueschl C., Kluger B., Lemmens M., Adam G., Wiesenberger G., Maschietto V., Marocco A., Strauss J., Bödi S., Thallinger G. G., Krska R., and Schuhmacher R. (2014) A novel stable isotope labelling assisted workflow for improved untargeted LC-HRMS based metabolomics research. Metabolomics 10, 754–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bueschl C., Kluger B., Berthiller F., Lirk G., Winkler S., Krska R., and Schuhmacher R. (2012) MetExtract: a new software tool for the automated comprehensive extraction of metabolite-derived LC/MS signals in metabolomics research. Bioinformatics 28, 736–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reinders J., Zahedi R. P., Pfanner N., Meisinger C., and Sickmann A. (2006) Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics. J. Proteome Res. 5, 1543–1554 [DOI] [PubMed] [Google Scholar]
- 23. Sickmann A., Reinders J., Wagner Y., Joppich C., Zahedi R., Meyer H. E., Schönfisch B., Perschil I., Chacinska A., Guiard B., Rehling P., Pfanner N., and Meisinger C. (2003) The proteome of Saccharomyces cerevisiae mitochondria. Proc. Natl. Acad. Sci. U.S.A. 100, 13207–13212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Emanuelsson O., Nielsen H., Brunak S., and von Heijne G. (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 300, 1005–1016 [DOI] [PubMed] [Google Scholar]
- 25. Huck J. H., Roos B., Jakobs C., van der Knaap M. S., and Verhoeven N. M. (2004) Evaluation of pentitol metabolism in mammalian tissues provides new insight into disorders of human sugar metabolism. Mol. Genet. Metab. 82, 231–237 [DOI] [PubMed] [Google Scholar]
- 26. Elsinghorst E. A., and Mortlock R. P. (1988) d-Arabinose metabolism in Escherichia coli B: induction and cotransductional mapping of the l-fucose-d-arabinose pathway enzymes. J. Bacteriol. 170, 5423–5432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heuel H., Shakeri-Garakani A., Turgut S., and Lengeler J. W. (1998) Genes for d-arabinitol and ribitol catabolism from Klebsiella pneumoniae. Microbiology 144, 1631–1639 [DOI] [PubMed] [Google Scholar]
- 28. Neuberger M. S., Hartley B. S., and Walker J. E. (1981) Purification and properties of d-ribulokinase and d-xylulokinase from Klebsiella aerogenes. Biochem. J. 193, 513–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Overbeek R., Begley T., Butler R. M., Choudhuri J. V., Chuang H. Y., Cohoon M., de Crécy-Lagard V., Diaz N., Disz T., Edwards R., Fonstein M., Frank E. D., Gerdes S., Glass E. M., Goesmann A., et al. (2005) The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33, 5691–5702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Capra J. A., and Singh M. (2008) Characterization and prediction of residues determining protein functional specificity. Bioinformatics 24, 1473–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yeh J. I., Charrier V., Paulo J., Hou L., Darbon E., Claiborne A., Hol W. G., and Deutscher J. (2004) Structures of enterococcal glycerol kinase in the absence and presence of glycerol: correlation of conformation to substrate binding and a mechanism of activation by phosphorylation. Biochemistry 43, 362–373 [DOI] [PubMed] [Google Scholar]
- 32. Kameyama T., and Shimazono N. (1962) Enzymic phosphorylation of d-ribulose in guinea-pig liver. Biochim. Biophys. Acta 64, 180–181 [DOI] [PubMed] [Google Scholar]
- 33. Kameyama T., and Shimazono N. (1965) Studies on ribulokinase from liver. J. Biochem. 57, 339–345 [DOI] [PubMed] [Google Scholar]
- 34. Stayton M. M., and Fromm H. J. (1979) Purification, properties, and kinetics of d-ribulokinase from Aerobacter aerogenes. J. Biol. Chem. 254, 3765–3771 [PubMed] [Google Scholar]
- 35. Lee L. V., Gerratana B., and Cleland W. W. (2001) Substrate specificity and kinetic mechanism of Escherichia coli ribulokinase. Arch. Biochem. Biophys. 396, 219–224 [DOI] [PubMed] [Google Scholar]
- 36. Reiner A. M. (1975) Genes for ribitol and d-arabitol catabolism in Escherichia coli: their loci in C strains and absence in K-12 and B strains. J. Bacteriol. 123, 530–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kagawa Y., Kameyama T., Mano Y., and Shimazono N. (1960) Formation of d-ribulose from d-gluconate in guinea-pig liver. Biochim. Biophys. Acta 44, 205–206 [PubMed] [Google Scholar]
- 38. Smiley J. D., and Ashwell G. (1961) Purification and properties of β-l-hydroxy acid dehydrogenase. J. Biol. Chem. 236, 357–364 [Google Scholar]
- 39. Ishikura S., Usami N., Araki M., and Hara A. (2005) Structural and functional characterization of rabbit and human l-gulonate 3-dehydrogenase. J. Biochem. 137, 303–314 [DOI] [PubMed] [Google Scholar]
- 40. Winkelman J., and Ashwell G. (1961) Enzymic formation of l-xylulose from β-keto-l-gulonic acid. Biochim. Biophys. Acta 52, 170–175 [DOI] [PubMed] [Google Scholar]
- 41. McCorkindale J., and Edson N. L. (1954) Polyol dehydrogenases. 1. The specificity of rat-liver polyol dehydrogenase. Biochem. J. 57, 518–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hollmann S., and Touster O. (1957) The l-xylulose-xylitol enzyme and other polyol dehydrogenases of guinea pig liver mitochondria. J. Biol. Chem. 225, 87–102 [PubMed] [Google Scholar]
- 43. Canh D. S., Horak J., Kotyk A., and Rihova L. (1975) Transport of acyclic polyols in Saccharomyces cerevisiae. Folia Microbiol. 20, 320–325 [DOI] [PubMed] [Google Scholar]
- 44. Brown A. D. (1974) Microbial water relations: features of the intracellular composition of sugar-tolerant yeasts. J. Bacteriol. 118, 769–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bernard E. M., Christiansen K. J., Tsang S. F., Kiehn T. E., and Armstrong D. (1981) Rate of arabinitol production by pathogenic yeast species. J. Clin. Microbiol. 14, 189–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wong B., Murray J. S., Castellanos M., and Croen K. D. (1993) d-Arabitol metabolism in Candida albicans: studies of the biosynthetic pathway and the gene that encodes NAD-dependent d-arabitol dehydrogenase. J. Bacteriol. 175, 6314–6320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Quong M. W., Miyada C. G., Switchenko A. C., and Goodman T. C. (1993) Identification, purification, and characterization of a d-arabinitol-specific dehydrogenase from Candida tropicalis. Biochem. Biophys. Res. Commun. 196, 1323–1329 [DOI] [PubMed] [Google Scholar]
- 48. Linster C. L., Van Schaftingen E., and Hanson A. D. (2013) Metabolite damage and its repair or pre-emption. Nat. Chem. Biol. 9, 72–80 [DOI] [PubMed] [Google Scholar]
- 49. Van Schaftingen E., Rzem R., Marbaix A., Collard F., Veiga-da-Cunha M., and Linster C. L. (2013) Metabolite proofreading, a neglected aspect of intermediary metabolism. J. Inherit. Metab. Dis. 36, 427–434 [DOI] [PubMed] [Google Scholar]
- 50. Kuznetsova E., Nocek B., Brown G., Makarova K. S., Flick R., Wolf Y. I., Khusnutdinova A., Evdokimova E., Jin K., Tan K., Hanson A. D., Hasnain G., Zallot R., de Crécy-Lagard V., Babu M., et al. (2015) Functional diversity of haloacid dehalogenase superfamily phosphatases from Saccharomyces cerevisiae: biochemical, structural, and evolutionary insights. J. Biol. Chem. 290, 18678–18698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ingram J. M., and Wood W. A. (1965) Enzymatic basis for d-arabitol production by Saccharmyces rouxii. J. Bacteriol. 89, 1186–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Futterman S., and Roe J. H. (1955) The identification of ribulose and l-xylulose in human and rat urine. J. Biol. Chem. 215, 257–262 [PubMed] [Google Scholar]
- 53. Huck J. H., Verhoeven N. M., Struys E. A., Salomons G. S., Jakobs C., and van der Knaap M. S. (2004) Ribose-5-phosphate isomerase deficiency: new inborn error in the pentose phosphate pathway associated with a slowly progressive leukoencephalopathy. Am. J. Hum. Genet. 74, 745–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Verhoeven N. M., Huck J. H., Roos B., Struys E. A., Salomons G. S., Douwes A. C., van der Knaap M. S., and Jakobs C. (2001) Transaldolase deficiency: liver cirrhosis associated with a new inborn error in the pentose phosphate pathway. Am. J. Hum. Genet. 68, 1086–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Berg J. M., Tymoczko J. L., and Stryer L. (2007) Biochemistry, 6th Ed, pp. 467–468, W. H. Freeman and Co., New York [Google Scholar]
- 56. Riemersma M., Froese D. S., van Tol W., Engelke U. F., Kopec J., van Scherpenzeel M., Ashikov A., Krojer T., von Delft F., Tessari M., Buczkowska A., Swiezewska E., Jae L. T., Brummelkamp T. R., Manya H., et al. (2015) Human ISPD is a cytidyltransferase required for dystroglycan O-mannosylation. Chem. Biol. 22, 1643–1652 [DOI] [PubMed] [Google Scholar]
- 57. Kanagawa M., Kobayashi K., Tajiri M., Manya H., Kuga A., Yamaguchi Y., Akasaka-Manya K., Furukawa J., Mizuno M., Kawakami H., Shinohara Y., Wada Y., Endo T., and Toda T. (2016) Identification of a post-translational modification with ribitol-phosphate and its defect in muscular dystrophy. Cell Rep. 14, 2209–2223 [DOI] [PubMed] [Google Scholar]
- 58. Gerin I., Ury B., Breloy I., Bouchet-Seraphin C., Bolsée J., Halbout M., Graff J., Vertommen D., Muccioli G. G., Seta N., Cuisset J. M., Dabaj I., Quijano-Roy S., Grahn A., Van Schaftingen E., and Bommer G. T. (2016) ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto α-dystroglycan. Nat. Commun. 7, 11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Brockington M., Blake D. J., Prandini P., Brown S. C., Torelli S., Benson M. A., Ponting C. P., Estournet B., Romero N. B., Mercuri E., Voit T., Sewry C. A., Guicheney P., and Muntoni F. (2001) Mutations in the Fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin α2 deficiency and abnormal glycosylation of α-dystroglycan. Am. J. Hum. Genet. 69, 1198–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hewitt J. E. (2009) Abnormal glycosylation of dystroglycan in human genetic disease. Biochim. Biophys. Acta 1792, 853–861 [DOI] [PubMed] [Google Scholar]
- 61. Follens A., Veiga-da-Cunha M., Merckx R., van Schaftingen E., and van Eldere J. (1999) acs1 of Haemophilus influenzae type a capsulation locus region II encodes a bifunctional ribulose 5-phosphate reductase-CDP-ribitol pyrophosphorylase. J. Bacteriol. 181, 2001–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gibney P. A., Lu C., Caudy A. A., Hess D. C., and Botstein D. (2013) Yeast metabolic and signaling genes are required for heat-shock survival and have little overlap with the heat-induced genes. Proc. Natl. Acad. Sci. U.S.A. 110, E4393–E4402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Treusch S., Albert F. W., Bloom J. S., Kotenko I. E., and Kruglyak L. (2015) Genetic mapping of MAPK-mediated complex traits across S. cerevisiae. PLoS Genet. 11, e1004913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. de Koning W., and van Dam K. (1992) A method for the determination of changes of glycolytic metabolites in yeast on a subsecond time scale using extraction at neutral pH. Anal. Biochem. 204, 118–123 [DOI] [PubMed] [Google Scholar]
- 65. Creek D. J., Jankevics A., Breitling R., Watson D. G., Barrett M. P., and Burgess K. E. (2011) Toward global metabolomics analysis with hydrophilic interaction liquid chromatography-mass spectrometry: improved metabolite identification by retention time prediction. Anal. Chem. 83, 8703–8710 [DOI] [PubMed] [Google Scholar]
- 66. Pluskal T., Castillo S., Villar-Briones A., and Oresic M. (2010) MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xia J., Sinelnikov I. V., Han B., and Wishart D. S. (2015) MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic Acids Res. 43, W251–W257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Price M. N., Dehal P. S., and Arkin A. P. (2010) FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jones D. T., Taylor W. R., and Thornton J. M. (1992) The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282 [DOI] [PubMed] [Google Scholar]
- 71. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Eisenberg D., Lüthy R., and Bowie J. U. (1997) VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 277, 396–404 [DOI] [PubMed] [Google Scholar]
- 73. Zhang Y. (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schwede T., Kopp J., Guex N., and Peitsch M. C. (2003) SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Peng J., and Xu J. (2011) RaptorX: exploiting structure information for protein alignment by statistical inference. Proteins 79, 161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pieper U., Eswar N., Davis F. P., Braberg H., Madhusudhan M. S., Rossi A., Marti-Renom M., Karchin R., Webb B. M., Eramian D., Shen M. Y., Kelly L., Melo F., and Sali A. (2006) MODBASE: a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 34, D291–D295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 78. Kramer B., Rarey M., and Lengauer T. (1999) Evaluation of the FLEXX incremental construction algorithm for protein-ligand docking. Proteins 37, 228–241 [DOI] [PubMed] [Google Scholar]
- 79. Schneider N., Hindle S., Lange G., Klein R., Albrecht J., Briem H., Beyer K., Claussen H., Gastreich M., Lemmen C., and Rarey M. (2012) Substantial improvements in large-scale redocking and screening using the novel HYDE scoring function. J. Comput. Aided Mol. Des. 26, 701–723 [DOI] [PubMed] [Google Scholar]
- 80. Glaab E. (2016) Building a virtual ligand screening pipeline using free software: a survey. Brief. Bioinform. 17, 352–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.