

Abstract
Cardiolipin (CL), the signature phospholipid of mitochondrial membranes, plays an important role in mitochondrial processes and bioenergetics. CL is synthesized de novo and undergoes remodeling in the mitochondrial membranes. Perturbation of CL remodeling leads to the rare X-linked genetic disorder Barth syndrome, which shows disparities in clinical presentation. To uncover biochemical modifiers that exacerbate CL deficiency, we carried out a synthetic genetic array screen to identify synthetic lethal interactions with the yeast CL synthase mutant crd1Δ. The results indicated that crd1Δ is synthetically lethal with mutants in pyruvate dehydrogenase (PDH), which catalyzes the conversion of pyruvate to acetyl-CoA. Acetyl-CoA levels were decreased in the mutant. The synthesis of acetyl-CoA depends primarily on the PDH-catalyzed conversion of pyruvate in the mitochondria and on the PDH bypass in the cytosol, which synthesizes acetyl-CoA from acetate. Consistent with perturbation of the PDH bypass, crd1Δ cells grown on acetate as the sole carbon source exhibited decreased growth, decreased acetyl-CoA, and increased intracellular acetate levels resulting from decreased acetyl-CoA synthetase activity. PDH mRNA and protein levels were up-regulated in crd1Δ cells, but PDH enzyme activity was not increased, indicating that PDH up-regulation did not compensate for defects in the PDH bypass. These findings demonstrate for the first time that CL is required for acetyl-CoA synthesis, which is decreased in CL-deficient cells as a result of a defective PDH bypass pathway.
Keywords: acetyl coenzyme A (acetyl-CoA), cardiolipin, energy metabolism, pyruvate dehydrogenase complex (PDC), yeast genetics
Introduction
Cardiolipin (CL)4 is a unique phospholipid with dimeric structure that constitutes about 15% of the total phospholipid in mitochondria (1–4). CL is synthesized de novo in the inner mitochondrial membrane (5) and undergoes remodeling in which saturated fatty acyl chains are replaced with unsaturated fatty acids (6, 7). CL plays an important role in maintenance of mitochondrial structure, interaction with mitochondrial membrane proteins, respiration and stability of respiratory chain supercomplexes (8–10), and other mitochondrial functions such as protein import and maintenance of the membrane potential (11–13). Loss of CL perturbs mitochondrial bioenergetics and decreases ATP synthesis (12, 14–16). Aberrant CL remodeling due to mutation of tafazzin, the transacylase that remodels CL, leads to the severe genetic disorder Barth syndrome (BTHS) (17). Loss of tafazzin results in decreased total CL, increased monolysocardiolipin, and aberrant CL species (18–20). In yeast, tafazzin mutant phenotypes are not due to aberrant CL species but more likely result from decreased total CL/increased monolysocardiolipin (21, 22). The clinical presentation of BTHS includes cardio- and skeletal myopathy, neutropenia, 3-methylglutaconic aciduria, growth retardation, abnormal mitochondria, and defective oxidative phosphorylation (23). However, disparities in the clinical manifestation are characteristic of the disorder (24–26), indicating that physiological modifiers affect the phenotype associated with CL deficiency.
To identify modifiers that exacerbate CL deficiency, we carried out a synthetic genetic array (SGA) screen with the yeast CL synthase mutant crd1Δ, which lacks CL. A striking finding was that crd1Δ cells exhibited genetic interaction with mutants of pyruvate dehydrogenase (PDH), which converts pyruvate to acetyl-CoA (27). PDH null mutants are viable because acetyl-CoA can be synthesized by the cytosolic PDH bypass pathway. This pathway culminates in the acetyl-CoA synthetase-catalyzed conversion of acetate to acetyl-CoA, which is shuttled into the mitochondria. Other routes to acetyl-CoA synthesis include oxidative degradation of amino acids (28), decarboxylation of pyruvate (29), and oxidation of fatty acids (30). Synthetic lethality with PDH mutants could be explained by a requirement for CL in one or more alternate routes to acetyl-CoA synthesis. In this study, we demonstrate that crd1Δ cells have reduced acetyl-CoA levels, diminished ability to utilize acetate, and decreased activity of acetyl-CoA synthetase, consistent with a defective PDH bypass pathway. Interestingly, gene expression and protein levels of PDH were increased, but PDH enzyme activity remained unaltered, indicating that up-regulation of PDH expression is unable to compensate for defects in the PDH bypass in CL-deficient cells. These results show for the first time that CL is required for synthesis of acetyl-CoA.
Results
Genome-wide Synthetic Lethal SGA Screen with crd1Δ
The genome-wide synthetic lethality screen (31) was performed using SGA methodology by mating the query strain (BY4742 MATα can1Δcrd1Δ) to the yeast deletion set and selecting double mutants at two different temperatures (30 and 37 °C). In the query strain, the crd1Δ mutation was linked to the dominant selectable marker URA3 and the reporter construct MFA1pr-HIS3, which is expressed only in MATa cells. This strain (MATα) was separately crossed with the array of all 4800 deletion mutants in the MATa background, in which the deletion is linked to the dominant selectable marker for geneticin resistance, KanMX. The MATα strain was lys− met+, and the MATa strain was met− lys+. Diploids were selected on plates lacking both lysine and methionine, and sporulation was induced. Haploid spore progeny were transferred to the synthetic medium lacking histidine, which allows for selective germination of MATa cells. Following two rounds of selection, the MATa cells were transferred to medium lacking uracil and containing geneticin. Synthetic interaction was indicated by decreased growth of the haploid progeny.
As expected, the SGA screen identified a large number of potential synthetic interactions, including 105 synthetic lethal interactions at 30 °C and 65 lethal interactions at 37 °C. These were grouped based on biological function (Fig. 1A and Tables 1 and 2). Validation of the screen is reflected in the identification of mutants previously shown to be synthetically lethal with crd1Δ, including psd1Δ (32), tom5Δ (13), and get3Δ (33). Synthetic lethal mutants were also identified in cellular functions previously shown to require CL, including cell wall biogenesis, Fe-S biogenesis, mitochondrial protein import, morphology, and supercomplex formation (Fig. 1B).
FIGURE 1.

Functional classification of genes exhibiting synthetic lethality with crd1Δ. A, synthetic lethal interactions identified by the SGA screen were classified by biological process based on gene ontology. B, synthetic lethal partners of crd1Δ identified eight biological processes. Genes that are functionally related radiate from a central point.
TABLE 1.
Synthetic interaction with crd1Δ at 30 °C
Genes were identified as synthetically lethal with the crd1Δ mutant at 30 °C. The identified genes are grouped based on biological function (using GO SLIM Mapper from Saccharomyces Genome Database).
| GO term | Frequency | Gene(s) |
|---|---|---|
| Molecular function unknown | 19 out of 105 genes,18.1% | ERP2, ERP1, VBA4, NBP2, MIT1, RMD6, HMF1, CGR1, SOH1, MDS3, EMC5, HOS4, RAX2, SPH1, NKP2, ESC8, APM4, SGO1, TCO89 |
| Hydrolase activity | 15 out of 105 genes,14.3% | RRT12, DYN2, BNA7, PTC2, RSR1, VMA16, MYO3, OCT1, DBR1, DNM1, ELP6, MSG5, RHO2, ARL3, KRE6 |
| Transferase activity | 13 out of 105 genes,12.4% | SAS4, BUD16, UTR2, GUP1, NMA2, KTR7, TPK3, RTT109, RCK2, KTR5, MET2, MUM3 |
| Structural molecule activity | 11 out of 105 genes,10.5% | RPL21A, IMG2, STE5, RSM24, RPL22B, CHC1, CLC1, RPL27A, PIR3, RPS12, RTC6 |
| Ion binding | 10 out of 105 genes, 9.5% | SHS1, BMH2, STE5, GIC2, RTF1, ATG27, ISA1, ROM2, ISA2 |
| Structural constituent of ribosome | 7 out of 105 genes, 6.7% | RPL21A, IMG2, RSM24, RPL22B, RPL27A, RPS12, RTC6 |
| Transmembrane transporter activity | 7 out of 105 genes, 6.7% | FCY22, VPS73, VMA16, JEN1, COX7, YVC1, TOM5 |
| RNA binding | 7 out of 105 genes, 6.7% | SRO9, RMD9, RTF1, RIM4, ELP6, THP1, CBC2 |
| Oxidoreductase activity | 6 out of 105 genes, 5.7% | SDH4, ERG4, SOD2, COX7, GRX5 |
| DNA binding | 6 out of 105 genes, 5.7% | RRN10, BMH2, ECM22, THP1, TYE7, DIG1 |
| Lipid binding | 6 out of 105 genes, 5.7% | RVS161, STE5, GIC2, ATG27, ROM2, TCB1 |
| GTPase activity | 4 out of 105 genes, 3.8% | RSR1, DNM1, RHO2, ARL3 |
| mRNA binding, peptidase activity | 4 out of 105 genes, 3.8% | SRO9, RMD9, RRT12, OCT1 |
| ATPase activity, signal transducer activity | 4 out of 105 genes, 3.8% | VMA16, ELP6, RSR1, ROM2 |
| Cytoskeletal protein binding | 3 out of 105 genes, 2.9% | RVS161, BUD6, TPM1 |
| Enzyme regulator activity | 3 out of 105 genes, 2.9% | GIC2, BUD6, IRA2 |
| Small conjugating protein binding | 3 out of 105 genes, 2.9% | SLA1, STP22, VPS36 |
| Kinase activity | 3 out of 105 genes, 2.9% | BUD16, TPK3, RCK2 |
| Protein binding transcription factor activity | 3 out of 105 genes, 2.9% | SSN2, RTF1, STB1 |
| Nucleic acid binding transcription factor activity | 3 out of 105 genes, 2.9% | RRN10, ECM22, TYE7 |
| Lyase activity | 3 out of 105 genes, 2.9% | ARO10, ENO1, PSD1 |
| Transferase activity, transferring glycosyl groups | 3 out of 105 genes, 2.9% | UTR2, KTR7, KTR5 |
| Transcription factor binding | 2 out of 105 genes, 1.9% | STB1, DIG1 |
| Phosphatase activity | 2 out of 105 genes, 1.9% | PTC2, MSG5 |
| Nuclease activity, ligase activity | 2 out of 105 genes, 1.9% | DBR1, ARG1 |
| Enzyme binding, chromatin binding | 2 out of 105 genes, 1.9% | GIC2, TOS4 |
| Hydrolase activity, acting on glycosyl bonds | 1 out of 105 genes, 1% | KRE6 |
| Protein transporter activity | 1 out of 105 genes, 1% | TOM5 |
| Protein binding, bridging | 1 out of 105 genes, 1% | SLA1 |
| Nucleotidyltransferase activity | 1 out of 105 genes, 1% | NMA2 |
| Unfolded protein binding | 1 out of 105 genes, 1% | SSQ1 |
| Guanyl-nucleotide exchange factor activity | 1 out of 105 genes, 1% | ROM2 |
TABLE 2.
Synthetic interaction with crd1Δ at 37 °C
Genes were identified as synthetically lethal with the crd1Δ mutant at 37 °C. The identified genes are grouped based on biological function (using GO SLIM Mapper from Saccharomyces Genome Database).
| GO term | Frequency | Gene(s) |
|---|---|---|
| Molecular function unknown | 24 out of 65 genes, 36.9% | DEP1, PRM9, FIG2, HBT1, RTN1, PMP3, RAD34, EAF1, ITC1, EMP24, OST5, TED1, MAD2, ABM1, BUD28, COQ9, REC102, PAU4, AIM34, MDM12, MAM3, TMA16, PRM3, NCA2 |
| Transferase activity | 14 out of 65 genes, 21.5% | DPB3, GRX1, SAT4, PAA1, DBF2, MET14, ELM1, TGL4, ERG6, GTO3, PSK2, PSH1, TUM1, NAT5 |
| Kinase activity | 5 out of 65 genes, 7.7% | SAT4, DBF2, MET14, ELM1, PSK2 |
| Oxidoreductase activity | 4 out of 65 genes, 6.2% | PDB1, GRX1, TSA1, GCY1 |
| Hydrolase activity | 4 out of 65 genes, 6.2% | GET3, TIF2, CPS1, TGL4 |
| RNA binding | 4 out of 65 genes, 6.2% | EFT2, TIF2, GCY1, LEO1 |
| Structural constituent of ribosome | 3 out of 65 genes, 4.6% | RPL27A, RPS24B, RPS4A |
| DNA binding | 3 out of 65 genes, 4.6% | DPB3, BDF2, MIG2 |
| Lipid binding | 3 out of 65 genes, 4.6% | SWH1, TCB2, YPR097W |
| Transcription factor binding | 3 out of 65 genes, 4.6% | BDF2, GAL80, SIN4 |
| Structural molecule activity | 3 out of 65 genes, 4.6% | RPL27A, RPS24B, RPS4A |
| Ion binding | 3 out of 65 genes, 4.6% | SWH1, LEO1, YPR097W |
| Translation factor activity, nucleic acid binding | 2 out of 65 genes, 3.1% | EFT2, TIF2 |
| Enzyme regulator activity | 2 out of 65 genes, 3.1% | GAL80, CLN2 |
| ATPase activity | 2 out of 65 genes, 3.1% | GET3, TIF2 |
| Transmembrane transporter activity | 2 out of 65 genes, 3.1% | FLC2, HUT1 |
| mRNA binding, peptidase activity | 2 out of 65 genes, 3.1% | GCY1, CPS1 |
| Protein transporter activity, protein binding transcription | 2 out of 65 genes, 3.1% | KAP114, LEO1 |
| Nucleic acid binding transcription factor activity, nucleotidyltransferase activity | 2 out of 65 genes, 3.1% | MIG2, DPB3 |
| Lyase activity/ligase activity | 2 out of 65 genes, 3.1% | DSD1, ADE6 |
| Methyltransferase activity, histone binding | 2 out of 65 genes, 3.1% | ERG6, BDF2 |
| Helicase activity | 1 out of 65 genes, 1.5% | TIF2 |
| Small conjugating protein binding | 1 out of 65 genes, 1.5% | GGA1 |
| Protein binding transcription factor activity | 1 out of 65 genes, 1.5% | LEO1 |
| Protein binding, bridging | 1 out of 65 genes, 1.5% | INP1 |
| Unfolded protein binding | 1 out of 65 genes, 1.5% | TSA1 |
| Guanyl-nucleotide exchange factor activity | 1 out of 65 genes, 1.5% | GET3 |
Cell wall biogenesis was previously shown to require a functional CL pathway (34, 35). CL mutants exhibit temperature-sensitive growth that is associated with defects in the cell wall (34, 35). Previous studies reported that the PKC-Slt2 cell integrity pathway and glucan synthase activity require the synthesis of mitochondrial phospholipids phosphatidylglycerol and/or CL (35). pgs1Δ cells, which are blocked in the first step of CL synthesis, have reduced glucan synthase activity and decreased protein levels of the glucan synthase catalytic subunit. In addition, activation of Slt2, the downstream effector of the PKC-activated cell integrity pathway, is defective in the mutant. In this light, it was interesting that the SGA screen identified KRE6, which encodes an integral membrane protein required for β-1,6-glucan biosynthesis (36). Disruption of KRE6 results in perturbation of cell wall biogenesis and decreased growth on fermentable carbon source. Mutants of other genes involved in the cell integrity pathway were also identified in the screen, including MSG5, PIR3, RHO2, ROM2, and UTR2.
The synthetic lethality screen also identified mutants in mitochondrial processes that have been shown to require CL (Fig. 1B). Previous studies indicated that the loss of CL leads to perturbation of mitochondrial and cellular iron homeostasis (37). This study identified synthetic lethal interaction with ISA1 and ISA2, which encode a protein required for maturation of mitochondrial Fe-S proteins, and with GRX5 and SSQ1, which are required for assembly of Fe-S clusters. These findings support a role for CL in Fe-S cluster formation and/or transfer of Fe-S clusters to apoproteins. crd1Δ was also synthetically lethal with a mutant in SOD2, which codes for mitochondrial manganese superoxide dismutase, an enzyme that protects cells against oxygen toxicity (38, 39). Disruption of SOD2 in the CL mutant may lead to lethality due to severe oxidative stress or accumulation of free radicals. The crd1Δ mutant exhibits increased reactive oxygen species, and SOD2 may play a pivotal role in scavenging free radicals in the mutant (40). Synthetic lethality was also observed with OCT1, which encodes the mitochondrial intermediate peptidase required for processing inside mitochondria. This enzyme indirectly plays a role in mitochondrial iron homeostasis (41–43).
The SGA screen identified synthetic lethal interaction with TOM5, which is required for import of mitochondrial proteins. Genetic interaction of crd1Δ with TOM5 was shown in previous studies and is consistent with findings of decreased import of proteins into mitochondria in crd1Δ cells (12, 13). The mitochondrial distribution and morphology complex in the outer mitochondrial membrane are required for protein import, maintenance of mitochondrial DNA and morphology, and exchange of phospholipids between the ER and mitochondrial membrane through the ER-mitochondria encounter structure complex (44). The SGA screen identified synthetic lethality with MDM12, consistent with the previous finding (33). Mutants of other genes involved in mitochondrial morphology were also identified, including DNM1 and MIG1. Taken together, these interactions support a role for CL in tethering of the mitochondrial membrane and ER (33).
The screen also determined that deletion of transporters of lactate, pyruvate, or acetate, including JEN1 and ESBP6, causes a growth defect in crd1Δ cells. The transfer of soluble molecules and substrates across mitochondrial membranes requires anionic phospholipid, especially CL (45). A possible explanation for the observed synthetic lethal interactions between transporters and crd1Δ is that CL is required for association of these transporters with the membrane.
The stabilization of supercomplexes was previously shown to require CL (10, 48). CL mutants exhibit decreased activity of ADP/ATP carrier protein and decreased association of this enzyme with supercomplexes (12, 16). Interestingly, this study determined that mutants of COX7, MGR1, and NCA2 synthetically interact with crd1Δ. Cox7 is a subunit of cytochrome c oxidase (complex IV); Mgr1 is a subunit of the mitochondrial i-AAA protease, and Nca2 regulates expression of ATP synthase. These proteins play an important role in electron transport and ATP synthesis. Synthetic interaction with crd1Δ supports a role for CL in assembly and stabilization of supercomplexes and association with ATP synthase. In Saccharomyces cerevisiae, with electron transport chain complex deficiency is not lethal because cells can grow on glucose by fermentation and synthesize ATP by substrate level phosphorylation (50). Synthetic lethality with electron transport chain and ATP synthesis mutants suggests that CL mutants exhibit defects in fermentative metabolism. Consistent with this, we have shown previously that the loss of mitochondrial DNA (and hence elimination of mitochondrial respiration) in crd1Δ cells leads to decreased growth on glucose as well as activation of the retrograde pathway, consistent with mitochondrial dysfunction (51).
Loss of CL Leads to Decreased Acetyl-CoA Synthesis
The SGA screen revealed that crd1Δ is synthetically lethal with pdb1Δ at 37 °C (the E1β subunit of PDH), as shown in Fig. 2A. This finding was striking, as it suggested for the first time that CL may play a role in the synthesis of acetyl-CoA. We screened all mutants of the PDH complex for synthetic lethality with crd1Δ. In S. cerevisiae, PDH is an ∼8-MDa multienzyme complex (52) consisting of multiple copies of the following three enzymes: pyruvate dehydrogenase (E1) (Pda1p, and Pdb1p) (53, 54); dihydrolipoamide acetyltransferase (E2) (Lat1p) (55); and dihydrolipoamide dehydrogenase (E3) (Lpd1p) (56). A fourth component, protein X (Pdx1p), does not appear to have a catalytic function but is probably involved in assembly of the complex (57). Double mutants (crd1ΔpdhΔ) were tested for growth at elevated temperatures. The crd1Δ mutant in this genetic background (BY4741) can grow at 37 °C but not at 39 °C (58). With the exception of E3, all PDH mutants were synthetically lethal with crd1Δ, including mutants of E1 and E2 subunits and protein X of the PDH complex (Fig. 2B). Because acetyl-CoA synthesis depends primarily on pyruvate utilization by PDH in the mitochondria and the PDH bypass in the cytosol, decreased growth of the double mutants suggested that crd1Δ cells exhibit defective synthesis of acetyl-CoA by the PDH bypass.
FIGURE 2.
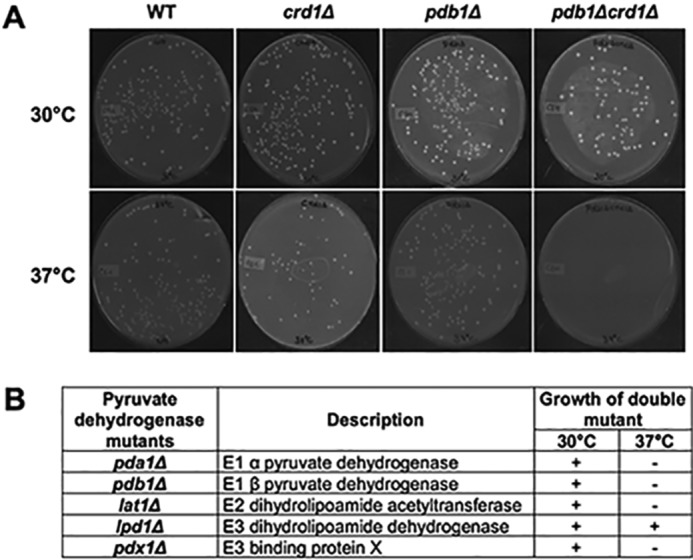
Genetic interaction of PDH mutants with crd1Δ. A, cells were pre-cultured in YPD overnight, diluted, plated in equal number on YNBD, and incubated at 30 or 37 °C for 3–5 days. B, synthetic interaction between CRD1 and PDH mutants was determined by examining growth of the double mutant compared with isogenic parental strains and wild type.
Acetyl-CoA levels were measured in wild-type and crd1Δ cells at optimal (30 °C) and elevated (35, 37, and 39 °C) temperatures. The acetyl-CoA levels in crd1Δ were decreased at elevated temperatures relative to levels in wild-type cells. At 39 °C, an 80% reduction was observed in crd1Δ cells (Fig. 3). Previous studies reported that the PDH bypass pathway predominates over the mitochondrial PDH pathway during fermentative growth (59–64), suggesting that decreased acetyl-CoA levels in crd1Δ cells are likely due to perturbation of this pathway.
FIGURE 3.
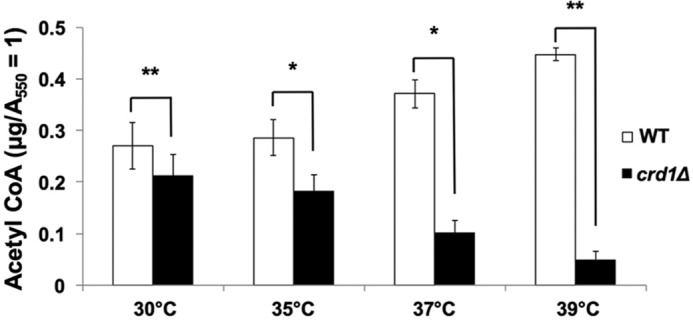
Decreased acetyl-CoA levels in crd1Δ cells. Wild-type and crd1Δ cells were grown at the indicated temperatures in YPD until the cells reached an A550 of 1. Cells were pelleted, and acetyl-CoA was extracted using glutaryl-CoA as an internal standard. The clarified extract (10 μl) was analyzed by HPLC-MS/MS. Data shown are mean ± S.D. (n = 6) (*, p < 0.05; **, p < 0.01).
PDH Bypass Deficiencies in crd1Δ
During fermentative growth, the PDH bypass in the cytoplasm converts pyruvate to acetaldehyde, which is converted to acetate. In the final step, acetyl-CoA synthetase catalyzes the conversion of acetate to acetyl-CoA (27, 65–68). We explored the possibility that decreased acetyl-CoA levels in the crd1Δ mutant resulted from perturbation of the PDH bypass. Consistent with a defective PDH bypass pathway (69–71), crd1Δ cells exhibited decreased growth (Fig. 4A) and an increased doubling time (Fig. 4B) on medium containing acetate as the sole carbon source. Decreased growth was likely not due to defective uptake of acetate, as intracellular acetate levels were significantly increased in crd1Δ cells relative to wild type (Fig. 5). The accumulation of acetate suggested that cells were defective in the conversion of acetate to acetyl-CoA. In agreement with this, crd1Δ cells grown on acetate medium exhibited an ∼50% decrease in acetyl-CoA levels (Fig. 6A) and an ∼45% decrease in acetyl-CoA synthetase activity (Fig. 6B). These results indicate that CL deficiency leads to a decreased synthesis of acetyl-CoA by the PDH bypass route as a result of defective acetyl-CoA synthetase activity.
FIGURE 4.

Decreased growth of crd1Δ cells on acetate. A, cells were pre-cultured overnight in YNBD and diluted, plated in equal number on YNBD and YNBA, and incubated at 30 °C for 3–5 days. B, growth in liquid YNBA was determined by measuring A550. The growth curve values are an average of at least three experiments ± S.D., n ≥3.
FIGURE 5.
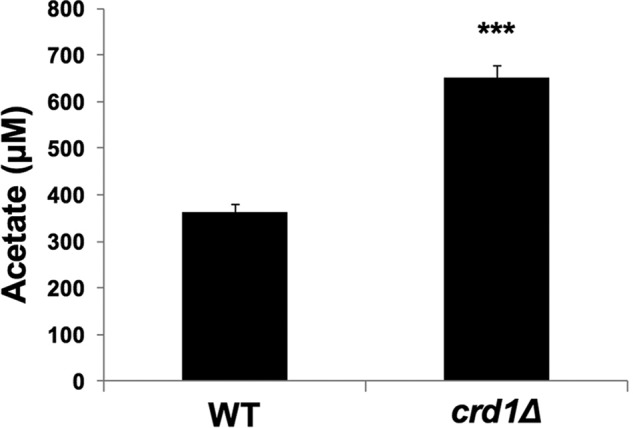
Increased acetate levels in crd1Δ cells. Wild-type and crd1Δ cells were grown in YNBA at 30 °C, and intracellular acetate levels were determined as described under “Experimental Procedures.” Data shown are mean ± S.D. (n = 6) (***, p < 0.001).
FIGURE 6.

Perturbation of PDH bypass in crd1Δ cells. Wild-type and crd1Δ cells were grown in YNBA at 30 °C to the logarithmic phase. A, acetyl-CoA levels were determined as described under “Experimental Procedures.” Data shown are mean ± S.D. (n = 6) (**, p < 0.01). B, enzyme activity (units/mg) of acetyl-CoA synthetase was assayed as described under “Experimental Procedures.” Data shown are mean ± S.D. (n = 6) (***, p < 0.001).
Up-regulation of PDH Expression Does Not Compensate for Defective PDH Bypass in crd1Δ
PDH and PDH bypass null mutants are viable, indicating that either pathway suffices to synthesize acetyl-CoA. Published studies have shown that cells compensate for a defective PDH bypass by up-regulating PDH gene expression and increasing PDH protein levels (59, 71–73). The crd1Δ mutant exhibited more than 2-fold increased mRNA levels of PDH genes, including PDA1, PDB1, LAT1, LPD1, and PDX1 (Fig. 7A). Protein levels of Pdb1, Lat1, Lpd1, and Pdx1 were also elevated in crd1Δ cells, although levels of Pda1 were not (Fig. 7B). However, despite increased PDH mRNA and protein, the enzyme activity of PDH was not increased in crd1Δ cells (Fig. 7C). One possible explanation for this is that CL is required for optimal PDH enzyme activity. Consistent with this, PDH enzyme activity is increased when reconstituted in the presence of CL (Fig. 7D). Taken together, these experiments indicate that PDH does not compensate for defects in the PDH bypass in CL-deficient cells.
FIGURE 7.

PDH activity is not increased in crd1Δ cells. Cells were pre-cultured in YPD overnight, diluted, plated in equal numbers on YNBD, and incubated at 30 or 37 °C for 3–5 days. A, mRNA levels of PDH genes from cells grown in YPD at 30 °C to the logarithmic phase were quantified by qPCR. Values are reported as fold change in expression compared with wild type. Expression was normalized to the mRNA levels of the internal control ACT1. Data shown are mean ± S.D. (n = 6) (*, p < 0.05; **, p < 0.01; ***, p < 0.001). B, anti-HA antibody was used to detect HA-tagged proteins in cell lysates by Western blotting analysis. 50 μg of total protein was loaded for each sample, and α-tubulin was used as an internal control. The molecular masses are as follows: tubulin, 50 kDa; PDA1, 39.4 kDa; PDB1, 43.3 kDa; LAT1, 69 kDa; LPD1, 54 kDa; and PDX1, 37 kDa (top). The levels of PDH protein were quantified using ImageJ software (bottom). Data shown are mean ± S.D. (n = 3) (*, p < 0.05; **, p < 0.01). C, enzyme activity (units/mg) of PDH was assayed in cell extracts as described under “Experimental Procedures.” Data shown are mean ± S.D. (n = 6). D, enzyme activity (units/mg) of PDH was assayed in the presence or absence of CL as described under “Experimental Procedures.” Data shown are mean ± S.D. (n = 6) (*, p < 0.05; ***, p < 0.001).
Discussion
In this study, we identified mutants that are synthetically lethal with the CL mutant crd1Δ, which may thus identify physiological modifiers of CL deficiency. The most striking finding was synthetic lethality of crd1Δ and PDH mutants, as this suggested for the first time that CL is required for the synthesis of acetyl-CoA. In agreement with this, acetyl-CoA levels were decreased in CL-deficient cells (Fig. 3). The likely mechanism underlying this defect is perturbation of the PDH bypass, as crd1Δ cells grown on medium containing acetate as the sole carbon source exhibited decreases in growth, acetyl-CoA levels, and activity of acetyl-CoA synthetase (Figs. 4 and 6). Acetyl-CoA synthetase is localized in the outer mitochondrial membrane. Optimal activity of the enzyme may be dependent on interaction with CL, which is present in small amounts in the outer membrane (13, 74, 75).
Consistent with previous studies showing increased PDH expression in response to perturbation of the PDH bypass, PDH mRNA and protein levels were increased in crd1Δ cells. However, this response did not lead to increased PDH enzyme activity. Several possibilities could account for this finding. Import of PDH into the mitochondria may be decreased in CL-deficient cells, which exhibit decreased import of some mitochondrial proteins (12, 13). Alternatively, PDH stability and/or enzymatic activity may require association with CL in the mitochondrial membrane (75). This is supported by our finding that in vitro activity of PDH is increased when reconstituted in the presence of CL (Fig. 7D).
Based on these results, we propose the following model to account for decreased acetyl-CoA synthesis in crd1Δ cells (Fig. 8). The two common routes for acetyl-CoA synthesis are through direct conversion of pyruvate, either by the PDH bypass under fermentative conditions (76) or PDH under respiratory conditions (29). These routes complement each other when either pathway is defective. Under fermentative conditions, CL-deficient cells cannot convert acetate to acetyl-CoA, resulting in decreased acetyl-CoA levels. The cellular response to decreased acetyl-CoA in crd1Δ cells is increased PDH gene expression and protein synthesis. However, in the absence of CL, PDH enzyme activity is not increased, resulting in decreased acetyl-CoA levels.
FIGURE 8.

Model, perturbation of acetyl-CoA synthesis in crd1Δ cells. In the proposed model, loss of CL leads to decreased acetyl-CoA synthetase enzyme activity, resulting in accumulation of acetate and decreased synthesis of acetyl-CoA by the PDH bypass. To compensate for defects in the PDH bypass, PDH gene expression and protein synthesis are increased in CL-deficient cells. However, PDH activity is not increased, possibly because mitochondrial import, enzymatic activity, or assembly of PDH inside the mitochondria requires CL. Thus, a defective PDH bypass pathway results in decreased synthesis of acetyl-CoA, which is not compensated by PDH.
The synthetic interactions identified by the SGA screen provided additional support for the role of CL in Fe-S biogenesis and cell wall synthesis. Studies have shown that crd1Δ cells exhibit defective Fe-S biogenesis resulting in perturbation of iron homeostasis (37). Defective Fe-S biogenesis leads to decreased activity of Fe-S enzymes, including the TCA cycle enzymes aconitase and succinate dehydrogenase, which in turn leads to deficiencies in the TCA cycle. This study identified genes that are involved in assembly of the Fe-S cluster and maturation of mitochondrial Fe-S proteins, further supporting a role for CL in Fe-S biogenesis. The CL pathway was also shown to be required for cell wall synthesis (34, 35). The cell wall integrity mitogen-activated protein kinase Slt2 is not phosphorylated in the pgs1Δ mutant, which is blocked in the first step of the CL pathway, synthesis of phosphatidylglycerol phosphate. Perturbation of the PKC pathway in pgs1Δ leads to the breakdown of glucan and chitin in the cell wall. This study identified synthetic lethality of crd1Δ with a mutant in the cell wall biogenesis gene KRE6, further supporting the role of CL in maintaining cell integrity. In addition, the screen also identified genes involved in mitochondrial protein import, mitochondrial morphology, and transport of metabolites, which supports previous findings that CL is required for these cellular processes.
Is the role of CL in acetyl-CoA synthesis relevant to BTHS and other cardiac disorders? Acetyl-CoA interconnects metabolic pathways that are crucial for cardiac energy metabolism (77). Mutations in the human E1-α subunit of the PDH complex, which is homologous to yeast PDA1, lead to Leigh's syndrome, which is characterized by hypertrophic cardiomyopathy, among other phenotypes (78).5 Mutations in mitochondrial acetyl-CoA synthetase (AceCS1 and AceCS2) impair acetyl-CoA synthesis and induce cardiac hypertrophy (75, 80). Cardiomyopathy and heart failure are generally caused by depletion of cardiomyocyte ATP and impaired energy homeostasis (79, 81). Elucidating the role of CL in acetyl-CoA synthesis may shed light on the wide disparities in clinical phenotypes observed in patients with BTHS and other cardiac disorders.
Experimental Procedures
Yeast Strains, Plasmids, and Growth Media
The yeast S. cerevisiae strains used in this study are listed in Table 3. Single deletion mutants were obtained from the yeast knock-out deletion collection (Invitrogen). Double mutants were obtained by tetrad dissection. Synthetic complete (YNB) medium contained adenine (20.25 mg/liter), arginine (20 mg/liter), histidine (20 mg/liter), leucine (60 mg/liter), lysine (200 mg/liter), methionine (20 mg/liter), threonine (300 mg/liter), tryptophan (20 mg/liter), uracil (20 mg/liter), yeast nitrogen base without amino acids (Difco), and glucose (2%) (YNBD) or sodium acetate (2%) (YNBA). Synthetic dropout medium contained all ingredients mentioned above but lacked specific compounds required for selection. Sporulation medium contained potassium acetate (1%), glucose (0.05%), and the essential amino acids. Complex medium contained yeast extract (1%), peptone (2%), and glucose (2%) (YPD). Solid medium was prepared by adding 2% agar. To construct the crd1Δcan1Δ starting strain, a 1.8-kb CAN1 deletion cassette was amplified from Y5563 by sense primer (5′-TAGGGCGAACTTGAAGAATAACC-3′) and antisense primer (5′-ACGAAAAATGAGTAAAAATTATCTT-3′) and inserted into the genome of BY4741 (MATa) with the disruption confirmed by PCR. The can1Δ mutant was then crossed to BY4742 (MATα) to obtain an MATα can1Δ strain by tetrad analysis. The CRD1 gene in the can1Δ mutant (MATα) was disrupted by a 1.8-kb URA3 fragment released from the PUC19 plasmid digested using PvuII. The disruption of CRD1 was confirmed by PCR, Southern blotting, and phospholipid analysis.
TABLE 3.
Yeast strains used in this study
| Strains | Genotype | Source or Ref. |
|---|---|---|
| Y5563 | MATα, his3Δ1, leu2Δ0, ura3Δ0, met15Δ0, LYS2+, lyp1Δ, can1Δ::MFA1pr-HIS3 | Tong et al. (31) |
| BY4741 | MATα, his3Δ1, leu2Δ0, ura3Δ0, met15Δ0 | Invitrogen |
| BY4742 | MATα, his3Δ1, leu2Δ0, ura3Δ0, lys2Δ0 | Invitrogen |
| crd1Δcan1Δ | MATα, his3Δ1, leu2Δ0, lys2Δ0, crd1Δ::URA3, can1Δ::MFA1pr-HIS3 | This study |
| VGY1 | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, crd1Δ::URA3 | Gohil et al. (32) |
| CG922-a | MATa, lys2-801, ade2-101, trp1Δ1, his3Δ200, leu2Δ1, crd1Δ::URA3 | Invitrogen |
| CG923-α | MATα, lys2-801, ade2-101, trp1Δ1, his3Δ200, leu2Δ1, crd1Δ::URA3 | Invitrogen |
| pda1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, pda1Δ::KanMX4 | Invitrogen |
| pdb1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, pdb1Δ::KanMX4 | Invitrogen |
| lat1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, lat1Δ::KanMX4 | Invitrogen |
| lpd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, lpd1Δ::KanMX4 | Invitrogen |
| pdx1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, pdx1Δ::KanMX4 | Invitrogen |
| acs1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, acs1Δ::KanMX4 | Invitrogen |
| acs2Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, acs2Δ::KanMX4 | Invitrogen |
| pda1Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, pda1Δ::KanMX4, crd1Δ::KanMX4 | This study |
| pdb1Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, pdb1Δ::KanMX4, crd1Δ::KanMX4 | This study |
| lat1Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, lat1Δ::KanMX4, crd1Δ::KanMX4 | This study |
| lpd1Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, lpd1Δ::KanMX4, crd1Δ::KanMX4 | This study |
| pdx1Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, pdx1Δ::KanMX4, crd1Δ::KanMX4 | This study |
| acs1Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, acs1Δ::KanMX4, crd1Δ::KanMX4 | This study |
| acs2Δcrd1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, acs2Δ::KanMX4, crd1Δ::KanMX4 | This study |
SGA
The MATα can1Δcrd1Δ mutant was crossed to the array of deletion mutants in the MATa background, in which the deletions are linked to the dominant selectable marker for geneticin resistance, KanMX4. Diploids were selected, and sporulation was induced. Haploid spore progeny were transferred to synthetic medium lacking histidine, which allowed for selective germination of MATa cells. Following two rounds of selection in His− synthetic medium, double mutant MATa progeny were selected in Ura− medium supplemented with geneticin. Independent confirmation of synthetic lethality was carried out by tetrad analysis to rule out false positives and false negatives (31). Synthetic interaction between CL and deletion mutants was determined by examining growth of the double mutant compared with single mutants and wild type on YPD.
Acetyl-CoA Determination
Cells were grown to the logarithmic growth phase, and samples (3 ml) of the culture were centrifuged at 1700 × g. Pellets were resuspended in 1 ml of double distilled H2O and centrifuged for 1 min at 9300 × g. The resulting cell pellets were lysed and rapidly quenched with 130 μl of 45:45:10 acetonitrile/methanol/H2O + 0.1% glacial acetic acid and 10 μmol liter−1 glutaryl-CoA as an internal standard. The resuspended extracts were incubated on ice with intermittent vortexing for 15 min. An equal molar volume of ammonium hydroxide was added postincubation to neutralize the acetic acid, and each extract was centrifuged for 3 min at 15,700 × g, transferred to a new 1.5-ml microcentrifuge tube, and centrifuged for 5 min at 15,700 × g. The clarified extract (10 μl) was injected for HPLC-MS/MS analysis (46, 47).
Acetate Measurement
Cells were grown to the logarithmic phase, and samples (1 ml) of the culture were centrifuged at 5000 × g. Pellets were resuspended in 1 ml of double distilled H2O and centrifuged for 5 min at 5000 × g. The resulting cell pellets were lysed. The levels of intracellular acetate in lysed samples were determined spectrophotometrically by monitoring the conversion of ATP to AMP, coupled to the reduction of a reporter dye to yield a colored reaction product with strong absorbance at 450 nm (BioVision kit).
Acetyl-CoA Synthetase Activity
Acetyl-CoA synthetase was measured in cell lysates as described previously (49). The reaction mixture contained 100 mm Tris-HCl (pH 7.8), 5 mm dl-malate, 1 mm ATP, 2.5 mm MgCl2, 0.1 mm coenzyme A, 3 mm NAD+, 2.5 units/ml malate dehydrogenase, 1.25 units/ml citrate synthase, and 100 mm sodium acetate. The reaction mixture was incubated at 37 °C for 10 min. Acetyl-CoA synthetase activity was measured spectrophotometrically by determining the reduction of NAD+ to NADH by recording the continuous increase in absorbance at 340 nm at 37 °C. Total protein was determined using a Bradford assay kit (Pierce) with BSA as the standard. Acetyl-CoA synthetase enzyme activity was expressed as units per mg of total protein.
Quantitative PCR (qPCR)
Cultures (10 ml) were grown to the logarithmic growth phase; cells were harvested, and total RNA was isolated using the RNeasy Plus mini kit from Qiagen. Complementary DNA (cDNA) was synthesized using a transcriptor first strand cDNA synthesis kit (Roche Applied Science) according to the manufacturer's manual. qPCR was performed in a 25-μl volume using Brilliant III Ultra-Faster SYBR Green qPCR Master Mix (Agilent Technologies, Santa Clara, CA). The primers for qPCR are listed in Table 4. PCRs were initiated at 95 °C for 10 min for denaturation followed by 40 cycles consisting of 30 s at 95 °C and 60 s at 55 °C. RNA levels were normalized to ACT1. Relative values of mRNA transcripts are shown as fold change relative to indicated controls. Primer sets were validated according to the Methods and Applications Guide from Agilent Technologies. Optimal primer concentrations were determined, and primer specificity of a single product was monitored by a melt curve following the amplification reaction. All primers were validated by measurement of PCR efficiency and have calculated reaction efficiencies between 95 and 105%.
TABLE 4.
Primers used for qPCR analyses
| Gene | Primer sequence | |
|---|---|---|
| ACT1 | Forward | ACGTTCCAGCCTTCTACGTTTCCA |
| Reverse | CGTGAGTAACACCATCACCGGAA | |
| PDA1 | Forward | ATTGATGGGTAGAAGAGCCGGTGT |
| Reverse | AGGCGTCCTCGTTCTTGTATTGGT | |
| PDB1 | Forward | TCCCATCATTTGGTGTTGGTGCTG |
| Reverse | TGGAACATCGGCACCAGTAACTCT | |
| LAT1 | Forward | AGGAACTAGTCAAGCGTGCCAGAA |
| Reverse | TCCACAGCGACCCTTTCAACAGTA | |
| LPD1 | Forward | AGAGCCAAGACCAACCAAGACACT |
| Reverse | CATTTCACCGGCATTTGGACCGAT | |
| PDX1 | Forward | GCAGCCAAGCCAATCTTGAACAGA |
| Reverse | TTGGAACCAGATGGCGCAATTTCC | |
Gene Tagging, SDS-PAGE, and Western Blotting Analysis
Strains containing C-terminal HA-tagged Pda1, Pdb1, Lat1, Lpd1, and Pdx1 were derived from the BY4742 wild-type and crd1Δ strains by transforming with a PCR product amplified from pFA6–3HA-TRP1 (TRP marker). The primers used for tagging are listed in Table 5. Proteins were extracted from cells grown to an A550 of 0.5, separated by 8% SDS-PAGE, transferred to a PVDF membrane, and analyzed using primary antibodies to the HA tag (1:1000) and to α-tubulin (1:1000) (Santa Cruz Biotechnology). Proteins were visualized using appropriate secondary antibody conjugated to horseradish peroxidase (1:3000) and detected using the ECL chemiluminescence system (GE Healthcare).
TABLE 5.
Primers used for C-terminal tagging of proteins
| 5′ F2-pda1-HA tag | TCCCTGAAGATACTTGGGACTTCAAAAAGCAAGGTTTTGCCTCTAGGGAT CGGATCCCCGGGTTAATTAA |
|---|---|
| 3′ R1-pda1-HA tag | GAATATCATGCGATCACAGCACTATTATTTTATTTTTCCTTACGATTTAA GAATTCGAGCTCGTTTAAAC |
| 5′ F2-pdb1-HA tag | CTGATACTCCAACCATCGTTAAAGCTGTCAAAGAAGTCTTGTCAATTGAA CGGATCCCCGGGTTAATTAA |
| 3′ F2-pdb1-HA tag | AAAGTTCCCTATCTCCTTCTTTCTCTCCTTCCTATTGGATTGAAGTTTAT GAATTCGAGCTCGTTTAAAC |
| 5′ F2-lat1-HA tag | TCATGAAGGAATTGAAAACTGTTATTGAAAATCCTTTGGAAATGCTATTGCGGATCCCCGGGTTAATTAA |
| 3′ F2-lat1-HA tag | ATTTTCTCCAAGATACGCATTTACTGGCGAATTTTATTTTCATTCTAACCGAATTCGAGCTCGTTTAAAC |
| 5′ F2-lpd1-HA tag | AAGCATTTAAGGAAGCTAACATGGCTGCCTATGATAAAGCTATTCATTGT CGGATCCCCGGGTTAATTAA |
| 3′ F2-lpd1-HA tag | AAGCGGTTCTTCATAAATATATATACTATACTGTTTATTATTTCCTGTTT GAATTCGAGCTCGTTTAAAC |
| 5′ F2-pdx1-HA tag | AGGCCAAGGCAAAAAGATTCCTTGATTACGTAAGGGAGTTAGAATCATTT CGGATCCCCGGGTTAATTAA |
| 3′ F2-pdx1-HA tag | ATGGCAGTATTGATAATGATAAACTCGAACTGAAAAAGCGTGTTTTTTAT GAATTCGAGCTCGTTTAAAC |
| 5′ Forward HA tag | CGGATCCCCGGGTTAATTAA |
| 3′ Reverse HA tag | GAATTCGAGCTCGTTTAAAC |
| 3′ HA-KMX-D-general | GGTAGAGGTGTGGTCAATAAGAGC |
| 3′ HA tag-Diag | GGGACGTCATACGGATAGCC |
Pyruvate Dehydrogenase (PDH) Activity
PDH activity in cell extracts was measured spectrophotometrically by determining the reduction of NAD+ to NADH, coupled to the reduction of a reporter dye to yield a colored reaction product with an increase in absorbance at 450 nm at 37 °C (BioVision kit). PDH enzyme activity was expressed in units per mg of total protein.
To determine the effect of CL on PDH activity, aliquots of sedimented yeast mitochondria (400 μg of protein) were suspended in 40 μl of 50 mm NaCl and 50 mm imidazole-HCl (pH 7.0). 2 μl of CL (from a stock solution containing 20 mg/ml in 5% digitonin and 50% ethanol) and 1 μl of digitonin (10% in water) were added to obtain a CL/protein ratio of 1:10 (g/g) and a digitonin/protein ratio of 0.5 (g/g). After 2 h of incubation on ice, 10 μl of digitonin (10% stock solution in water) was added to obtain a digitonin/protein ratio of 3 (g/g), which was sufficient for quantitative solubilization. The samples were used directly in PDH activity assays.
Author Contributions
V. R. and M. L. G. designed the research, analyzed the data, and wrote the paper; V. R. conducted all of the experiments; A. S. J. assisted in growth experiments; G. L. constructed the SGA starting strain; K. R. M. carried out the mass spectrometry.
Acknowledgments
We thank Dr. Charles Boone for the Y5563 yeast strain and Dr. Athar Ansari for the pFA6–3HA-TRP1 plasmid.
This work was supported by National Institutes of Health Grant HL117880, the Barth Syndrome Foundation (to M. L. G.), and the Graduate School of Wayne State University (to V. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
R. E. Frye and P. J. Benke (2008) Pyruvate dehydrogenase complex deficiency, personal communication.
- CL
- cardiolipin
- PDH
- pyruvate dehydrogenase
- BTHS
- Barth syndrome
- SGA
- synthetic genetic array
- qPCR
- quantitative PCR
- ER
- endoplasmic reticulum.
References
- 1. Pangborn M. C. (1947) The composition of cardiolipin. J. Biol. Chem. 168, 351–361 [PubMed] [Google Scholar]
- 2. Schlame M., and Ren M. (2009) The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim. Biophys. Acta 1788, 2080–2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schlame M., Rua D., and Greenberg M. L. (2000) The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 39, 257–288 [DOI] [PubMed] [Google Scholar]
- 4. Jakovcic S., Getz G. S., Rabinowitz M., Jakob H., and Swift H. (1971) Cardiolipin content of wild type and mutant yeasts in relation to mitochondrial function and development. J. Cell Biol. 48, 490–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gallet P. F., Petit J. M., Maftah A., Zachowski A., and Julien R. (1997) Asymmetrical distribution of cardiolipin in yeast inner mitochondrial membrane triggered by carbon catabolite repression. Biochem. J. 324, 627–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beranek A., Rechberger G., Knauer H., Wolinski H., Kohlwein S. D., and Leber R. (2009) Identification of a cardiolipin-specific phospholipase encoded by the gene CLD1 (YGR110W) in yeast. J. Biol. Chem. 284, 11572–11578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xu Y., Kelley R. I., Blanck T. J., and Schlame M. (2003) Remodeling of cardiolipin by phospholipid transacylation. J. Biol. Chem. 278, 51380–51385 [DOI] [PubMed] [Google Scholar]
- 8. Cruciat C. M., Brunner S., Baumann F., Neupert W., and Stuart R. A. (2000) The cytochrome bc1 and cytochrome c oxidase complexes associate to form a single supracomplex in yeast mitochondria. J. Biol. Chem. 275, 18093–18098 [DOI] [PubMed] [Google Scholar]
- 9. Zhang M., Mileykovskaya E., and Dowhan W. (2002) Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J. Biol. Chem. 277, 43553–43556 [DOI] [PubMed] [Google Scholar]
- 10. Pfeiffer K., Gohil V., Stuart R. A., Hunte C., Brandt U., Greenberg M. L., and Schägger H. (2003) Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880 [DOI] [PubMed] [Google Scholar]
- 11. Schlame M., and Ren M. (2006) Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 580, 5450–5455 [DOI] [PubMed] [Google Scholar]
- 12. Jiang F., Ryan M. T., Schlame M., Zhao M., Gu Z., Klingenberg M., Pfanner N., and Greenberg M. L. (2000) Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J. Biol. Chem. 275, 22387–22394 [DOI] [PubMed] [Google Scholar]
- 13. Gebert N., Joshi A. S., Kutik S., Becker T., McKenzie M., Guan X. L., Mooga V. P., Stroud D. A., Kulkarni G., Wenk M. R., Rehling P., Meisinger C., Ryan M. T., Wiedemann N., Greenberg M. L., and Pfanner N. (2009) Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: implications for Barth syndrome. Curr. Biol. 19, 2133–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koshkin V., and Greenberg M. L. (2000) Oxidative phosphorylation in cardiolipin-lacking yeast mitochondria. Biochem. J. 347, 687–691 [PMC free article] [PubMed] [Google Scholar]
- 15. Koshkin V., and Greenberg M. L. (2002) Cardiolipin prevents rate-dependent uncoupling and provides osmotic stability in yeast mitochondria. Biochem. J. 364, 317–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Claypool S. M., Oktay Y., Boontheung P., Loo J. A., and Koehler C. M. (2008) Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J. Cell Biol. 182, 937–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schlame M., Ren M., Xu Y., Greenberg M. L., and Haller I. (2005) Molecular symmetry in mitochondrial cardiolipins. Chem. Phys. Lipids 138, 38–49 [DOI] [PubMed] [Google Scholar]
- 18. Schlame M., Towbin J. A., Heerdt P. M., Jehle R., DiMauro S., and Blanck T. J. (2002) Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann. Neurol. 51, 634–637 [DOI] [PubMed] [Google Scholar]
- 19. Valianpour F., Mitsakos V., Schlemmer D., Towbin J. A., Taylor J. M., Ekert P. G., Thorburn D. R., Munnich A., Wanders R. J., Barth P. G., and Vaz F. M. (2005) Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. J. Lipid Res. 46, 1182–1195 [DOI] [PubMed] [Google Scholar]
- 20. Schlame M., Kelley R. I., Feigenbaum A., Towbin J. A., Heerdt P. M., Schieble T., Wanders R. J., DiMauro S., and Blanck T. J. (2003) Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 42, 1994–1999 [DOI] [PubMed] [Google Scholar]
- 21. Ye C., Lou W., Li Y., Chatzispyrou I. A., Hüttemann M., Lee I., Houtkooper R. H., Vaz F. M., Chen S., and Greenberg M. L. (2014) Deletion of the cardiolipin-specific phospholipase Cld1 rescues growth and life span defects in the tafazzin mutant: implications for Barth syndrome. J. Biol. Chem. 289, 3114–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baile M. G., Sathappa M., Lu Y. W., Pryce E., Whited K., McCaffery J. M., Han X., Alder N. N., and Claypool S. M. (2014) Unremodeled and remodeled cardiolipin are functionally indistinguishable in yeast. J. Biol. Chem. 289, 1768–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barth K. H., Brusilow S. W., Kaufman S. L., and Ferry F. T. (1981) Percutaneous transluminal angioplasty of homograft renal artery stenosis in a 10-year-old girl. Pediatrics 67, 675–677 [PubMed] [Google Scholar]
- 24. Johnston J., Kelley R. I., Feigenbaum A., Cox G. F., Iyer G. S., Funanage V. L., and Proujansky R. (1997) Mutation characterization and genotype-phenotype correlation in Barth syndrome. Am. J. Hum. Genet. 61, 1053–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bleyl S. B., Mumford B. R., Brown-Harrison M. C., Pagotto L. T., Carey J. C., Pysher T. J., Ward K., and Chin T. K. (1997) Xq28-linked noncompaction of the left ventricular myocardium: prenatal diagnosis and pathologic analysis of affected individuals. Am. J. Med. Genet. 72, 257–265 [PubMed] [Google Scholar]
- 26. Barth P. G., Wanders R. J., Vreken P., Janssen E. A., Lam J., and Baas F. (1999) X-linked cardioskeletal myopathy and neutropenia (Barth syndrome) (MIM 302060). J. Inherit. Metab. Dis. 22, 555–567 [DOI] [PubMed] [Google Scholar]
- 27. Pronk J. T., Yde Steensma H., and Van Dijken J. P. (1996) Pyruvate metabolism in Saccharomyces cerevisiae. Yeast 12, 1607–1633 [DOI] [PubMed] [Google Scholar]
- 28. Vorapreeda T., Thammarongtham C., Cheevadhanarak S., and Laoteng K. (2012) Alternative routes of acetyl-CoA synthesis identified by comparative genomic analysis: involvement in the lipid production of oleaginous yeast and fungi. Microbiology 158, 217–228 [DOI] [PubMed] [Google Scholar]
- 29. Guest J. R., Angier S. J., and Russell G. C. (1989) Structure, expression, and protein engineering of the pyruvate dehydrogenase complex of Escherichia coli. Ann. N.Y. Acad. Sci. 573, 76–99 [DOI] [PubMed] [Google Scholar]
- 30. Trotter P. J. (2001) The genetics of fatty acid metabolism in Saccharomyces cerevisiae. Annu. Rev. Nutr. 21, 97–119 [DOI] [PubMed] [Google Scholar]
- 31. Tong A. H., Evangelista M., Parsons A. B., Xu H., Bader G. D., Pagé N., Robinson M., Raghibizadeh S., Hogue C. W., Bussey H., Andrews B., Tyers M., and Boone C. (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294, 2364–2368 [DOI] [PubMed] [Google Scholar]
- 32. Gohil V. M., Thompson M. N., and Greenberg M. L. (2005) Synthetic lethal interaction of the mitochondrial phosphatidylethanolamine and cardiolipin biosynthetic pathways in Saccharomyces cerevisiae. J. Biol. Chem. 280, 35410–35416 [DOI] [PubMed] [Google Scholar]
- 33. Joshi A. S., Fei N., and Greenberg M. L. (2016) Get1p and Get2p are required for maintenance of mitochondrial morphology and normal cardiolipin levels. FEMS Yeast Res. 16, fow019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhong Q., Gvozdenovic-Jeremic J., Webster P., Zhou J., and Greenberg M. L. (2005) Loss of function of KRE5 suppresses temperature sensitivity of mutants lacking mitochondrial anionic lipids. Mol. Biol. Cell 16, 665–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhong Q., Li G., Gvozdenovic-Jeremic J., and Greenberg M. L. (2007) Up-regulation of the cell integrity pathway in Saccharomyces cerevisiae suppresses temperature sensitivity of the pgs1Δ mutant. J. Biol. Chem. 282, 15946–15953 [DOI] [PubMed] [Google Scholar]
- 36. Roemer T., and Bussey H. (1991) Yeast β-glucan synthesis: KRE6 encodes a predicted type II membrane protein required for glucan synthesis in vivo and for glucan synthase activity in vitro. Proc. Natl. Acad. Sci. U.S.A. 88, 11295–11299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Patil V. A., Fox J. L., Gohil V. M., Winge D. R., and Greenberg M. L. (2013) Loss of cardiolipin leads to perturbation of mitochondrial and cellular iron homeostasis. J. Biol. Chem. 288, 1696–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saffi J., Sonego L., Varela Q. D., and Salvador M. (2006) Antioxidant activity of l-ascorbic acid in wild-type and superoxide dismutase deficient strains of Saccharomyces cerevisiae. Redox Rep. 11, 179–184 [DOI] [PubMed] [Google Scholar]
- 39. van Loon A. P., Pesold-Hurt B., and Schatz G. (1986) A yeast mutant lacking mitochondrial manganese-superoxide dismutase is hypersensitive to oxygen. Proc. Natl. Acad. Sci. U.S.A. 83, 3820–3824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen S., He Q., and Greenberg M. L. (2008) Loss of tafazzin in yeast leads to increased oxidative stress during respiratory growth. Mol. Microbiol. 68, 1061–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Branda S. S., Yang Z. Y., Chew A., and Isaya G. (1999) Mitochondrial intermediate peptidase and the yeast frataxin homolog together maintain mitochondrial iron homeostasis in Saccharomyces cerevisiae. Hum. Mol. Genet. 8, 1099–1110 [DOI] [PubMed] [Google Scholar]
- 42. Cavadini P., Adamec J., Taroni F., Gakh O., and Isaya G. (2000) Two-step processing of human frataxin by mitochondrial processing peptidase. Precursor and intermediate forms are cleaved at different rates. J. Biol. Chem. 275, 41469–41475 [DOI] [PubMed] [Google Scholar]
- 43. Knight S. A., Sepuri N. B., Pain D., and Dancis A. (1998) Mt-Hsp70 homolog, Ssc2p, required for maturation of yeast frataxin and mitochondrial iron homeostasis. J. Biol. Chem. 273, 18389–18393 [DOI] [PubMed] [Google Scholar]
- 44. Kornmann B., Currie E., Collins S. R., Schuldiner M., Nunnari J., Weissman J. S., and Walter P. (2009) An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325, 477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bisaccia F., and Palmieri F. (1984) Specific elution from hydroxylapatite of the mitochondrial phosphate carrier by cardiolipin. Biochim. Biophys. Acta 766, 386–394 [DOI] [PubMed] [Google Scholar]
- 46. Bennett B. D., Kimball E. H., Gao M., Osterhout R., Van Dien S. J., and Rabinowitz J. D. (2009) Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 5, 593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Armando J. W., Boghigian B. A., and Pfeifer B. A. (2012) LC-MS/MS quantification of short-chain acyl-CoA's in Escherichia coli demonstrates versatile propionyl-CoA synthetase substrate specificity. Lett. Appl. Microbiol. 54, 140–148 [DOI] [PubMed] [Google Scholar]
- 48. Zhang M., Mileykovskaya E., and Dowhan W. (2005) Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J. Biol. Chem. 280, 29403–29408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brown T. D., Jones-Mortimer M. C., and Kornberg H. L. (1977) The enzymic interconversion of acetate and acetyl-coenzyme A in Escherichia coli. J. Gen. Microbiol. 102, 327–336 [DOI] [PubMed] [Google Scholar]
- 50. Ephrussi B., and Slonimski P. P. (1955) Subcellular units involved in the synthesis of respiratory enzymes in yeast. Nature 176, 1207–1208 [DOI] [PubMed] [Google Scholar]
- 51. Chen S., Liu D., Finley R. L. Jr., and Greenberg M. L. (2010) Loss of mitochondrial DNA in the yeast cardiolipin synthase crd1 mutant leads to up-regulation of the protein kinase Swe1p that regulates the G2/M transition. J. Biol. Chem. 285, 10397–10407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Uhlinger D. J., Yang C. Y., and Reed L. J. (1986) Phosphorylation-dephosphorylation of pyruvate dehydrogenase from bakers' yeast. Biochemistry 25, 5673–5677 [DOI] [PubMed] [Google Scholar]
- 53. Steensma H. Y., Holterman L., Dekker I., van Sluis C. A., and Wenzel T. J. (1990) Molecular cloning of the gene for the E1 α subunit of the pyruvate dehydrogenase complex from Saccharomyces cerevisiae. Eur. J. Biochem 191, 769–774 [DOI] [PubMed] [Google Scholar]
- 54. Miran S. G., Lawson J. E., and Reed L. J. (1993) Characterization of PDHβ1, the structural gene for the pyruvate dehydrogenase β subunit from Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 90, 1252–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Niu X. D., Browning K. S., Behal R. H., and Reed L. J. (1988) Cloning and nucleotide sequence of the gene for dihydrolipoamide acetyltransferase from Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 85, 7546–7550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dickinson J. R., Roy D. J., and Dawes I. W. (1986) A mutation affecting lipoamide dehydrogenase, pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase activities in Saccharomyces cerevisiae. Mol. Gen. Genet. 204, 103–107 [DOI] [PubMed] [Google Scholar]
- 57. Lawson J. E., Behal R. H., and Reed L. J. (1991) Disruption and mutagenesis of the Saccharomyces cerevisiae PDX1 gene encoding the protein X component of the pyruvate dehydrogenase complex. Biochemistry 30, 2834–2839 [DOI] [PubMed] [Google Scholar]
- 58. Zhong Q., Gohil V. M., Ma L., and Greenberg M. L. (2004) Absence of cardiolipin results in temperature sensitivity, respiratory defects, and mitochondrial DNA instability independent of pet56. J. Biol. Chem. 279, 32294–32300 [DOI] [PubMed] [Google Scholar]
- 59. Boubekeur S., Bunoust O., Camougrand N., Castroviejo M., Rigoulet M., and Guérin B. (1999) A mitochondrial pyruvate dehydrogenase bypass in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 274, 21044–21048 [DOI] [PubMed] [Google Scholar]
- 60. Ciriacy M. (1975) Genetics of alcohol dehydrogenase in Saccharomyces cerevisiae. II. Two loci controlling synthesis of the glucose-repressible ADH II. Mol. Gen. Genet. 138, 157–164 [DOI] [PubMed] [Google Scholar]
- 61. Paquin C. E., and Williamson V. M. (1986) Ty insertions at two loci account for most of the spontaneous antimycin A resistance mutations during growth at 15°C of Saccharomyces cerevisiae strains lacking ADH1. Mol. Cell. Biol. 6, 70–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Briquet M. (1977) Transport of pyruvate and lactate in yeast mitochondria. Biochim. Biophys. Acta 459, 290–299 [DOI] [PubMed] [Google Scholar]
- 63. Nałecz M. J., Nałecz K. A., and Azzi A. (1991) Purification and functional characterisation of the pyruvate (monocarboxylate) carrier from baker's yeast mitochondria (Saccharomyces cerevisiae). Biochim. Biophys. Acta 1079, 87–95 [DOI] [PubMed] [Google Scholar]
- 64. Johnston M. (1999) Feasting, fasting and fermenting. Glucose sensing in yeast and other cells. Trends Genet. 15, 29–33 [DOI] [PubMed] [Google Scholar]
- 65. Dickinson F. M. (1996) The purification and some properties of the Mg2+-activated cytosolic aldehyde dehydrogenase of Saccharomyces cerevisiae. Biochem. J. 315, 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. van den Berg M. A., de Jong-Gubbels P., Kortland C. J., van Dijken J. P., Pronk J. T., and Steensma H. Y. (1996) The two acetyl-coenzyme A synthetases of Saccharomyces cerevisiae differ with respect to kinetic properties and transcriptional regulation. J. Biol. Chem. 271, 28953–28959 [DOI] [PubMed] [Google Scholar]
- 67. Van den Berg M. A., and Steensma H. Y. (1995) ACS2, a Saccharomyces cerevisiae gene encoding acetyl-coenzyme A synthetase, essential for growth on glucose. Eur. J. Biochem. 231, 704–713 [DOI] [PubMed] [Google Scholar]
- 68. Meaden P. G., Dickinson F. M., Mifsud A., Tessier W., Westwater J., Bussey H., and Midgley M. (1997) The ALD6 gene of Saccharomyces cerevisiae encodes a cytosolic, Mg2+-activated acetaldehyde dehydrogenase. Yeast 13, 1319–1327 [DOI] [PubMed] [Google Scholar]
- 69. De Virgilio C., Bürckert N., Barth G., Neuhaus J. M., Boller T., and Wiemken A. (1992) Cloning and disruption of a gene required for growth on acetate but not on ethanol: the acetyl-coenzyme A synthetase gene of Saccharomyces cerevisiae. Yeast 8, 1043–1051 [DOI] [PubMed] [Google Scholar]
- 70. Remize F., Andrieu E., and Dequin S. (2000) Engineering of the pyruvate dehydrogenase bypass in Saccharomyces cerevisiae: role of the cytosolic Mg2+ and mitochondrial K+ acetaldehyde dehydrogenases Ald6p and Ald4p in acetate formation during alcoholic fermentation. Appl. Environ. Microbiol. 66, 3151–3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kozak B. U., van Rossum H. M., Luttik M. A., Akeroyd M., Benjamin K. R., Wu L., de Vries S., Daran J. M., Pronk J. T., and van Maris A. J. (2014) Engineering acetyl coenzyme A supply: functional expression of a bacterial pyruvate dehydrogenase complex in the cytosol of Saccharomyces cerevisiae. MBio 5, e01696–01614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wei Y., Lin M., Oliver D. J., and Schnable P. S. (2009) The roles of aldehyde dehydrogenases (ALDHs) in the PDH bypass of Arabidopsis. BMC Biochem. 10, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Avidan O., and Pick U. (2015) Acetyl-CoA synthetase is activated as part of the PDH-bypass in the oleaginous green alga Chlorella desiccata. J. Exp. Bot. 66, 7287–7298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fujino T., Kondo J., Ishikawa M., Morikawa K., and Yamamoto T. T. (2001) Acetyl-CoA synthetase 2, a mitochondrial matrix enzyme involved in the oxidation of acetate. J. Biol. Chem. 276, 11420–11426 [DOI] [PubMed] [Google Scholar]
- 75. Campuzano V., Montermini L., Lutz Y., Cova L., Hindelang C., Jiralerspong S., Trottier Y., Kish S. J., Faucheux B., Trouillas P., Authier F. J., Dürr A., Mandel J. L., Vescovi A., Pandolfo M., and Koenig M. (1997) Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 6, 1771–1780 [DOI] [PubMed] [Google Scholar]
- 76. Shiba Y., Paradise E. M., Kirby J., Ro D. K., and Keasling J. D. (2007) Engineering of the pyruvate dehydrogenase bypass in Saccharomyces cerevisiae for high-level production of isoprenoids. Metab. Eng. 9, 160–168 [DOI] [PubMed] [Google Scholar]
- 77. Abo Alrob O., and Lopaschuk G. D. (2014) Role of CoA and acetyl-CoA in regulating cardiac fatty acid and glucose oxidation. Biochem. Soc. Trans. 42, 1043–1051 [DOI] [PubMed] [Google Scholar]
- 78. Stacpoole P. W., Barnes C. L., Hurbanis M. D., Cannon S. L., and Kerr D. S. (1997) Treatment of congenital lactic acidosis with dichloroacetate. Arch. Dis. Child. 78, 535–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Neubauer S. (2007) The failing heart–an engine out of fuel. N. Engl. J. Med. 356, 1140–1151 [DOI] [PubMed] [Google Scholar]
- 80. Ellis J. M., Mentock S. M., Depetrillo M. A., Koves T. R., Sen S., Watkins S. M., Muoio D. M., Cline G. W., Taegtmeyer H., Shulman G. I., Willis M. S., and Coleman R. A. (2011) Mouse cardiac acyl coenzyme a synthetase 1 deficiency impairs fatty acid oxidation and induces cardiac hypertrophy. Mol. Cell. Biol. 31, 1252–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ingwall J. S. (2009) Energy metabolism in heart failure and remodelling. Cardiovasc. Res. 81, 412–419 [DOI] [PMC free article] [PubMed] [Google Scholar]