

Abstract
Eph receptors, the largest subfamily of transmembrane tyrosine kinase receptors, have been increasingly implicated in various physiologic and pathologic processes, and the roles of the Eph family members during tumorigenesis have recently attracted growing attentions. In the present study, we explored the function of EphB3, one member of Eph family, in papillary thyroid cancer (PTC). We found that the expression of EphB3 was significantly elevated in PTC. Either overexpression of EphB3 or activation of EphB3 by EfnB1-Fc/EfnB2-Fc stimulated in vitro migration of PTC cells. In contrast, siRNA-mediated knockdown of EphB3 or EphB3-Fc treatment, which only blocked EphB3-mediated forward signaling, inhibited migration and metastasis of PTC cells. A mechanism study revealed that EphB3 knockdown led to suppressed activity of Rac1 and enhanced activity of RhoA. Moreover, we found that Vav2, an important regulator of Rho family GTPases, was activated by EphB3 in a kinase-dependent manner. Altogether, our work suggested that EphB3 acted as a tumor promoter in PTC by increasing the in vitro migration as well as the in vivo metastasis of PTC cells through regulating the activities of Vav2 and Rho GTPases in a kinase-dependent manner.
Keywords: cancer biology, cell migration, focal adhesion, metastasis, Rho (Rho GTPase), EphB3, Vav2, papillary thyroid cancer
Introduction
Thyroid cancer (TC)3 is the most common endocrine malignancy, and its incidence is rising rapidly. The main type of TC is papillary thyroid cancer (PTC), which accounts for >81% of the new TC cases (1, 2). The treatment for PTC generally benefits the majority of the patients with well differentiated PTC; however, up to 10% of patients eventually die of the disease, and many more patients have the risk of recurrences (3–5). Clinical investigations reveal that patients diagnosed with PTC at an early stage usually have excellent prognosis, whereas individuals with large, invasive tumors and/or distant metastases have a low survival rate (6–8). Genetically, PTC is frequently associated with the oncogenic conversion of receptor-tyrosine kinases (RTKs), thus clarifying the role of RTKs in PTC metastasis, and targeting of specific RTKs will benefit the treatment of thyroid cancer (9).
The Eph receptor family, known as the largest receptor-tyrosine kinase superfamily, contains 14 distinct members, and its ligand ephrin family has 9 members identified to date (10). Either Eph receptors or ephrin ligands are classified as type A or type B, according to the sequence homology and binding specificity. All Eph receptors are single transmembrane proteins with intrinsic tyrosine kinase activity. As for ephrin ligands, ephrin-Bs are also single transmembrane proteins with intrinsic tyrosine kinase domain, whereas ephrin-As are linked into membrane by a glycosylphosphatidylinositol anchor without transmembrane and endocytotic domain (11, 12). When activated by the cognate ephrin, Eph receptor can transduce intracellular signals, which are involved in the regulation of various biological processes, including axon guidance, neural crest cell migration, hindbrain segmentation, somite formation, and vasculogenesis (11, 12).
Apart from development, accumulating evidence suggests involvement of Ephs/ephrins in cell migration and metastasis in several cancers. It is reported that loss of expression of EphB1 in serous carcinoma of ovary is corrected with metastasis and poor survival (13), and enhanced expression of ephrinB1 is associated with lymph node metastasis and poor prognosis in breast cancer (14). Knockdown of EphA1 promotes adhesion and motility of colorectal carcinoma cells (15), whereas EphA2 promotes epithelial-mesenchymal transition in gastric cancer cells (16). These studies suggested that the effect of Ephs/ephrins on migration and metastasis is divergent, dependent on cancer types and stages.
Despite increasing studies of Ephs/ephrins in cancer biology, their function in thyroid cancer remains largely unknown. In a previous study, EphA2 and EphA4 expression in human benign and malignant thyroid lesions were examined by immunohistochemistry, and it is found that overexpression of EphA2, but not EphA4, may be associated with malignant transformation of thyroid neoplasia (17). Another study using a loss-of-function genetic screening has identified novel mediators of thyroid cancer cell viability, including EphA2, A7, B2, and B6 (18). However, there have been no published reports investigating the role and mechanism of Ephs/ephrins on migration and metastasis of thyroid cancer cells to date, which is urgently required to improve therapeutic strategies for this malignancy.
In the present study, we reported that EphB3 effectively regulated either in vitro migration or in vivo metastasis of PTC via modulating the activities of Vav2 and Rho family GTPases in a kinase-dependent manner. Our study will not only expand the understanding of Ephs/ephrins in cancer biology but also indicate potential therapeutic strategy for PTC treatment.
Results
The Expression Level of EphB3 Was Elevated in PTC Tissues
To determine the expression pattern of EphB3 in primary PTC, we first quantified the mRNA level of EphB3 in 15 pairs of primary PTC samples and the matched normal thyroid tissue samples by real-time PCR. The mRNA level of β-actin was used to normalize EphB3 expression. Up-regulation of EphB3 occurred in 10 of 15 (67%) of the PTC samples compared with the normal counterparts (Fig. 1A). Furthermore, immunostaining of EphB3 in three pairs of PTC tissues and the matched normal thyroid tissues was shown in Fig. 1B. Strong staining of EphB3 was observed in the PTC tissues, whereas the normal thyroid tissues showed negative/weak staining of EphB3 (Fig. 1B). Moreover, evaluation of a published microarray data set of seven paired PTC samples and normal thyroid samples (GDS1732) also revealed significantly elevated expression of EphB3 in PTC samples (Fig. 1C, p = 0.0141). Therefore, the expression of EphB3 was increased in PTCs, indicating its role as a tumor promoter in PTC.
FIGURE 1.

Expression level of EphB3 was increased in clinical samples of PTC. A, relative expression of EphB3 mRNA in 15 pairs of PTC and their matched normal thyroid tissue samples. B, immunohistochemistry survey of EphB3 expression in three pairs of PTC and the matched normal thyroid tissues. Scale bar, 100 μm. C, mRNA expression of EphB3 was examined by microarray assay in seven papillary thyroid carcinoma samples and seven paired normal thyroid tissue samples (GDS1732).
EphB3 Regulated Both in Vitro Migration and in Vivo Metastasis of PTC Cells in a Kinase-dependent Manner
To explore the role of EphB3 in PTC, we introduced wild-type EphB3 and kinase dead mutant of EphB3 (EphB3-KD) (Fig. 2A) into the papillary thyroid cancer cell line BHP17, which showed a low endogenous expression level of EphB3 (Fig. 2B). We found that overexpression of EphB3 and EphB3-KD had no effect on the growth of BHP17 cells, which was examined by either MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay or soft agar assay (data not shown). However, the migration ability of BHP17/EphB3 cells was significantly increased compared with empty-vector transfected BHP17 (BHP17/v), but EphB3-KD had little effect on BHP17 migration (Fig. 2C). The promoting effect of EphB3 overexpression on cell migration was not only observed in the resting state but also in activation state with EfnB1/EfnB2-Fc stimulation (Fig. 2D). In contrast, knockdown of EphB3 expression by siRNA in BHP17 cells decreased the migration ability of BHP17 cells compared with control cells (BHP17/EphB3 icon) either in resting state or under stimulating condition (supplemental Fig. 1). Taken together, EphB3 could promote PTC cell migration in a kinase-dependent manner.
FIGURE 2.

EphB3 promoted PTC cell migration in a kinase-dependent manner. A, schematic description of EphB3 and EphB3-KD. B, BHP17 cells were transfected with empty vector and constructs expressing EphB3 and EphB3-KD, respectively. The expression of EphB3 was examined by Western blot. v represents empty vector, EphB3-1 was a positive EphB3-overxpressing clone, EphB3-2 was a pool of cells overexpressing EphB3, and EphB3-KD was a cell pool overexpressing EphB3-KD. C, the migration capability of control cell (v), EphB3-overexpressing cells (EphB3-1 and EphB3-2), and EphB3-KD-overexpressing cells was examined by Boyden chamber assay. The bar graph (right) represented the mean ± S.D. number of the migrated cells from four randomly selected fields. ***, p < 0.001 for EphB3-1/EphB3-2 versus v, unpaired t test. Scale bar, 100 μm. D, migration of BHP17/v and BHP17/EphB3 cells were examined when treated with Fc, EfnB1-Fc, EfnB2-Fc, and EphB3-Fc. The bar graph (right) represents the mean ± S.D. number of the migrated cells from four randomly selected fields. For BHP17/v cells: p = 0.0381 (*) for EfnB1-Fc versus Fc, and p = 0.0105 (*) for EfnB2-Fc versus Fc. For BHP17/EphB3 cells: p < 0.001 (***) for EfnB1/EfnB2/EphB3-Fc versus Fc, unpaired t test. Scale bar, 100 μm.
Because EphB3 was able to regulate the migration of BHP17 cells, we wondered whether it could also influence metastasis of PTC cells in vivo. We constructed a BHP17-Luc2 cell that was stably transfected with a firefly luciferase gene, then the BHP17-Luc2 cell was infected with either vectors overexpressing EphB3/EphB3-KD or EphB3 siRNA-expressing viruses and their respective control. BHP17-Luc2/v/EphB3/EphB3-KD cells were injected into the left ventricles of nude mice, respectively. As shown in Fig. 3A, metastasis development in BHP17-Luc2/EphB3 group was dramatically faster than BHP17-Luc2/v and the BHP17-Luc2/EphB3-KD group. Moreover, metastatic foci in BHP17-Luc2/EphB3 group were much bigger than those of the other two groups. BHP17-Luc2/EphB3-KD cells generated comparable metastatic foci to control cells, which was similar to the result of migration analysis. In contrast, BHP17-Luc2/EphB3 i2 and i3 cells failed to develop any foci at 4 weeks after initial injection, whereas EphB3 icon cells generated visible metastatic foci in the meantime. These results indicated that EphB3 promoted PTC cell metastasis in a kinase-dependent manner, whereas loss of EphB3 inhibited metastasis of BHP17 cells in vivo, which was consistent with migration data in vitro.
FIGURE 3.
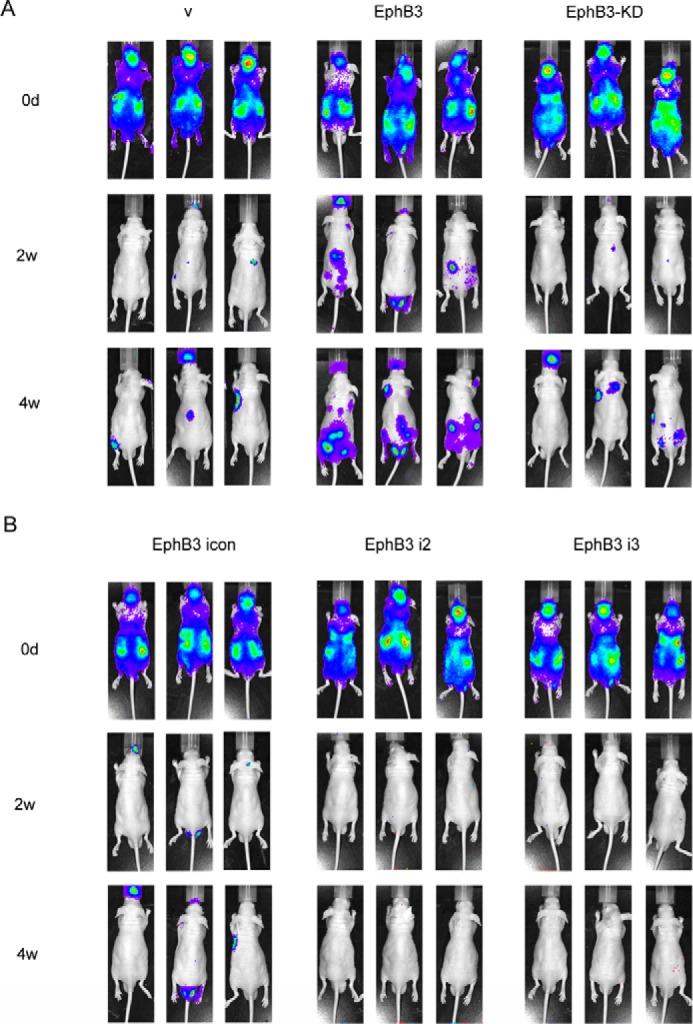
Effect of EphB3 on in vivo metastasis of BHP17 cells. A, BHP17-Luc2/v, BHP17-Luc2/EphB3, and BHP17-Luc2/EphB3-KD cells were injected into the left ventricles of 6-week-old nude mice, respectively. The in vivo metastasis was examined once a week after injection by IVIS imaging System, and representative images are shown. v represents empty vector. B, BHP17-Luc2/EphB3 icon cells and BHP17-Luc2/EphB3 RNAi cells were injected into the left ventricles of 6-week old nude mice, respectively. The in vivo metastasis was examined once a week after injection by IVIS imaging System, and representative images are shown.
Ligand-activated Forward Signaling Was Responsible for the Promotive Effect of EphB3 on PTC Cell Migration Exerted by EphB3
Bidirectional signaling induced upon ligation of Eph receptor and ephrin ligand is a particular characteristic of Ephs/ephrins. EphB3 may exert its promotive effect on PTC cell migration through either the forward signaling or the reverse signaling. In the previous report, recombinant EphB-Fc soluble monomer interfered with the interaction between normal EphB3 and its ligands and acted as a receptor antagonist (11, 12), whereas clustered ephrinB-Fc ligand could artificially function as an agonist (19). EphrinB1 and ephrinB2 have been reported as the cognate ligands of EphB3 (20, 21); therefore, we used EfnB1/EfnB2-Fc consisting of the extracellular domain of human ephrinB1/ephrinB2 and the Fc region of human IgG to activate EphB3-mediated forward signaling. On the other hand, we utilized EphB3-Fc, which consists of the extracellular domain of EphB3 and the Fc region to inhibit the forward signaling and suppress EphB3 activity. We found that both EfnB1-Fc and EfnB2-Fc treatment stimulated BHP17/v and BHP17/EphB3 cell migration compared with Fc control. However, due to EphB3 overexpression, the stimulative effect was more significant in BHP17/EphB3 cells. In contrast, EphB3-Fc treatment remarkably reduced cell migration in BHP17/EphB3 cells but showed little effect on BPH17/v cells, most likely due to the low expression level of endogenous EphB3 in BPH17/v cells (Fig. 2D). Therefore, the ligand-induced forward signaling contributed to the promotive effect on PTC cell migration exerted by EphB3.
FAK and Paxillin Contributed to EphB3-mediated Alteration in Cell Migration, Adhesion, and Spreading
To further explore the underlying mechanism, we examined the expression of two important molecules involved in migration, paxillin and FAK by immunostaining. As shown in Fig. 4A, the signals of paxillin and FAK, which indicated the assembly of focal adhesions (22, 23), were much stronger in BHP17/EphB3 RNAi cells compared with BHP17/EphB3 icon cells. It has been reported that the increase of focal adhesion often promotes the adhesion of host cells with ECM, and loss of polar distribution of focal adhesion inhibits cell mobility (24, 25). Thus the increase of focal adhesion assembly in EphB3 knockdown cells was consistent with its impaired mobility. Furthermore, assembly of F-actin was dramatically increased in BHP17/EphB3 RNAi cells compared with BHP17/EphB3 icon cells (Fig. 4A), which also implied the inhibition of cell migration. In addition, we measured the adhesion capability of BHP17 cells by cell adhesion assay. The results demonstrated that cell adhesion was obviously increased in EphB3 knockdown cells (Fig. 4B). Moreover, we also examined cell spreading capability and found that BHP17/EphB3 RNAi cells spread more efficiently and firmly on fibronectin-coated plate than BHP17/EphB3 icon cells (Fig. 4C). These results suggested that cell-matrix adhesion molecules, including FAK and paxillin, contributed to EphB3-induced alteration in migration, adhesion, and cell spreading.
FIGURE 4.

FAK and Paxillin contributed to EphB3-mediated alteration in cell migration, adhesion, and spreading. A, the immunofluorescent staining of nucleus (DAPI, blue), paxillin (green), FAK (red), and actin stress fiber (green) in BHP17/EphB3 icon cells and BHP17/EphB3 RNAi cells. Scale bar, 100 μm. B, the graph demonstrated the quantitative results of cell adhesion of BHP17/EphB3 icon and BHP17/EphB3 RNAi cells, and the mean ± S.D. was indicated. ***, p < 0.001 for EphB3 RNAi versus EphB3 icon, unpaired t test. C, the representative images (left, on fibronectin-coated plate) and graph (right) demonstrated different cell spreading of BHP17/EphB3 icon and BHP17/EphB3 RNAi cells on fibronectin-coated plate, and bars represent the mean ± S.D. *, p = 0.0109 for EphB3 RNAi versus EphB3 icon (30 min); *, p = 0.0149 for EphB3 RNAi versus EphB3 icon (60min), unpaired t test.
Inhibition of EphB3 Expression Influenced the Activity of RhoA and Rac1
It has been known that actin assembly and disassembly provide the driving force for cell migration, and Rho family GTPases play key roles in cell migration by regulating actin dynamics (26, 27). Rho mediates stress fiber formation, and Rac1 is involved in membrane ruffling and the formation of lamellipodia (28). Therefore, the alteration of actin assembly in BHP17 cells induced by EphB3 knockdown promoted us to examine whether Rho family GTPases were involved in the regulation of migration by EphB3. To measure Rho GTPase activity, active Rac1 was pulled down by GST-PAK-PBD, and active RhoA was pulled down by GST-Rhotekin-RBD. As expected, RhoA activity was increased in the BHP17/EphB3 RNAi cells, whereas Rac1 activity was decreased in the BHP17/EphB3 RNAi cells compared with BHP17/EphB3 icon cells (Fig. 5, A and B). In addition, treatment of BHP17/EphB3 RNAi cells with Y27632, an inhibitor of ROCK1, one downstream effector of RhoA (26, 27), led to elimination of the assembly of F-actin in EphB3 knockdown cells, indicating that the accumulation of dense F-actin stress fibers in BHP17/EphB3 RNAi cells were attributed to increased activity of RhoA (Fig. 5C). The decrease in Rac1 activity and increase in RhoA activity led to an altered balance between these GTPases in favor of RhoA, which has been previously linked to inhibition of cell motility (29–31). These results thereby suggested that the predominance of RhoA signaling and inhibition of Rac1 activity contributed to suppressed cell migration induced by EphB3 knockdown.
FIGURE 5.

Inhibition of EphB3 expression influenced the activity of Rac1 and RhoA. A, the activity of Rac1 in BHP17/EphB3 icon and BHP17/EphB3 i1, i2, and i3 cells was examined by a GST pulldown assay using GST-PAK-PBD. The bar graph (right) represented the relative intensity of the band signals measured by Image-J software (Active Rac1/Total Rac1 for each cell). ***, p < 0.001 for i1/i2/i3 versus icon, unpaired t test. B, the activity of RhoA in BHP17/EphB3 icon and BHP17/EphB3 i1, i2, and i3 cells was examined by a GST pulldown assay using GST-Rhotekin-RBD. The bar graph (right) represented the relative intensity of the band signals measured by Image-J software (Active RhoA/Total RhoA for each cell). ***, p < 0.001 for i1/i2/i3 versus icon, unpaired t test. C, immunofluorescent staining of nucleus (DAPI, blue), paxillin (red), and actin stress fiber (green) showed the effect of ROCK inhibitor Y-27632 on focal adhesions and actin fibers in both BHP17/EphB3 icon and BHP17/EphB3 RNAi cells, DMSO is used as a control. Scale bar, 100 μm.
Vav2-mediated EphB3-stimulated Migration in PTC Cells
It has been established that Vav2 is a pivotal regulator for Rac1 and RhoA (32–34), and previous studies indicated that Eph receptors could interact with Vav2 to regulate cell migration (35, 36). Therefore, we wondered whether Vav2 mediated the influence on cell migration by EphB3 in BHP17 PTC cells. By simultaneous overexpression of Vav2 and EphB3, we found an obvious interaction between Vav2 and EphB3 by co-immunoprecipitation (Fig. 6A). Additionally, endogenous Vav2 was also found to interact with EphB3 in BHP17/EphB3 cells (Fig. 6B). Moreover, we found that the interaction between Vav2 and EphB3 was dependent on the kinase activity of EphB3, because EphB3-KD could not interact with Vav2 (Fig. 6A), which was consistent with the previous reports that Vav2 binds to the kinase domain of Eph receptors (35, 36).
FIGURE 6.

Vav2 interacted with EphB3 and acted downstream of EphB3 to regulate PTC cell migration. A, Vav2, EphB3, and EphB3-KD was co-expressed in 293T cells as indicated, and their interaction was examined by immunoprecipitation (IP). B, interaction between EphB3 and endogenous Vav2 was examined by immunoprecipitation in BHP17/EphB3 cells. C, BHP17/EphB3 cells were treated with Fc, EfnB1-Fc, and EfnB2-Fc, respectively, then total Vav2 and p-Vav2 were examined by Western blot. D, Vav2 was knocked down by RNAi in BHP17/EphB3 cells, and i1, i2, and i3 represent three different siRNA sequences targeting Vav2. E, migration of BHP17/v, BHP17/EphB3+Vav2 icon, and BHP17/EphB3+Vav2 i1/i2 cells were examined by Boyden chamber assay. Scale bar, 100 μm. The bar graph (right) represents the mean ± S.D. number of the migrated cells from four randomly selected fields. ***, p < 0.001 for EphB3+Vav2 icon versus EphB3+Vav2 i1/i2, unpaired t test. v represents empty vector.
It has been reported that Vav2 is activated by receptor-tyrosine kinase-mediated tyrosine phosphorylation (35–40), including Eph receptors (11, 12), and the above results also identified that the interaction between Vav2 and EphB3 relied on the kinase domain of EphB3. Therefore, we explored whether activation of EphB3 could phosphorylate Vav2. As shown in Fig. 6C, both EfnB1-Fc and EfnB2-Fc treatment led to Vav2 phosphorylation.
To further confirm the involvement of Vav2 in EphB3-promoted PTC cell migration, the expression of Vav2 was knocked down by RNAi in BHP17/EphB3 cells (Fig. 6D). As shown in Fig. 6E, knockdown of Vav2 in BHP17/EphB3 cells effectively inhibited EphB3-promoted cell migration. Therefore, Vav2 directly interacted with EphB3 and was responsible for EphB3-induced PTC cell migration. Activated EphB3 might recruit and phosphorylate Vav2 for regulating BHP17 cell motility.
Discussion
The role of EphB3 has been explored in several types of cancers. Clinical investigation indicates that overexpression of EphB3 is associated with a poor outcome in ependymomas (41). EphB3 is also found overexpressed in pancreatic cancer cell lines and may be a potential target for gene therapy of pancreatic cancer (42). In esophageal squamous cell carcinoma, EphB3 is positively related with cancer cell proliferation (43). Overexpression of EphB3 also occurs in human non-small-cell lung cancer (44). However, EphB3 is suggested to play an inhibitory role in colorectal cancer (45). The interaction between EphB3 and its ligands is able to restrict the spreading of EphB3-expressing tumor cells into ephrinB1-positive territories of colons through a mechanism dependent on E-cadherin-mediated adhesion, which turns to impede the progression of colorectal cancer (45). In our study, we found that EphB3 positively regulated both in vitro migration and in vivo metastasis of PTC cells, demonstrating a tumor-promoting effect of EphB3 in PTC. Taken together, the function of EphB3 in cancer is complex and dependent on cancer types.
The ligation of EphB3 and its ligands usually elicit bidirectional signals (11, 12). In our study we observed that EfnB1/EfnB2-Fc treatment, which was able to block the reverse signal while activating the forward signal (11, 12), significantly promoted PTC cell migration. On the other hand, EphB3-Fc treatment, which eliminated the forward signaling decreased cell migration. Thus, ephrin-activated forward signaling is responsible for EphB3-stimulated PTC cell migration.
In the nervous system, it has been established that Eph receptors and ephrins play important roles in neuron migration by regulating actin assembly and disassembly (11, 12). As for cancers, accumulating evidence has also suggested the involvement of Ephs and ephrins in cell migration (46–48). For example, ephrinB3 could promote glioma cell migration through activation of Rac1, whereas ephrinB2 overexpression enhances B16 melanoma cell migration through regulating ECM attachment ability (49). Interaction of EphA2 and ephrinA1 activated a Src/FAK-mediated motility response leading to Rho-dependent actino/myosin contractility in prostatic carcinoma cells (50). Eph-ephrin interaction was also observed in the migration of glioma cells (51) and metastasis of mammary tumor cells (52).
Our study revealed that EphB3-stimulated migration was attributed to the altered activity of Rac1 and RhoA, two members of Rho GTPase family (27). The Rho GTPase family has been well demonstrated to regulate actin dynamics and plays key roles in regulating cell migration (27). Rac1 and Cdc42 stimulate the formation of lamellipodia and filopodia at the leading edge of migrating cells, respectively, whereas RhoA stimulates the formation of focal adhesion and stress fibers. It is believed that the balance between Rac1/Cdc42 and RhoA is a key determinant in the regulation of cell motility, i.e. the dominance of Rac1/Cdc42 signaling promotes cell migration, whereas enhanced RhoA activity inhibits migration (29–31). Consistent with these reports, our results showed that knockdown of EphB3 suppressed Rac1 activity but enhanced RhoA activity, leading to inhibition of PTC cell migration.
The three Rho GTPase family members, Cdc42, Rac1, and RhoA, have been previously proven to be downstream targets for Eph/ephrin proteins (11, 12); however, the mediators in the pathways remain largely unknown. Vav2 is one of the guanine nucleotide exchange factors for Rho GTPase family members, including Rac1 and RhoA, and functions as an important mediator for the signal transduction from cell surface receptors to Rho GTPases-induced actin dynamic changes (35–40). Vav2 has been found necessary for rearrangement of actin cytoskeleton, which is closely involved in cell spreading and migration (32–34). It is reported that the binding of Vav2 to activated EphA4 promotes receptor endocytosis and is pivotally involved in the axon guidance (35). Another work discloses that in response to ephrinA1 stimulation, Vav2 is recruited to activated EphA2 receptor in mammalian cells and regulates endothelial cell migration and assembly (36). In our study we found that Vav2 interacted with and was phosphorylated by activated EphB3 in PTC cells, indicating that EphB3 could activate Vav2 in PTC cells. Therefore, it is possible that Vav2 plays as a general effector downstream of Eph receptors and is required for regulating the activity of Rho GTPases as well as cell migration by Eph receptors.
In summary, our work reported that EphB3 was overexpressed in PTC and positively regulated both in vitro migration and in vivo metastasis of PTC cells. Moreover, we found that EphB3 could activate Vav2 in a kinase-dependent manner and subsequently regulate the activity of Rac1 and RhoA, which led to altered migration ability of PTC cells. Our work not only improves the understanding of the role of EphB3 in PTC but also provides indications for therapeutic strategies for PTC.
Experimental Procedures
PTC Tissue Samples and Cell Line
Paraffin-embedded thyroid carcinoma specimens from 12 patients and 15 pairs of primary PTC samples and their corresponding normal tissues were obtained from PTC patients treated at the First Affiliated Hospital of Zhengzhou University (Henan, China) from 2002 to 2005 after their written informed consent, and none of the patients received any neo-adjuvant therapy. All specimens were frozen at once in liquid nitrogen after surgical excision and stored at −80 °C until use. Our work was approved with the Institutional Review Board of the Institute for Nutritional Sciences, Chinese Academy of Sciences. The PTC cell line BHP17 was purchased from the American Type Culture Collection (Manassas, VA) and cultured in RPMI 1640, supplemented with 10% fetal bovine serum, 10 units/ml penicillin, and 10 units/ml streptomycin at 37 °C in a humidified atmosphere containing 5% CO2.
Reagents
Anti-EphB3 antibody (ab66337) was purchased from Abcam. Anti-β-actin (sc-47778), anti-α-tubulin (sc-32293), anti-RhoA (sc-418), and anti-p-Vav2 (Tyr-172) (sc-16409-R) antibodies were obtained from Santa Cruz Biotechnology Inc. Anti-paxillin (610051), anti-FAK (610087), and anti-Rac1 (610650) antibodies were from BD Transduction Laboratories. Phosphotyrosine antibody (Tyr(P)-100) (#9411) was purchased from Cell Signaling Technology. The secondary antibodies for immunofluorescence (A21422 and A21202) were from Molecular Probe Inc. Y-27632 (Y0503) was purchased from Sigma. Alexa Fluor (R) 488 phalloidin (8878S) was purchased from Cell Signaling.
Real-time PCR Analysis
Primers for the genes tested in the present experiments were designed using software PRIMER5. The primers for amplifying human EphB3 gene were as follows: forward (5′-TCGTGGTCATCGCTATCGTCT-3′) and reverse (5′-AAACTCCCGAACAGCCTCATT-3′). The primers for β-actin were: forward (5′-GATCATTGCTCCTCCTGAGC-3′) and reverse (5′-ACTCCTGCTTGCTGATCCAC-3′). Amplification reactions were done in 20 μl of the LightCycler-DNA Master SYBR Green I mix (Roche Applied Science) with 10 pmol of primer, 2 mmol/liter MgCl2, a 200 μmol/liter deoxynucleotide triphosphate mixture, 0.5 units of TaqDNA polymerase, and universal buffer. All of the reactions were done in triplicate in an iCycler iQ system (Bio-Rad), and the thermal cycling conditions were as follows: 95 °C for 3 min; 40 cycles of 95 °C for 30 s, 58 °C for 20 s, and 72 °C for 30 s; 72 °C for 10 min.
Reactions were characterized at the point during cycling when amplification of the PCR product was first detected after a fixed number of cycles, which is defined as Ct. The target message in unknown samples was quantified by measuring the Ct value, and the Ct value of β-actin was also measured as the endogenous RNA control. The levels of EphB3 expression in each sample were normalized on the basis of its β-actin content through the formula LevelEphB3/Levelβ-actin = 2(Ctβ-actin − CtEphB3). The relative EphB3 expression level of each pair of PTC samples was calculated as EphB3tumor/EphB3normal when EphB3tumor > EphB3normal, and the values were counted as −(EphB3normal/EphB3tumor). Paired t test was applied to statistical analysis. The statistical results were considered significant at p < 0.05. All data analyses were done using the program SPSS for Windows.
RNAi of EphB3
To silence endogenous EphB3, we selected three siRNA hairpin sequences against EphB3 mRNA as follows that were designed using the software from Ambion web: 5′-CTCTACTGCAACGGCGAT-3′, 5′-CTGCAGCAGTACATTGCT-3′, and 5′-TACCTGTCCGAGATGAAC-3′. These sequences were cloned to the FG12 vector and produced RNAi cell lines as described previously (53). For knockdown of Vav2, we employed pLKO.1 vector, and the target sequences 5′-GCCACGATAAATTTGGATTAA-3′, 5′-CAAGTGAAACTGGAGGAATTT-3′, and 5′-CCCGAGATATGAGGGAGCTTT-3′.
Constructs of EphB3
Full-length cDNA of either EphB3-WT or EphB3-KD was subcloned from pCMV-EphB3-WT and pCMV-EphB3-KD, respectively, which were kindly provided by Dr. Bingcheng Wang, and the fragment of EphB3-WT or EphB3-KD was inserted into pcDNA3.1 vectors (Invitrogen) at the appropriate sites. By stable transfection, we established control cells (BHP17/v) and the cell lines overexpressing EphB3-WT or EphB3-KD. We also used a modified lentivirus-based FG12 vector with a CMV promoter (53) to produce viruses overexpressing EphB3/EphB3-KD. After infection, positive cells were sorted and enriched by FACS using the coexpressed GFP as a marker.
Boyden Chamber Assay
Boyden chamber (8-μm pore size polycarbonate membrane) was obtained from Neuro Probe Corp. Cells (2 × 105) in 0.05 ml of medium with 1% FBS were placed in the upper chamber, and the lower chamber was loaded with cell culture medium containing 10% FBS. Cells that migrated to the lower surface of filters were detected with traditional H&E staining, and multiple fields of each well were counted after 4–8 h of incubation at 37 °C with 5% CO2. Three wells were examined for each condition and cell type, and the experiments were repeated thrice.
Cell Adhesion Assay
96-Well plates were coated with fibronectin overnight at 4 °C followed by a blocking step with heat-denatured BSA for 1 h at 37 °C. Cells were trypsinized and resuspended, and 2 × 104 cells were plated into each fibronectin-coated well and allowed to adhere for 30 min at 37 °C. Every condition was done in quadruplicate. After 30 min, non-adherent cells were removed, and adherent cells were washed once with prewarmed phosphate buffer saline (PBS). The adherent cells were stained with trypan blue and manually counted under the microscope in multiple different fields.
Cell Spreading Assay
Cells were seeded into a 24-well coated with fibronectin at a density of 1 × 105 cells per well and fixed at the indicated time points. Spread and non-spread cells were counted in five representative high power fields. Non-spread cells were defined as small, round cells with little or no membrane protrusions, whereas spread cells were defined as large cells with extensive visible lamellipodia. Results reveal the number of spread cells in five high power fields. The data are presented as the average of the results from three independent experiments.
Measurement of Rac1 Activity
The Rac1 activity assay was performed by pulldown using GST fusion protein containing the protein binding domain of p21-activated kinase (GST-PAK-PBD). Briefly, cells were plated onto the plate at the density of ∼2 × 106 cells/10-cm dish. When grown to 60–70% confluence, cells were washed twice with PBS and lysed with 750 μl of ice-cold eukaryotic lysis buffer (25 mm Tris, pH 7.4, 1 mm EDTA, 5 mm MgCl2, 1 mm DTT, 0.1 mm EGTA, 100 mm NaCl, 1% Nonidet P-40, 5% glycerol, 1 mm PMSF, 1 μg/ml aprotinin,1 μg/ml leupeptin, and 1 μg/ml pestatin) on ice and then incubated with GST-PAK-PBD fusion protein that had been adsorbed to glutathione agarose beads for 30 min. After that, the beads were washed 3 times with 500 μl of cold lysis buffer then resuspended in 10 μl of reducing electrophoresis sample buffer (2% SDS, 10% glycerol, 80 mm Tris, pH 6.8, 2 mm EDTA, 100 mm DTT, and 0.1% bromphenol blue) and analyzed by SDS-PAGE. After electrophoresis, samples were transferred to nitrocellulose membrane and immunoblotted with mouse anti-human Rac1 antibody at a dilution of 1:1000.
Measurement of RhoA Activity
RhoA activity was measured in a pulldown assay using the Rho binding domain from Rhotekin, similar to the Rac1 activity assay.
Fluorescence Microscopy
Cells were fixed for 10 min in 4% paraformaldehyde in PBS and permeabilized for 10 min with 0.01% Triton X-100 in PBS. Cover slides were blocked in 10% goat serum at room temperature for 40 min and then stained for 60 min in a 1:10 dilution of FITC-phalloidin (Sigma). Cover slides were then stained with anti-FAK (1:250) or anti-paxillin (1:1000) antibodies followed by incubation with fluorescent secondary antibody at 1:1000 for 60 min, then rinsed with PBS several times. Finally, the cover slides were mounted in Immuno-fluore mounting medium and viewed using a ZEISS laser scanning confocal microscope.
In Vivo Bioluminescence Imaging of Tumor Cells
BHP17 cells were transfected with plasmids expressing firefly luciferase (PGL4-CMV-Luc2, Longmed Corp., Beijing, China) using Lipofectamine 2000 reagent, and G418 resistant clones were selected. The surviving colonies were screened for bioluminescence in complete media supplemented with 150 μg/ml D-luciferin by in vitro imaging using the IVIS-Imaging System (Xenogen, Alameda, CA). Bioluminescent, antibiotic-resistant clones were amplified in culture and characterized for stable luminescence in vitro and tumorigenic potential in vivo. One positive clone, BHP17-Luc2, was selected for further studies. BHP17-Luc2 cells were infected with control virus (v) and viruses overexpressing EphB3 and EphB3-KD, respectively, as well as control siRNA virus (BHP17-Luc2/icon) and EphB3 siRNA viruses (BHP17-Luc2/EphB3 RNAi). Pools of BHP17-Luc2/v, BHP17-Luc2/EphB3, BHP17-Luc2/EphB3-KD, BHP17-Luc2/EphB3 icon and BHP17-Luc2/EphB3 RNAi cells were obtained after FACS sorting. They were injected into the left ventricles of nude mice. The mice were examined weekly for metastasis after injection following the Xenogen protocol using a cooled CCD camera system (IVIS-100, Xenogen) and Living Image software (Xenogen).
Author Contributions
J.-J. L., Z.-J. S., and D. X. conceived the idea of the project. J.-J. L. and Z.-J. S. performed most of the experiments. Y.-M. Y., F.-F. Y., Y.-G. B., L.-Y. L., and X.-L. Z. helped with data acquisition and provided critical materials. J.-J. L. wrote the manuscript. D. X. provided most of the resources and funding and revised the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
This work was supported by the National Basic Research Program of China (2010CB912102), Ministry of Science and Technology Key Program (2012ZX10002009-017), the National Natural Science Foundation of China (81230058, 81321062, 30930023, 31100551, 31201046, 81021002, 31520103907), CAS/SAFEA International Partnership Program for Creative Research Teams, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (SIBS2012004), and Technology Commission of Shanghai Municipality (12XD1405600) (to D. X.). This work was also supported by Young Scientist Fund of National Science Foundation of China (31201046) and Sanofi-Aventis-Sibs Scholarship Program (2011) (to J.-J. L.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Fig. 1.
- TC
- thyroid cancer
- PTC
- papillary thyroid cancer
- PBD
- protein binding domain
- RBD
- Rho binding domain
- PAK
- p21-activated kinase
- ECM
- extracellular matrix.
References
- 1. Gimm O. (2001) Thyroid cancer. Cancer Lett. 163, 143–156 [DOI] [PubMed] [Google Scholar]
- 2. Hundahl S. A., Cady B., Cunningham M. P., Mazzaferri E., McKee R. F., Rosai J., Shah J. P., Fremgen A. M., Stewart A. K., and Hölzer S. (2000) Initial results from a prospective cohort study of 5583 cases of thyroid carcinoma treated in the united states during 1996: U.S. and German Thyroid Cancer Study Group. An American College of Surgeons Commission on Cancer Patient Care Evaluation study. Cancer 89, 202–217 [DOI] [PubMed] [Google Scholar]
- 3. Lang B. H., Lo C. Y., Chan W. F., Lam K. Y., and Wan K. Y. (2007) Prognostic factors in papillary and follicular thyroid carcinoma: their implications for cancer staging. Ann. Surg. Oncol. 14, 730–738 [DOI] [PubMed] [Google Scholar]
- 4. Mazzaferri E. L., and Kloos R. T. (2001) Clinical review 128: Current approaches to primary therapy for papillary and follicular thyroid cancer. J. Clin. Endocrinol. Metab. 86, 1447–1463 [DOI] [PubMed] [Google Scholar]
- 5. Eustatia-Rutten C. F., Corssmit E. P., Biermasz N. R., Pereira A. M., Romijn J. A., and Smit J. W. (2006) Survival and death causes in differentiated thyroid carcinoma. J. Clin. Endocrinol. Metab. 91, 313–319 [DOI] [PubMed] [Google Scholar]
- 6. Sherman S. I., Brierley J. D., Sperling M., Ain K. B., Bigos S. T., Cooper D. S., Haugen B. R., Ho M., Klein I., Ladenson P. W., Robbins J., Ross D. S., Specker B., Taylor T., and Maxon H. R. 3rd. (1998) Prospective multicenter study of thyroiscarcinoma treatment: initial analysis of staging and outcome. National Thyroid Cancer Treatment Cooperative Study Registry Group. Cancer 83, 1012–1021 [DOI] [PubMed] [Google Scholar]
- 7. Kondo T., Ezzat S., and Asa S. L. (2006) Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat. Rev. Cancer 6, 292–306 [DOI] [PubMed] [Google Scholar]
- 8. Dulgeroff A. J., and Hershman J. M. (1994) Medical therapy for differentiated thyroid carcinoma. Endocr. Rev. 15, 500–515 [DOI] [PubMed] [Google Scholar]
- 9. Castellone M. D., Carlomagno F., Salvatore G., and Santoro M. (2008) Receptor tyrosine kinase inhibitors in thyroid cancer. Best Pract. Res. Clin. Endocrinol. Metab. 22, 1023–1038 [DOI] [PubMed] [Google Scholar]
- 10. (1997) Unified nomenclature for Eph family receptors and their ligands, the ephrins: Eph Nomenclature Committee. Cell 90, 403–404 [DOI] [PubMed] [Google Scholar]
- 11. Wilkinson D. G. (2001) Multiple roles of EPH receptors and ephrins in neural development. Nat. Rev. Neurosci. 2, 155–164 [DOI] [PubMed] [Google Scholar]
- 12. Palmer A., and Klein R. (2003) Multiple roles of ephrins in morphogenesis, neuronal networking, and brain function. Genes Dev. 17, 1429–1450 [DOI] [PubMed] [Google Scholar]
- 13. Wang H., Wen J., Wang H., Guo Q., Shi S., Shi Q., Zhou X., Liu Q., Lu G., and Wang J. (2014) Loss of expression of EphB1 protein in serous carcinoma of ovary associated with metastasis and poor survival. Int. J. Clin. Exp. Pathol 7, 313–321 [PMC free article] [PubMed] [Google Scholar]
- 14. Yin H., Lu C., Tang Y., Wang H., Wang H., and Wang J. (2013) Enhanced expression of EphrinB1 is associated with lymph node metastasis and poor prognosis in breast cancer. Cancer Biomark. 13, 261–267 [DOI] [PubMed] [Google Scholar]
- 15. Wu B. O., Jiang W. G., Zhou D., and Cui Y. X. (2016) Knockdown of EPHA1 by CRISPR/CAS9 promotes adhesion and motility of HRT18 colorectal carcinoma cells. Anticancer Res. 36, 1211–1219 [PubMed] [Google Scholar]
- 16. Huang J., Xiao D., Li G., Ma J., Chen P., Yuan W., Hou F., Ge J., Zhong M., Tang Y., Xia X., and Chen Z. (2014) EphA2 promotes epithelial-mesenchymal transition through the Wnt/β-catenin pathway in gastric cancer cells. Oncogene 33, 2737–2747 [DOI] [PubMed] [Google Scholar]
- 17. Karidis N. P., Giaginis C., Tsourouflis G., Alexandrou P., Delladetsima I., and Theocharis S. (2011) Eph-A2 and Eph-A4 expression in human benign and malignant thyroid lesions: an immunohistochemical study. Med. Sci. Monit. 17, BR257–BR265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cantisani M. C., Parascandolo A., Perälä M., Allocca C., Fey V., Sahlberg N., Merolla F., Basolo F., Laukkanen M. O., Kallioniemi O. P., Santoro M., and Castellone M. D. (2016) A loss-of-function genetic screening identifies novel mediators of thyroid cancer cell viability. Oncotarget 7, 28510–28522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davis S., Gale N. W., Aldrich T. H., Maisonpierre P. C., Lhotak V., Pawson T., Goldfarb M., and Yancopoulos G. D. (1994) Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science 266, 816–819 [DOI] [PubMed] [Google Scholar]
- 20. Brambilla R., Br uuml, ckner K., Orioli D., Bergemann A. D., Flanagan J. G., and Klein R. (1996) Similarities and differences in the way transmembrane-type ligands interact with the Elk subclass of Eph receptors. Mol. Cell. Neurosci. 8, 199–209 [PubMed] [Google Scholar]
- 21. Brambilla R., Schnapp A., Casagranda F., Labrador J. P., Bergemann A. D., Flanagan J. G., Pasquale E. B., and Klein R. (1995) Membrane-bound LERK2 ligand can signal through three different Eph-related receptor tyrosine kinases. EMBO J. 14, 3116–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burridge K., and Chrzanowska-Wodnicka M. (1996) Focal adhesions, contractility, and signaling. Annu. Rev. Cell Dev. Biol. 12, 463–518 [DOI] [PubMed] [Google Scholar]
- 23. Miyamoto S., Teramoto H., Coso O. A., Gutkind J. S., Burbelo P. D., Akiyama S. K., and Yamada K. M. (1995) Integrin function: molecular hierarchies of cytoskeletal and signaling molecules. J. Cell Biol. 131, 791–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carragher N. O., and Frame M. C. (2004) Focal adhesion and actin dynamics: a place where kinases and proteases meet to promote invasion. Trends Cell Biol. 14, 241–249 [DOI] [PubMed] [Google Scholar]
- 25. Hood J. D., and Cheresh D. A. (2002) Role of integrins in cell invasion and migration. Nat. Rev. Cancer 2, 91–100 [DOI] [PubMed] [Google Scholar]
- 26. Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., and Horwitz A. R. (2003) Cell migration: integrating signals from front to back. Science 302, 1704–1709 [DOI] [PubMed] [Google Scholar]
- 27. Etienne-Manneville S., and Hall A. (2002) Rho GTPases in cell biology. Nature 420, 629–635 [DOI] [PubMed] [Google Scholar]
- 28. Nobes C. D., and Hall A. (1995) Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53–62 [DOI] [PubMed] [Google Scholar]
- 29. Nimnual A. S., Taylor L. J., and Bar-Sagi D. (2003) Redox-dependent downregulation of Rho by Rac. Nat. Cell Biol. 5, 236–241 [DOI] [PubMed] [Google Scholar]
- 30. Arthur W. T., and Burridge K. (2001) RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Mol. Biol. Cell 12, 2711–2720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Caron E. (2003) Rac signalling: a radical view. Nat. Cell Biol. 5, 185–187 [DOI] [PubMed] [Google Scholar]
- 32. Sastry S. K., Rajfur Z., Liu B. P., Cote J. F., Tremblay M. L., and Burridge K. (2006) PTP-PEST couples membrane protrusion and tail retraction via VAV2 and p190RhoGAP. J. Biol. Chem. 281, 11627–11636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heo J., Thapar R., and Campbell S. L. (2005) Recognition and activation of Rho GTPases by Vav1 and Vav2 guanine nucleotide exchange factors. Biochemistry 44, 6573–6585 [DOI] [PubMed] [Google Scholar]
- 34. Wennerberg K., Ellerbroek S. M., Liu R. Y., Karnoub A. E., Burridge K., and Der C. J. (2002) RhoG signals in parallel with Rac1 and Cdc42. J. Biol. Chem. 277, 47810–47817 [DOI] [PubMed] [Google Scholar]
- 35. Cowan C. W., Shao Y. R., Sahin M., Shamah S. M., Lin M. Z., Greer P. L., Gao S., Griffith E. C., Brugge J. S., and Greenberg M. E. (2005) Vav family GEFs link activated Ephs to endocytosis and axon guidance. Neuron 46, 205–217 [DOI] [PubMed] [Google Scholar]
- 36. Hunter S. G., Zhuang G., Brantley-Sieders D., Swat W., Cowan C. W., and Chen J. (2006) Essential role of Vav family guanine nucleotide exchange factors in EphA receptor-mediated angiogenesis. Mol. Cell. Biol. 26, 4830–4842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garrett T. A., Van Buul J. D., and Burridge K. (2007) VEGF-induced Rac1 activation in endothelial cells is regulated by the guanine nucleotide exchange factor Vav2. Exp. Cell Res. 313, 3285–3297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Marcoux N., and Vuori K. (2003) EGF receptor mediates adhesion-dependent activation of the Rac GTPase: a role for phosphatidylinositol 3-kinase and Vav2. Oncogene 22, 6100–6106 [DOI] [PubMed] [Google Scholar]
- 39. Tamás P., Solti Z., Bauer P., Illés A., Sipeki S., Bauer A., Faragó A., Downward J., and Buday L. (2003) Mechanism of epidermal growth factor regulation of Vav2, a guanine nucleotide exchange factor for Rac. J. Biol. Chem. 278, 5163–5171 [DOI] [PubMed] [Google Scholar]
- 40. Tamás P., Solti Z., and Buday L. (2001) Membrane-targeting is critical for the phosphorylation of Vav2 by activated EGF receptor. Cell. Signal. 13, 475–481 [DOI] [PubMed] [Google Scholar]
- 41. Lukashova-v Zangen I., Kneitz S., Monoranu C. M., Rutkowski S., Hinkes B., Vince G. H., Huang B., and Roggendorf W. (2007) Ependymoma gene expression profiles associated with histological subtype, proliferation, and patient survival. Acta Neuropathol. 113, 325–337 [DOI] [PubMed] [Google Scholar]
- 42. Farivar R. S., Gardner-Thorpe J., Ito H., Arshad H., Zinner M. J., Ashley S. W., and Whang E. E. (2003) The efficacy of tyrosine kinase inhibitors on human pancreatic cancer cell lines. J. Surg. Res. 115, 219–225 [DOI] [PubMed] [Google Scholar]
- 43. Nemoto T., Ohashi K., Akashi T., Johnson J. D., and Hirokawa K. (1997) Overexpression of protein tyrosine kinases in human esophageal cancer. Pathobiology 65, 195–203 [DOI] [PubMed] [Google Scholar]
- 44. Ji X. D., Li G., Feng Y. X., Zhao J. S., Li J. J., Sun Z. J., Shi S., Deng Y. Z., Xu J. F., Zhu Y. Q., Koeffler H. P., Tong X. J., and Xie D. (2011) EphB3 is overexpressed in non-small-cell lung cancer and promotes tumor metastasis by enhancing cell survival and migration. Cancer Res. 71, 1156–1166 [DOI] [PubMed] [Google Scholar]
- 45. Cortina C., Palomo-Ponce S., Iglesias M., Fernández-Masip J. L., Vivancos A., Whissell G., Humà M., Peiró N., Gallego L., Jonkheer S., Davy A., Lloreta J., Sancho E., and Batlle E. (2007) EphB-ephrin-B interactions suppress colorectal cancer progression by compartmentalizing tumor cells. Nat. Genet. 39, 1376–1383 [DOI] [PubMed] [Google Scholar]
- 46. Wimmer-Kleikamp S. H., and Lackmann M. (2005) Eph-modulated cell morphology, adhesion and motility in carcinogenesis. IUBMB Life 57, 421–431 [DOI] [PubMed] [Google Scholar]
- 47. Poliakov A., Cotrina M., and Wilkinson D. G. (2004) Diverse roles of eph receptors and ephrins in the regulation of cell migration and tissue assembly. Dev. Cell 7, 465–480 [DOI] [PubMed] [Google Scholar]
- 48. Surawska H., Ma P. C., and Salgia R. (2004) The role of ephrins and Eph receptors in cancer. Cytokine Growth Factor Rev. 15, 419–433 [DOI] [PubMed] [Google Scholar]
- 49. Meyer S., Hafner C., Guba M., Flegel S., Geissler E. K., Becker B., Koehl G. E., Orsó E., Landthaler M., and Vogt T. (2005) Ephrin-B2 overexpression enhances integrin-mediated ECM-attachment and migration of B16 melanoma cells. Int. J. Oncol. 27, 1197–1206 [PubMed] [Google Scholar]
- 50. Parri M., Buricchi F., Giannoni E., Grimaldi G., Mello T., Raugei G., Ramponi G., and Chiarugi P. (2007) EphrinA1 activates a Src/focal adhesion kinase-mediated motility response leading to rho-dependent actino/myosin contractility. J. Biol. Chem. 282, 19619–19628 [DOI] [PubMed] [Google Scholar]
- 51. Liu D. P., Wang Y., Koeffler H. P., and Xie D. (2007) Ephrin-A1 is a negative regulator in glioma through down-regulation of EphA2 and FAK. Int. J. Oncol. 30, 865–871 [PubMed] [Google Scholar]
- 52. Brantley-Sieders D. M., Fang W. B., Hwang Y., Hicks D., and Chen J. (2006) Ephrin-A1 facilitates mammary tumor metastasis through an angiogenesis-dependent mechanism mediated by EphA receptor and vascular endothelial growth factor in mice. Cancer Res. 66, 10315–10324 [DOI] [PubMed] [Google Scholar]
- 53. Qin X. F., An D. S., Chen I. S., and Baltimore D. (2003) Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc. Natl. Acad. Sci. U.S.A. 100, 183–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.