

Abstract
T-cell cross-reactivity is essential for effective immune surveillance but has also been implicated as a pathway to autoimmunity. Previous studies have demonstrated that T-cell receptors (TCRs) that focus on a minimal motif within the peptide are able to facilitate a high level of T-cell cross-reactivity. However, the structural database shows that most TCRs exhibit less focused antigen binding involving contact with more peptide residues. To further explore the structural features that allow the clonally expressed TCR to functionally engage with multiple peptide-major histocompatibility complexes (pMHCs), we examined the ILA1 CD8+ T-cell clone that responds to a peptide sequence derived from human telomerase reverse transcriptase. The ILA1 TCR contacted its pMHC with a broad peptide binding footprint encompassing spatially distant peptide residues. Despite the lack of focused TCR-peptide binding, the ILA1 T-cell clone was still cross-reactive. Overall, the TCR-peptide contacts apparent in the structure correlated well with the level of degeneracy at different peptide positions. Thus, the ILA1 TCR was less tolerant of changes at peptide residues that were at, or adjacent to, key contact sites. This study provides new insights into the molecular mechanisms that control T-cell cross-reactivity with important implications for pathogen surveillance, autoimmunity, and transplant rejection.
Keywords: peptides, surface plasmon resonance (SPR), telomerase, tumor immunology, X-ray crystallography, T cell receptor, T cell degeneracy, T cells
Introduction
Recognition of peptide-major histocompatibility complexes (pMHCs)5 by the clonally expressed αβ T-cell receptor (TCR) mediates T-cell immunity. Although TCRs generally interact with pMHC via a conserved binding mode, with the TCRα chain positioned over the MHCα1 domain and the TCRβ chain positioned over the MHCα2 domain, the TCR complementarity-determining region (CDR) loops can use a variety of mechanisms to probe both the MHC surface and bound peptide (1). This flexible binding probably mediates the ability of a single TCR to interact productively with a large range of different epitopes (2–6). Thus, TCR degeneracy enables the approximately 25 million distinct TCR clonotypes expressed by an individual host (7) to have the potential to recognize the entire theoretical peptide universe that could be presented by MHC (2, 8), minimizing the likelihood of pathogens escaping immune surveillance. Given the highly diverse number of TCR-pMHC binding modes seen to date, it is reasonable to predict that different TCRs will exhibit distinct levels of cross-reactivity, depending on the chemical characteristics of their CDR loops and how they interact with pMHC. Such distinctions could determine whether certain TCRs are more likely to offer sufficient protection against hypervariable pathogens, such as human immunodeficiency virus type 1, hepatitis B virus, hepatitis C virus, and influenza, or conversely to trigger autoimmune disease.
Although new quantitative information on the extent of T-cell cross-reactivity has recently come to light (3, 5, 9), the molecular rules that determine this important facet of cellular adaptive immunity remain unclear. Understanding TCR binding degeneracy, and the ensuing T-cell cross-reactivity it enables, is of emerging importance given the increasing use of T-cell therapies using modified TCRs, one of which has already demonstrated the dangers of unintentional T-cell cross-reactivity with self-ligands (10–12). Currently, there are few examples of TCR-pMHC complex structures for which the cross-reactivity profiles of the corresponding T-cell clone have also been determined (3, 13, 14). In a previous study, we demonstrated that an insulin-reactive human CD8+ T-cell clone (1E6) could recognize upward of one million unique peptide ligands. The structure of the 1E6 TCR with its cognate ligand revealed focused TCR-peptide binding with the interaction of only two TCR residues and two adjacent peptide residues accounting for the majority of the binding interface. We speculated that this focused binding might enable the 1E6 TCR to tolerate changes outside of the core motif, mediating the high level of degeneracy. In support of this, another study recently demonstrated that a high level of cross-reactivity was mediated by similar focused TCR-peptide binding by an MHC class II-restricted TCR (13). However, whether TCRs must exhibit focused peptide binding to cross-react remains unclear. This is an important question because, unlike the two examples of focused TCR-peptide binding mentioned above, most TCRs that have been studied structurally to date make more comprehensive interactions with the pMHC surface. Thus, whether a “typical” TCR binding footprint can underpin T-cell cross-reactivity remains unknown.
Here, we used a well characterized CD8+ T-cell clone (ILA1) (15) that responds to residues 540–548 (sequence, ILAKFLHWL) of human telomerase reverse transcriptase to further investigate the structural basis of TCR degeneracy. We have previously characterized a limited number of altered peptide ligands (APLs) for the ILA1 T-cell clone that exhibit different potencies in terms of T-cell activation (9, 15), corresponding to a wide range of binding affinities with the ILA1 TCR (15–17). These previous findings clearly demonstrate that the ILA1 T-cell clone can recognize multiple different peptide ligands. Here, we solved the structure of the ILA1 TCR in complex with HLA-A*0201-ILAKFLHWL (A2-ILA) and several previously defined APLs. Combined with biophysical analysis, we demonstrate the molecular mechanism for antigen discrimination by the ILA1 TCR and model the mode of cross-reactivity with these APLs. In addition, we used our previously published peptide sampling approach (3) to estimate the number of pMHCI molecules that could be recognized by the ILA1 TCR. These data offer novel insight into the molecular factors that determine T-cell cross-reactivity, extending our understanding of the nature of T-cell antigen discrimination.
Results
The ILA1 TCR Makes a Broad Contact Network with A2-ILA
We solved the structure of the ILA1 TCR-A2-ILA complex at 2.8 Å in space group P 1 21 1 with crystallographic Rwork/Rfree ratios within accepted limits as shown by the theoretically expected distribution (18) (Table 1). The electron density was high quality throughout, represented by an omit map analysis of the ILA peptide (Fig. 1A). The ILA1 TCR utilized a canonical binding mode to engage A2-ILA (Fig. 1A) with a buried surface area (2507.2 Å2) and surface complementarity (TCR-pMHC = 0.641) within the normal range (Table 2) (1). The TCR α-chain was orientated over the MHCI α1 helices, and the TCR β-chain was oriented over the MHCI α2 helices (Fig. 1B), positioning the CDR loops of the ILA1 TCR over the central portion of the peptide, enabling contacts with 4 of the 9 peptide residues (Fig. 1C). Peptide residues Lys-4 and Trp-8 engaged in a complex network of contacts with the TCR CDR1/3α loops and CDR1/3β loops, respectively, contributing 39 of the 49 total peptide contacts (Table 2). Binding to Lys-4 involved a tight ball-and-socket interaction with 7 TCR residues, whereas contacts with Trp-8 were less restrictive, involving only TCR β-chain residues Glu-30 and Gln-96 (Table 3 and Fig. 1C). The 49 TCR interactions with peptide were supported by 58 contacts with MHC, involving 12 TCR α-chain residues and 4 TCR β-chain residues (Fig. 1D), contributing to the slightly TCR α-chain-skewed binding mode (α, 58%; β, 42% of total contacts). Notably, the TCR α-chain residue Arg-68 in the framework region loop (outside of the traditional CDR loops) formed two salt bridges with MHC residue Glu-166, and all of the MHC restriction triad residues (Arg-65, Ala-69, and Gln-155) (19) interacted with the ILA1 TCR (Table 3).
TABLE 1.
Data collection and structure refinement statistics
Values in parentheses refer to the highest resolution bin. One crystal was used for solving each structure. DLS, Diamond Light Source; r.m.s., root mean square; n/a, not applicable; CC1/2, correlation coefficient.
| ILA1-A2-ILA | A2-ILA3G8R | A2-ILA3G | A2-ILA8T | A2-ILA8E | |
|---|---|---|---|---|---|
| Data collection | |||||
| Protein Data Bank code | 5MEN | 5MEO | 5MEP | 5MEQ | 5MER |
| Space group | P 1 21 1 | P 21 21 21 | P 21 21 21 | P 1 21 1 | P 21 21 21 |
| Beamline | DLS I24 | DLS I04 | DLS I02 | DLS I04 | DLS I02 |
| Cell dimensions | |||||
| a (Å) | 93.2 | 49.2 | 119.4 | 53.34 | 45.7 |
| b (Å) | 48.7 | 74.9 | 169.6 | 81.44 | 119.0 |
| c (Å) | 118.1 | 125.8 | 47.1 | 56.77 | 170.3 |
| α (°) | 90 | 90 | 90 | 90 | 90 |
| β (°) | 108.2 | 90 | 90 | 113.5 | 90 |
| γ (°) | 90 | 90 | 90 | 90 | 90 |
| Resolution maximum (Å) | 2.81 (2.88–2.81) | 1.77 (1.82–1.77) | 2.71 (2.78–2.71) | 2.27 (2.33–2.27) | 1.88 (1.93–1.88) |
| Rmerge (%) | 0.100 (0.73) | 0.105 (0.718) | 0.105 (.831) | 0.086 (0.632) | 0.098 (0.621) |
| Total measurements | 91,454 (6,913) | 332,927 (22,417) | 195,005 (13,976) | 74,664 (5,741) | 545,975 (42,439) |
| Unique reflections | 25,031 (1,865) | 46,011 (3,362) | 26,894 (1,974) | 20,523 (1,527) | 76,630 5,603 |
| I/σI | 9.2 (1.9) | 10.5 (2.5) | 14.1 (2.3) | 12.8 (1.9) | 10.8 (3.2) |
| CC1/2 | n/a | n/a | 0.997 (0.714) | 0.997 (0.842) | 0.997 (0.913) |
| Completeness (%) | 99.7 (99.8) | 100 (100) | 100 (100) | 99.3 (98.9) | 99.9 (100) |
| Multiplicity | 3.7 (3.7) | 7.2 (6.7) | 7.3 (7.1) | 3.5 (3.8) | 7.1 (7.6) |
| Refinement | |||||
| Resolution (Å) | 56.11–2.81 | 48.16–1.77 | 42.39–2.71 | 48.90–2.27 | 34.06–1.9 |
| No. reflections in work set | 23,666 | 43,626 | 25,511 | 19,484 | 72,747 |
| No. reflections in Rfree set | 1,267 | 2,319 | 1,333 | 1,023 | 3,797 |
| Rwork/Rfree (%) | 18.9/27.2 | 17.1/21.4 | 18.2/23.9 | 20.3/26.4 | 18.4/22.4 |
| Mean B value (Å2) | 57.1 | 27.7 | 52.2 | 37.7 | 33.0 |
| Wilson B-factor (Å2) | 70.5 | 21.9 | 39.6 | 32.97 | 28.6 |
| Overall coordinate error (Å) | 0.377 | 0.082 | 0.252 | 0.232 | 0.098 |
| r.m.s. deviations | |||||
| Bond lengths (Å) | 0.017 | 0.019 | 0.013 | 0.019 | 0.019 |
| Bond Angles (°) | 2.025 | 1.983 | 1.658 | 1.976 | 1.967 |
| Ramachandran plot statistics | |||||
| Most favored region (%) | 92.27 | 97.85 | 97.23 | 96.54 | 98.12 |
| Allowed region (%) | 6.38 | 2.15 | 2.77 | 3.19 | 1.88 |
| Outliers (%) | 1.35 | 0 | 0 | 0.27 | 0 |
FIGURE 1.

The ILA1 TCR uses a broad binding footprint to engage A2-ILA. A, the overall binding mode of ILA1 TCR (blue and cyan schematic; CDR loops shown in multicolored schematic) in complex with A2-ILA (gray schematic and orange sticks). The box shows the observed map (top) at 1.0 σ and an omit maps (below) in which the model was refined in the absence of the ILA peptide with difference density contoured at 3.0 σ; positive contours are shown in green, and negative contours are shown in red. B, position of the ILA1 TCR CDR loops (multicolored sticks) with the ILA peptide (orange sticks) is shown in the HLA-A*0201 binding groove (gray surface). The crossing angle of the ILA1 TCR (black line) was calculated using previously published parameters (37). Briefly, this crossing angle represents the angle between a best fit straight line through the Cα atoms from the two MHC helices and a line that links the disulfide bond in the TCR α-chain variable region to the disulfide bond in the TCR β-chain variable region. C, interaction between residues in the ILA1 TCR CDR loops (multicolored sticks) and the ILA1 peptide (orange sticks) with the MHC α1 helix shown as a gray schematic. D, the ILA1 TCR residues in the CDR loops that contact the MHC surface are shown in multicolored schematic, and the surface with the MHC binding groove is shown in gray schematic and surface. CDR loops are colored as follows throughout: CDR1α, red; CDR2α, green; CDR3α, blue; CDR1β, yellow; CDR2β, purple; and CDR3β, cyan.
TABLE 2.
ILA1-A2-ILA contact summary
Buried surface area, 2507.2 Å2; surface complementarity for TCR-MHC, 0.635; surface complementarity for TCR-peptide, 0.707; surface complementarity for TCR-pMHC, 0.641; crossing angle, 51.8°, calculated as described previously (37). vdW, van der Waals; FW, framework region.
| vdW (≤4 Å) | H-bonds (≤3.4 Å) | Salt bridges (≤3.4 Å) | |
|---|---|---|---|
| MHC | 58 | 6 | 2 |
| Peptide | 49 | 4 | 1 |
| Peptide Lys-4 | 20 | 2 | 1 |
| Peptide Trp-8 | 19 | 2 | 0 |
| TCRα | 62 | 7 | 3 |
| CDR1α | 16 | 2 | 0 |
| CDR2α/FW | 9 | 2 | 2 |
| CDR3α | 37 | 3 | 1 |
| TCRα Asp-97 | 6 | 2 | 0 |
| TCRα Arg-68 | 4 | 0 | 2 |
| TCRβ | 45 | 3 | 0 |
| CDR1β | 13 | 0 | 0 |
| CDR2β | 7 | 0 | 0 |
| CDR3β | 25 | 3 | 0 |
| TCRβ Gln-96 | 18 | 3 | 0 |
| Total contacts | 107 | 10 | 3 |
TABLE 3.
ILA1-A2-ILA contacts
A 3.4 Å cutoff was used for H-bonds and salt bridges, and a 4 Å cutoff was used for van der Waals (vdW). FW, framework region.
| CDR loop | Gene usage | TCR residue | Peptide residue | MHC residue | vdW (≤4 Å) | H-bonds (≤3.4 Å) |
|---|---|---|---|---|---|---|
| CDR1α | TRAV22 | Asp-27Oδ2 | Thr-163Oγ1 | 4 | 1 | |
| TRAV22 | Asp-27Oδ1 | Glu-166Oϵ2 | 2 | 1 | ||
| TRAV22 | Ser-28 | Ala-158 | 1 | |||
| TRAV22 | Val-29 | Gln-155 | 4 | |||
| TRAV22 | Val-29 | Lys-4 | 4 | |||
| TRAV22 | Asn-30 | Gln-155 | 1 | |||
| CDR2α | TRAV22 | Tyr-48 | Glu-154 | 1 | ||
| TRAV22 | Tyr-48 | Gln-155 | 2 | |||
| TRAV22 | Ser-51O | Glu-154Oδ | 1 | 1 | ||
| TRAV22 | Ser-51 | Arg-157 | 1 | |||
| TRAV22 | Gln-55Oϵ1 | Glu-154Oϵ2 | 1 | |||
| FWα | TRAV22 | Arg-68NH1/NH2 | Glu-166Oϵ1/Oϵ2 | 4 | 2 salt bridges | |
| CDR3α | TRAJ40 | Asp-91Oδ1 | Lys-4Nζ | 3 | 1 salt bridge | |
| TRAJ40 | Ser-92O | Lys-4Nζ | 2 | 1 | ||
| TRAJ40 | Ala-93 | Lys-66 | 1 | |||
| TRAJ40 | Ala-93 | Thr-163 | 2 | |||
| TRAJ40 | Ala-93 | Lys-4 | 2 | |||
| TRAJ40 | Thr-94O | Lys-66Nζ | 3 | 1 | ||
| TRAJ40 | Thr-94 | Trp-167 | 3 | |||
| TRAJ40 | Ser-95 | Lys-4 | 4 | |||
| TRAJ40 | Gly-96 | Arg-65 | 3 | |||
| TRAJ40 | Thr-97O | Lys-4Nζ | 3 | 1 | ||
| TRAJ40 | Tyr-98 | Lys-66 | 3 | |||
| TRAJ40 | Tyr-98 | Ala-69 | 2 | |||
| TRAJ40 | Tyr-98 | Lys-4 | 2 | |||
| TRAJ40 | Tyr-98 | Leu-6 | 4 | |||
| CDR1β | TRBV6 | Glu-30 | Trp-8 | 13 | ||
| CDR2β | TRBV6 | Val-50 | Val-152 | 1 | ||
| TRBV6 | Val-50 | Leu-6 | 1 | |||
| TRBV6 | Val-50 | Trp-8 | 1 | |||
| TRBV6 | Ile-54 | Gln-72 | 4 | |||
| CDR3β | TRBJ1-1 | Tyr-95 | Lys-146 | 5 | ||
| TRBJ1-1 | Tyr-95 | Ala-150 | 2 | |||
| TRBJ1-1 | Gln-96 | Lys-146 | 2 | |||
| TRBJ1-1 | Gln-96Oϵ1 | Trp-147Nϵ1 | 4 | 1 | ||
| TRBJ1-1 | Gln-96 | Ala-150 | 1 | |||
| TRBJ1-1 | Gln-96 | Val-152 | 1 | |||
| TRBJ1-1 | Gln-96 | Leu-6 | 2 | |||
| TRBJ1-1 | Gln-96 | His-7 | 3 | |||
| TRBJ1-1 | Gln-96Oϵ1 | Trp-8N/O | 5 | 2 |
We have shown previously that the ILA1 TCR binds with a moderate/weak affinity (KD = 34 μm) to A2-ILA (20), consistent with the observation that TCRs specific for self-pMHCIs usually bind at the lower end of the TCR-pMHC affinity scale (21, 22). We performed a thermodynamic analysis of the ILA1 TCR-A2-ILA interaction by measuring binding using surface plasmon resonance at a range of temperatures (5–37 °C) (Fig. 2A). These analyses demonstrated that the weak affinity was not temperature-dependent, ranging from KD = 34 (25 °C) to 74 μm (5 °C). At physiological temperature (37 °C), the affinity was in the middle of this range (KD = 48 μm). The energetic analysis (Fig. 2B) revealed that the ILA1 TCR-A2-ILA interaction was driven entropically (TΔS = 5.86 kcal/mol) with only a minor change in enthalpy (ΔH = −0.16 kcal/mol). These values indicate almost no net loss, or gain, in electrostatic interactions during complex formation, indicative of structural reordering of the TCR and/or pMHC when binding. The entropic contribution suggested that ordered water molecules are squeezed out at the interface as the TCR and pMHC engage. Overall, these analyses demonstrated that the ILA1 TCR utilizes a relatively broad binding footprint, contacting spatially distant regions on the peptide and involving 21 different TCR residues contacting 17 A2-ILA residues (4 peptide and 13 MHC residues), contributing to an entropically driven, moderate-to-weak affinity interaction.
FIGURE 2.

Thermodynamic analysis of the ILA1 TCR with A2-ILA. A, eight to ten serial dilutions of the ILA1 TCR were injected in duplicate over A2-ILA at 5, 13, 21, 25, 29, and 37 °C. The equilibrium binding constants (KD) were calculated using a non-linear curve fit (y = (P1x)/P2 + x)), and kinetic and affinity parameters are shown in the table. B, the binding free energies, ΔG0 (ΔG0 = RT ln KD), were plotted against temperature (K) using non-linear regression to fit the three-parameter van 't Hoff equation (RT ln KD = ΔH0 − TΔS0 + ΔCp0(T − T0) − TΔCp0 ln(T/T0) with T0 = 298 K).
APLs Guide ILA1 Antigen Recognition through Structural Alterations in Peptide Conformation
We have previously characterized a number of APLs that alter the T-cell activation profile and TCR binding affinity of the ILA1 T-cell clone (9, 15, 16). To investigate how these ligands adjust TCR interactions to tune affinity, we solved the structure of four APLs (A2-ILA3G8R, ILGKFLHRL; A2-ILA3G, ILGKFLHWL; A2-ILA8T,ILAKFLHTL; and A2-ILA8E, ILAKFLHEL), included our previously published APL structure (A2-ILA8R, ILAKFLHRL) (23), and used the ILA1-A2-ILA complex as a model. The electron density was high quality throughout, represented by an omit map analysis of the ILA peptide variants (Fig. 3, A–D), and B-factor analysis indicated that there were no major differences in peptide mobility across the peptide variants (Fig. 3, E–J). Thermal stability analysis demonstrated that most of the APLs had a similar apparent Tm value (the term “apparent Tm” is used here because the protein irreversibly aggregates at high temperature) of around 55 °C with extremes in the range of 50 to 61 °C (Fig. 4). These similar stabilities are consistent with our previous observation that all these APLs bind equally to HLA-A2 on the cell surface (15). As we have shown previously in other systems (24), apparent Tm values correlated poorly with antigen potency (for instance, A2-ILA3G had the lowest apparent Tm value but was a potent activator of the ILA1 T-cell clone), suggesting that different pMHC cell surface expression levels were a minor factor in T-cell recognition. The A2-ILA3G structure was determined at 2.7 Å resolution, and the other APL structures were determined at resolutions between at 1.9 and 1.8 Å with crystallographic Rwork/Rfree ratios within accepted limits as shown by the theoretically expected distribution (18) (Table 1). The overall conformations of A2-ILA3G8R, A2-ILA3G, and A2-ILA8R were virtually identical to A2-ILA (Fig. 5, A–D) with Lys-4, Leu-6, and Trp-8 pointing up and away from the MHC binding groove and Leu-2, Phe-5, His-7, and Leu-9 acting as primary and secondary anchors, indicating that a molecular mimicry mechanism underpins ILA1 TCR recognition of these APLs. In contrast, in the A2-ILA8T and A2-ILA8E structures, peptide residues 5–7 were flipped so that Leu-6 acted as a secondary anchor and Phe-5 and His-7 were in more solvent-exposed positions (Fig. 5, E and F). In both peptide variants, the mutated residue was at position 8, distal from this structural rearrangement. Closer inspection of the structures did not reveal an obvious mechanism for this indirect effect on peptide conformation.
FIGURE 3.

Density plot, omit map, and B-factor analysis of ILA1 peptide variants. A–D, top, the observed map at 1.0 σ is shown. Bottom, omit maps are shown in which the model was refined in the absence of the ILA peptide variants with difference density contoured at 3.0 σ; positive contours are shown in green, and negative contours are shown in red. A, A2-ILGKFLHRL (purple sticks). B, A2-ILGKFLHWL (green sticks). C, A2-ILAKFLHTL (sand sticks). D, A2-ILAKFLHEL (black sticks). A2-ILAKFLHRL was solved previously (23). E–J, each APL is colored by B-factor with light blue representing a low B-factor and red representing a high B-factor. The conformation of each APL (sticks) with arrows indicating the direction of each residue in the peptide (solvent-exposed, MHC anchor, or in between) with the MHC α1 helix shown as a gray schematic. Residues in red indicate differences from the index sequence. An up arrow indicates solvent-exposed, a down arrow indicates anchor position, and no arrow indicates an intermediate position. E, A2-ILAKFLHWL. F, A2-ILGKFLHRL. G, A2-ILGKFLHWL. H, A2-ILAKFLHRL. I, A2-ILAKFLHTL. J, A2-ILAKFLHEL.
FIGURE 4.
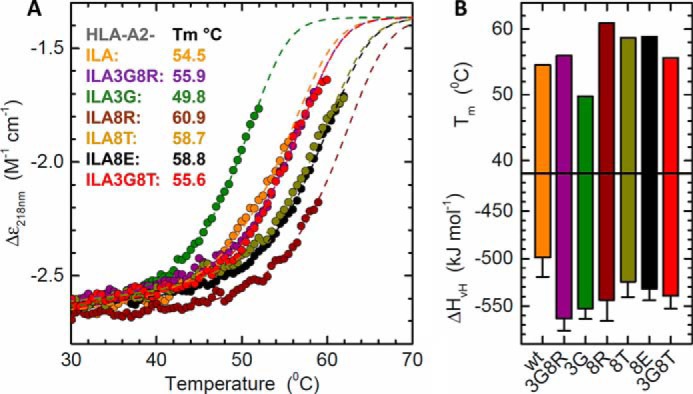
Stability of HLA-A2-ILA variants using circular dichroism. A, CD thermal denaturation curves recorded at 218 nm are shown for selected peptide-HLA class I samples. Dots represent measured values fitted assuming a two-state trimer-to-monomer transition (dashed lines) as described under “Experimental Procedures.” B, bar graphs of the thermal stability with respect to melting temperature (upper panel) and van 't Hoff's enthalpy of unfolding (lower panel). Error bars represent S.D. resulting from the multivariable curve fitting.
FIGURE 5.

Structural analysis of ILA1 TCR ligands. Shown is the conformation of each APL (sticks) demonstrating the direction of each residue in the peptide (solvent-exposed, MHC anchor, or in between) with the MHC α1 helix shown as a gray schematic. Residues in red indicate differences from the index sequence. An up arrow indicates solvent-exposed, a down arrow indicates anchor position, and no arrow indicates an intermediate position. A, A2-ILAKFLHWL (orange sticks). B, A2-ILGKFLHRL (purple sticks). C, A2-ILGKFLHWL (green sticks). D, A2-ILAKFLHRL (brown sticks) (reproduced from Ref. 23). E, A2-ILAKFLHTL (sand sticks). F, A2-ILAKFLHEL (black sticks). The circled residues in E and F face in different directions as compared with the index telomerase sequence (ILAKFLHWL) in A.
We next explored the binding affinity of the ILA1 TCR for the APLs included in this study using previously published (9, 15, 16) and new data (Fig. 6, A–E, and Table 4). Despite the relatively weak affinity between the ILA1 TCR and the natural A2-ILA ligand (KD = 34 μm), the ILA1 TCR could recognize A2-ILA3G8R and A2-ILA3G with antiviral-like affinities (KD = 1.0 and 3.7 μm, respectively). Both of these ligands included substitution of peptide residue 3 from Ala to Gly, a substitution that was clearly indicated in our previously published unbiased combinatorial peptide library screening using the ILA1 T-cell clone (9). Structural modeling of the ILA1 TCR with these ligands indicated that interaction with the N-terminal portion of the peptide was likely to be very similar for both ligands (Fig. 6, F and G). However, for A2-ILA3G8R, a major reorientation of TCR β-chain residue Gln-96 would be required to tolerate Arg at position 8 in the peptide. For both of these ligands, the extra flexibility, afforded at the N terminus of the peptide by the substitution of Gly compared with Ala, may enable the ILA1 TCR to establish enhanced contacts with peptide residue Lys-4. This represents a likely mechanism for the stronger affinity, supported further by the observation that all of the other APLs that did not include Gly at position 3 were bound by ILA1 with weaker affinity compared with the ILA1-A2-ILA interaction (Fig. 6, C–E). Structural modeling of ILA1 in complex with A2-ILA8R (Fig. 6H) revealed the same potential for steric hindrance between the TCR β-chain residues Glu-30 and Gln-96 but without the compensatory substitution at position 3. The structural rearrangement that would be required for ILA1 to bind to A2-ILA8R (KD = 151 μm) was reflected by a much weaker affinity compared with ILA1-A2-ILA (KD = 34 μm). Both A2-ILA8T and A2-ILA8E ligands underwent a conformational transition compared with the other APLs (Fig. 5). Modeling demonstrated that this alteration could lead to a steric conflict between TCR β-chain residue Gln-96 and the C-terminal residues of the peptide (Fig. 6, I and J). However, the position of peptide residue Phe-5 in A2-ILA8T may allow for compensatory interactions with TCR α-chain residues Asp-91 and Tyr-98, which are not present with A2-ILA8E. Furthermore, the smaller Thr-8 side chain in A2-ILA8T, compared with Glu-8 in A2-ILA-8E, would require smaller modifications in TCR docking. Taken together, these structural distinctions may explain the extremely weak affinity observed for ILA1 binding to A2-ILA8E compared with A2-ILA8T (KD values >500 μm and 28 μm, respectively).
FIGURE 6.
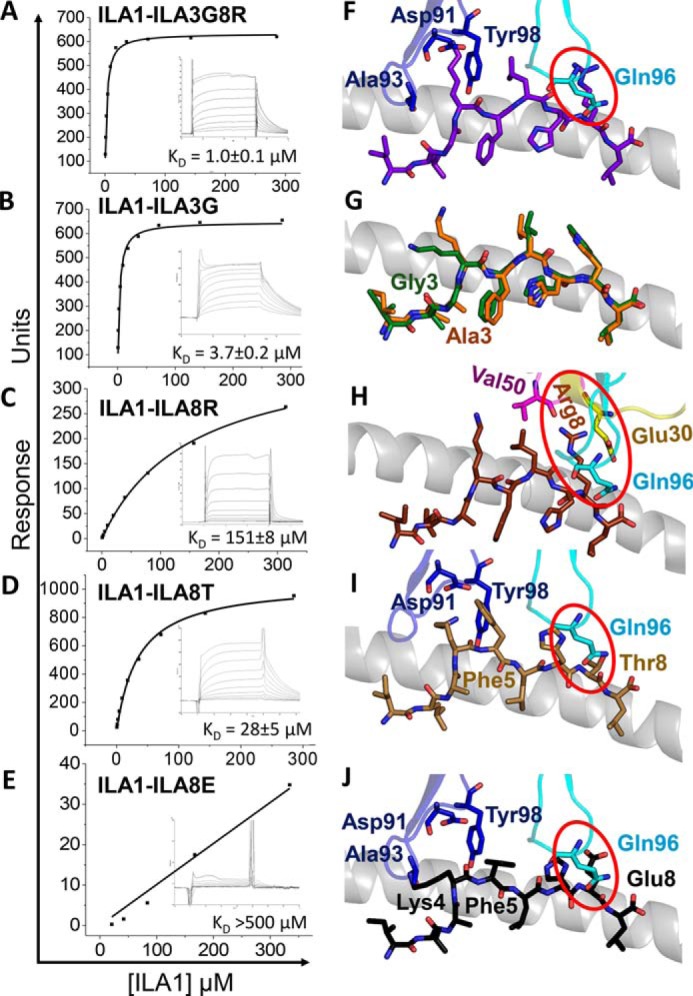
Equilibrium binding analysis and structural modeling of the ILA1 TCR interaction with the APLs. Binding affinity of the ILA1 TCR interaction with different APLs at 25 °C is shown. Eight to ten serial dilutions of the ILA1 TCR were injected over A2-ILA8R and A2-ILA3G8R, and representative data from three independent experiments are plotted after deducting binding to a control sample (HLA-A*0201-ALWGPDPAAA). The equilibrium binding constants (KD) were calculated using a non-linear curve fit (y = (P1x)/P2 + x)). A, ILA1-A2-ILA3G8R. B, ILA1-A2-ILA3G (reproduced from Ref. 15). C, ILA1-A2-ILA8R. D, ILA1-A2-ILA8T (reproduced from Ref. 15). E, ILA1-A2-ILA8E (reproduced from Ref. 15). The ILA1 TCR was modeled with each of the APL ligands by aligning the uncomplexed APLs with A2-ILA in the ILA1-A2-ILA complex structure (HLA-A*0101 α1 helix shown in gray schematic in F–J). Potential steric clashes are highlighted in red circles. F, A2-ILA3G8R (peptide in purple) showing modeled positions of the TCR CDR3α and -β loops (blue and cyan, respectively). G, A2-ILA3G (peptide in green) superposed with the A2-ILA peptide (orange). H, A2-ILA8R (peptide in brown) showing modeled positions of the TCR CDR1, -2, and -3 α/β loops (yellow, pink, and cyan, respectively). I, A2-ILA8T (peptide in sand) showing modeled positions of the TCR CDR3α and -β loops (blue and cyan, respectively). J, ILA1-A2-ILA8E (peptide in black) showing modeled positions of the TCR CDR3α and -β loops (blue and cyan, respectively).
TABLE 4.
ILA1 TCR binding affinity to peptide variants
KD was calculated from equilibrium binding experiments. n/m, kinetics were too fast to accurately measure.
| HLA-A*0201-ILA variant | kon | koff | χ2 for kon and koff | KD |
|---|---|---|---|---|
| m−1 s−1 | s−1 | μm | ||
| HLA-A*0201-ILGKFLHRL | 3 × 104 | 0.16 | 4.2 | 1.0 ± 0.1 |
| HLA-A*0201-ILGKFLHWL (39) | 1.6 × 104 | 0.05 | 3.6 | 3.7 ± 0.2 |
| HLA-A*0201-ILGKFLHTL (17) | 1.95 × 104 | 0.05 | 2.5 | 2.5 ± 0.5 |
| HLA-A*0201-ILAKFLHYL (39) | n/m | n/m | n/m | 22.6 ± 2.1 |
| HLA-A*0201-ILAKFLHWL (20) | 4.5 × 103 | 0.15 | 4.3 | 34 ± 2 |
| HLA-A*0201-ILAKFLHTL (39) | 2.2 × 103 | 0.08 | 1.9 | 28 ± 5 |
| HLA-A*0201-ILAKFLYWL (39) | n/m | n/m | n/m | 82 ± 8 |
| HLA-A*0201-ILALFLHWL (16) | 1.7 × 103 | 0.2 | 4.2 | 117 ± 6 |
| HLA-A*0201-ILAKFLHRL | n/m | n/m | n/m | 151 ± 8 |
| HLA-A*0201-ILAKYLHWL (17) | 1.3 × 103 | 0.32 | 3.5 | 242 ± 20 |
| HLA-A*0201-ILAKFLHEL (39) | n/m | n/m | n/m | >500 |
Quantification of ILA1 TCR Degeneracy
Our previous investigations have demonstrated that even single residue substitutions outside of the two main peptide interaction zones (Lys-4 and Trp-8) could have a substantial impact on the ILA1 TCR-A2-ILA complex, reflected by the different binding affinities and antigen potencies shown here and published previously (9, 15, 16). We next generated a degeneracy curve for the ILA1 TCR using our previously described approach that quantifies TCR cross-reactivity (3).
Combinatorial peptide library scan data were used to design four different motif-restricted peptide sets (I, XLGXXXXRL (total set size, 195); II, XLXKFLXXL (total set size, 194); III, XLG(K/L)F(L/I)(M/F/Y/N/H)(R/T/Y/K/S/F/H/I/L/M/Q/V/G/N)(L/V) (total set size, 10,640); and IV, (A/I/L/M/P/Q/W)L(G/A)(K/L)F(L/I)(N/H)(F/K/N/Q/T/Y)(L/V) (total set size, 1344) where X denotes any of the 19 proteogenic amino acids excluding cysteine). Between 19 and 30 peptides were sampled at random from each of these motif-restricted peptides cohorts. In addition, we performed importance sampling where 20 peptides were sampled from an effective sample size of 1.5 × 107. The pEC50 for each peptide was estimated by simultaneous curve fitting (Fig. 7), and these values were used to construct a degeneracy curve for the ILA1 TCR (Fig. 8A). These analyses indicated that ILA1 could recognize ∼2 × 103 peptides with a functional sensitivity at least as high as the functional sensitivity of the optimal agonist. At 2 orders of magnitude from the optimum (i.e. peptides ranging from of the optimal agonist to the optimal agonist) ∼4 × 104 peptides could be recognized by the ILA1 T-cell clone.
FIGURE 7.

pEC50 values for all peptide ligands tested. Simultaneous curve fitting was used to estimate functional sensitivity measured as pEC50 for peptides sampled from the following sets: I, XLGXXXXRL (set size, 195; 30 peptides sampled at random); II, XLXKFLXXL (set size, 194; 30 peptides sampled at random); III, XLG(K/L)F(L/I)(M/F/Y/N/H)(R/T/Y/K/S/F/H/I/L/M/Q/V/G/N)(L/V) (set size, 10,640; 30 peptides sampled at random); IV, (A/I/L/M/P/Q/W)L(G/A)(K/L)F(L/I)(N/H)(F/K/N/Q/T/Y)(L/V); and V, replicate of a biased sampling of each set (20 peptides sampled from an effective sample size of 1.5 × 107).
FIGURE 8.

TCR binding footprint contributes to T-cell cross-reactivity. A, peptide recognition degeneracy for ILA1. The degeneracy curve plots the estimated number of peptides that have a functional sensitivity at least as strong as abscissa. The highest value of abscissa corresponds to inferred functional sensitivity of optimal peptide, whereas the lowest value of abscissa lies 10 orders of magnitude below this optimum. Bias, degeneracy curve based on agonist-biased importance sampling. Curves I–IV are motif-based, i.e. sampled from subsets of the entire peptide universe, and therefore lie below the degeneracy curve. I, XLGXXXXRL (set size, 195; 30 peptides sampled at random); II, XLXKFLXXL (set size, 194; 30 peptides sampled at random); III, XLG(K/L)F(L/I)(M/F/Y/N/H)(R/T/Y/K/S/F/H/I/L/M/Q/V/G/N)(L/V) (set size, 10,640; 30 peptides sampled at random); IV, (A/I/L/M/P/Q/W)L(G/A)(K/L)F(L/I)(N/H)(F/K/N/Q/T/Y)(L/V) (set size, 1344; 19 peptides sampled at random); X denotes any of the 19 amino acids excluding cysteine. Cross-reactivity of the 1E6 (B) and ILA1 (C) CD8+ T-cell clones was estimated by the number of residues recognized in a combinatorial peptide library screen generating responses over >0.5 ng/ml MIP-1β. Bars that represent positions in the peptide that are anchor residues are colored gray. Bars that represent residues that are main contact sites for each respective TCR are colored red.
This analysis suggests that the ILA1 TCR can cross-react with a diverse peptide universe. Although smaller than the estimated number of peptides recognized by the 1E6 T-cell clone (∼106), it should be noted that 1E6 recognizes a 10-mer peptide, whereas ILA1 recognizes a 9-mer. Thus, the peptide universe under consideration for 1E6 is 20 times larger than that for ILA1. Although it is unknown how this difference in peptide length affects the comparison between the two degeneracy profiles, this difference may partly explain why ILA1 appears to be less cross-reactive than 1E6. This analysis also demonstrated a different pattern of cross-reactivity between the 1E6 and ILA1 T-cell clones that was consistent with the respective binding footprints of their TCRs. The 1E6 T-cell clone could recognize a large number of sublibraries (46 in total) outside of the central binding zone (residues 4–6) (Fig. 8B). In contrast, the ILA1 T-cell clone was generally more sensitive across the peptide backbone, reflecting a more globally coordinated interaction between the ILA1 TCR and the antigenic peptide (Fig. 8C). Taken together, these results are broadly consistent with the idea that different TCR binding footprints (i.e. TCRs that focus on a minimal peptide motif compared with TCRs that make contacts across the peptide backbone) can enable T-cell cross-reactivity, adding further support to the general idea that T-cells must be cross-reactive to fully protect us against a highly variable pathogen universe (for a review, see Ref. 8).
Discussion
To mount effective immune responses in the face of a diverse antigenic milieu using a limited set of TCRs (estimated ∼25 million distinct clonotypes in an individual), each T-cell must be able to interact productively with a vast array of different antigens (2, 8). Indeed, recent experimental evidence supports this notion, including our own study demonstrating that a single T-cell clone can recognize over one million different peptides at physiologically relevant concentrations (3). However, structural investigations of TCR-pMHC interactions have demonstrated that the TCR can establish a distinct and highly specific interaction with both peptide and MHC. In keeping with such specificity of binding requirements, even single mutations at key residues in the TCR, peptide, or MHC that are involved in the binding interface have been shown to abrogate antigen recognition (25–29). The importance of T-cell cross-reactivity is manifest not only in effective immune surveillance (2, 8) but also in autoreactivity (30) and the design of therapeutics (10, 11). Structural rules must therefore exist that allow cross-reactivity to take place; here, we report early steps toward deconstructing these rules.
We investigated a TCR isolated from ILA1, a well characterized HLA-A*0201-restricted, telomerase-specific, CD8+ T-cell clone. Our previous work has shown that ILA1 cross-reacts with an array of APLs with different potencies, tuned by the CD8 co-receptor, and that antigen “potency” generally correlates directly with the affinity of TCR binding (9, 15). Here, we solved the complex structure of the ILA1 TCR with the natural index ligand (A2-ILA) and used the structures of a number of unligated APLs to model the mode of APL recognition and determine the structural basis for ILA1 cross-reactivity.
We have demonstrated previously that the ILA1 T-cell clone is particularly sensitive to APLs with modifications at peptide residues 3 and 8 (15). The unligated structures of five APLs with alterations in these positions demonstrated that the overall conformation of the Cα peptide backbone could be altered by introduction of Thr or Glu at position 8, possibly explaining the weaker binding affinity between ILA1 and A2-ILA8R and between ILA1 and A2-ILA8E. In contrast, substitution of Ala to Gly at peptide residue 3 enhanced recognition, and Gly at this position was strongly recognized in combinatorial peptide library screens (9). Our structural analysis indicated that the surmised extra flexibility afforded to the N terminus of the peptide mediated by substitution at position 3 with Gly would likely enable more favorable interactions with Lys-4, which made a network of contacts with the ILA1 TCR through a “ball-and-socket”-like interaction. In fact, substitution at position 3 with Gly could override the negative impact of modifications to peptide residue 8, revealed by the enhanced binding affinity of the ILA1-A2-ILA3G8R interaction (KD = 1.0 μm) compared with the index peptide (KD = 34 μm) and ILA8R (KD = 151 μm). Our structural analysis demonstrated that, again, even single peptide substitutions outside of the main interaction interface could have a substantial impact on TCR binding affinity and T-cell antigen potency, consistent with our previous data (9, 15, 16). These observations add further evidence to our recent findings (19, 31–34) that peptide presentation by MHCI can be dynamic and difficult to predict.
Recent reports have demonstrated that TCRs using focused TCR-peptide binding can be highly cross-reactive (6, 36). However, this binding footprint is not representative of most TCRs described in the literature. Indeed, on average, TCR-peptide binding is spread out over ∼60% of the peptide backbone for MHCI-restricted epitopes, often including contacts with both the N- and C-terminal regions of the peptide (1, 37). This interconnected binding network between the TCR and the peptide may not allow a high degree of cross-reactivity because most peptide modifications could impact binding. Unlike the focused TCR-peptide binding utilized by the 1E6 (6) and 42F3 (38) TCRs, the ILA1 TCR utilized a more representative binding footprint. This was reflected by a larger buried surface area value (2540 Å for ILA1 compared with 1640 Å for 1E6) and a binding motif that included contacts spread out over peptide residues 4–8. Thus, we explored the consequences of the ILA1 TCR binding footprint on ILA1 T-cell cross-reactivity using our previously published methodology (3). Despite the broader peptide contact zone utilized by the ILA1 TCR, the ILA1 T-cell clone was still able to recognize ∼4 × 104 peptides with equal or greater sensitivity compared with the index peptide and many more at lower potency. These data suggest that although the TCR binding footprint is very likely to tune T-cell cross-reactivity to some degree the ability of T-cells to recognize a vast array of different peptides is likely to be commonplace.
In summary, we demonstrate that the interaction between a TCR from a human CD8+ T-cell clone that recognizes a peptide sequence from an important tumor antigen contacts the peptide at spatially distant sites along the peptide backbone. The affinity of this TCR can be tuned by various peptide modifications through both direct and indirect effects, demonstrating the dynamic nature of the interaction among TCR, peptide, and MHC. Even though the ILA1 T-cell clone was sensitive to modifications along the peptide backbone, consistent with its broad binding interface, it was still able to cross-react with a vast array of different peptides. These data demonstrate that focused TCR-peptide binding is not a requirement for T-cell degeneracy. Indeed, a broader binding footprint, as observed in most TCR-pMHC structures reported to date, is also likely to facilitate T-cell cross-reactivity. These results have important implications for immune surveillance, i.e. how a limited set of TCRs can recognize all potential antigens variants, and the complex mechanisms that may lead to autoreactivity mediated by molecular mimicry.
Experimental Procedures
T-cells and Target Cells
The ILA1 CD8+ T-cell clone is specific for the HLA-A*0201-restricted human telomerase reverse transcriptase-derived epitope ILAKFLHWL (residues 540–548) (39), and the 1E6 T-cell clone is specific for the human leukocyte antigen HLA-A*0201-restricted autoantigen preproinsulin epitope ALWGPDPAAA (residues 15–24) (40). CD8+ T-cell clones were maintained in RPMI 1640 medium (Life Technologies) containing 100 units/ml penicillin (Life Technologies), 100 mg/ml streptomycin (Life Technologies), 2 mm l-glutamine (Life Technologies), and 10% heat-inactivated FCS (Life Technologies) (R10) supplemented with 2.5% Cellkines (Helvetica Healthcare, Geneva, Switzerland), 200 IU/ml IL-2 (PeproTech, Rocky Hill, NJ), and 25 ng/ml IL-15 (PeproTech). Hmy.2 C1R B-cells expressing full-length HLA-A*0201 were generated as described previously (41).
Protein Expression, Refolding, and Purification
The ILA1 TCR, HLA-A*0201 α-chain, and human β2-microglobulin chain sequences were generated as described previously (20) and cloned into separate pGMT7 expression plasmids under the control of the T7 promoter. The ILA1 TCR and HLA-A*0201 in complex with various different peptide variants (as indicated) were refolded and purified as described previously (14). Biotinylated pMHCI was prepared as described previously (42).
pMHC Stability Assays
Thermal stability of the HLA-A*0201-peptide complexes was assessed by circular dichroism spectroscopy, monitoring the change in ellipticities at 218 nm upon heating as described (31). Briefly, samples were prepared in PBS at a concentration of ∼3 μm and measured in 0.1-cm quartz cells. Melting curves were analyzed assuming a two-state trimer-to-monomer transition from the native to unfolded conformation and fitted as described (43). As all protein complexes aggregated to various degrees upon unfolding, the ellipticity of the unfolded state was set as a constant of −1.35 m−1 cm−1 (44).
Surface Plasmon Resonance Analysis
Binding analysis was performed in duplicate using a BIAcoreT200TM equipped with a CM5 sensor chip as described previously (45). Approximately 200–500 response units of HLA-A*0201-ILGKFLHRL or HLA-A*0201-ILAKFLHRL peptide complex was attached to the CM5 sensor chip at a slow flow rate of 10 μl/min to ensure uniform distribution on the chip surface. HLA-A*0201-ILAKFLHWL was used as a positive control as the binding affinity with the ILA1 TCR has been published previously (15, 20). The ILA1 TCR was purified and concentrated to ∼300 μm on the same day of surface plasmon resonance analysis. For equilibrium analysis, 10 serial dilutions were prepared in duplicate for each sample and injected over the relevant sensor chips at 25 °C. TCR was injected over the chip surface using kinetic injections at a flow rate of 45 μl/min using HLA-A*0201-ALWGPDPAAA as a negative control surface on flow cell 1. Results were analyzed using BIAevaluationTM 3.1, Excel, and Origin 6.0 software. The equilibrium dissociation constant (KD) values were calculated assuming a 1:1 interaction by plotting specific equilibrium binding responses against protein concentrations followed by non-linear least square fitting of the Langmuir binding equation. For kinetics analysis, the kon and koff values were calculated assuming 1:1 Langmuir binding, and the data were analyzed using a global fit algorithm (BIAevaluation 3.1).
Crystal Structure Determination
All protein crystals were grown at 18 °C by vapor diffusion via the sitting drop technique. 200 nl of each pMHCI (10 mg/ml) in crystallization buffer (10 mm Tris, pH 8.1, and 10 mm NaCl) was added to 200 nl of reservoir solution. ILA1-HLA-A*0201-ILAKFLHWL (ILA1-A2-ILA) and HLA-A*0201-ILAKFLHTL (A2-ILA8T) crystals were grown in 0.2 m ammonium sulfate, 0.1 m HEPES, pH 7, and 20% PEG 8000 (46); HLA-A*0201-ILGKFLHRL (A2-ILA3G8R) crystals were grown in 0.2 m ammonium sulfate, 0.1 m Tris, pH 7.5, and 25% PEG 8000 (46); HLA-A*0201-ILGKFLHWL (A2-ILA3G) crystals were grown in 0.2 m ammonium sulfate, 0.1 m MES, pH 7, and 15% PEG 8000; and HLA-A*0201-ILAKFLHEL (A2-ILA8E) crystals were grown in 0.2 m ammonium sulfate, 0.1 m MES, pH 7, and 25% PEG 8000 (46). Crystallization screens were conducted using an Art-Robbins Phoenix dispensing robot (Alpha Biotech Ltd., UK), and data were collected at 100 K at the Diamond Light Source, Oxfordshire, UK, at a wavelength of 0.98 Å using an Area Detector Systems Corp. Q315 charge-coupled device detector. Reflection intensities were estimated using XIA2 (47), and the data were analyzed with SCALA and the CCP4 package (48). Structures were solved with molecular replacement using Phaser (49). Sequences were adjusted with Coot (50), and the models were refined with REFMAC5. Graphical representations were prepared with PyMOL (35). The reflection data and final model coordinates were deposited with the Protein Data Bank under codes 5MEN (ILA1-A2-ILA), 5MEO (A2-ILA3G8R), 5MEP (A2-ILA3G), 5MEQ (A2-ILA8T), and 5MER (A2-ILA8E).
CD8+ T-cell Effector Function Assays: MIP1β ELISA
6 × 104 C1R-A2 cells were incubated with peptide at various concentrations in duplicate for 2 h at 37 °C. Subsequently, 3 × 104 ILA1 CD8+ T-cells were added, and the assay was incubated overnight at 37 °C. The supernatant was harvested and assayed for MIP1β by ELISA according to the manufacturer's instructions (R&D Systems). Functional sensitivity of individual peptides was expressed as the pEC50 of each peptide, which is defined as −1 × the base 10 logarithm (p) of the 50% efficacy concentration (EC50).
Quantification of ILA1 TCR Degeneracy
The degeneracy of the ILA1 TCR was estimated as described previously (3). Briefly, the degeneracy at ω, defined as the number of peptides whose functional sensitivity is at least as large as ω, was estimated directly using importance sampling based on the combinatorial peptide library scan and bounded below by sampling from motif-based subsets of the peptide universe. The degeneracy is reported by plotting this quantity as a function of ω where the functional sensitivity ω was scaled relative to a clone-specific reference peptide (the “index”).
Author Contributions
H. A. v. d. B., A. L., M. D. C., K. B., J. E.-M., J. J. M., A. M. B., G. D., A. J. S., A. W., A. F., M. C., B. L., P. J. R., L. W., and D. K. C. performed experiments and analyzed the data. A. K. S., L. W., and D. K. C. wrote the manuscript. A. K. S., L. W., H. A. v. d. B., and D. K. C. conceived and directed the study. A. K. S., L. W., and D. K. C. funded the study. All authors contributed to discussions.
Acknowledgment
We thank the staff at Diamond Light Source for providing facilities and support.
This work was supported in part by United Kingdom Biotechnology and Biological Sciences Research Council Grant BB/H001085/1 and Wellcome Trust Intermediate Clinical Fellowship WT079848MA (to L. W.). The authors declare that they have no conflicts of interest with the contents of this article.
- pMHC
- peptide-major histocompatibility complex
- TCR
- T-cell receptor
- CDR
- complementarity-determining region
- APL
- altered peptide ligand
- A2-ILA
- HLA-A*0201-ILAKFLHWL
- EC50
- 50% efficacy concentration.
References
- 1. Rossjohn J., Gras S., Miles J. J., Turner S. J., Godfrey D. I., and McCluskey J. (2015) T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 33, 169–200 [DOI] [PubMed] [Google Scholar]
- 2. Mason D. (1998) A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol. Today 19, 395–404 [DOI] [PubMed] [Google Scholar]
- 3. Wooldridge L., Ekeruche-Makinde J., van den Berg H. A., Skowera A., Miles J. J., Tan M. P., Dolton G., Clement M., Llewellyn-Lacey S., Price D. A., Peakman M., and Sewell A. K. (2012) A single autoimmune T cell receptor recognizes more than a million different peptides. J. Biol. Chem. 287, 1168–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wilson D. B., Wilson D. H., Schroder K., Pinilla C., Blondelle S., Houghten R. A., and Garcia K. C. (2004) Specificity and degeneracy of T cells. Mol. Immunol. 40, 1047–1055 [DOI] [PubMed] [Google Scholar]
- 5. Birnbaum M. E., Mendoza J. L., Sethi D. K., Dong S., Glanville J., Dobbins J., Ozkan E., Davis M. M., Wucherpfennig K. W., and Garcia K. C. (2014) Deconstructing the peptide-MHC specificity of T cell recognition. Cell 157, 1073–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cole D. K., Bulek A. M., Dolton G., Schauenberg A. J., Szomolay B., Rittase W., Trimby A., Jothikumar P., Fuller A., Skowera A., Rossjohn J., Zhu C., Miles J. J., Peakman M., Wooldridge L., et al. (2016) Hotspot autoimmune T cell receptor binding underlies pathogen and insulin peptide cross-reactivity. J. Clin. Investig. 126, 2191–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arstila T. P., Casrouge A., Baron V., Even J., Kanellopoulos J., and Kourilsky P. (1999) A direct estimate of the human αβ T cell receptor diversity. Science 286, 958–961 [DOI] [PubMed] [Google Scholar]
- 8. Sewell A. K. (2012) Why must T cells be cross-reactive? Nat. Rev. Immunol. 12, 669–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wooldridge L., Laugel B., Ekeruche J., Clement M., van den Berg H. A., Price D. A., and Sewell A. K. (2010) CD8 controls T cell cross-reactivity. J. Immunol. 185, 4625–4632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Linette G. P., Stadtmauer E. A., Maus M. V., Rapoport A. P., Levine B. L., Emery L., Litzky L., Bagg A., Carreno B. M., Cimino P. J., Binder-Scholl G. K., Smethurst D. P., Gerry A. B., Pumphrey N. J., Bennett A. D., et al. (2013) Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122, 863–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cameron B. J., Gerry A. B., Dukes J., Harper J. V., Kannan V., Bianchi F. C., Grand F., Brewer J. E., Gupta M., Plesa G., Bossi G., Vuidepot A., Powlesland A. S., Legg A., Adams K. J., et al. (2013) Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 5, 197ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Raman M. C., Rizkallah P. J., Simmons R., Donnellan Z., Dukes J., Bossi G., Le Provost G. S., Todorov P., Baston E., Hickman E., Mahon T., Hassan N., Vuidepot A., Sami M., Cole D. K., et al. (2016) Direct molecular mimicry enables off-target cardiovascular toxicity by an enhanced affinity TCR designed for cancer immunotherapy. Sci. Rep. 6, 18851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Adams J. J., Narayanan S., Birnbaum M. E., Sidhu S. S., Blevins S. J., Gee M. H., Sibener L. V., Baker B. M., Kranz D. M., and Garcia K. C. (2016) Structural interplay between germline interactions and adaptive recognition determines the bandwidth of TCR-peptide-MHC cross-reactivity. Nat. Immunol. 17, 87–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bulek A. M., Cole D. K., Skowera A., Dolton G., Gras S., Madura F., Fuller A., Miles J. J., Gostick E., Price D. A., Drijfhout J. W., Knight R. R., Huang G. C., Lissin N., Molloy P. E., et al. (2012) Structural basis for the killing of human beta cells by CD8+ T cells in type 1 diabetes. Nat. Immunol. 13, 283–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Laugel B., van den Berg H. A., Gostick E., Cole D. K., Wooldridge L., Boulter J., Milicic A., Price D. A., and Sewell A. K. (2007) Different T cell receptor affinity thresholds and CD8 coreceptor dependence govern cytotoxic T lymphocyte activation and tetramer binding properties. J. Biol. Chem. 282, 23799–23810 [DOI] [PubMed] [Google Scholar]
- 16. Melenhorst J. J., Scheinberg P., Chattopadhyay P. K., Lissina A., Gostick E., Cole D. K., Wooldridge L., van den Berg H. A., Bornstein E., Hensel N. F., Douek D. C., Roederer M., Sewell A. K., Barrett A. J., and Price D. A. (2008) Detection of low avidity CD8+ T cell populations with coreceptor-enhanced peptide-major histocompatibility complex class I tetramers. J. Immunol. Methods 338, 31–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van den Berg H. A., Ladell K., Miners K., Laugel B., Llewellyn-Lacey S., Clement M., Cole D. K., Gostick E., Wooldridge L., Sewell A. K., Bridgeman J. S., and Price D. A. (2013) Cellular-level versus receptor-level response threshold hierarchies in T-cell activation. Front. Immunol. 4, 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tickle I. J., Laskowski R. A., and Moss D. S. (2000) Rfree and the Rfree ratio. II. Calculation of the expected values and variances of cross-validation statistics in macromolecular least-squares refinement. Acta Crystallogr. D Biol. Crystallogr. 56, 442–450 [DOI] [PubMed] [Google Scholar]
- 19. Tynan F. E., Burrows S. R., Buckle A. M., Clements C. S., Borg N. A., Miles J. J., Beddoe T., Whisstock J. C., Wilce M. C., Silins S. L., Burrows J. M., Kjer-Nielsen L., Kostenko L., Purcell A. W., McCluskey J., et al. (2005) T cell receptor recognition of a “super-bulged” major histocompatibility complex class I-bound peptide. Nat. Immunol. 6, 1114–1122 [DOI] [PubMed] [Google Scholar]
- 20. Cole D. K., Pumphrey N. J., Boulter J. M., Sami M., Bell J. I., Gostick E., Price D. A., Gao G. F., Sewell A. K., and Jakobsen B. K. (2007) Human TCR-binding affinity is governed by MHC class restriction. J. Immunol. 178, 5727–5734 [DOI] [PubMed] [Google Scholar]
- 21. Bridgeman J. S., Sewell A. K., Miles J. J., Price D. A., and Cole D. K. (2012) Structural and biophysical determinants of αβ T-cell antigen recognition. Immunology 135, 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aleksic M., Liddy N., Molloy P. E., Pumphrey N., Vuidepot A., Chang K.-M., and Jakobsen B. K. (2012) Different affinity windows for virus and cancer-specific T-cell receptors: implications for therapeutic strategies. Eur. J. Immunol. 42, 3174–3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ekeruche-Makinde J., Miles J. J., van den Berg H. A., Skowera A., Cole D. K., Dolton G., Schauenburg A. J., Tan M. P., Pentier J. M., Llewellyn-Lacey S., Miles K. M., Bulek A. M., Clement M., Williams T., Trimby A., et al. (2013) Peptide length determines the outcome of TCR/peptide-MHCI engagement. Blood 121, 1112–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miles K. M., Miles J. J., Madura F., Sewell A. K., and Cole D. K. (2011) Real time detection of peptide-MHC dissociation reveals that improvement of primary MHC-binding residues can have a minimal, or no, effect on stability. Mol. Immunol. 48, 728–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cole D. K., Miles K. M., Madura F., Holland C. J., Schauenburg A. J., Godkin A. J., Bulek A. M., Fuller A., Akpovwa H. J., Pymm P. G., Liddy N., Sami M., Li Y., Rizkallah P. J., Jakobsen B. K., et al. (2014) T-cell Receptor (TCR)-peptide specificity overrides affinity-enhancing TCR-major histocompatibility complex interactions. J. Biol. Chem. 289, 628–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Borg N. A., Ely L. K., Beddoe T., Macdonald W. A., Reid H. H., Clements C. S., Purcell A. W., Kjer-Nielsen L., Miles J. J., Burrows S. R., McCluskey J., and Rossjohn J. (2005) The CDR3 regions of an immunodominant T cell receptor dictate the “energetic landscape” of peptide-MHC recognition. Nat. Immunol. 6, 171–180 [DOI] [PubMed] [Google Scholar]
- 27. Madura F., Rizkallah P. J., Miles K. M., Holland C. J., Bulek A. M., Fuller A., Schauenburg A. J., Miles J. J., Liddy N., Sami M., Li Y., Hossain M., Baker B. M., Jakobsen B. K., Sewell A. K., et al. (2013) T-cell receptor specificity maintained by altered thermodynamics. J. Biol. Chem. 288, 18766–18775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gras S., Chen Z., Miles J. J., Liu Y. C., Bell M. J., Sullivan L. C., Kjer-Nielsen L., Brennan R. M., Burrows J. M., Neller M. A., Khanna R., Purcell A. W., Brooks A. G., McCluskey J., Rossjohn J., et al. (2010) Allelic polymorphism in the T cell receptor and its impact on immune responses. J. Exp. Med. 207, 1555–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burrows S. R., Chen Z., Archbold J. K., Tynan F. E., Beddoe T., Kjer-Nielsen L., Miles J. J., Khanna R., Moss D. J., Liu Y. C., Gras S., Kostenko L., Brennan R. M., Clements C. S., Brooks A. G., et al. (2010) Hard wiring of T cell receptor specificity for the major histocompatibility complex is underpinned by TCR adaptability. Proc. Natl. Acad. Sci. U.S.A. 107, 10608–10613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sethi D. K., Gordo S., and Schubert D. A., and Wucherpfennig K. W. (2013) Crossreactivity of a human autoimmune TCR is dominated by a single TCR loop. Nat. Commun. 4, 2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kløverpris H. N., Cole D. K., Fuller A., Carlson J., Beck K., Schauenburg A. J., Rizkallah P. J., Buus S., Sewell A. K., and Goulder P. (2015) A molecular switch in immunodominant HIV-1-specific CD8 T-cell epitopes shapes differential HLA-restricted escape. Retrovirology 12, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Madura F., Rizkallah P. J., Holland C. J., Fuller A., Bulek A., Godkin A. J., Schauenburg A. J., Cole D. K., and Sewell A. K. (2015) Structural basis for ineffective T-cell responses to MHC anchor residue-improved “heteroclitic” peptides. Eur. J. Immunol. 45, 584–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Motozono C., Pearson J. A., De Leenheer E., Rizkallah P. J., Beck K., Trimby A., Sewell A. K., Wong F. S., and Cole D. K. (2015) Distortion of the major histocompatibility complex class I binding groove to accommodate an insulin-derived 10-mer peptide. J. Biol. Chem. 290, 18924–18933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miles J. J., Elhassen D., Borg N. A., Silins S. L., Tynan F. E., Burrows J. M., Purcell A. W., Kjer-Nielsen L., Rossjohn J., Burrows S. R., and McCluskey J. (2005) CTL recognition of a bulged viral peptide involves biased TCR selection. J. Immunol. 175, 3826–3834 [DOI] [PubMed] [Google Scholar]
- 35. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Version 1, Schrödinger LLC, New York [Google Scholar]
- 36. Degano M., Garcia K. C., Apostolopoulos V., Rudolph M. G., Teyton L., and Wilson I. (2000) A functional hot spot for antigen recognition in a superagonist TCR/MHC complex. Immunity 12, 251–261 [DOI] [PubMed] [Google Scholar]
- 37. Rudolph M. G., Stanfield R. L., and Wilson I. A. (2006) How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 24, 419–466 [DOI] [PubMed] [Google Scholar]
- 38. Adams J. J., Narayanan S., Liu B., Birnbaum M. E., Kruse A. C., Bowerman N. A., Chen W., Levin A. M., Connolly J. M., Zhu C., Kranz D. M., and Garcia K. C. (2011) T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity 35, 681–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Laugel B., Price D. A., Milicic A., and Sewell A. K. (2007) CD8 exerts differential effects on the deployment of cytotoxic T lymphocyte effector functions. Eur. J. Immunol. 37, 905–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Skowera A., Ellis R. J., Varela-Calviño R., Arif S., Huang G. C., Van-Krinks C., Zaremba A., Rackham C., Allen J. S., Tree T. I., Zhao M., Dayan C. M., Sewell A. K., Unger W. W., Drijfhout J. W., et al. (2008) CTLs are targeted to kill β cells in patients with type 1 diabetes through recognition of a glucose-regulated preproinsulin epitope. J. Clin. Investig. 118, 3390–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Purbhoo M. A., Boulter J. M., Price D. A., Vuidepot A. L., Hourigan C. S., Dunbar P. R., Olson K., Dawson S. J., Phillips R. E., Jakobsen B. K., Bell J. I., and Sewell A. K. (2001) The human CD8 coreceptor effects cytotoxic T cell activation and antigen sensitivity primarily by mediating complete phosphorylation of the T cell receptor ζ chain. J. Biol. Chem. 276, 32786–32792 [DOI] [PubMed] [Google Scholar]
- 42. Cole D. K., Rizkallah P. J., Boulter J. M., Sami M., Vuidepot A. L., Glick M., Gao F., Bell J. I., Jakobsen B. K., and Gao G. F. (2007) Computational design and crystal structure of an enhanced affinity mutant human CD8 αα coreceptor. Proteins Struct. Funct. Genet. 67, 65–74 [DOI] [PubMed] [Google Scholar]
- 43. Greenfield N. J. (2004) Analysis of circular dichroism data. Methods Enzymol. 383, 282–317 [DOI] [PubMed] [Google Scholar]
- 44. Venyaminov S. Yu., Baikalov I. A., Shen Z. M., Wu C. S., and Yang J. T. (1993) Circular dichroic analysis of denatured proteins: inclusion of denatured proteins in the reference set. Anal. Biochem. 214, 17–24 [DOI] [PubMed] [Google Scholar]
- 45. Cole D. K., Dunn S. M., Sami M., Boulter J. M., Jakobsen B. K., and Sewell A. K. (2008) T cell receptor engagement of peptide-major histocompatibility complex class I does not modify CD8 binding. Mol. Immunol. 45, 2700–2709 [DOI] [PubMed] [Google Scholar]
- 46. Bulek A. M., Madura F., Fuller A., Holland C. J., Schauenburg A. J., Sewell A. K., Rizkallah P. J., and Cole D. K. (2012) TCR/pMHC optimized protein crystallization screen. J. Immunol. Methods 382, 203–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Winter G., Lobley C. M., and Prince S. M. (2013) Decision making in xia2. Acta Crystallogr. D Biol. Crystallogr. 69, 1260–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Collaborative Computational Project, Number 4 (1994) 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 49. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]