

Abstract
The metalloproteinase anthrax lethal factor (LF) is secreted by Bacillus anthracis to promote disease virulence through disruption of host signaling pathways. LF is a highly specific protease, exclusively cleaving mitogen-activated protein kinase kinases (MKKs) and rodent NLRP1B (NACHT leucine-rich repeat and pyrin domain-containing protein 1B). How LF achieves such restricted substrate specificity is not understood. Previous studies have suggested the existence of an exosite interaction between LF and MKKs that promotes cleavage efficiency and specificity. Through a combination of in silico prediction and site-directed mutagenesis, we have mapped an exosite to a non-catalytic region of LF. Mutations within this site selectively impair proteolysis of full-length MKKs yet have no impact on cleavage of short peptide substrates. Although this region appears important for cleaving all LF protein substrates, we found that mutation of specific residues within the exosite differentially affects MKK and NLRP1B cleavage in vitro and in cultured cells. One residue in particular, Trp-271, is essential for cleavage of MKK3, MKK4, and MKK6 but dispensable for targeting of MEK1, MEK2, and NLRP1B. Analysis of chimeric substrates suggests that this residue interacts with the MKK catalytic domain. We found that LF-W271A blocked ERK phosphorylation and growth in a melanoma cell line, suggesting that it may provide a highly selective inhibitor of MEK1/2 for use as a cancer therapeutic. These findings provide insight into how a bacterial toxin functions to specifically impair host signaling pathways and suggest a general strategy for mapping protease exosite interactions.
Keywords: anthrax toxin, bacterial pathogenesis, host-pathogen interaction, macrophage, melanoma, protein kinase, proteinase, NACHT leucine-rich repeat and pyrin domain containing protein (NLRP), mitogen-activated protein kinase kinase
Introduction
Infectious bacteria often produce protein toxins that promote virulence, by facilitating evasion of the host immune system, by promoting bacterial dissemination, or by otherwise contributing to disease pathology (1). So-called “A-B toxins” enter host cells by means of a targeting subunit, allowing an enzymatic subunit to act on host substrates. Several well characterized A-B toxins are proteases that disable host cell function by cleaving intracellular proteins. One might expect that a nonspecific digestive protease, delivered intracellularly, would compromise cell function. In practice, however, protease toxins typically act with exquisite specificity. For example, botulinum and tetanus neurotoxins exclusively cleave substrates involved in synaptic vesicle fusion, promoting paralysis that is a characteristic disease symptom (2, 3). Understanding how these proteases selectively cleave an extremely limited substrate repertoire therefore provides basic insight into how bacterial toxins function in disease.
Anthrax lethal factor (LF)2 is a metalloproteinase produced by the Gram-positive bacterium Bacillus anthracis (4). LF is the enzymatic component of anthrax lethal toxin (LeTx), a classic A-B toxin that includes the protein PA (protective antigen). PA facilitates LF delivery into the host cell cytosol through an endocytic route that has been characterized in detail (5). As with other protease toxins, LF cleaves a small number of proteins in the host cell with a high degree of specificity. The major targets of LF in humans are the mitogen-activated protein kinase kinases (MKKs). LF cleaves six MKKs (MEK1/2 and MKK3/4/6/7) within sites that directly interact with their cognate MAPK substrates (4, 6). LF cleavage thereby functionally inactivates the MKKs, terminating signaling through the three major MAPK pathways: the ERK1/2, p38, and JNK pathways (4, 7).
MAPK pathways mediate responses to diverse extracellular signals in many cell types (8). MAPK function in cells of the innate immune system is particularly important in the context of anthrax infection (4). For example, neutrophil chemotaxis and antimicrobial activity are dependent on p38 and ERK signaling (9, 10). In addition, ERK-dependent transcriptional induction and p38-dependent translational up-regulation are essential for inflammatory cytokine production by activated macrophages (11, 12). Furthermore, activated macrophages are protected from apoptosis by p38-dependent induction of pro-survival genes (13). Multiple MAPK pathways are also important for proliferation of cells of the adaptive immune system such as T and B lymphocytes (4). Consequently, it is thought that an important function of LF is to help to establish infection by impairing host defense. LeTx also causes calcium dysregulation and impairs contractile function in cardiovascular muscle cells, possibly by inhibiting JNK, a phenomenon that likely contributes to fatality in late stage anthrax (14–16).
In addition to its MKK substrates, LF also cleaves NLRP1B, a rodent-specific paralog of the NOD-like inflammasome protein NLRP1 (17, 18). Cleavage of NLRP1B triggers inflammasome activation, caspase-1 activation, and rapid pyroptotic death of macrophages and dendritic cells from inbred strains of mice and rats harboring specific Nlrp1b alleles (17–19). This phenomenon appears to have evolved in rodents as a defense mechanism against anthrax infection to limit B. anthracis dissemination but is not known to be relevant to human disease.
LF cleaves both MKKs and NLRP1B site-specifically, exclusively at one or two sites close to their N termini (4, 18). The primary sequences surrounding cleavage sites in MKKs and NLRP1B conform to a consensus sequence characterized by basic residues at multiple positions upstream of the cleavage site, a basic residue, or Pro immediately upstream of the cleavage site (the P1 position) and hydrophobic residues positioned two residues upstream (P2) and immediately downstream (P1′) of the cleavage site. These features are also found in LF substrates cleaved in random peptide libraries, likely explaining why LF chooses particular sites in its substrate proteins (20). However, because many proteins having these sequences features are present in mammalian proteomes, the established LF cleavage site motif cannot explain why LF only cleaves MKKs and NLRP1B. Multiple lines of evidence suggest the presence of an exosite, or non-catalytic site, interaction between LF and its substrates that is required for efficient proteolysis. For example, MEK2 constructs lacking the N-terminal LF cleavage sequence interacted with LF in a yeast two-hybrid assay (21). Furthermore, deletion analysis of MEK1 identified a region within its catalytic domain distal from the cleavage site that was required for efficient proteolysis by LF, and deletion of the MKK6 catalytic domain greatly impairs cleavage by LF (22, 23). In addition, we have recently identified small molecules that competitively inhibit LF cleavage of MKKs while having no effect on cleavage of short peptides (23). These observations suggest that a physical interaction between LF and the catalytic domain of MKKs is required for efficient cleavage.
While there is evidence for an interaction between LF and the MKK catalytic domain, the location of the putative exosite on LF has not been mapped. LF is a 90-kDa protein comprising four domains (24). The C-terminal catalytic domain (domain IV) contains the signature HEXXH motif including two zinc-binding His residues and the catalytic base Glu residue found in many groups of zinc-binding metalloproteases. The LF catalytic domain is most closely related to MtfA, a putative intracellular aminopeptidase widespread in Gram-negative bacteria, and a group of secreted metalloproteinases found in some species of pathogenic Gram-positive bacteria, represented by Zmp1 from Clostridium difficile (25, 26). These proteases share with LF a conserved zinc-binding Glu residue and an additional active site Tyr residue important for catalysis. LF has been co-crystallized with both peptide substrates and substrate-based inhibitors, which has provided insight into its cleavage site specificity (20, 24, 27). Outside of the catalytic domain, the N-terminal domain I binds PA and is necessary and sufficient for PA-mediated uptake of LF into cells (28). Domain II shares its fold with a family of bacterial ADP ribosyltransferases but lacks necessary catalytic residues and is thought to be inactive. Domain III is a small helical bundle inserted into domain II. Domains II and III together form the central portion of the protein and have been hypothesized to regulate substrate access to the active site in domain IV (24, 29). This region has also been proposed to mediate substrate recruitment, yet details of these interactions remain obscure (30). Here, we identify and characterize in detail an LF exosite in domain II that is required for the efficient cleavage of MKK substrates.
Results
In Silico Prediction of Protein Interaction Sites on LF
To identify potential LF residues that interact with MKK substrates, we used CPORT, a server that combines results from multiple structure-based protein interface prediction algorithms (31–34). We submitted Protein Data Bank files corresponding to all reported X-ray crystal structures of full-length LF for CPORT analysis and prioritized investigation of residues that were identified in three or more of the eight analyses (Fig. 1 and supplemental Table S1). Interestingly, CPORT analysis identified residues at two established interaction sites: the PA binding site (including His-229, Leu-235, and Tyr-236) and the peptide substrate binding cleft (including Gly-657, Leu-658, Tyr-659, His-690, Tyr-728, and Glu-735). In addition to these known sites, CPORT also identified a continuous patch of residues spanning the non-catalytic domains I and II of LF as a predicted protein interaction interface. Based on these predictions we hypothesized that domain I of LF could be important for protease activity against protein substrates. We tested this hypothesis by examining the proteolytic activity of an N-terminally truncated form of LF, LF-CT, comprising domains II–IV (Fig. 2A). We found that LF-CT cleaved a short peptide substrate with similar kinetics to full-length LF (Fig. 2B). Strikingly, however, we found that concentrations of LF and LF-CT displaying equivalent activity on the peptide substrate had substantially different activity on a full-length MKK6 substrate, with LF-CT being ∼10-fold less active than full-length LF (Fig. 2C). These results suggest that in addition to its well characterized role in mediating LF uptake into cells, domain I is also required for optimal cleavage of MKK substrates. We therefore proceeded to test the role of individual residues in the putative interaction surface spanning domains I and II.
FIGURE 1.
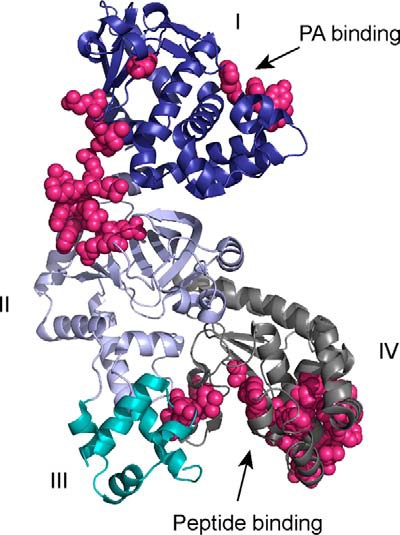
LF structure showing protein interface residues predicted by CPORT. The four domains of LF as described in the text are labeled and color-coded blue (domain I), lavender (domain II), cyan (domain III), and gray (domain IV). Residues predicted by CPORT to be involved in protein interaction interfaces are shown in magenta. The figure was made from Protein Data Bank entry 1J7N using PyMOL (54).
FIGURE 2.
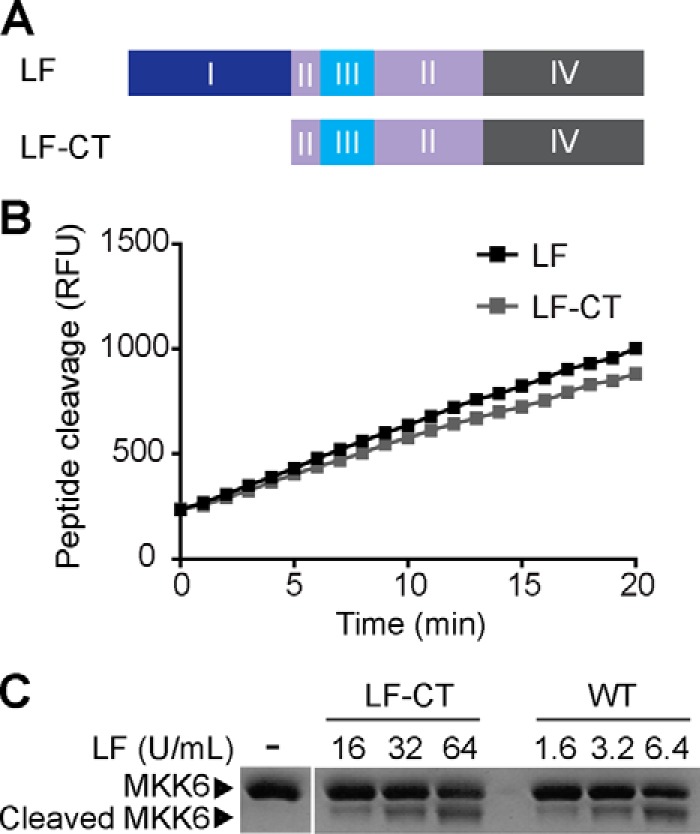
Removal of domain I specifically reduces cleavage of a full-length MKK substrate by LF. A, scheme showing LF-CT protein, an LF construct lacking the N-terminal domain I. B, peptide cleavage assay comparing full-length LF with LF-CT. C, MKK6 cleavage assay comparing full-length LF with LF-CT. Recombinant MKK6 protein substrate was subjected to proteolysis by the indicated units of LF and LF-CT as defined by activity against the peptide substrate. MKK6 cleavage was visualized by SDS-PAGE followed by Coomassie staining.
Specific Residues in LF Domain II Are Required for Efficient Substrate Proteolysis
To evaluate the role of predicted residues in LF-substrate interactions, we generated a series of LF point mutants and examined their cleavage of purified MKKs in vitro. To ensure that these mutations did not cause global destabilization or unfolding of LF, each mutant was first evaluated in peptide cleavage assays. A protease inactive LF mutant (E687A) had no detectable activity using the peptide substrate (not shown), indicating that peptide cleavage activity in our preparations was not due to contaminating proteases. Although we found no consistent decrease in peptide cleavage rate for any of the mutants tested, we did observe some variability in activity between LF preparations, including WT LF. To control for such variability, we defined units of LF based on activity on the peptide substrate and included equivalent units in MKK cleavage reactions. Of 11 mutants tested, we found that 6 of them had significantly reduced activity with MKK6 as the substrate (Fig. 3, A and B). The most dramatic effect was observed with LF-W271A, which had no detectable activity against MKK6 despite being fully active against the peptide substrate. Two additional mutants, LF-M264A and LF-Y268A, had substantially (7- and 12-fold, respectively) reduced protease activity against MKK6 compared with WT LF, whereas the other mutants (L259A, L265A, and R491E) had more modestly reduced activity (2–3-fold). Notably, the three residues that appeared to be most important for MKK6 cleavage (Met-264, Tyr-268, and Trp-271) form a continuous patch located in the first α-helix of domain II (Fig. 3C), suggesting that this region is a hotspot for the interaction between LF and MKK6.
FIGURE 3.

MKK cleavage assays with LF mutants. A, recombinant MKK6 or MEK1 substrate was incubated with increasing concentrations of WT LF or the indicated LF point mutants for 30 min at room temperature. Reactions were fractionated by SDS-PAGE followed by Coomassie staining. A representative gel from at least three separate experiments is shown. B, apparent catalytic efficiencies were calculated from quantified data corresponding to the experiment shown in A and its replicates. The error bars show S.E. (n ≥ 3). Statistical significance was determined by testing for differences with a one-way analysis of variance, followed by Fisher's least significant difference post hoc test to compare with WT. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. C, residues mutated that had a statistically significant impact on LF cleavage of MKK6 are shown in space fill representation: Trp-271 (red), Met-264 and Tyr-268 (bright pink), and Leu-259 and Arg-491 (light pink).
To examine whether these mutations affect MKK cleavage in general, we next examined activity of the LF mutants using purified MEK1 as the substrate (Fig. 3, A and B). We found that most mutations that reduced cleavage of MKK6 also impacted cleavage of MEK1 but to a lesser extent. For example, LF-M264A and LF-Y268A cleaved MEK1 2.5- and 6-fold less efficiently than WT LF, respectively. Other mutations (L259A and R491E) affected MEK1 cleavage ∼2-fold, and the L265A mutation did not significantly impact on MEK1 cleavage. Most notably, the W271A mutation that completely abolishes cleavage of MKK6 had no effect on the ability of LF to cleave MEK1. These observations suggest that this region of LF is generally important for maximal cleavage of both MKK6 and MEK1 but that Trp-271 is uniquely essential for its interaction with MKK6.
The Kinase Domain of MEK1 Confers Cleavage by LF-W271A
MKKs consist of a short unstructured N-terminal region harboring the LF cleavage site and a C-terminal kinase catalytic domain. To determine which region of an MKK permits cleavage by LF-W271A, we generated chimeric MEK1/MKK6 proteins in which we exchanged their N-terminal sequences (Fig. 4A). For these experiments, WT or chimeric MKKs were overexpressed in HEK293T cells, and cleavage was assessed in cell lysates. In this system, WT LF efficiently cleaved both MEK1 and MKK6, whereas as expected LF-W271A only cleaved MEK1 (Fig. 4, B and C). We found that the chimera in which the N terminus of MKK6 was fused to the kinase domain of MEK1 was cleavable by LF-W271A, albeit with slightly lower efficiency than WT LF. The analogous chimera harboring the N terminus of MEK1 and the MKK6 catalytic domain expressed poorly and was not cleaved by either WT LF or LF-W271A, suggesting that this fusion protein was non-functional (data not shown). These results suggest that the failure of LF-W271A to cleave MKK6 is due to loss of a critical interaction with its kinase domain and not with the N-terminal cleavage sequence of MKK6.
FIGURE 4.
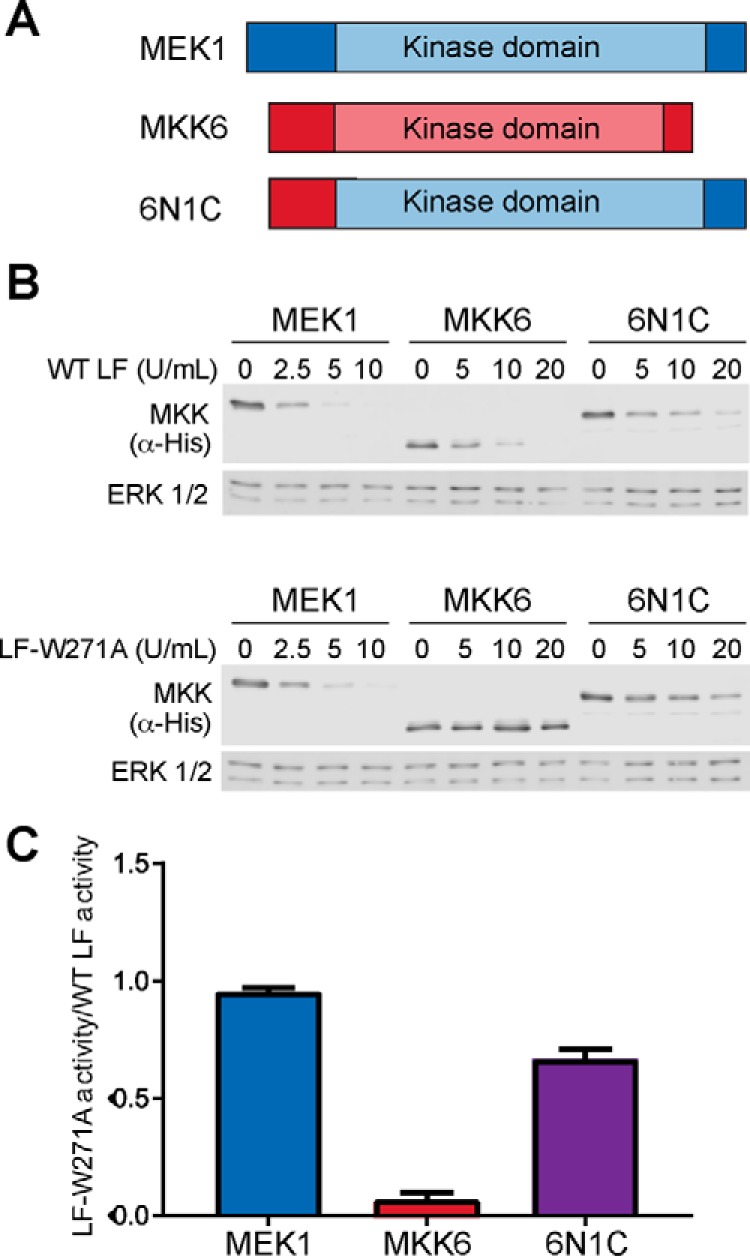
Cleavage of a chimeric MEK1/MKK6 substrate by LF. A, schematic showing MEK1 (blue), MKK6 (red), and the chimeric construct 6N1C. B, WT LF and LF-W271A cleavage of MEK1, MKK6, and 6N1C in mammalian cell lysates. A representative immunoblot from one of three replicates is shown. C, the relative activity of LF-W271A as a fraction of WT LF cleavage of the same substrate was calculated from the experiment shown in B and replicates. The error bars show S.E. (n = 3).
LF Functions as a Monomer
Although LF is typically described as being monomeric, full-length LF has been crystallized in both monomeric and dimeric forms. Because several of the residues important for MKK cleavage, in particular Met-264 and Tyr-268, map to the dimer interface observed in some crystal structures, we considered whether LF cleavage of MKKs might require dimerization. We first examined whether we could detect formation of dimeric LF in solution by size exclusion chromatography with multiangle light scattering (SEC-MALS) analysis. We found that LF is almost entirely (98%) monomeric in solution, with an observed mass of 96.8 ± 0.4 kDa, with the remainder appearing as early eluting higher order multimers, suggestive of aggregation (Fig. 5A). Importantly, we did not detect any dimeric LF by this analysis. This observation indicates that at the low nanomolar LF concentrations used in our cleavage reactions, at most only a very small fraction could be dimeric. To further test the possibility that LF acts as a dimer, we examined the concentration dependence of LF activity. Because LF is predominantly monomeric in our cleavage reactions, if LF functions as a dimer, we would expect its activity to increase exponentially with enzyme concentration because of higher levels of the dimeric form. Instead, we found that the amount of product formed varied linearly with concentration at low LF concentrations and began to plateau at higher concentrations because of substrate depletion (Fig. 5B). Taken together, these results indicate that LF is unlikely to function as an obligate dimer and suggest that residues in domain II required for efficient proteolysis are more likely to interact directly with MKKs.
FIGURE 5.
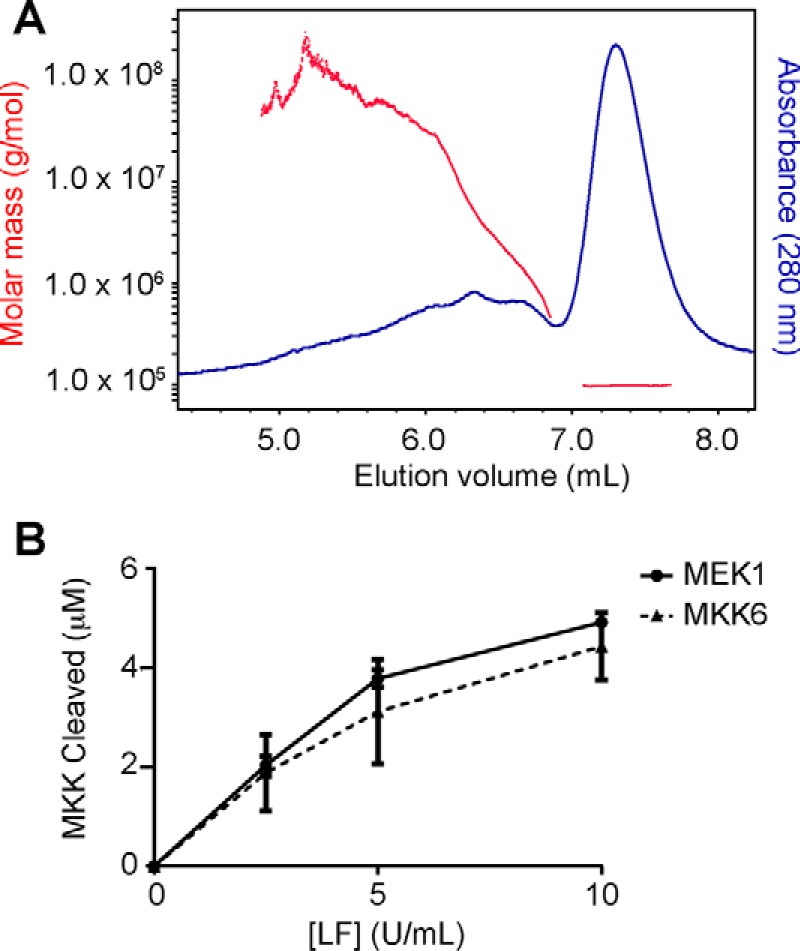
LF shows no evidence of dimerization or dimerization-dependent proteolytic activity. A, WT LF was subjected to SEC, and the molecular weight of protein in the eluate was calculated by MALS analysis. B, titration of LF using MEK1 and MKK6 substrates indicates that proteolytic activity does not increase exponentially with LF concentration.
LF Mutations Impact Cleavage of Endogenous MKKs in Cultured Cells
To determine whether residues that affect MKK cleavage in vitro are also important for LF function in cells, we initially examined cleavage of endogenous MKKs in J774A.1 cells, a mouse macrophage cell line that is susceptible to LeTx-induced pyroptosis. Because endogenous, untagged MEK1 undergoes a very small change in molecular weight upon LF cleavage, for these experiments we instead examined cleavage of its closest homolog, MEK2, using an antibody directed against its N terminus that does not react with its LF cleavage product. Likewise, we were unable to detect endogenous MKK6 in these cells, so we instead examined cleavage of MKK3. In addition to MEK2 and MKK3, we also assayed cleavage of MKK4. J774A.1 cells were treated with a fixed concentration of PA and varying concentrations of WT LF or those LF mutants that had the largest effect on MKK cleavage in vitro, and cleavage of MEK2, MKK3 and MKK4 was analyzed by immunoblotting.
As observed for cleavage of MKK6 in vitro, LF-W271A was completely impaired in its ability to cleave MKK3 in cells (Fig. 6A). Likewise, LF-Y268A displayed a marked reduction in MKK3 cleavage, whereas LF-M264A and LF-L265A mutants had a comparatively smaller effect on MKK3 cleavage in cells than on MKK6 cleavage in vitro. These differences may reflect variability between MKK3 and MKK6 as substrates for LF but could also reflect effects of these mutations on cellular stability or uptake of LF. Interestingly, as for MKK3, the proteolytic activity of LF-W271A on MKK4 in cells is also completely abolished (Fig. 6B), suggesting that Trp-271 is involved in an exosite interaction required for targeting of MKK3, MKK4, and MKK6. We also observed a notable reduction in cleavage of MKK4 by LF-Y268A, whereas LF-M264A, LF-L265A, and LF-E269A mutants were not significantly less effective than WT LF. Lastly, in keeping with in vitro assays performed with MEK1, we found that mutation of Met-264 and Tyr-268 had the largest effect on MEK2 cleavage in cells, although LF-Y268A appeared comparatively more active on MEK2. Lastly, as for cleavage of MEK1 in vitro, LF-W271A retains complete WT level proteolytic activity against MEK2 in cells (Fig. 6C). Taken together, these results indicate that residues in domain II have differential roles in cleavage of MKKs in cultured cells as well as in vitro.
FIGURE 6.

LF exosite mutants differentially impact cleavage of MKKs in in cultured macrophage cells. J774A.1 mouse macrophage cells were treated with 0.5 μg/ml PA and increasing concentrations of WT or mutant LF for 90 min. The cells were harvested, and cleavage of MKK3 (A), MKK4 (B), and MEK2 (C) was assessed by immunoblotting of cell lysates. Representative immunoblots are shown at left, and quantified results are reported as percentages of uncleaved MKK remaining. The error bars show S.E. (n = 3). Statistical significance was calculated as in Fig. 3 comparing the value corresponding to the highest concentration of a given mutant to that of WT LF. ns, not significant; *, p < 0.05; ***, p < 0.001; ****, p < 0.0001.
Domain II Mutations Differentially Affect LF Cleavage of NLRP1B and Macrophage Pyroptosis
Susceptibility of rodent macrophages to LF-induced cytolysis is thought to be independent of MKK cleavage, instead being triggered by LF cleavage of the inflammasome protein NLRP1B. To examine whether our MKK-targeting exosite is more generally important for LF function, we initially examined the effect of domain II mutations on NLRP1B cleavage in vitro. For these experiments we overexpressed N-terminally GFP-tagged NLRP1B in HEK293T cells and performed cleavage assays in cell lysates. Interestingly, the effect of domain II mutations on GFP-NLRP1B cleavage in vitro followed a pattern distinct from any of the MKKs. Most mutations that decreased MKK cleavage had little impact on NLRP1B cleavage, with the exception of LF-M264A, which is completely inactive with NLRP1B as a substrate (Fig. 7A). We next examined the activity of the same panel of mutants co-administered with PA in J774A.1 cell killing assays. In keeping with its effect on NLRP1B cleavage in vitro, LF-M264A was completely unable to kill J774A.1 cells (Fig. 7B). In contrast, MKK cleavage defective mutants such as LF-Y268A and LF-W271A were fully active in inducing cell death. These results suggest that NLRP1B interacts differently with the LF exosite in comparison with MKKs, and furthermore supports a role for NLRP1B rather than MKK cleavage in mediating LeTx-induced macrophage death.
FIGURE 7.
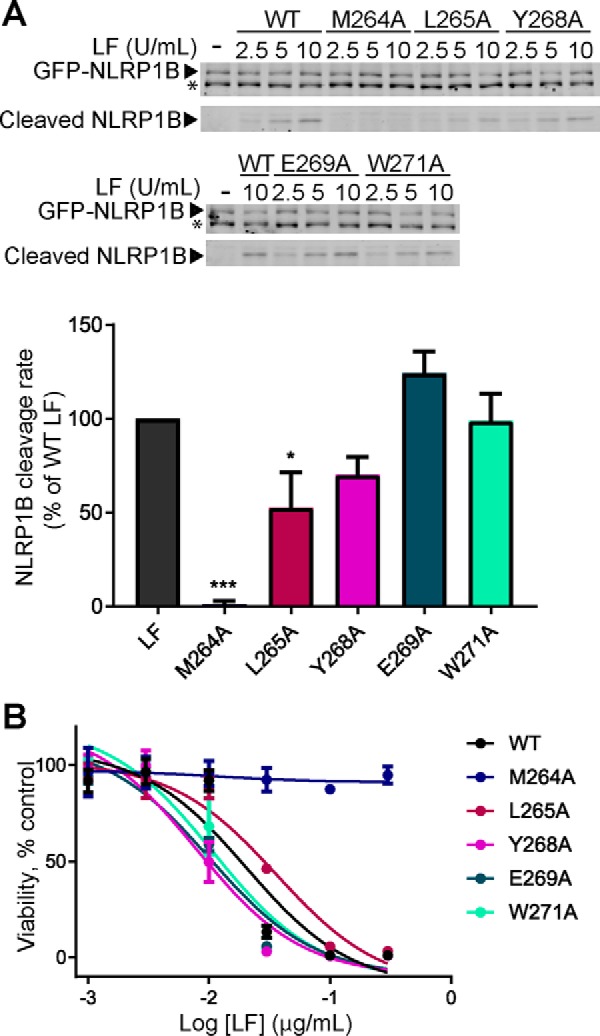
Activity of LF exosite mutants in cleaving NLRP1B and causing macrophage death. A, HEK293T cell lysates containing exogenously expressed GFP-NLRP1B were incubated with WT or mutant LF for 30 min at room temperature. A representative immunoblot (top panel) shows the levels of uncleaved GFP-NLRP1B and GFP-tagged cleavage product. The asterisk indicates a cross-reacting background band. B, activity of each mutant is shown as a percentage of WT LF activity. The error bars show S.E. (n = 3). B, J774A.1 cells were subjected to the indicated concentrations WT and mutant LF in combination with 0.5 μg/ml PA for 4 h, and cell viability was measured by resazurin assay. Results show cell viability as a percentage of untreated cells. The error bars show S.E. (n = 3). Statistical significance was calculated as in Fig. 3. *, p < 0.05; ***, p < 0.001.
LF-W271A Cleavage of MEK1/2 Suppresses Growth of BRAF-driven Melanoma Cells
Melanomas are characterized by hyperactivation of the ERK MAPK signaling pathway, most commonly because of recurrent activating mutations in the MEK1/2 kinase BRAF (35). By virtue of its ability to inactivate MEK1 and MEK2, LeTx can suppress growth of human melanoma cell lines in culture and in mouse xenografts (36–38). These observations have led to the proposal that LeTx could be used therapeutically to treat melanoma. Because domain II mutations affect the activity of LF in a substrate-selective manner, we examined the activity of our panel of mutants in growth assays using the LeTx-sensitive human melanoma cell line A375, which harbors the BRAF-V600E activating mutation (36). We quantified A375 cell growth following treatment with a fixed concentration of PA and increasing concentrations of WT or mutant LF for 3 days. Consistent with their reduced ability to cleave MEK1 in vitro, we observed impaired growth inhibition by LF-M264A, LF-L265A, and LF-Y268A (Fig. 8, A and B). In separate assays, we found that protease inactive mutant LF-E687A did not detectably inhibit growth at these concentrations (data not shown). The reduced activity in blocking cell growth observed for the LF exosite mutants correlated with maintenance of phosphorylated ERK1/2, a measure of MEK1/2 activity, in these cells (Fig. 7, C and D). As anticipated from its capacity to cleave MEK1 in vitro, LF-W271A was fully active in suppressing ERK phosphorylation and growth in A375 cells (Fig. 8, A–D). LF-W271A, however, failed to cleave MKK3 appreciably and did not block phosphorylation of its substrate p38 MAPK. These observations indicate that unlike WT LF, which acts as a pan-inhibitor of MAPK signaling through the ERK, p38 MAPK, and JNK pathways, LF-W271A specifically impairs ERK signaling.
FIGURE 8.

Effect of exosite mutations on LF activity in melanoma cells. A, A375 cells were treated with 0.5 μg/ml PA and the indicated concentrations of WT or mutant LF for 72 h, and cell viability was measured by resazurin assay. Measurements from three independent experiments are plotted as percentages of cell number in the untreated control group. The error bars indicate S.E. B, IC50 values calculated from A375 cell growth assays. The means and S.E. are shown (n = 3). C–E. A375 cells were treated with 0.5 μg/ml PA and 0.2 ng/ml LF or LF mutant for 16 h, and cells were harvested. Cell lysates were prepared and immunoblotted to detect phospho-ERK1/2 (C), phospho-p38 (D), and MKK3 (E). Representative immunoblots are shown at left, and mean quantified signals relative to the untreated control are shown at right. The error bars indicate standard errors (n = 3). Statistical significance was calculated as for Fig. 3. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Discussion
Protease exosites have previously been identified and characterized through a variety of methods including protease-substrate co-crystallography, investigation of evolutionarily conserved surfaces, mutational analyses, and the use of small molecule or peptide probes (22, 39–41). Mapping the LF exosite has proven challenging. Aside from very closely related homologs from pathogenic strains of Bacillus cereus, LF lacks orthologs from other bacterial species (42). Thus, it is difficult to infer functionally important regions of LF by sequence conservation. Furthermore, although LF has been successfully crystallized in complex with short peptides, no structure of LF bound to a full-length MKK substrate has been reported (20, 24, 27, 43, 44). The availability of numerous X-ray crystal structures of full-length LF allowed us to predict in silico residues likely to be located at protein interfaces. These predictions allowed us to take a directed approach to identifying residues involved in the exosite interaction.
Through biochemical characterization of truncation and point mutants of LF, we have identified an LF exosite that is required for efficient proteolysis of MKK substrates both in vitro and in cultured cells. The patch of residues that had the largest effect on substrate cleavage (Met-264, Tyr-268, and Trp-271) reside in domain II close to its junction with domain I. Domain I has been thought to function exclusively in toxin uptake (45, 46), yet we found that its removal substantially reduces cleavage of MKK6. Although our domain I deletion construct begins with Met-264, a likely possibility is that the exosite region becomes at least partly unstructured when domain I is removed. Thus, whereas domain I of LF is primarily involved in toxin uptake, our studies suggest its presence is also important for promoting LF protease activity, possibly by maintaining the local structure of the LF exosite.
We observed that mutation of specific residues within the exosite had varying effects on cleavage of the different substrates of LF. For example, mutation of Met-264 and Tyr-268 reduced cleavage of all MKKs examined, but mutation of Trp-271 abolished cleavage of MKK3, MKK4, and MKK6 only. Furthermore, LF-M264A appeared completely unable to cleave NLRP1B, while maintaining some activity against MKKs. These results suggest that although all LF substrates likely interact with this exosite, the specific modes of interaction vary from substrate to substrate. Mogridge and co-workers (47) previously reported that an LF double mutant (LF-K518E/E682G), although fully active in cleaving MEK1, MKK3, and MKK4, was impaired in cleavage of MEK2 and MKK6. This mutant was also unable to mediate inflammasome activation and subsequent pyroptosis in a macrophage cell line, suggesting that it was also incapable of cleaving NLRP1B. Lys-518 is located in domain II ∼9 Å from Trp-271, suggesting that it could form part of the exosite defined in the present work. However, Glu-682 is located within the S1′ pocket of the LF catalytic domain, and thus the double mutant could selectively impair substrate cleavage through differential interactions with residues proximal to the cleavage site.
The differential activity of LF-W271A on MEK1 versus MKK6 permitted us to assess which portion of the MKK protein is targeted by the domain II exosite. From LF cleavage assays with a MEK1/MKK6 chimera, it appears that the kinase domain of MEK1 is primarily responsible for its susceptibility to cleavage by this mutant. This observation suggests that Trp-271, and possibly other nearby residues in domain II, directly interact with residues in the kinase domain of MKKs. This finding is consistent with the reported LF-interacting region mapping to residues within helix αG of MKKs (22). Interestingly, this region is also important for interaction of MKKs with the Yersinia pestis virulence factor YopJ, which inhibits MKKs by acetylation of residues required for kinase activation (48). Furthermore, RAF kinases interact directly with this area of MEK1, and LF can compete with RAF for interaction with MEK1 (22, 49). These observations suggest that B. anthracis and other pathogenic microbes have evolved strategies to exploit regions of key signaling molecules that are essential for their function in the host. Doing so may provide an evolutionary competitive advantage for the microbes, because it would be difficult for the host to adapt through mutation of the target.
The ability of LF-W271 to cleave only MEK1 and MEK2 among MKKs is of particular interest given the involvement of MEK1/2 signaling in BRAF driven cancers (36, 37). LeTx has been proposed as a potential therapeutic for melanoma by virtue of its ability to inactivate MEK1/2. However, potential therapeutic utility of LF would likely be hampered by off target inhibition of other MKKs. Indeed, blockade of p38 and JNK signaling through inhibition of the MKK kinase ZAK has been implicated in off target toxicity of the clinical BRAF inhibitor vemurafenib (50). LF-W271A has the potential to offer the advantages of a biological therapeutic in its exquisite specificity for its targets, while being uniquely capable of targeting intracellular proteins. Thus, in addition to providing basic insight into how a pathogenic microbe targets host signaling pathways for its own benefit, these studies may provide a useful therapeutic agent for treating human cancer. Future studies will be aimed at further examining the therapeutic value of the LF-W271A mutant.
Experimental Procedures
Plasmids
Bacterial expression vectors for MBP-MEK1 (a gift from Jesse Rinehart) and GST-MKK6 were previously described (23, 51). The expression vector for His6-tagged full-length LF (pET-15b LF WT) was a gift from John Collier (Addgene plasmid no. 11076). All LF point mutants were generated on this vector background using the QuikChange protocol (Stratagene). The His6-tagged LF-CT expression construct was generated by subcloning the coding sequence for residues 264–776 of LF plus a stop codon into the BamHI site of pET-28a. The mammalian expression vector for MEK1 was generated by subcloning the entire expression cassette encoding His6-tagged human MEK1 from pRSET-MEK1 (a gift from Natalie Ahn) into pcDNA3 (52). The corresponding mammalian expression vector for MKK6 was generated by excising the MEK1 sequence with BamHI and XbaI and inserting the entire human MKK6 coding sequence generated by PCR using pCMV-Sp6-MKK6 (GenBankTM accession no. BC012009.1, obtained from ATCC) as a template. The human MKK6-MEK1 chimera (6N1C), encoding residues 1–46 of MKK6 fused to residues 83–414 of MEK1 in the same vector background, was generated by overlap extension PCR. A full-length cDNA corresponding to the Balb/c allele of NLRP1B was assembled from the coding regions of exon I and exon II (PCR amplified from RAW 264.7 cell genomic DNA), a synthetic fragment encompassing nucleotides 1820–2510 (GenScript), and 5′ sequence amplified from a mouse spleen cDNA library (Biochain). The expression vector for the NLRP1B N-terminal GFP fusion was generated by subcloning the entire coding sequence into pEGFP-C3 (Clontech).
Antibodies
Penta-His (34660) antibody was obtained from Qiagen. Rabbit polyclonal antibodies against MEK2 (N-20), MKK3 (C-19), and MEK4 (C-20) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal vinculin (V-9131) antibody was obtained from Sigma-Aldrich. Phospho-ERK1/2 (D13.14.4E, 4370), total ERK1/2 (9102), phospho-p38 MAPK (9211), and total p38 MAPK (9212) antibodies were obtained from Cell Signaling Technology (Danvers, MA). Polyclonal GFP (600-101-215) antibody was obtained from Rockland Immunochemicals, Inc. (Limerick, PA). Secondary antibodies (IRDye® 800CW-conjugated goat anti-mouse IgG, IRDye® 680RD-conjugated donkey anti-goat IgG, and IRDye® 680LT-conjugated goat anti-rabbit IgG) were obtained from LI-COR Biosciences (Lincoln, NE).
CPORT Protein Interface Residue Prediction
All full-length structures of LF available in the RCSB Protein Data Bank (1J7N, 1JKY, 1PWV, 1ZXV, 1PWP, 1PWW, 1PWQ, and 1PWU) were submitted to the CPORT server for protein interface prediction analysis using the “very sensitive” setting. Residues that scored as predicted to be at protein-protein interfaces by one or more of the algorithms PIER, SPPIDER, and PINUP are reported in supplemental Table S1 (31–34).
Protein Expression and Purification
WT and mutant His6-tagged LF, MBP-His6-MEK1, and GST-MKK6 were expressed in BL21(DE3) E. coli by overnight induction with 0.2 mm isopropyl β-d-thiogalactopyranoside at 16 °C. The cells were harvested by centrifugation, washed with PBS, subjected to flash freeze and thaw, and suspended in 20 ml of resuspension buffer (20 mm Tris-HCl, pH 7.5, 140 mm NaCl, 13 mm MgCl2, 1 mm PMSF, 10 μg/ml leupeptin, 10 μg/μl pepstatin A, 0.2 mg/ml lysozyme, 30 units/μl DNase I, 0.01% Igepal CA630, and 1 mm DTT for MKK6) per liter of original culture volume. Lysates were then subjected to five 10-s cycles of sonication at level 3.5 using a VirTis VirSonic 600 cell disrupter. Lysates were centrifuged for 30 min at 22,000 × g and 4 °C. For purification of His6-tagged protein, supernatants were applied to 1.25 ml of TALON resin (Clontech) by tumbling for 1.5 h at 4 °C. The bead suspension was loaded onto a column, washed with 10 ml of PBS + 0.5% Igepal CA630 followed by 10 ml of wash buffer (20 mm Tris-HCl, pH 7.5, 500 mm NaCl, and 10 mm imidazole, pH 7.4), and then protein was eluted in 1-ml fractions with elution buffer (20 mm Tris-HCl, pH 7.5, 100 mm NaCl, 250 mm imidazole, pH 7.4, and 10 μg/μl leupeptin). Fractions containing protein were dialyzed overnight into dialysis buffer (10 mm HEPES, pH 7.4, 100 mm NaCl, 10% glycerol), snap frozen in aliquots, and stored at −80 °C. GST-MKK6 was applied to 670 μl of glutathione-Sepharose 4B resin (GE Healthcare Life Sciences) for 1.5 h at 4 °C, and beads were washed twice in batch with PBS + 0.5% Igepal CA630 and once with GSH wash buffer (50 mm Tris-HCl, pH 8.0, 50 mm NaCl, 1 mm DTT, 0.01% Igepal CA630, and 10% glycerol). MKK6 was eluted from the resin by cleavage with 4 units of thrombin (GE Healthcare) overnight at 4 °C in 1.7 ml of thrombin buffer (GSH wash buffer containing 2.5 mm CaCl2 and 5 mm MgCl2). Thrombin was then removed from solution by mixing 30 min at 4 °C with 50 μl of p-aminobenzamidine agarose (Sigma-Aldrich) followed by centrifugation (3300 × g for 4 min at 4 °C). Both MEK1 and MKK6 were concentrated with Amicon Ultra 4 Ultracel 10K centrifugal filters to a volume of 0.5 ml and flash frozen in aliquots for storage at −80 °C.
Peptide Cleavage Assays
Cleavage of LF (WT or mutant) was assayed using the optimized fluorescent LF substrate Mca-AKVYPYPME-Dnp (20). LF (15 nm WT or mutant) was incubated with 8 μm peptide in reaction buffer (40 mm HEPES, pH 7.4, 0.1 mg/ml BSA, 100 mm NaCl, and 1 mm DTT). Mca fluorescence was monitored continuously at 1-min intervals over 20 min at an excitation wavelength of 325 nm and an emission wavelength of 393 nm at ambient temperature. The assays were performed in triplicate. The cleavage rates were calculated from the initial rate period of the reaction. One unit of LF was defined as the quantity having peptide cleavage activity equivalent to 1 pmol of WT LF measured concurrently.
Recombinant Protein Cleavage Assays
MEK1 or MKK6 (8 μm) was incubated with varying concentrations (2.5, 5, and 10 units/ml) of WT or mutant LF at room temperature for 30 min in 50-μl reactions consisting of 33.15 μl of reaction buffer (40 mm HEPES, pH 7.4, 0.1 mg/ml BSA, 100 mm NaCl, and 1 mm DTT), 3.4 μl of thrombin buffer, and 13.45 μl of dialysis buffer. The reactions were stopped by adding 17 μl of 4× SDS-PAGE loading buffer and boiling for 5 min. The samples were then separated by SDS-PAGE (12% acrylamide) followed by Coomassie staining. The gels were scanned at 700 nm with a LiCor Odyssey CLx imager, and Coomassie densitometry-based quantification of cleavage was carried out within the LiCor Image Studio 4.0 software suite.
To calculate an apparent catalytic efficiency for WT LF and LF mutants, we fit the results to the integrated, closed form of the Michaelis-Menten equation (53),
| (Eq. 1) |
where W is the omega function. Using this equation, we fit each experimental replicate for kcat/Km, which we label kcat/Km,app to acknowledge the uncertainty in the proportion of correctly folded and accessible substrate in our assays. The fits were performed using the FindFit function in Wolfram Mathematica 9.0.1.0 via the NMinimize method with a working precision >6 digits and up to 10,000 iterations. These fits were averaged and are reported as means ± S.E. for each LF mutant-substrate pair. Each of these values represents at least three independent experiments and fits.
For LF cleavage assays performed in cell lysates, His6-MEK1, His6-MKK6, or GFP-NLRP1B was expressed in HEK293T cells. Cells at ∼80% confluence were transfected in serum- and antibiotic-free DMEM with 8 μg of polyethylenimine and 2 μg of plasmid DNA per 10-cm dish. After incubation for 5 h at 37 °C, the medium was aspirated and replaced with fresh, complete DMEM (including 10% FBS and penicillin-streptomycin). The cells were washed with cold PBS and harvested 48 h post-transfection in 1.5 ml of lysis buffer (20 mm HEPES, pH 7.4, 100 mm NaCl, 1 mm DTT, 0.5% Igepal CA630, 5% glycerol, 1 mm PMSF, 10 μg/ml aprotinin, 2 μg/ml pepstatin A, and 10 μg/ml leupeptin). Cell lysates were flash frozen and stored at −80 °C. Cleavage reactions included either 5.2 μl of lysis buffer containing equivalent amounts of MKK1, MKK6, or 6N1C protein or 12 μl of GFP-NLRP1B lysate and were brought to a final volume of 25 μl with simple LF reaction buffer (40 mm HEPES, pH 7.4, and 100 mm NaCl) containing a final concentration of 2.5, 5, 10, and 20 units/ml of LF. After 30 min of incubation at room temperature, 8 μl of 4× SDS-PAGE loading buffer was added to quench the reaction. The samples were boiled for 5 min and subjected to SDS-PAGE followed by immunoblotting with either α-His6 antibody (1:10,000) or α-GFP antibody (1:10,000) and the corresponding secondary antibody (1:10,000). Western blots were imaged with a LiCor Odyssey CLx imager, and densitometry-based quantification of cleavage was carried out with LiCor Image Studio 4.0.
SEC-MALS Analysis of LF
For SEC-MALS analysis, LF prepared and centrifugally concentrated to 1 ml as described above was subjected to gel filtration chromatography on an Åkta FPLC with a Superdex 200 column using 10 mm HEPES, pH 7.4, 100 mm NaCl as the running buffer. Fractions corresponding to the major protein peak were pooled and flash frozen for storage (−80 °C). Just prior to analysis, fractions were thawed, concentrated to 100 μl with a final concentration of ∼2 mg/ml, and centrifuged (16,000 × g, 4 °C, 5 min) to remove particulates. Samples (LF or BSA standard) were run on an Agilent 1260 HPLC system with a WTC-015S5 SEC column and inline DAWN HELEOS II MALS detector (Wyatt) in 10 mm HEPES, pH 7.4, 100 mm NaCl, and 0.02% NaN3 running buffer. The data were processed using Astra6 software (Wyatt).
Analysis of MKK Cleavage in Cultured Cells
The LF-susceptible mouse macrophage cell line J774A.1 was cultured in DMEM with 10% FBS and penicillin-streptomycin. The cells (0.5 × 106 per well) were plated in 6-well dishes and allowed to recover overnight. The cells were exchanged into fresh medium and incubated with 0.25 μg/ml PA and varying concentrations of LF for 90 min at 37 °C. The cells were then washed with ice-cold PBS and harvested in 200 μl of boiling 1× SDS-PAGE loading buffer. The samples were then fractionated by SDS-PAGE (12% acrylamide), transferred to PVDF membrane, and probed with α-MEK2 (1:1000), α-MKK3 (1:1000), or α-MEK4 (1:1000) antibody and α-vinculin (1:10,000) antibody and the corresponding secondary antibodies (1:10,000).
A375 melanoma cells were cultured in Opti-MEM with 10% FBS. The cells (0.3 × 106/well) were plated in 6-well dishes and allowed to recover overnight. After exchanging to fresh medium containing 0.5 μg/ml PA and 0.2 ng/ml LF (WT or mutant), the cells were incubated for 16 h at 37 °C. Samples to be analyzed for p38 phosphorylation were incubated 10 μg/ml anisomycin 30 min prior to lysis. The cells were washed once with ice-cold PBS and extracted into 100 μl of A375 cell lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na2VO4, 1 μg/ml leupeptin, 1 mm PMSF). After 5 min of incubation on ice, lysates were scraped into 1.5-ml microcentrifuge tubes and centrifuged at 16,000 × g for 10 min. The supernatant was added to 40 μl of 4× SDS-PAGE loading buffer and heated to 95 °C for 5 min. Samples were subjected to SDS-PAGE, transferred to PVDF, and probed with α-phospho-ERK (1:2000), α-ERK (1:1000), α-phospho-p38 (1:1000), α-p38 (1:1000), α-MKK3 (1:1000), or α-vinculin (1:10,000) and corresponding secondary antibodies (1:10,000). The results are reported as the means of three experiments ± S.E.
LF Cytotoxicity Assays in J774A.1 Cells
J774A.1 cells (7.5 × 104 cells/well) were plated in 96-well dishes at and allowed to recover overnight. The cells were exchanged into 100 μl of fresh complete medium containing 0.5 μg/ml PA and the indicated concentrations of LF (6 wells/condition). The cells were incubated at 37 °C for 4 h, exchanged into 100 μl of medium containing 44 μm resazurin, and incubated for 2 h in the dark at 37 °C. Fluorescence (excitation, 560 nm; emission, 590 nm) was measured in a plate reader.
A375 Cell Growth Assays
A375 cells were seeded in 96-well plates at 1200 cells per well and permitted to recover overnight. After exchanging to fresh medium containing 0.5 μg/ml PA and the indicated concentrations of LF (6 wells/concentration), the cells were incubated for 72 h at 37 °C. Cell viability was assessed using the resazurin assay described above for J774A.1 cells. At the time that LF treatment was initiated, a starting time signal was obtained on a separate plate containing untreated cells. Starting point readings were subtracted from the 72-h readings to measure overall growth inhibition. IC50 values were calculated by fitting to a sigmoidal curve with Prism 6 (GraphPad).
Author Contributions
A. B. G. performed all experiments unless noted otherwise, analyzed data, and co-wrote the paper. E. C. performed and analyzed data for experiments in A375 melanoma cells. C. J. M. provided mathematical modeling of enzyme kinetic data. H. J. L. generated plasmid constructs used for analysis of MKK chimeras. B. E. T. conceived the project, analyzed data, and co-wrote the paper.
Supplementary Material
Acknowledgments
We thank James Murphy (Yale University) for assistance in conducting SEC-MALS experiments. We thank Jesse Rinehart (Yale University) for providing the bacterial expression construct for MEK1 and Natalie Ahn (University of Colorado, Boulder) for the MEK1 clone. We thank Emily Jarrell, Grace Jeschke, and Sirlester Parker (Turk laboratory) for generation of plasmid constructs. We thank Eugene Douglass for advice on quantitative analysis of LF cleavage assay data.
This work was supported by National Institutes of Health Grants R01 GM105947, R01 GM104047, and T32 GM007324. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Table S1.
- LF
- lethal factor
- PA
- protective antigen
- MKK
- MAP kinase kinase
- LeTx
- lethal toxin
- Mca
- 7-methoxycoumarin-4-acetic acid
- SEC-MALS
- size exclusion chromatography with multiangle light scattering.
References
- 1. Alouf J. E. (2000) Bacterial protein toxins: an overview. Methods Mol. Biol. 145, 1–26 [DOI] [PubMed] [Google Scholar]
- 2. Blasi J., Chapman E. R., Link E., Binz T., Yamasaki S., De Camilli P., Südhof T. C., Niemann H., and Jahn R. (1993) Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature 365, 160–163 [DOI] [PubMed] [Google Scholar]
- 3. Schiavo G., Benfenati F., Poulain B., Rossetto O., Polverino deLaureto P., DasGupta B. R., and Montecucco C. (1992) Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 359, 832–835 [DOI] [PubMed] [Google Scholar]
- 4. Turk B. E. (2007) Manipulation of host signalling pathways by anthrax toxins. Biochem. J. 402, 405–417 [DOI] [PubMed] [Google Scholar]
- 5. Brossier F., Lévy M., Landier A., Lafaye P., and Mock M. (2004) Functional analysis of Bacillus anthracis protective antigen by using neutralizing monoclonal antibodies. Infect. Immun. 72, 6313–6317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bardwell A. J., Abdollahi M., and Bardwell L. (2004) Anthrax lethal factor-cleavage products of MAPK (mitogen-activated protein kinase) kinases exhibit reduced binding to their cognate MAPKs. Biochem. J. 378, 569–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tonello F., and Montecucco C. (2009) The anthrax lethal factor and its MAPK kinase-specific metalloprotease activity. Mol. Aspects Med. 30, 431–438 [DOI] [PubMed] [Google Scholar]
- 8. Keshet Y., and Seger R. (2010) The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods. Mol. Biol. 661, 3–38 [DOI] [PubMed] [Google Scholar]
- 9. Cara D. C., Kaur J., Forster M., McCafferty D. M., and Kubes P. (2001) Role of p38 mitogen-activated protein kinase in chemokine-induced emigration and chemotaxis in vivo. J. Immunol. 167, 6552–6558 [DOI] [PubMed] [Google Scholar]
- 10. Brown G. E., Stewart M. Q., Bissonnette S. A., Elia A. E., Wilker E., and Yaffe M. B. (2004) Distinct ligand-dependent roles for p38 MAPK in priming and activation of the neutrophil NADPH oxidase. J. Biol. Chem. 279, 27059–27068 [DOI] [PubMed] [Google Scholar]
- 11. Guha M., and Mackman N. (2001) LPS induction of gene expression in human monocytes. Cell. Signal. 13, 85–94 [DOI] [PubMed] [Google Scholar]
- 12. Tiedje C., Holtmann H., and Gaestel M. (2014) The role of mammalian MAPK signaling in regulation of cytokine mRNA stability and translation. J. Interferon Cytokine Res. 34, 220–232 [DOI] [PubMed] [Google Scholar]
- 13. Park J. M., Greten F. R., Li Z. W., and Karin M. (2002) Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science 297, 2048–2051 [DOI] [PubMed] [Google Scholar]
- 14. Liu S., Zhang Y., Moayeri M., Liu J., Crown D., Fattah R. J., Wein A. N., Yu Z. X., Finkel T., and Leppla S. H. (2013) Key tissue targets responsible for anthrax-toxin-induced lethality. Nature 501, 63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kandadi M. R., Hua Y., Ma H., Li Q., Kuo S. R., Frankel A. E., and Ren J. (2010) Anthrax lethal toxin suppresses murine cardiomyocyte contractile function and intracellular Ca2+ handling via a NADPH oxidase-dependent mechanism. PLoS One 5, e13335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Golden H. B., Watson L. E., Nizamutdinov D., Feng H., Gerilechaogetu F., Lal H., Verma S. K., Mukhopadhyay S., Foster D. M., Dillmann W. H., and Dostal D. E. (2013) Anthrax lethal toxin induces acute diastolic dysfunction in rats through disruption of the phospholamban signaling network. Int. J. Cardiol. 168, 3884–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hellmich K. A., Levinsohn J. L., Fattah R., Newman Z. L., Maier N., Sastalla I., Liu S., Leppla S. H., and Moayeri M. (2012) Anthrax lethal factor cleaves mouse Nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PLoS One 7, e49741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chavarría-Smith J., and Vance R. E. (2013) Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog. 9, e1003452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Levinsohn J. L., Newman Z. L., Hellmich K. A., Fattah R., Getz M. A., Liu S., Sastalla I., Leppla S. H., and Moayeri M. (2012) Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 8, e1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Turk B. E., Wong T. Y., Schwarzenbacher R., Jarrell E. T., Leppla S. H., Collier R. J., Liddington R. C., and Cantley L. C. (2004) The structural basis for substrate and inhibitor selectivity of the anthrax lethal factor. Nat. Struct. Mol. Biol. 11, 60–66 [DOI] [PubMed] [Google Scholar]
- 21. Vitale G., Pellizzari R., Recchi C., Napolitani G., Mock M., and Montecucco C. (1998) Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem. Biophys. Res. Commun. 248, 706–711 [DOI] [PubMed] [Google Scholar]
- 22. Chopra A. P., Boone S. A., Liang X., and Duesbery N. S. (2003) Anthrax lethal factor proteolysis and inactivation of MAPK kinase. J. Biol. Chem. 278, 9402–9406 [DOI] [PubMed] [Google Scholar]
- 23. Bannwarth L., Goldberg A. B., Chen C., and Turk B. E. (2012) Identification of exosite-targeting inhibitors of anthrax lethal factor by high throughput screening. Chem. Biol. 19, 875–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pannifer A. D., Wong T. Y., Schwarzenbacher R., Renatus M., Petosa C., Bienkowska J., Lacy D. B., Collier R. J., Park S., Leppla S. H., Hanna P., and Liddington R. C. (2001) Crystal structure of the anthrax lethal factor. Nature 414, 229–233 [DOI] [PubMed] [Google Scholar]
- 25. Xu Q., Göhler A. K., Kosfeld A., Carlton D., Chiu H. J., Klock H. E., Knuth M. W., Miller M. D., Elsliger M. A., Deacon A. M., Godzik A., Lesley S. A., Jahreis K., and Wilson I. A. (2012) The structure of Mlc titration factor A (MtfA/YeeI) reveals a prototypical zinc metallopeptidase related to anthrax lethal factor. J. Bacteriol. 194, 2987–2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cafardi V., Biagini M., Martinelli M., Leuzzi R., Rubino J. T., Cantini F., Norais N., Scarselli M., Serruto D., and Unnikrishnan M. (2013) Identification of a novel zinc metalloprotease through a global analysis of Clostridium difficile extracellular proteins. PLoS One 8, e81306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shoop W. L., Xiong Y., Wiltsie J., Woods A., Guo J., Pivnichny J. V., Felcetto T., Michael B. F., Bansal A., Cummings R. T., Cunningham B. R., Friedlander A. M., Douglas C. M., Patel S. B., Wisniewski D., et al. (2005) Anthrax lethal factor inhibition. Proc. Natl. Acad. Sci. U.S.A. 102, 7958–7963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rabideau A. E., and Pentelute B. L. (2016) Delivery of non-native cargo into mammalian cells using anthrax lethal toxin. ACS Chem. Biol. 11, 1490–1501 [DOI] [PubMed] [Google Scholar]
- 29. Maize K. M., Kurbanov E. K., De La Mora-Rey T., Geders T. W., Hwang D.-J., Walters M. A., Johnson R. L., Amin E. A., and Finzel B. C. (2014) Anthrax toxin lethal factor domain 3 is highly mobile and responsive to ligand binding. Acta Crystallogr. D 70, 2813–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liang X., Young J. J., Boone S. A., Waugh D. S., and Duesbery N. S. (2004) Involvement of domain II in toxicity of anthrax lethal factor. J. Biol. Chem. 279, 52473–52478 [DOI] [PubMed] [Google Scholar]
- 31. de Vries S. J., and Bonvin A. M. (2011) CPORT: a consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PLoS One 6, e17695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khashan R., Zheng W., and Tropsha A. (2012) Scoring protein interaction decoys using exposed residues (SPIDER): a novel multibody interaction scoring function based on frequent geometric patterns of interfacial residues. Proteins 80, 2207–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kufareva I., Budagyan L., Raush E., Totrov M., and Abagyan R. (2007) PIER: protein interface recognition for structural proteomics. Proteins 67, 400–417 [DOI] [PubMed] [Google Scholar]
- 34. Liang S., Zhang C., Liu S., and Zhou Y. (2006) Protein binding site prediction using an empirical scoring function. Nucleic Acids Res. 34, 3698–3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsao H., Chin L., Garraway L. A., and Fisher D. E. (2012) Melanoma: from mutations to medicine. Genes Dev. 26, 1131–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abi-Habib R. J., Urieto J. O., Liu S., Leppla S. H., Duesbery N. S., and Frankel A. E. (2005) BRAF status and mitogen-activated protein/extracellular signal-regulated kinase kinase 1/2 activity indicate sensitivity of melanoma cells to anthrax lethal toxin. Mol. Cancer Ther. 4, 1303–1310 [DOI] [PubMed] [Google Scholar]
- 37. Lee C.-S., Dykema K. J., Hawkins D. M., Cherba D. M., Webb C. P., Furge K. A., and Duesbery N. S. (2011) MEK2 is sufficient but not necessary for proliferation and anchorage-independent growth of SK-MEL-28 melanoma cells. PLoS One 6, e17165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Koo H. M., VanBrocklin M., McWilliams M. J., Leppla S. H., Duesbery N. S., and Vande Woude G. F. (2002) Apoptosis and melanogenesis in human melanoma cells induced by anthrax lethal factor inactivation of mitogen-activated protein kinase kinase. Proc. Natl. Acad. Sci. U.S.A. 99, 3052–3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hardy J. A., Lam J., Nguyen J. T., O'Brien T., and Wells J. A. (2004) Discovery of an allosteric site in the caspases. Proc. Natl. Acad. Sci. U.S.A. 101, 12461–12466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jabaiah A. M., Getz J. A., Witkowski W. A., Hardy J. A., and Daugherty P. S. (2012) Identification of protease exosite-interacting peptides that enhance substrate cleavage kinetics. Biol. Chem. 393, 933–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dumas J. J., Kumar R., Seehra J., Somers W. S., and Mosyak L. (2003) Crystal structure of the GpIbα-thrombin complex essential for platelet aggregation. Science 301, 222–226 [DOI] [PubMed] [Google Scholar]
- 42. Hoffmaster A. R., Hill K. K., Gee J. E., Marston C. K., De B. K., Popovic T., Sue D., Wilkins P. P., Avashia S. B., Drumgoole R., Helma C. H., Ticknor L. O., Okinaka R. T., and Jackson P. J. (2006) Characterization of Bacillus cereus isolates associated with fatal pneumonias: strains are closely related to Bacillus anthracis and harbor B. anthracis virulence genes. J. Clin. Microbiol. 44, 3352–3360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Forino M., Johnson S., Wong T. Y., Rozanov D. V., Savinov A. Y., Li W., Fattorusso R., Becattini B., Orry A. J., Jung D., Abagyan R. A., Smith J. W., Alibek K., Liddington R. C., Strongin A. Y., et al. (2005) Efficient synthetic inhibitors of anthrax lethal factor. Proc. Natl. Acad. Sci. U.S.A. 102, 9499–9504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Panchal R. G., Hermone A. R., Nguyen T. L., Wong T. Y., Schwarzenbacher R., Schmidt J., Lane D., McGrath C., Turk B. E., Burnett J., Aman M. J., Little S., Sausville E. A., Zaharevitz D. W., Cantley L. C., et al. (2004) Identification of small molecule inhibitors of anthrax lethal factor. Nat. Struct. Mol. Biol. 11, 67–72 [DOI] [PubMed] [Google Scholar]
- 45. Arora N., Klimpel K. R., Singh Y., and Leppla S. H. (1992) Fusions of anthrax toxin lethal factor to the ADP-ribosylation domain of Pseudomonas exotoxin A are potent cytotoxins which are translocated to the cytosol of mammalian cells. J. Biol. Chem. 267, 15542–15548 [PubMed] [Google Scholar]
- 46. Arora N., and Leppla S. H. (1993) Residues 1–254 of anthrax toxin lethal factor are sufficient to cause cellular uptake of fused polypeptides. J. Biol. Chem. 268, 3334–3341 [PubMed] [Google Scholar]
- 47. Ngai S., Batty S., Liao K.-C., and Mogridge J. (2010) An anthrax lethal factor mutant that is defective at causing pyroptosis retains proapoptotic activity. FEBS J. 277, 119–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hao Y.-H., Wang Y., Burdette D., Mukherjee S., Keitany G., Goldsmith E., and Orth K. (2008) Structural requirements for Yersinia YopJ inhibition of MAP kinase pathways. PLoS One 3, e1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Haling J. R., Sudhamsu J., Yen I., Sideris S., Sandoval W., Phung W., Bravo B. J., Giannetti A. M., Peck A., Masselot A., Morales T., Smith D., Brandhuber B. J., Hymowitz S. G., and Malek S. (2014) Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell 26, 402–413 [DOI] [PubMed] [Google Scholar]
- 50. Vin H., Ojeda S. S., Ching G., Leung M. L., Chitsazzadeh V., Dwyer D. W., Adelmann C. H., Restrepo M., Richards K. N., Stewart L. R., Du L., Ferguson S. B., Chakravarti D., Ehrenreiter K., Baccarini M., et al. (2013) BRAF inhibitors suppress apoptosis through off-target inhibition of JNK signaling. eLife 2, e00969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Park H. S., Hohn M. J., Umehara T., Guo L. T., Osborne E. M., Benner J., Noren C. J., Rinehart J., and Söll D. (2011) Expanding the genetic code of Escherichia coli with phosphoserine. Science 333, 1151–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mansour S. J., Resing K. A., Candi J. M., Hermann A. S., Gloor J. W., Herskind K. R., Wartmann M., Davis R. J., and Ahn N. G. (1994) Mitogen-activated protein (MAP) kinase phosphorylation of MAP kinase kinase: determination of phosphorylation sites by mass spectrometry and site-directed mutagenesis. J. Biochem. 116, 304–314 [DOI] [PubMed] [Google Scholar]
- 53. Schnell S., and Mendoza C. (1997) Closed form solution for time-dependent enzyme kinetics. J. Theor. Biol. 187, 207–212 [Google Scholar]
- 54. DeLano W. L. (2015) The PyMOL Molecular Graphics System, version 1.5.0.8, Schroedinger, LLC, New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.