

Abstract
The emergence of avian H7N9 influenza A virus in humans with associated high mortality has highlighted the threat of a potential pandemic. Fatal H7N9 infections are characterized by hyperinflammation and increased cellular infiltrates in the lung. Currently there are limited therapies to address the pathologies associated with H7N9 infection and the virulence factors that contribute to these pathologies. We have found that PB1-F2 derived from H7N9 activates the NLRP3 inflammasome and induces lung inflammation and cellular recruitment that is NLRP3-dependent. We have also shown that H7N9 and A/Puerto Rico/H1N1 (PR8)PB1-F2 peptide treatment induces significant mitochondrial reactive oxygen production, which contributes to NLRP3 activation. Importantly, treatment of cells or mice with the specific NLRP3 inhibitor MCC950 significantly reduces IL-1β maturation, lung cellular recruitment, and cytokine production. Together, these results suggest that PB1-F2 from H7N9 avian influenza A virus may be a major contributory factor to disease pathophysiology and excessive inflammation characteristic of clinical infections and that targeting the NLRP3 inflammasome may be an effective means to reduce the inflammatory burden associated with H7N9 infections.
Keywords: inflammasome, inflammation, influenza, IL-1, NLRP3
Introduction
Since 2013, novel avian influenza A viruses (IAVs)5 of the H7N9 subtype have caused sporadic infections in humans, with over 800 laboratory cases now confirmed in China and a mortality rate of ∼40% (1, 2). The potential of H7N9 IAV to cause a pandemic poses a constant and realistic threat to global health (3), particularly as many isolates have been identified to carry genetic modifications that confer antiviral resistance. Understanding the mechanisms involved in the induction of immunopathology and fatal disease is imperative to allow the development of improved and better targeted treatments to reduce the mortality associated with pathogenic IAV.
The NLRP3 inflammasome is an oligomeric innate immune intracellular signaling complex that senses many pathogen-, host-, and environment-derived factors (4). Inflammasome-induced cytokine release requires two signals: up-regulation of components of the NLRP3 inflammasome and synthesis of pro-IL-1β through activation of the prototypic inflammatory transcription factor NF-κB and inflammasome formation that results in IL-1β maturation and secretion. Following activation, NLRP3 binds to the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC). ASC further recruits the enzyme caspase-1 to form the inflammasome complex, initiating autocatalytic cleavage of caspase-1. The NLRP3 inflammasome is now recognized as a major route by which the innate immune system recognizes and responds during IAV infection (4). To date, IAV single-stranded RNA and proton flux via the IAV-encoded matrix-2 (M2) ion channel have been shown to activate the NLRP3 inflammasome (5, 6), which is important in the development of adaptive immune responses to IAV (7). Studies utilizing mice lacking components of the NLRP3 inflammasome have demonstrated its importance in eliciting rapid protective responses following infection with the mouse-adapted A/Puerto Rico/8/34 strain (PR8, H1N1) (6, 8). In these studies, inflammasome-deficient mice were highly susceptible to both low and high inoculum doses of PR8, and the NLRP3 inflammasome was shown to be required for production of pyrogenic IL-1β and IL-18 in the airways, cellular infiltration, and lung immunopathology. These findings suggested that NLRP3-induced responses were crucial to host protection via recruitment of effector cells and the induction of tissue repair. Additionally, a lack of the IL-1 receptor has been shown to increase mortality but reduce lung immunopathology following infection with PR8 H1N1 or avian H5N1, suggesting that IL-1 receptor signaling may increase damage to the lung (9, 10). Critically, we recently identified that the NLRP3 inflammasome plays both a protective and detrimental role during pathogenic IAV challenge through temporal inhibition of NLRP3 function (11). This therefore suggests that excessive or prolonged NLRP3 inflammasome activation contributes to the hyperinflammatory phenotype associated with pathogenic infections.
Fatal IAV infections correlate with production of excessive levels of proinflammatory cytokines and chemokines (termed a “cytokine storm”) and excessive cellular inflammation. PB1-F2 protein is encoded by a +1 alternate reading frame of the PB1 gene, and protein expression in cells has been linked to cell death, viral replication, and virulence, although these properties can differ between virus strains (12). Crucially, we have shown that PB1-F2 proteins derived from 20th century pandemic and highly pathogenic IAV strains, but not mildly virulent IAVs, activate the NLRP3 inflammasome, inducing IL-1β production and pulmonary inflammation (13). PB1-F2 triggers excessive cellular recruitment and hyperinflammatory responses in the lungs of infected mice (14, 15). Specific PB1-F2 amino acids that enhance inflammation and are linked to virulence have been identified at the C terminus and map to residues Leu-62, Arg-75, Arg-79, and Leu-82, termed the four inflammatory residues (16).
In this study, we have identified that PB1-F2 from H7N9 is a potent activator of the NLRP3 inflammasome. We also identified that inflammasome activation was related to PB1-F2-induced mitochondrial reactive oxygen species (mtROS) and that targeting mtROS with the specific inhibitor MitoTempo inhibited PB1-F2 peptide-induced inflammasome activation and IL-1β secretion and maturation. PB1-F2 peptide-mediated inflammasome activation and inflammation were phagocytosis-dependent and inhibited by the small-molecule NLRP3 inhibitor MCC950 in vitro and in vivo. This study identifies H7N9 PB1-F2 as an activator of the NLRP3 inflammasome and suggests that PB1-F2 may contribute to the hyperinflammation associated with current human H7N9 infections. Our identification of MCC950 as a potent inhibitor of H7N9 PB1-F2-induced lung inflammation and cellular influx suggests that novel therapies that specifically target the NLRP3 inflammasome may therefore provide an effective treatment to reduce the mortality associated with pathogenic H7N9 IAV and offset the ineffectiveness of current antiviral treatments in the later stages of infection.
Results
H7N9-derived PB1-F2 Peptide Induces IL-1β in Murine Macrophages and Human PBMCs
We have previously demonstrated that aggregated PB1-F2 derived from the PR8 strain of IAV (H1N1) activates the NLRP3 inflammasome, contributing to disease pathophysiology (13). We examined the inflammatory potential of aggregated PB1-F2 derived from the recently identified novel avian-origin H7N9 IAV. Using aggregated peptides derived from the C termini of PB1-F2 from both PR8 and H7N9 (the latter being A/Shanghai/02/2013), we show that >85% of both peptides form similar concentrations of protein aggregates between 0.5 and 2.5 μm (supplemental Fig. 1, A–C), a particle size found previously to be optimal for NLRP3 inflammasome activation by other aggregates (17).
To examine whether H7N9 PB1-F2 aggregated peptide could induce IL-1β secretion, we challenged LPS-primed WT immortalized bone marrow-derived macrophage (iBMDMs) from mice with H7N9 PB1-F2 peptide and analyzed IL-1β secretion by ELISA. We have previously demonstrated the requirement for LPS priming prior to PB1-F2 peptide challenge to induce up-regulation of inflammasome components, including pro-IL-1β and pro-caspase-1 (13). As seen in Fig. 1A, H7N9 PB1-F2 peptide induced IL-1β secretion by macrophages in a dose-dependent manner, similar to that observed with PR8 PB1-F2. Furthermore, H7N9 PB1-F2 peptide corresponding to the sequence changes associated with crossover from avian to human (G70E and S77L) also induced IL-1β secretion, whereas substitution of the four critical amino acids associated with PB1-F2 inflammation with the equivalent non-inflammatory Wuhan sequence (L62P, R75H, R79Q, and L82S) was ineffective (supplemental Fig. S2A). H7N9 PB1-F2 peptide inflammasome activation was further validated by dose-dependent secretion of IL-1β in primary murine BMDMs challenged with H7N9 PB1-F2 peptide (supplemental Fig. 2B). We determined the ability of H7N9 PB1-F2 to induce caspase-1 cleavage into the p20 subunit and the subsequent processing of IL-1β to its mature form, p17, by immunoblotting. Wild-type primary BMDMs primed with LPS were challenged with either PR8- or H7N9-derived PB1-F2. H7N9 PB1-F2 induced proteolytic cleavage of caspase-1 in a dose-dependent manner (Fig. 1B, top panel) with concomitant processing of IL-1β p17 (Fig. 1B, bottom panel). Caspase-1 p20 and IL-1β p17 were also detected following stimulation with PR8 PB1-F2 and silica (Fig. 1B, top and bottom panels, respectively).
FIGURE 1.

H7N9 PB1-F2 induces caspase-1-dependent release of IL-1β in macrophages and PBMCs. iBMDMs or primary BMDMs were primed with LPS (100 ng/ml) for 3 h and then stimulated with a dose range of either PR8 or H7N9 PB1-F2 peptide (5–200 μg/ml) or silica (150 μg/ml) or left unstimulated (NS) in triplicate for 6 h. A, cellular supernatants from iBMDMs were analyzed for secreted IL-1β by ELISA. Results are representative of three independent experiments, and data represent the mean ± S.E. B–E, protein in primary BMDM supernatants were separated by 4–12% SDS-PAGE and immunoblotted (IB) for the indicated antibodies. The result represents one of three independent experiments. iBMDMs were primed with LPS (100 ng/ml) for 3 h and then pretreated with the inhibitors latrunculin A (Lat A, C), bafilomycin (BAF, D), or YVAD (E) at the indicated doses 40 min prior to treatment with H7N9 PB1-F2 peptide (100 μg/ml) or left unstimulated for 6 h. The levels of IL-1β in cell supernatants were determined by ELISA. Results are the mean ± S.E. MW, molecular weight. F, PBMCs were primed in LPS (50 pg/ml) for 3 h prior to stimulation with an H7N9 PB1-F2 peptide (10–200 μg/ml), silica (150 μg/ml), or nigericin (1 μm) or left unstimulated for 6 h. G, primed PBMCs were pretreated with YVAD for 40 min and then stimulated with H7N9 PB1-F2 peptide (100 μg/ml) or silica (150 μg/ml) or left unstimulated for 6 h. Cultured supernatants were assayed for IL-1β by ELISA. Results are the mean ± S.E. of three independent donors. **, p < 0.01; ***, p < 0.001; one-way ANOVA.
The uptake of infection- and disease-related protein aggregates by phagocytes and subsequent lysosomal destabilization are known triggers of inflammasome activation (17). In our experiments, inhibition of phagocytosis with latrunculin A, prevention of phagolysosomal maturation with bafilomycin A, and inhibition of caspase-1 activity with Z-YVAD reduced IL-1β secretion in a concentration-dependent manner in both iBMDMs and primary BMDMs stimulated with H7N9 PB1-F2 peptide (Fig. 1, C–E, and supplemental Fig. 2, C and D), respectively.
To evaluate whether H7N9 derived PB1-F2 protein may also enhance inflammatory events in human cells, we examined cellular responses in LPS-primed human peripheral blood mononuclear cells (hPBMCs) exposed to H7N9 PB1-F2. H7N9 PB1-F2 peptide induced robust secretion of IL-1β from hPBMCs (Fig. 1F) (17), which was inhibited via treatment with the caspase-1 inhibitor Z-YVAD (Fig. 1G). Taken together, these data suggest that H7N9-derived PB1-F2 aggregate induces IL-1β secretion in murine macrophages and human PBMCs in manner that is dependent on phagocytosis and caspase-1 activity.
H7N9 PB1-F2 Induces ASC Speck Formation and IL-1β Secretion via the NLRP3 Inflammasome
We next confirmed that phagocytosis of H7N9 PB1-F2 is required for inflammasome activation or ASC speck formation by bioimaging. H7N9 PB1-F2 was labeled with the pH-sensitive dye pHrodo, which dramatically increases in fluorescence intensity in the acidic environment of the phagolysosomal compartment. NLRP3-deficient iBMDMs stably reconstituted with cerulean-tagged ASC and NLRP3-FLAG, which obviates the need for priming, were used to visualize inflammasome activation via speck formation. ASC-cerulean macrophages were challenged with labeled H7N9-pHrodo peptide, and ASC speck formation was visualized as a well characterized marker of inflammasome formation and activation of caspase-1 (18).
As observed in Fig. 2 and the live-cell imaging in supplemental Movies 1 and 2, pHrodo-labeled H7N9 PB1-F2 is phagocytosed rapidly into the lysosomal pathway, as indicated by increased PB1-F2 red fluorescence within cellular vesicles (Fig. 2, 120 min). This event is followed by ASC speck formation (Fig. 2, top panels, 200 min). Notably, treatment of ASC-cerulean cells with latrunculin A prior to challenge with pHrodo-labeled H7N9 PB1-F2 blocked cellular uptake of the peptide aggregate, prohibiting ASC speck formation (supplemental Movie 3).
FIGURE 2.
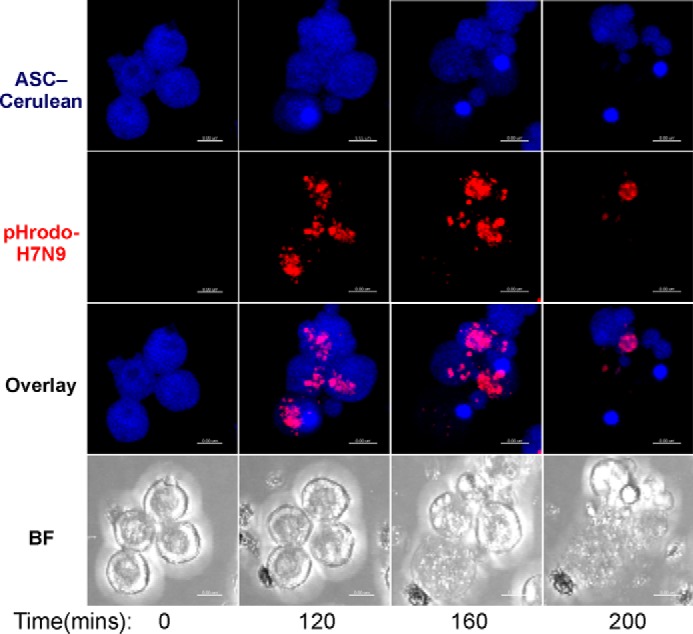
The H7N9 PB1-F2 peptide induces ASC speck formation in macrophages. NLRP3-deficient iBMDMs stably reconstituted with ASC-cerulean (blue) and NLRP3-FLAG were stimulated with pHrodo-labeled (red) H7N9 PB1-F2 peptide at 0 min. Live-cell imaging was performed for 200 min to visualize the uptake of pHrodo-labeled H7N9 PB1-F2 peptide (30 μg/ml) by the macrophages and subsequent formation of ASC specks. Representative images shown are maximum intensity projections of 3D deconvoluted z stacks using Imaris imaging software. Scale bars = 8 μm. BF, Brightfield.
To confirm that H7N9 PB1-F2 activates the NLRP3 inflammasome complex, we compared iBMDMs derived from mice deficient in the inflammasome components caspase-1/11, ASC, or NLRP3 with iBMDMs from WT mice (17). In WT iBMDMs, H7N9 PB1-F2 induced IL-1β secretion in a dose-dependent manner at levels comparable with the NLRP3 and AIM2 inflammasome activators silica and poly(dA-dT) (Fig. 3A). ASC- (Fig. 3B) and NLRP3-deficient (Fig. 3C) iBMDMs failed to secrete IL-1β in response to H7N9 PB1-F2 peptide. Notably, the dependence on caspase-1 as seen in our earlier experiments was supported by the abrogation of IL-1β secretion in caspase-1-deficient iBMDMs challenged with H7N9 PB1-F2 (Fig. 3D). Priming of the inflammasome in all iBMDM lines was confirmed by disrupting cellular membranes of LPS-stimulated cells by repeated freeze-thawing and analysis of cellular lysate IL-1β concentrations (Fig. 3, A–D, Lysate). Validating these data, and consistent with earlier reports (17), cells exposed to silica displayed ASC-, NLRP3-, and caspase-1-dependent responses (Fig. 3, B–D) whereas those transfected with poly(dA-dT) showed only a requirement for caspase-1 (Fig. 3D) and ASC (Fig. 3B). Using primary BMDMs generated from WT and NLRP3-deficient mice, we confirmed that NLRP3 is required for H7N9 PB1-F2 peptide-induced IL-1β secretion in (supplemental Fig. 3A).
FIGURE 3.

H7N9 PB1-F2 induced IL-1β maturation is NLRP3 inflammasome- and caspase-1-dependent. A–D, iBMDMs (wild-type, A; ASC−/−, B; NLRP3−/−, C; and caspase-1−/−, D) were primed with LPS (100 ng/ml) for 3 h, followed by stimulation with H7N9 PB1-F2 peptide (25–100 μg/ml), silica (150 μg/ml), or poly(dA-dT) (500 ng/ml) or left unstimulated (NS) in triplicate for a further 6 h. LPS priming of cells was confirmed by disrupting cellular membranes of primed but unstimulated cells by repeated freeze-thawing and analysis of cellular lysates. Supernatants were analyzed for IL-1β by ELISA. The results are representative of three independent experiments and the mean ± S.E. E, primary WT and Nlrp3−/− BMDMs were primed with LPS (100 ng/ml) for 3 h and then exposed to either the PR8 or H7N9 PB1-F2 peptides (both at 200 μg/ml), silica (150 μg/ml), or poly(dA-dT) (1000 ng/ml) or left unstimulated for a further 6 h. Proteins from BMDM supernatants were separated by 4–12% SDS-PAGE and immunoblotted (IB) with the indicated antibodies. The result is representative of three independent experiments.
The requirement of NLRP3 for H7N9 PB1-F2-induced IL-1β production was further verified in primary BMDMs from WT and NLRP3−/− mice by immunoblot. BMDMs were primed with LPS, challenged with either PR8- or H7N9-derived PB1-F2, and assessed for levels of caspase-1 p20 and IL-1β p17 in cell supernatants. Proteolytic cleavage of caspase-1 was detected following stimulation of WT BMDMs with PR8 PB1-F2, H7N9 PB1-F2, silica or poly(dA-dT) (Fig. 3E, top panels). In agreement with this, processing of IL-1β into mature and bioactive p17 was detected in these samples (Fig. 3E, bottom panels). Neither caspase-1 p20 nor IL-1 p17 were detected in PR8 PB1-F2-, H7N9 PB1-F2-, or silica-challenged NLRP3−/− BMDMs (Fig. 3E, top and bottom panels). Caspase-1 p20 and IL-1β p17 were detected in NLRP3−/− BMDMs stimulated with poly(dA-dT), indicating the presence of a fully functional and activated AIM2 inflammasome in these cells. This result was validated by the analysis of IL-1β levels in these samples by ELISA supplemental Fig. 3B).
PB1-F2 derived from H1N1 strains has been described previously as containing a mitochondrial targeting sequence (19, 20) that disrupts mitochondrial function commensurate with the C-terminal domain that, as we have found, activates the NLRP3 inflammasome. Given that mtROS have been found previously to activate the NLRP3 inflammasome, we examined whether H7N9 and PR8 PB1-F2 peptide could induce mtROS. ASC-cerulean iBMDMs were pretreated with MitoSOX Red, a mitochondrially localized fluorogenic dye selective for mitochondrial superoxide in live cells (21), prior to challenge with H7N9 and PR8 PB1-F2 peptides. As can be observed in supplemental Movies 4 and 5, respectively, live cell imaging of H7N9 and PR8 PB1-F2-treated iBMDMs demonstrated increased mitochondrial fluorescence intensity of MitoSOX rapidly following treatment. Bafilomycin treatment of cells demonstrated that phagolysosomal disruption is required for PB1-F2-induced mtROS production (supplemental Movie 6). To examine whether PB1-F2 peptide-induced mtROS plays a role in PB1-F2-induced inflammasome activation, we next treated both iBMDMs (Fig. 4A) and human PBMCs (Fig. 4D) with micromolar concentrations of MitoTempo and examined PB1-F2 peptide-induced IL-1β secretion. We found that MitoTempo significantly inhibited PB1-F2-induced secretion in a dose-dependent manner. Furthermore, we found that MitoTempo inhibited the proteolytic maturation of both IL-1β and caspase-1 by PB1-F2 H7N9 and PR8 peptides (Fig. 4B) in primary BMDMs, in which secretion of IL-1β was also confirmed (Fig. 4C), and in human PBMCs (Fig. 4D). Taken together, these results suggest that, commensurate with previous studies, PB1-F2 localizes to the mitochondria to induce mtROS, which leads to activation of the inflammasome and IL-1β maturation and secretion.
FIGURE 4.
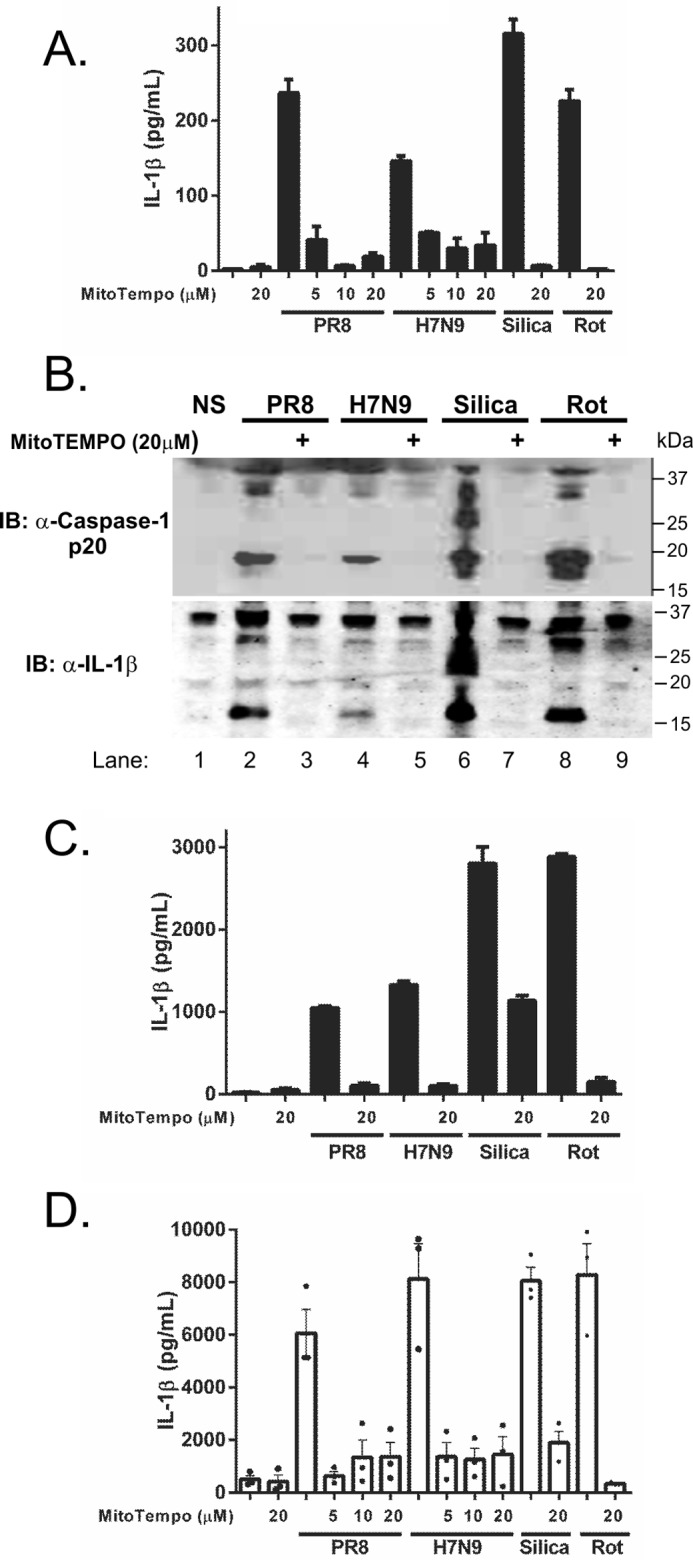
Inhibition of mtROS reduces IL-1β maturation and secretion induced by H7N9 PB1-F2 peptide. A–C, iBMDMs (A) or primary BMDMs (B and C) were primed with LPS (100 ng/ml) for 3 h and pretreated with a dose range of MitoTempo (5–20 μm) for 40 min prior to stimulation or no stimulation (NS) with PB1-F2 peptides derived from PR8, H7N9 (100 μg/ml respectively), silica (150 mg/ml), or rotenone (Rot, 50 nm). Cellular supernatants were analyzed for secreted IL-1β by ELISA (A and C). The results are representative of three independent experiments, and data represent the mean ± S.E. B, protein in primary BMDM supernatants was separated by 4–12% SDS-PAGE and immunoblotted (IB) for the indicated antibodies. The result represents one of three independent experiments. D, human PBMCs obtained from three individuals were primed in LPS (50 pg/ml) for 3 h and MitoTempo where indicated prior to stimulation with H7N9 or PR8 PB1-F2 peptide (100 μg/ml), silica, and rotenone for 6 h. Cultured supernatants were assayed for IL-1β by ELISA. Results are the mean ± S.E. of three independent donors.
In Vivo H7N9 PB1-F2-induced Inflammation Is NLRP3-dependent
We next confirmed the requirement for NLRP3 to elicit H7N9 PB1-F2-induced IL-1β secretion by examining inflammatory responses to the peptide in vivo. Neutrophils are known to be recruited in response to NLRP3-inflammasome activation in immune cells (4, 22). WT and NLRP3−/− mice were intranasally inoculated with the H7N9 PB1-F2 peptide. At 7 h following treatment, significant neutrophil influx was detected in bronchoalveolar lavage (BAL) of the airways of WT mice (Fig. 5A). Importantly, the cellular infiltrates observed in PB1-F2-treated NLRP3−/− mice were significantly reduced compared with WT mice (Fig. 5A). The difference in cellular infiltration observed between WT and NLRP3−/− mice treated with H7N9 PB1-F2 was reflected in significantly lower concentrations of IL-1β in BAL from NLRP3−/− mice compared with WT animals (Fig. 5B). Collectively, these data highlight the role of the NLRP3 inflammasome in the maturation of IL-1β in response to H7N9-derived PB1-F2 peptide both in vitro and in vivo.
FIGURE 5.

H7N9 PB1-F2 peptide induced neutrophil influx, and IL-1β secretion is NLRP3-dependent in vivo. A and B, groups of five WT and Nlrp3−/− mice were intranasally inoculated with PBS or H7N9 PB1-F2 peptide (50 μg). BAL was performed 7 h following challenge for detection of the number of infiltrating Ly6G+ neutrophils by flow cytometry (A) and the levels of IL-1β determined by ELISA (B). Data are expressed as mean ± S.E. and are representative of two independent experiments. **, p < 0.01; one-way ANOVA.
To explore whether H7N9-derived PB1-F2 protein expressed by virions during infection would induce NLRP3 inflammasome activation similar to that observed for the PR8 PB1-F2 protein, we attempted to use reverse genetics to generate IAVs expressing the PB1 gene derived from A/Anhui/1/2013 (H7N9) on the backbone of several well characterized laboratory strains. In parallel, to generate a mutant H7N9, we genetically modified the H7N9 PB1 plasmid to disrupt the PB1-F2 open reading frame and abrogate PB1-F2 expression, as demonstrated previously with PR8 (13). Using well established techniques, we attempted to reverse-engineer IAVs containing a wild-type or mutant H7N9 PB1 gene and the remaining seven viral genes derived from HKx31 (H3N2), A/Udorn/1972 (H3N2), or A/Puerto Rico/8/34 (PR8, H1N1). Despite multiple attempts, the infectious virus could not be rescued, suggesting that the H7N9 PB1 gene is incompatible for incorporation into viruses of either the H3N2 or H1N1 subtypes. The reasons for this unexpected outcome are currently being explored.
A Small-molecule Inhibitor of the NLRP3 Inflammasome Inhibits PB1-F2-induced IL-1β in Vitro
Recently, Coll et al. (23) described MCC950 as a potent (IC50, 7 nm) and specific diarylsulfonylurea-based inhibitor of NLRP3 inflammasome activity that acts by preventing ASC complex formation with potent activity in vivo. This compound represents both a potential NLPR3 therapeutic agent and also provides an important tool to dissect the temporal role of NLRP3 during infectious disease pathophysiology, which is not possible using gene-deficient mice (11).
We investigated the ability of MCC950 to inhibit NLRP3 activation in response to H7N9 PB1-F2 peptide in vitro. Macrophages were primed with LPS and then pretreated with MCC950 prior to challenge with PB1-F2 peptides from PR8 or H7N9 or with silica. Pretreatment with micromolar concentrations of MCC950 potently inhibited secretion of IL-1β by iBMDM (Fig. 6, A and B) and primary BMDMs (Fig. 6C) that were subsequently challenged with PR8 PB1-F2, H7N9 PB1-F2 or silica. It was also observed that ASC-cerulean cells pretreated with MCC950 were able to phagocytose pHrodo-labeled H7N9 PB1-F2 peptide but did not undergo ASC speck formation (supplemental Movies 7 and 8), suggesting that MCC950 acts prior to ASC oligomerization. Proteolytic cleavage of caspase-1 was detected in cell supernatants from BMDMs stimulated with either PR8- or H7N9-derived PB1-F2 or silica but not in supernatants from cells treated with MCC950 (Fig. 6D, top panel). Correlative maturation of bioactive IL-1β p17 was detected in supernatants of cells treated with either PR8- or H7N9-derived PB1-F2 or silica (Fig. 6D, bottom panel) but not in those treated with MCC950 (Fig. 6D, top panel). This result was reflected in the analysis of IL-1β levels in these samples by ELISA (supplemental Fig. 4). Importantly, MCC950 pretreatment of hPBMCs significantly reduced the levels of IL-1β in response to both PR8-derived (Fig. 6E) and H7N9-derived (Fig. 6F) PB1-F2 peptide.
FIGURE 6.

MCC950 inhibits H7N9 PB1-F2-induced inflammasome activation. A–C, iBMDMs (A and B) or primary BMDMs (C) were primed with LPS (100 ng/ml) for 3 h. Cells were then pretreated with MCC950 (an NLRP3 inhibitor, 12.5–50 μm) 40 min prior to exposure with either PR8 PB1-F2 (100 μg/ml, A), H7N9 PB1-F2 peptide (100 μg/ml, B), or PR8 and H7N9 PB1-F2 peptides (50–100 μg/ml as indicated, C) or left unstimulated (NS) for a further 6 h. The levels of IL-1β in cell supernatants were determined by ELISA. The results are representative of three independent experiments and are the mean ± S.E. D, primary BMDMs were primed with LPS (100 ng/ml). Cells were exposed to MCC950 (50 μm) 40 min prior to challenge with PR8 or H7N9 PB1-F2 peptides (50–100 μg/ml) or silica (150 μg/ml) or left unstimulated for 6 h. Supernatant proteins were separated by 4–12% SDS-PAGE and immunoblotted (IB) with the indicated antibodies. The result represents one of three experiments. E and F, PBMCs from three donors were primed with LPS and pretreated with MCC950 (10–50 μm) 40 min prior to challenge with PR8 PB1-F2 (100 μg/ml, E) or H7N9 PB1-F2 (100 μg/ml, F) and silica (150 mg/ml) for a further 6 h. Supernatants were assessed for IL-1β by ELISA. Results are the mean ± S.E. of three independent donors.
MCC950 Blocks PB1-F2-induced Inflammation in Vivo
Having established that MCC950 inhibits PB1-F2-induced IL-1β maturation in vitro, we next examined whether targeting inflammasome activation could reduce PB1-F2-induced inflammation in vivo. WT mice were treated intranasally with H7N9 PB1-F2 peptide in combination with MCC950 (5 mg/kg) or vehicle control (PBS). As seen in Fig. 7A, both PR8- and H7N9-derived PB1-F2 peptides induced significant neutrophil influx into the airways, and treatment with MCC950 potently inhibited cellular influx measured at 7 h following challenge. Concomitantly, IL-1β levels induced in response to both PR8 and H7N9 PB1-F2 peptides were significantly lower in BAL fluid from mice treated with MCC950 (Fig. 7B) than vehicle control. Together, these results clearly demonstrate that targeting NLRP3 inflammasome activation with a small-molecule inhibitor such as MCC950 ablates H7N9 PB1-F2-induced IL-1β and leads to reduced lung inflammation.
FIGURE 7.

MCC950 inhibits PB1-F2 peptide-induced airway neutrophil influx and IL-1β secretion. Groups of five wild-type C57BL/6 mice were intranasally inoculated with PR8 or H7N9 PB1-F2 (50 μg) alone or in combination with MCC950 (5 mg/kg). Uninfected mice treated with MCC950 were included for comparison. BAL was performed 7 h following challenge. A and B, the number of infiltrating Ly6G+ neutrophils in BAL determined by flow cytometry (A) and the levels of IL-1β by ELISA (B). Data the mean ± S.E. and are representative of two independent experiments. *, p < 0.05; ***, p < 0.001; one-way ANOVA.
Discussion
The strength of the innate immune response to IAV infection is a key determinant in clinical outcome. Excessive inflammation can cause death, particularly in the case of highly pathogenic IAV infections. It is well known that the three pandemics of the 20th century caused millions of deaths worldwide. Although a large proportion of deaths have been attributed to complications arising from secondary bacterial infections (24), the initial infection by the novel H1N1, H2N2, and H3N2 viruses caused remarkable inflammatory disease and contributed significantly to the hospitalization of patients presenting with pneumonia-like illness.
Common to 20th century pandemic IAVs is the direct reassortment of the HA and PB1 gene segments from avian IAV. The avian-derived PB1 gene segments in pandemic viruses all encoded a full-length PB1-F2 protein and contained the four proinflammatory amino acids identified previously (16). Conversely, the pandemic H1N1 IAV that emerged in 2009 contained a PB1 gene that had been circulating in swine IAV lineages and encoded a truncated and presumably non-functional PB1-F2 protein. Interestingly, in otherwise healthy individuals, infection with the 2009 pandemic IAV caused illness symptoms considered to be milder than those induced by the previous pandemic viruses and that were similar to those typically observed during a seasonal IAV infection, in which inflammatory disease is limited (25). Therefore, the contribution of the PB1-F2 protein toward enhancing the pathophysiology of IAV infections may have a significant impact on disease outcomes.
In this study, we have demonstrated that PB1-F2 peptide from H7N9 IAV activates an NLRP3-dependent inflammasome to induce the secretion of IL-1β. Analysis of PB1-F2 sequences from isolated human and avian H7N9 IAVs has identified only two minor amino acid changes from the avian to human isolate: G70E and S77L (3), both of which are found within the H7N9 peptide used in our study. Importantly, neither of these changes corresponds to the four amino acids that have been identified to be important for the pro-inflammatory properties of PB1-F2 (i.e. Leu-62, Arg-75, Arg-79, and Leu-82), nor are they expected to affect its aggregating potential (data not shown). Indeed, we compared the sequences of 89 human H7N9 virus isolates and found PB1-F2 to be highly conserved among all isolates (supplemental Fig. S5), particularly in the C-terminally aggregating peptide used in this study, where all four inflammatory amino acids are conserved throughout. Therefore, we would predict that, if H7N9 was to cause a pandemic, then it would likely contain a conserved, highly virulent PB1-F2 gene, further supporting that inhibition of the inflammasome is a potential strategy to reduce inflammation during infection.
In contrast to our findings demonstrating that IAV PB1-F2 peptide activates the NLRP3 inflammasome (Figs. 1–4 and Ref. 13), it was recently reported that PB1-F2 derived from PR8 and A/California(CA)/04/09 (H1N1) translocates to mitochondria, accelerating mitochondrial fragmentation to suppress inflammasome activation (26). That study found that reconstituting NLRP3 inflammasome activation in HEK293 cells by ectopically expressing NLRP3, ASC, caspase-1, and pro-IL-1β in the presence of PB1-F2 reduced IL-1β secretion. By contrast, in our previous study, we utilized reverse-engineered IAV in which PB1-F2 was absent to demonstrate the direct role of PB1-F2 in activating the inflammasome and inducing IL-1β secretion in vitro and in vivo in an NLRP3-dependent manner (13). Interestingly, in this study, we did observe that PB1-F2 peptide induced robust mtROS production, inhibition of which with the specific mtROS inhibitor MitoTempo reduced ROS production and inflammasome activation. These findings would therefore be consistent with PB1-F2-inducing dysregulation of the mitochondria, the result of which is the induction of the inflammasome and inflammation.
Two recent breakthrough studies also identified “prion-like” activities for ASC-containing inflammasomes whereby oligomeric inflammasome complexes released from cells following pyroptosis are engulfed by phagocytic cells to initiate inflammasome activation independent of stimuli, provided the cell had been already stimulated with signal 1 (18, 27). Within the confined lung environment, excessive ASC oligomerization and inflammasome activation may therefore induce a cellular hyperinflammatory state contributing to the cytokine storm and morbidity in the later stages of infection. Secondary bacterial infection is a leading cause of death during severe IAV infection and has been reported in human cases of H7N9 (28). Induction of inflammasome-driven pyroptosis by PB1-F2 may also lead to removal of alveolar macrophages and epithelial cell barriers, which may result in increased susceptibility to secondary bacterial infections causing pneumonia (29). We have demonstrated previously that PR8 or 1918 pandemic PB1-F2 proteins induce inflammatory disease that predisposes mice to secondary bacterial pneumonia (15). Indeed, IAV RNA and the IAV M2 ion channel protein are potential NLRP3 inflammasome activators that, during infection with IAV strains lacking full-length PB1-F2, may be responsible for initiating “protective” inflammasome activation, leading to protective immunity, increased disease tolerance through cellular recruitment, and induction of tissue repair (4, 5, 7).
The emergence of H7N9 avian IAV and the high levels of mortality associated with infections pose a considerable risk to human health. Recently, genetic analysis of H7N9 strains from human isolates and those isolated from poultry found considerable reassortment among viruses in the influenza ecosystem of China and concluded that it is reasonable to consider H7N9 a major candidate to emerge as a pandemic strain in humans and cause a substantial number of severe human infections (3). Given the potentially dire consequences of a virulent pandemic IAV outbreak, the emergence of viral resistance to existing antiviral therapies and the requirement to produce and disseminate a vaccine, there is a clear need to identify the molecular mechanisms of IAV-induced hyperinflammation and therapeutic strategies to reduce the inflammatory burden associated with pathogenic IAV infections.
Overall, our results suggest that H7N9 PB1-F2 represents a major virulence factor and mediator of excessive inflammation and that therapeutic targeting of the NLRP3 inflammasome may constitute a viable treatment strategy to decrease the excessive inflammation associated with pathogenic IAV, such as the H5N1 and H7N9 avian strains. Recently, we demonstrated that temporal administration of MCC950 to inhibit NLRP3 inflammasome activation delayed lethality associated with highly virulent PR8 challenge and reduced lung inflammation and cellular influx (11).
Given that we have now demonstrated the capacity of the human isolate H7N9 PB1-F2 peptide to induce NLRP3 inflammasome activation (Fig. 1) and amelioration of inflammation with MCC950 (Figs. 5 and 7), interventions with small-molecule NLRP3 inhibitors could be used therapeutically to augment current antiviral and vaccination strategies to potentially treat IAV outbreaks such as H7N9. Highly specific small-molecule inhibitors such as MCC950 have a relatively short half-life in vivo, and, therefore, the timing and dosing of treatments could be cost-effective and easily controlled (23). Given the need to stockpile antivirals for prophylactic protection and the limited therapeutic options available to infected individuals, our study not only identifies PB1-F2 as a major virulence factor in pandemic IAV infections but acknowledges that therapeutically targeting activation of the inflammasome may be an effective strategy to reduce IL-1β maturation in the lung to protect against the damaging hyperinflammatory effects characteristic of lethal infections.
Experimental Procedures
Cell Lines
Immortalized wild-type, NLRP3-, ASC-, and caspase-1/11-deficient, and ASC-cerulean-tagged C57BL/6 macrophages (17) were grown in DMEM (Gibco) supplemented with 10% heat-inactivated FBS and 2 mm glutamine. Bone marrow cells were isolated from 8-week-old C57BL/6 female mice and cultured in DMEM supplemented with 10% FCS, 2 mm glutamine, and 20% L929 conditioned medium (L-cell). Differentiated primary BMDMs were plated on day 7 for stimulation the following day. All cell cultures were maintained at 37 °C in a 5% CO2 incubator.
PB1-F2 Peptide Generation
Using the predicted amino acid sequence of the PB1-F2 protein from influenza virus strains A/Shanghai/02/2013 (H7N9) and A/Puerto Rico/8/34 (PR8, H1N1), peptides from amino acids 61–90 and 61–87, respectively, were synthesized by Genscript (H7N9, WLSLKNLTQGSLKTRVSKRWKLFSKQEWIN; H7N9 G70E, S77L, WLSLKNLTQESLKTRVLKRWKLFSKQEWIN; H7N9 four amino acids, WPSLKNLTQGSLKTHVSKQWKSFSKQEWIN; and PR8, WLSLRNPILVFLKTRVLKRWRLFSKHEWIN). Immediately prior to use, the H7N9 and PR8 peptides were resuspended in PBS and used at the indicated concentrations.
The NLRP3 Inhibitor MCC950
The NLRP3 inflammasome inhibitor MCC950 (N-((1,2,3,5,6,7-hexahydro-s-indacen-4-yl)carbamoyl)-4-(2-hydroxypropan-2-yl)furan-2-sulfonamide) was prepared as described previously (23).
Quantification of Cytokines
For the detection of IL-1β, cultured supernatants were collected and stored at −80 °C. Mouse IL-1β and human IL-1β were quantified by ELISA according to the instructions of the manufacturer (BD Biosciences). Levels of IL-1β in mouse BAL fluid were quantified by ELISA according to the instructions of the manufacturer.
Live-cell Bioimaging
NLRP3-deficient immortalized macrophages stably expressing ASC-cerulean and NLRP3 were seeded in 8-well Ibidi chamber slides 24 h prior to stimulation. Cells were pretreated with MitoTempo (20 μm, Sigma-Aldrich) for 10 min prior to challenge or stimulated with pHrodo-labeled H7N9 PB1-F2 peptide (100 μg/ml) where indicated at 37 °C. Imaging was performed on a Leica SP5 multichannel Acousto Optical Beam Splitter confocal laser-scanning microscope equipped with a temperature- and CO2-controlled sample chamber for live-cell imaging. Images are deconvoluted z stacks by overlapping tile scanning processed using Imaris software.
Caspase-1 and IL-1β Immunoblot
Primary BMDMs were plated into 6-well plates 24 h prior to priming with LPS (200 ng/ml, Enzo Life Sciences) for 3 h in serum-free medium. Macrophages were pretreated with inhibitors (as indicated) 40 min prior to stimulation with the indicated ligands or left unstimulated for a further 6 h. Protein lysates were boiled in SDS-PAGE sample buffer proteins while cultured supernatants were concentrated using Strataclean resin (Agilent). Concentrated supernatants were separated by 4–12% SDS-PAGE and transferred to PVDF membranes. Caspase-1 and IL-1β were imaged by immunoblotting with anti-mouse caspase-1 monoclonal antibody (AdipoGen Life Sciences) and anti-mouse IL-1β detection biotinylated IgG antibody (R&D Systems), respectively. Caspase-1 was imaged using chemiluminescence, and IL-1β was imaged using anti-streptavidin Alexa Fluor 680 conjugate (Life Technologies).
PB1-F2 Peptide Challenge and Influenza Virus Infection of Mice
6- to 8-week-old male or female C57BL/6 and NLRP3 knockout mice were maintained in the specific pathogen-free physical containment level 2 (PC2) animal research facility in the Monash Medical Centre animal facility. All experimental procedures were approved by the Monash Medical Centre Animal Ethics Committee.
Mice were anesthetized and inoculated with 50 μg of PR8 or H7N9 PB1-F2 peptides alone or in combination with MCC950 intranasally (5 mg/kg). Mice were euthanized 7 h following treatment. BAL was obtained from euthanized mice by flushing the lungs three times with 1 ml of PBS.
Recovery and Characterization of Leukocytes from Mice
For flow cytometric analysis, BAL cells were treated with red blood cell lysis buffer (Sigma-Aldrich), and cell numbers and viability were assessed via trypan blue exclusion using a hemacytometer. BAL cells were incubated with supernatants from hybridoma 2.4G2 to block Fc receptors. Neutrophils (Ly6G+) were quantified by flow cytometry as described previously (30). Living cells (propidium iodine-negative) were analyzed using a BD FACSCanto II flow cytometer (BD Biosciences), and total cell counts were calculated from viable cell counts performed via trypan blue exclusion.
Statistical Analysis
When comparing three or more sets of values, a one-way analysis of variance (ANOVA) was used with Tukey's post hoc analysis. Student's t test was used when comparing two values (two-tailed, two-sample equal variance). p < 0.05 was considered statistically significant. Survival proportions were compared using the Mantel-Cox log-rank test. p < 0.05 was considered statistically significant.
Author Contributions
A. M. and M. D. T. conceived and designed the experiments. A. P., J. K. D., N. J. B., J. L. M., M. D. T., C. R. S., and A. M. performed the experiments. A. P., J. K. D., J. L. M., A. M., and M. D. T. analyzed the data. J. L. M., G. R. D., E. L., A. A. B. R., M. C., and A. M. contributed reagents, materials, and analysis tools. A. P., J. K. D., J. L. M., G. R. D.,. M. A. C., M. D. T., and A. M. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank R. Smith for manuscript preparation.
This work was supported by the Victorian State Government Operational Infrastructure Scheme and by National Health and Medical Research Council of Australia Project Grants GNT1079924, GNT1062721, and GNT1062977 (to A. M., J. L. M., and G. D., respectively). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1–S4 and Movies 1–8.
- IAV
- Influenza A virus
- ASC
- apoptosis-associated speck-like protein containing a CARD
- mtROS
- mitochondrial reactive oxygen species
- iBMDM
- immortalized bone marrow-derived macrophage
- Z-YVAD
- benzyloxycarbonyl-YVAD
- hPBMC
- human peripheral blood mononuclear cell
- BAL
- bronchoalveolar lavage
- MCC950
- N-((1,2,3,5,6,7-hexahydro-s-indacen-4-yl)carbamoyl)-4-(2-hydroxypropan-2-yl)furan-2-sulfonamide
- ANOVA
- analysis of variance
- PR8
- A/Puerto Rico/8/34.
References
- 1. Gao R., Cao B., Hu Y., Feng Z., Wang D., Hu W., Chen J., Jie Z., Qiu H., Xu K., Xu X., Lu H., Zhu W., Gao Z., Xiang N., et al. (2013) Human infection with a novel avian-origin influenza A (H7N9) virus. N. Engl. J. Med. 368, 1888–1897 [DOI] [PubMed] [Google Scholar]
- 2. Normile D. (2015) Virology: bird flu virus's promiscuity raises red flags. Science 347, 1188. [DOI] [PubMed] [Google Scholar]
- 3. Lam T. T., Zhou B., Wang J., Chai Y., Shen Y., Chen X., Ma C., Hong W., Chen Y., Zhang Y., Duan L., Chen P., Jiang J., Zhang Y., Li L., et al. (2015) Dissemination, divergence and establishment of H7N9 influenza viruses in China. Nature 522, 102–105 [DOI] [PubMed] [Google Scholar]
- 4. Iwasaki A., and Pillai P. S. (2014) Innate immunity to influenza virus infection. Nat. Rev. Immunol. 14, 315–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ichinohe T., Pang I. K., and Iwasaki A. (2010) Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 11, 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thomas P. G., Dash P., Aldridge J. R. Jr., Ellebedy A. H., Reynolds C., Funk A. J., Martin W. J., Lamkanfi M., Webby R. J., Boyd K. L., Doherty P. C., and Kanneganti T. D. (2009) The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30, 566–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ichinohe T., Lee H. K., Ogura Y., Flavell R., and Iwasaki A. (2009) Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 206, 79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Allen I. C., Scull M. A., Moore C. B., Holl E. K., McElvania-TeKippe E., Taxman D. J., Guthrie E. H., Pickles R. J., and Ting J. P. (2009) The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30, 556–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmitz N., Kurrer M., Bachmann M. F., and Kopf M. (2005) Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J. Virol. 79, 6441–6448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Szretter K. J., Gangappa S., Lu X., Smith C., Shieh W. J., Zaki S. R., Sambhara S., Tumpey T. M., and Katz J. M. (2007) Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J. Virol. 81, 2736–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tate M. D., Ong J. D., Dowling J. K., McAuley J. L., Robertson A. B., Latz E., Drummond G. R., Cooper M. A., Hertzog P. J., and Mansell A. (2016) Reassessing the role of the NLRP3 inflammasome during pathogenic influenza A virus infection via temporal inhibition. Sci. Rep. 6, 27912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krumbholz A., Philipps A., Oehring H., Schwarzer K., Eitner A., Wutzler P., and Zell R. (2011) Current knowledge on PB1-F2 of influenza A viruses. Med. Microbiol. Immunol. 200, 69–75 [DOI] [PubMed] [Google Scholar]
- 13. McAuley J. L., Tate M. D., MacKenzie-Kludas C. J., Pinar A., Zeng W., Stutz A., Latz E., Brown L. E., and Mansell A. (2013) Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 9, e1003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McAuley J. L., Chipuk J. E., Boyd K. L., Van De Velde N., Green D. R., and McCullers J. A. (2010) PB1-F2 proteins from H5N1 and 20 century pandemic influenza viruses cause immunopathology. PLoS Pathog. 6, e1001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McAuley J. L., Hornung F., Boyd K. L., Smith A. M., McKeon R., Bennink J., Yewdell J. W., and McCullers J. A. (2007) Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2, 240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alymova I. V., Green A. M., van de Velde N., McAuley J. L., Boyd K. L., Ghoneim H. E., and McCullers J. A. (2011) Immunopathogenic and antibacterial effects of H3N2 influenza A virus PB1-F2 map to amino acid residues 62, 75, 79, and 82. J. Virol. 85, 12324–12333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hornung V., Bauernfeind F., Halle A., Samstad E. O., Kono H., Rock K. L., Fitzgerald K. A., and Latz E. (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Franklin B. S., Bossaller L., De Nardo D., Ratter J. M., Stutz A., Engels G., Brenker C., Nordhoff M., Mirandola S. R., Al-Amoudi A., Mangan M. S., Zimmer S., Monks B. G., Fricke M., Schmidt R. E., et al. (2014) The adaptor ASC has extracellular and “prionoid” activities that propagate inflammation. Nat. Immunol. 15, 727–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen C. J., Chen G. W., Wang C. H., Huang C. H., Wang Y. C., and Shih S. R. (2010) Differential localization and function of PB1-F2 derived from different strains of influenza A virus. J. Virol. 84, 10051–10062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen W., Calvo P. A., Malide D., Gibbs J., Schubert U., Bacik I., Basta S., O'Neill R., Schickli J., Palese P., Henklein P., Bennink J. R., and Yewdell J. W. (2001) A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 7, 1306–1312 [DOI] [PubMed] [Google Scholar]
- 21. Robinson K. M., Janes M. S., Pehar M., Monette J. S., Ross M. F., Hagen T. M., Murphy M. P., and Beckman J. S. (2006) Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc. Natl. Acad. Sci. U.S.A. 103, 15038–15043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grailer J. J., Canning B. A., Kalbitz M., Haggadone M. D., Dhond R. M., Andjelkovic A. V., Zetoune F. S., and Ward P. A. (2014) Critical role for the NLRP3 inflammasome during acute lung injury. J. Immunol. 192, 5974–5983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Coll R. C., Robertson A. A., Chae J. J., Higgins S. C., Muñoz-Planillo R., Inserra M. C., Vetter I., Dungan L. S., Monks B. G., Stutz A., Croker D. E., Butler M. S., Haneklaus M., Sutton C. E., Núñez G., et al. (2015) A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kash J. C., and Taubenberger J. K. (2015) The Role of viral, host, and secondary bacterial factors in influenza pathogenesis. Am. J. Pathol. 185, 1528–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dawood F. S., Iuliano A. D., Reed C., Meltzer M. I., Shay D. K., Cheng P. Y., Bandaranayake D., Breiman R. F., Brooks W. A., Buchy P., Feikin D. R., Fowler K. B., Gordon A., Hien N. T., Horby P., et al. (2012) Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect. Dis. 12, 687–695 [DOI] [PubMed] [Google Scholar]
- 26. Yoshizumi T., Ichinohe T., Sasaki O., Otera H., Kawabata S., Mihara K., and Koshiba T. (2014) Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 5, 4713. [DOI] [PubMed] [Google Scholar]
- 27. Baroja-Mazo A., Martín-Sánchez F., Gomez A. I., Martínez C. M., Amores-Iniesta J., Compan V., Barberà-Cremades M., Yagüe J., Ruiz-Ortiz E., Antón J., Buján S., Couillin I., Brough D., Arostegui J. I., and Pelegrín P. (2014) The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 15, 738–748 [DOI] [PubMed] [Google Scholar]
- 28. Yu L., Wang Z., Chen Y., Ding W., Jia H., Chan J. F., To K. K., Chen H., Yang Y., Liang W., Zheng S., Yao H., Yang S., Cao H., Dai X., et al. (2013) Clinical, virological, and histopathological manifestations of fatal human infections by avian influenza A(H7N9) virus. Clin. Infect. Dis. 57, 1449–1457 [DOI] [PubMed] [Google Scholar]
- 29. Coleman J. R. (2007) The PB1-F2 protein of influenza A virus: increasing pathogenicity by disrupting alveolar macrophages. Virol. J. 4, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tate M. D., Pickett D. L., van Rooijen N., Brooks A. G., and Reading P. C. (2010) Critical role of airway macrophages in modulating disease severity during influenza virus infection of mice. J Virol. 84, 7569–7580 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.