

Abstract
Clear cell renal cell carcinoma (CCRCC) is an incurable malignancy in advanced stages and needs newer therapeutic targets. Transcriptomic analysis of CCRCCs and matched microdissected renal tubular controls revealed overexpression of NOTCH ligands and receptors in tumor tissues. Examination of the TCGA RNA-seq data set also revealed widespread activation of NOTCH pathway in a large cohort of CCRCC samples. Samples with NOTCH pathway activation were also clinically distinct and were associated with better overall survival. Parallel DNA methylation and copy number analysis demonstrated that both genetic and epigenetic alterations led to NOTCH pathway activation in CCRCC. NOTCH ligand JAGGED1 was overexpressed and associated with loss of CpG methylation of H3K4me1-associated enhancer regions. JAGGED2 was also overexpressed and associated with gene amplification in distinct CCRCC samples. Transgenic expression of intracellular NOTCH1 in mice with tubule-specific deletion of VHL led to dysplastic hyperproliferation of tubular epithelial cells, confirming the procarcinogenic role of NOTCH in vivo. Alteration of cell cycle pathways was seen in murine renal tubular cells with NOTCH overexpression, and molecular similarity to human tumors was observed, demonstrating that human CCRCC recapitulates features and gene expression changes observed in mice with transgenic overexpression of the Notch intracellular domain. Treatment with the γ-secretase inhibitor LY3039478 led to inhibition of CCRCC cells in vitro and in vivo. In summary, these data reveal the mechanistic basis of NOTCH pathway activation in CCRCC and demonstrate this pathway to a potential therapeutic target.
Keywords: cancer biology, cancer therapy, DNA methylation, Notch pathway, renal physiology
Introduction
Advanced renal cell carcinoma (RCC)4 is an incurable disease and is associated with a rising incidence (1, 2) RCC comprises several histological subtypes, each with a different clinical phenotype and genetic abnormality. Clear cell renal cell Carcinoma (CCRCC) is the commonest subtype and has a high incidence of alterations on chromosome 3 affecting the VHL gene (1). CCRCC is generally resistant to chemotherapy and radiation therapy. Approved multikinase inhibitors have led to only minimal improvements in overall survival (3), thus necessitating novel therapeutic targets.
Recent genomic studies have shown that CCRCC is associated with mutations in chromatin modifying enzymes, such as PBRM1, BAP1, SETD2, and KDM5C, thus implying that epigenetic dysregulation plays a role in the pathogenesis of this malignancy (4–7). We developed an integrated epigenomics platform and used it to study CCRCC samples (8). Our studies showed that widespread DNA methylation changes could be seen in CCRCC and affect enhancer regions of the kidney genome. We also observed both novel and well characterized genomic copy numbers changes in these CCRCC samples (8) that were also seen in a large TCGA cohort of CCRCC samples. Analysis of differentially methylated regions in CCRCC in our study revealed enrichment for binding sites of HAIRY transcription factor. Because HAIRY is a downstream mediator of the NOTCH signaling pathway, we focused on analysis of this pathway in the present study. Furthermore, recent work has demonstrated that inhibition of NOTCH pathway can be efficacious in various malignancies. In fact, there have been reports that have suggested that components of the NOTCH pathway are activated in renal cell cancer and that targeting components such as DLL4 can have therapeutic efficacy in preclinical models (9–11). Still, not much is known about the mechanisms of NOTCH pathway activation in renal cell cancer.
In the current study, we analyzed genetic and epigenetic abnormalities related to the NOTCH pathway in CCRCC and determined that ligands JAGGED1 and JAGGED2 were overexpressed and associated with both genetic and epigenetic alterations. Widespread NOTCH activation was also seen in independent large TCGA data sets. Transgenic overexpression of NOTCH1 led to severely dysplastic and hyperproliferative tubules in vivo, demonstrating the procarcinogenic role of this pathway in RCC. Finally, treatment with a clinical inhibitor of the NOTCH pathway LY-3039478 led to increased survival in CCRCC xenografts, demonstrating this pathway as a therapeutic target in CCRCC.
Results
Notch Pathway Is Overexpressed in Cohorts of Primary CCRCC
We had conducted a recent study on integrated transcriptomic and epigenomic analysis of CCRCC and microdissected renal glomerular tissue from 13 primary samples (8). In this study we had observed that the binding site for NOTCH-driven transcription factor, HAIRY, was highly enriched in differentially methylated regions in CCRCC (8). Thus, we now analyzed gene expression data from these samples for numerous components of the NOTCH pathway, including ligands (JAGGED1), receptors (NOTCH 2-4) and downstream effectors (HEY1,2 and HES1) and found them to be overexpressed in CCRCC samples (Fig. 1A) when compared with controls. Next, we validated our findings by examining RNA-seq data from CCRCC samples in the TCGA portal (12). We examined a cohort of 405 RCC samples and 68 non-tumor kidney controls that included 66 matched CCRCC/control samples. Analysis of the complete cohort of matched and unmatched samples (n = 473) revealed widespread activation of NOTCH ligands and effectors in CCRCC (Fig. 1B) with up-regulation of JAG1, JAG2, DLL4, NOTCH 1,3,4, HEY1, HEY2, and HES5 genes. (p < 0.05, t test). To quantify the magnitude of up-regulation, we analyzed the matched pairs (n = 66) and determined that the ligands JAG1 and JAG2 were overactivated (>2-fold increase) in 36 and 67% cases, respectively (Fig. 1C). Interestingly, analysis of matched pairs also indicated the increased expression of another NOTCH ligand DLL4 in 85% of CCRCC cases.
FIGURE 1.
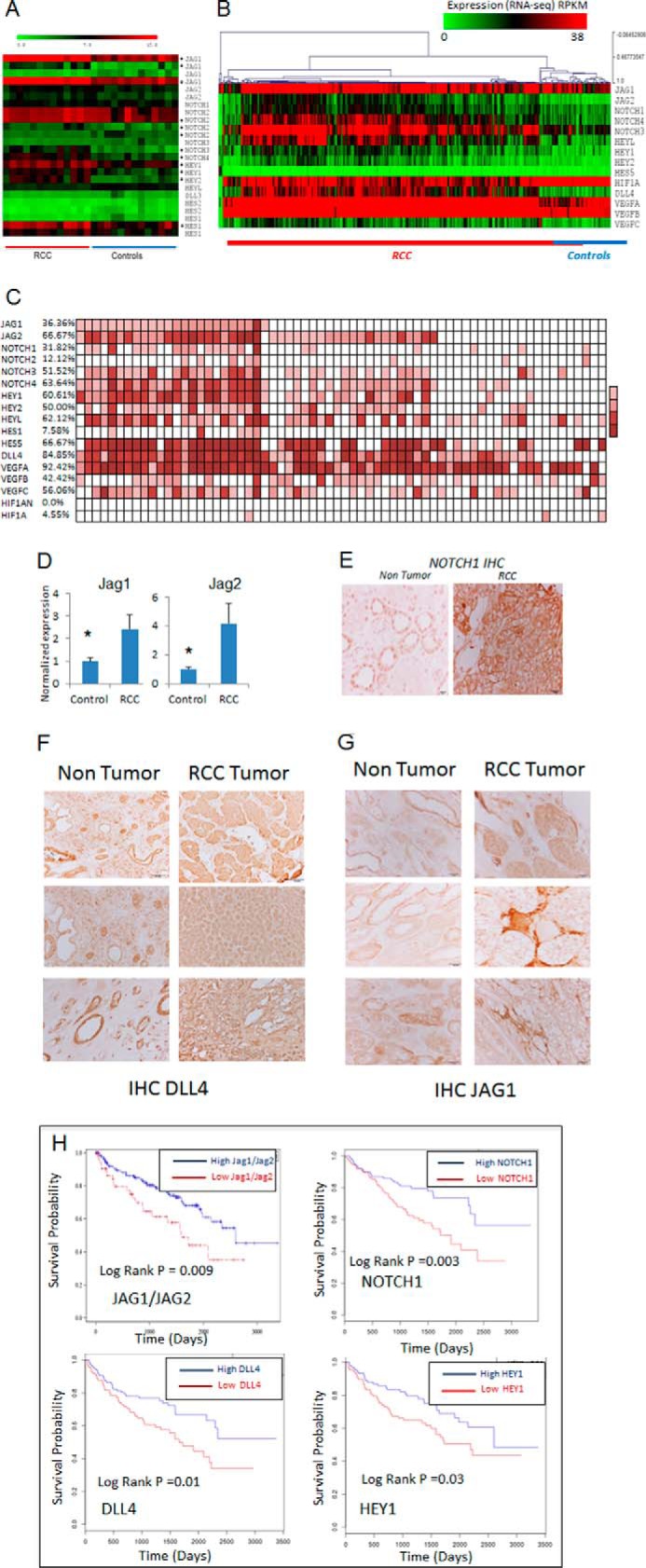
NOTCH pathway is overexpressed in independent cohorts of CCRCC and control samples. A, gene expression analysis of genes involved in the NOTCH pathway (heat map) shows the expression of ligands (JAG1 and JAG2), receptors (NOTCH 1–2) and downstream effectors (HEY1,2,L and HES1,2) in CCRCC and microdissected control samples. Significantly altered genes are marked with asterisks (*, p < 0.05). B, NOTCH pathway gene expression for complete cohort of 405 samples of RCC with 68 controls from TCGA RNA-seq data set are shown as a heat map. NOTCH pathway members with significant (P < 0.05) increased expression are shown. C, 66 RCC samples and matched normal controls were examined by RNA-seq in the TCGA database. Samples with more than 2-fold increase in expression in matched RCC sample are shown by colored boxes. Percentages of samples with overexpression are shown on the left. D, 7 sets of matched tumor and non-tumor control kidney samples were examined. qRT-PCR values show increased expression of JAG1 and JAG2 in RCC samples when compared with controls (means ± S.E., P value < 0.05, t test). E–G, immunohistochemical analysis of NOTCH1 (E), JAG1 (F), and DLL4 (G) for matched non-tumor kidney and CCRCC samples shows overexpression in RCC. Kaplan-Meir curves of 405 TCGA samples show that samples with JAG1 or JAG2 overexpression have a significantly better overall survival (log rank P value = 0.009). H, the blue lines show the top quartile of JAG1 or JAG2 expression, and the red lines show the bottom quartile of JAG1/JAG2 expressers. A similar pattern of survival is seen with NOTCH1, DLL4, and HEY1.
Next, we confirmed the overexpression of JAG1 and JAG2 seen by genome-wide analysis in an independent set of seven matched renal tumors and their non-tumor microdissected controls by qRT-PCR (Fig. 1D). Finally, the overexpression of NOTCH1, JAG1, and DLL4 was also corroborated by immunostaining. As shown on the representative figures, NOTCH1 and JAG1 were highly overexpressed in malignant CCRCC cells (Fig. 1, E and F), whereas DLL4 expression was overexpressed in the interstitial cells (Fig. 1G).
Next, we examined whether the JAGGED positive RCC samples are clinically different from those of low JAGGED expressing tumors. We found that the overall survival for cases with either JAG1 or JAG2 overactivation was significantly better than those with low JAGGED-expressing tumors (log rank p value = 0.0094). The median survival of the high JAG1/JAG2 RCC cases was 2600 days versus 1584 of the low JAG1/JAG2 cases (Fig. 1H). A similar prognostic impact was also seen in CCRCC cases with NOTCH1, DLL4, and HEY1 overexpression (Fig. 1H). In summary, these results demonstrate the increased activation of NOTCH pathway genes in independent cohorts of CCRCC and revealed that samples with NOTCH activation comprise a clinically distinct subgroup of tumors.
Integrative Analysis of Copy Number Alterations and Cytosine Methylation Reveals the Mechanistic Basis for Activation of NOTCH Pathway in RCC
Aberrant expression of genes in cancer can be accomplished via gene amplifications, as well as by aberrant epigenetic modifications (13). Analysis of our integrative CCRCC data set revealed that even though hypermethylation was the predominant epigenetic abnormality in CCRCC (8), JAG1 was one of the select genes that was aberrantly hypomethylated in CCRCC (supplemental Table S1). The loss of DNA methylation on the JAG1 locus coincided with H3K4me1 chromatin modification, and computational annotation indicated that these differentially hypomethylated regions are on an active enhancer regions (yellow, orange) (Fig. 2A). These aberrant differentially hypomethylated regions were validated the quantitatively by MassARRAY EpiTYPER analysis and demonstrated loss of methylation in tumors when compared with controls (Fig. 2B). To further establish that JAG1 is epigenetically regulated in kidney epithelia, we treated two sets of normal kidney tubular cells with the DNMT inhibitor decitabine in vitro and observed a significant increase in JAG1 expression in both cell lines (Fig. 2C). The increase was observed at both high and low doses of the inhibitor. Quantitative MassARRAY analysis of individual CpGs located in the differentially methylated JAG1 locus determined that significant demethylation of multiple CpGs occurred after decitabine treatment and correlated with the increase in expression (Fig. 2D). These results indicate that the hypomethylation of this enhancer region of JAG1 is associated with the increased ligand expression observed in CCRCC.
FIGURE 2.

Integrative analysis reveals that JAG1 and JAG2 are associated with DNA hypomethylation and gene amplification in CCRCC. A, loss of DNA methylation affecting the JAG1 gene locus was seen in CCRCC samples when compared with controls. Differentially hypomethylated intragenic regions in CCRCC samples for the JAG1 locus overlap with kidney specific H3K4me1 enhancer peaks. Methylation values generated by the HELP assay (log(HpaII/MspI) are shown as peaks (positive values corresponding with less methylation are shown in black, and negative values corresponding to increased methylation are shown in gray). Histone modification peaks are shown for adult kidney (BI27). Chromatin occupancy for other tissue types are shown at the bottom. The legend shows color coding for different regions with yellow regions representing enhancers. GM12878, lymphoblastoid cell; H1-hESC, embryonic stem cells; K562, leukemic cells; HepG2, hepatic cells; HUVEC, umbilical vein endothelial cells; HMEC, mammary epithelial cells; HSMM, skeletal muscle myoblasts; NHEK, epidermal keratinocytes; NHLF, lung fibroblasts. B, MassARRAY EpiTYPER analysis shows quantitative hypomethylation in CCRCC samples (n = 6) when compared with healthy controls (n = 6). C, treatment of non-transformed renal tubular cell lines HK2 and HKC8 with decitabine (DAC) results in increased expression of JAG1 by qRT-PCR (means ± S.E. of three independent experiments, P < 0.05, t test). D, MassARRAY EpiTYPER analysis of the JAG1 locus shows decrease in methylation after DAC treatment (means ± S.E. of three independent experiments, t test; P < 0.05 (*)). E, JAG2 lies on chromosome 14 (14q32). MspI representation identified 3 of 13 samples with JAG2 amplification. Representative sample is shown with JAG2 amplification indicated by the arrows.
The other NOTCH ligand, JAG2, was not affected by aberrant methylation but was found to be amplified in 3 of 13 cases of CCRCC (representative sample shown in Fig. 2E). These data revealed that both genetic and epigenetic mechanisms were up-regulating NOTCH receptor ligands in CCRCC.
Expression of NOTCH Leads to Dysplastic Changes in Vivo
To functionally test the role of NOTCH pathway activation as a driver of carcinogenesis in CCRCC, we generated a mouse model with tubule epithelial cell specific expression of the intracellular domain of NOTCH1. Because most human CCRCC cases occur in presence of deletion or mutations affecting the VHL gene, first we crossed the VHLflox/flox animals with the Kspcre (also known as Cdh16cre) animals. Kspcre mice express the cre recombinase in renal tubule epithelial cells (14) (Fig. 3A). As it has been described before, tubule-specific VHL null mice do not have an observable phenotype, which we confirmed (Fig. 3B). To test the effect of NOTCH activation, we crossed these animals with transgenic mice containing a sequence encoding an intracellular portion of the mouse NOTCH1 gene (amino acids 1749–2293) (5). Expression of the NOTCH1 fragment and GFP is blocked by a loxP-flanked STOP fragment placed between the coding sequence and the promoter. When crossed with the KspCre animals, the ICNOTCH1 is expressed constitutively in tubule epithelial cells (Fig. 3A). The truncated cytoplasmic fragment encoded by the NOTCH1 sequence causes constitutive signaling activity. Kidneys obtained from 8-week-old male double transgenic knock-out animals (Kspcre/ICNOTCH1/VHLflox/flox) were significantly larger than control animals (n = 9 from each genotype group) (Fig. 3, B and C). Kidney sections obtained from these animals showed striking dysplastic changes, consisting of nuclear enlargement and clearing, nucleolar prominence, microcystic change, and epithelial stratification (Fig. 3, B and E). Multiple cells layers of dysplastic cells were seen instead of a single layer of tubular cells observed in control animals.
FIGURE 3.

Tubule-specific expression of NOTCH leads to kidney dysplasia in vivo. A, schema of the transgenic model with deletion of VHL and expression of active Notch1 in renal tubular cells. B and C, representative images of Periodic acid Schiff-stained kidney sections of 8-week-old mice with tubule-specific VHL deletion with/without overexpression of intracellular domain of Notch1. D, renal enlargement in seen in VHL null mice following Notch overexpression. E, the red square shows a close-up image of dysplastic multilayered tubules with large nuclei.
Human CCRCC Recapitulates NOTCH-driven Expression Changes in Mice
Having seen that tubule-specific transgenic expression of Notch phenotypically recapitulates features of human CCRCC, we performed a comprehensive molecular transcript analysis to examine the molecular overlap between Notch target genes and genes differentially expressed in patients CCRCC. We obtained the list of tubule-specific Notch target genes from microarray studies performed in control and mice inducible expression of Notch intracellular domain in tubule epithelial cells using the Pax8rtTA TREICNotch1 model (15) (GEO GSE80384). Next, we examined the expression of the human orthologues of these kidney tubule-specific Notch target genes in human CCRCC samples. We observed that 380 genes that were regulated by Notch expression in mouse kidney tubules were also differentially expressed in human CCRCC. Human CCRCC samples recapitulated the regulation of close to 30% of differentially expressed Notch targets (Fig. 4A and supplemental Tables S2 and S3). As seen on the heat map analysis, the expression of these genes showed consistent patterns in numerous control and CCRCC samples and were able to distinctly cluster tumor from non-tumor samples in an unsupervised fashion (Fig. 4B). Next, we also compared the in vivo NOTCH signature to the human CCRCC signature using gene set enrichment analysis based on transcripts we identified in the Notch transgenic mice. When we examined the enrichment level of top 100 Notch target genes in CCRCC, there was a statistically significant enrichment for genes overexpressed in the Notch transgenic animals, and there was a statistically significant depletion for genes those expression decreased in tubule-specific Notch transgenic mice (Fig. 4, C and D).
FIGURE 4.
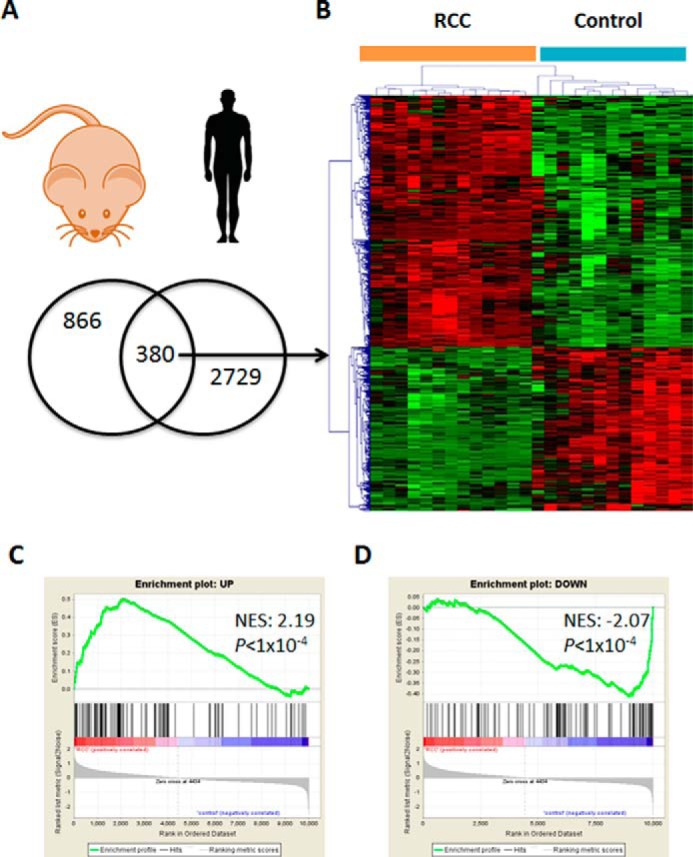
Human CCRCC gene expression is enriched for Notch target genes. A, genes with statistically significant differential expression in kidneys of mouse models with transgenic overexpression of Notch intracellular domain and genes with statistically significant enrichment in kidneys of patients with CCRCC. B, the common overlapping genes can segregate human CCRCC samples from controls using unsupervised cluster analysis. C and D, gene set enrichment analysis shows enrichment for genes with increased expression following overexpression of intracellular domain of Notch and corresponding depletion for the genes whose expression was decreased following Notch overexpression. NES, normalized enrichment score.
Functional pathway analysis of the common gene signature between NOTCH and human CCRCC revealed that cell cycle regulators were significantly represented in genes that were overexpressed (Fig. 5A and supplemental Table S4). Pathways involved in cell metabolism were also significantly represented in the commonly underexpressed genes (Fig. 5B). Having seen differential expression of genes involved in cell proliferative pathways, we examined the NOTCH overexpressing mice for changes in cell division. Expression of NOTCH1 in renal tubular cells was seen to be associated with an increase in Ki67 positive epithelial cell number, demonstrating increased epithelial cell proliferation (Fig. 5C). In summary, these data demonstrate molecular similarities between NOTCH-driven murine dysplasia and human CCRCC and show that a sizable proportion of dysregulated genes in CCRCC are driven by NOTCH pathway activation.
FIGURE 5.
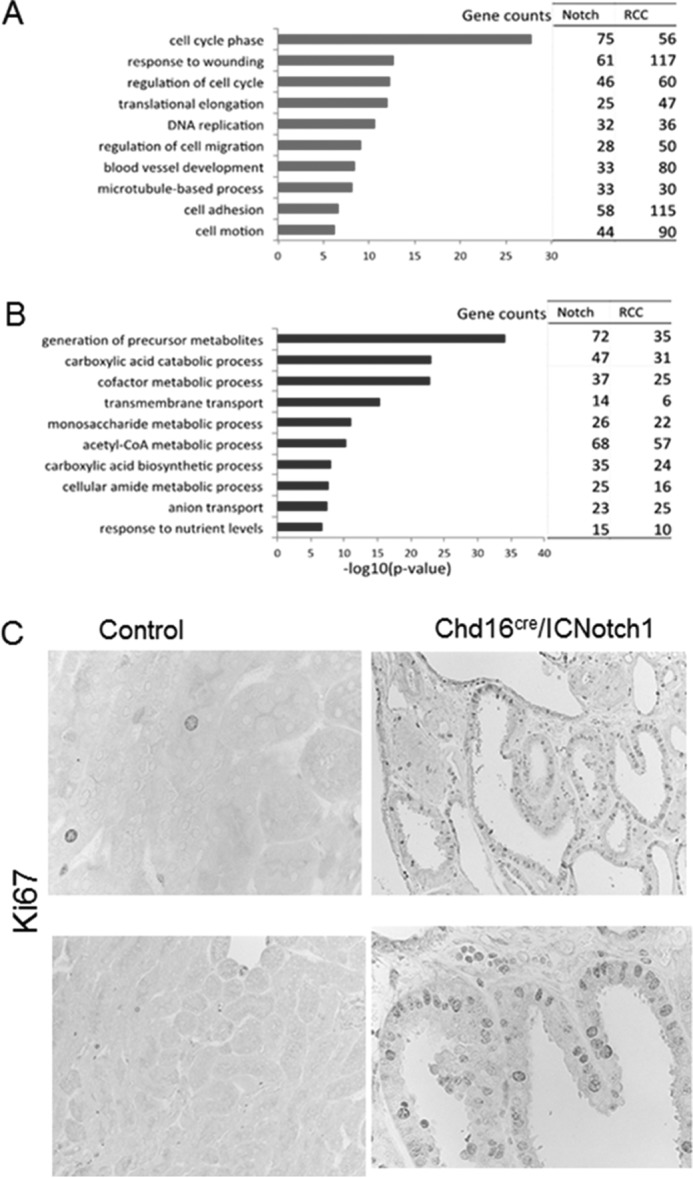
Cell proliferation genes are Notch targets of in renal tubules. A and B, gene ontology analysis of overlapping genes in Notch transgenic animals and human CCRCC samples; genes with increased expression (A) and genes with decreased expression (B). C, top significant GO terms were selected from each of 10 functional annotation clusters. Increased tubule proliferation was induced by Notch. Shown are representative images of Ki67 immunohistochemistry staining, with markers of proliferation in kidneys of control and Notch transgenic animals.
LY3039478 Is a Clinically Viable γ-Secretase Inhibitor That Can Inhibit CCRCC in Vitro and in Vivo
Having demonstrated overactivation of NOTCH pathway in CCRCC, we tested the effect of γ-secretase inhibition in representative human renal carcinoma cell lines. The γ-secretase complex is necessary for NOTCH cleavage, and this cleavage is essential for NOTCH activation. Treatment with a γ-secretase inhibitor that is being tested in clinical trials, LY3039478, significantly inhibited the growth of 2 CCRCC cell lines in a concentration-dependent manner (Fig. 6A). LY3039478 treatment also led to decreased expression of Myc and cyclin A1, two genes that were part of the NOTCH-driven proliferative signature in murine and human model systems (Fig. 6B). LY3039478 treatment also led to G0/G1 cell cycle arrest in CCRCC cells (Fig. 6C) Next, immunodeficient NOD-scid IL2Rγ null mice were xenografted with 769-P CCRCC cells, and mice were treated with 8 mg/kg of drug and vehicle control by oral gavage. LY3039478 treatment resulted in significantly increased survival and delayed tumor growth (Fig. 6, D and E) in independent cohorts of mice demonstrating in vivo efficacy in CCRCC.
FIGURE 6.

Pharmacologic inhibition of the NOTCH pathway leads to growth inhibition in CCRCC in vitro and in vivo. A, in vitro treatment with LY3039478 led to specific inhibition of viability in CCRCC cell lines Caki and 769-P (n = 3, p value < 0.05 shown with *, t test). B, treatment with LY3039478 (1 μm) led to decreased expression of Myc and CCNA1 as assessed by qRT-PCR with GAPDH as control (n = 2, p value < 0.05 shown with *, t test). C, treatment of Caki cell line with LY3039478 led to significant increase in cells in G0/G1 and decreased cells in S and G2/M phases of cell cycle as assessed by FACS (n = 2, p value < 0.05 shown with *, t test). D, dosing with LY3039478 at 8 mg/kg via oral gavage three times a week resulted in increased overall survival when compared with vehicle control in CCRCC xenografts (log rank P value = 0.003). E, LY3039478 treatment also led to decreased tumor growth in CCRCC xenografts in another cohort of mice (t test <0.05). Ctrl, control.
Discussion
Our work demonstrates that the NOTCH pathway is overexpressed in a large proportion of clear cell RCC cases. The NOTCH pathway has been shown to be an oncogenic pathway in T cell acute lymphoblastic leukemia and head and neck cancers (16–18), and activating NOTCH mutations have been shown to occur in some of these tumors. Components of the NOTCH signaling pathway have been shown to be active in RCC also (10, 19, 20), although not many NOTCH mutations have been seen in these tumors. Furthermore, even though mutations have been described in the NOTCH receptors in some tumor samples, they are not sufficient to explain the activation of this pathway in the majority of tumors. Our findings show that up-regulation of NOTCH ligands JAG1 and JAG2 occur via loss of DNA methylation and gene amplifications. In addition to our data, an analysis of the TCGA data shows that both JAG1 and JAG2 are amplified in CCRCC. The TCGA data also reveals that JAG2 is also hypomethylated in CCRCC samples. Thus, it is clear that both genetic and epigenetic alterations are leading to overexpression of these NOTCH ligands in CCRCC samples. JAG1 and JAG2 can bind to the NOTCH receptor and result in two proteolytic cleavage events in the receptor that lead to its activation. Once the intracellular domain of NOTCH is cleaved, it interacts with downstream proteins to promote transcription (15, 21). Thus, our findings demonstrate novel mechanisms for the activation of this pathway in CCRCC. Most importantly, the observation of both epigenetic and genetic hits leading to activation of the NOTCH pathway in different patients suggests the importance of this pathway in pathogenesis of CCRCC.
Activation of the NOTCH pathway mediates effects via various downstream mediators such as transcription factors belonging to the HAIRY (HES and HEY) family. In an earlier epigenomic study in CCRCC, we observed that differentially methylated sites in tumor tissues were enriched for binding sites for HAIRY (8), supporting the role of NOTCH activation in downstream epigenetic changes. In the present study we observed that Myc as well as cyclin A1 and cyclin D1 were overexpressed in both NOTCH-driven renal lesions as well as in human tumors. Myc is a known direct target of the NOTCH pathway and leads to procarcinogenic proliferations seen in these tumors. A recent study demonstrated that NOTCH can bind to Myc enhancers and lead to activation of Myc transcription in T cell acute lymphoblastic leukemia (22). We observed that pharmacologic inhibition of the NOTCH pathway in CCRCC cells led to a significant decrease in Myc and cyclin A1 expression, demonstrating on target effects on cell cycle proliferative pathways.
Furthermore, we observed that intracellular activation of NOTCH in murine renal tubular cells leads to severe dysplasia in the kidneys. These dysplastic cells acquire histologic characteristics of CCRCC and also contain molecular changes that mimic human tumors. We also observed that CCRCC tumors that overexpress NOTCH pathway have a distinct clinical outcome. The improved survival for the Jag1/Jag2 overexpressors was compared with the other cases of CCRCC (that have various other molecular alterations including PI3K pathway, chromatin, and metabolic mutations). It is possible that even though the NOTCH pathway activation is oncogenic as seen from our in vivo data, there are other pathways that are even more rapidly oncogenic in RCC. Furthermore, the in vivo data demonstrate that NOTCH activation in part leads to transcriptional changes seen in human CCRCC. Because of the lack of valid precancerous models of CCRCC pathogenesis, our findings suggest that our murine model can be used for future pathogenic and therapeutic studies in this malignancy.
The NOTCH pathway is comprised of numerous family members that include ligands (Jag1, Jag2, and DLL), receptors (NOTCH 1–4), and downstream effectors. Examination of the TCGA and our data reveal that numerous ligands and receptors are overexpressed in CCRCC samples, demonstrating the importance of this pathway in this malignancy. This raises the possibility that signaling downstream from the NOTCH receptors can differ depending on the type of receptor that is activated or the ligand that is overexpressed. This can also lead to subtle differences in outcomes and survival rates, even though we saw that overall the trends were similar with overexpression of various NOTCH components. These differences that can arise from the complexity of the pathway will be tested in subsequent studies, especially if NOTCH pathway inhibitors are translated into clinical trials in CCRCC.
NOTCH inhibition has been explored as a therapeutic strategy in various tumors. γ-Secretase is an internal protease that cleaves within the membrane-spanning domain of the NOTCH protein thus leading to its activation. Various γ-secretase inhibitors have been developed and are being tested in clinical trials. Adverse effects such as gastrointestinal toxicities have been seen with previous inhibitors thus limiting their clinical development. LY-3039478 is a novel γ-secretase inhibitor that has shown specific activity in inhibiting the NOTCH pathway (23) and is being tested in clinical trials. In the present study, we demonstrate significant in vivo activity of this compound and provide a preclinical rationale for the use of this and similar agents in future trials in CCRCC.
Experimental Procedures
Patient Samples, Microdissection, and Nucleic Acid Extraction
In total, 13 RCC samples and 13 control samples were collected from tumor nephrectomies performed at Montefiore Medical Center. Samples were collected under institutional review board protocol approved by the Albert Einstein College of Medicine (supplemental Table S5). Kidney samples were obtained from living allograft donors, surgical nephrectomies, and leftover portions of diagnostic kidney biopsies. Nephrectomies were anonymized with the corresponding clinical information and were collected by an individual who was not involved in the research protocol. These samples were collected without consent. For protocol allograft and kidney biopsies, informed consent was obtained from the donor. Tissue was placed into RNALater and manually microdissected at 4 °C for the tubular compartment (as renal cell cancer originates from tubular epithelial cells) (24). We used a Zeiss stereomicroscope under 60× magnification for the microdissection. Genomic DNA was isolated using the dialysis tubing method, as performed and described previously (13). RNA was extracted using Qiagen RNeasy mini kits. An additional seven non-tumor and tumor pairs were used for qRT-PCR-based confirmation studies.
Genome-wide DNA Methylation Analysis Using the HELP Assay
The HELP assay was carried out as previously published (8, 25).
Quantitative DNA Methylation Analysis by MassARRAY EpiTYPER
Validation of HELP microarray findings was carried out by MALDI-TOF mass spectrometry using EpiTYPER by MassARRAY (Sequenom, CA) on bisulfite-converted DNA as previously described (26, 27). MassARRAY primers were designed to cover the flanking HpaII sites for a given HAF, as well as any other HpaII sites found up to 2,000 bp upstream of the downstream site and up to 2,000 bp downstream of the upstream site, to cover all possible alternative sites of digestion. Jag1 intergenic regions were examined by MassARRAY in primary CCRCC samples, as well as in decitabine-treated human kidney-derived cell lines.
Gene Copy Number Analysis Using MspI Representations from the HELP Assay
The MspI representation in a HELP assay is not affected by cytosine methylation but is instead dependent on the amount of DNA available and thus was used to detect copy number variation, as described previously (8, 25).
qRT-PCR for Jagged Ligands
cDNA from was used for qRT-PCR using the SYBR Green method with gene-specific primers using the ABI 7900HT machine. The data were normalized using HPRT as housekeeping gene and the ddCT method.
Cell Lines
Normal human kidney tubular epithelial cell line HK2 and renal cell cancer cell lines 786-O, 769-P, and Caki were purchased from ATCC (with authentication). HKC-8 cells were kindly provided by Lorainne Racusen (Johns Hopkins University). The cells were cultured in DMEM/F12 medium supplemented with 2.5% fetal bovine serum, antibiotics, insulin, transferrin, and selenium.
Overlap with Histone Modification Studies
All the ChIP-seq data were downloaded from the Roadmap database with GEO accession numbers specified below (NCBI Build GRCh37/UCSC Build hg19). The following data sets were used adult kidney BI.27: “GEO accession: GSM670025” adult kidney input 27; AK H3K4me3 BI.27:“GEO accession: GSM621648” AK H3K9ac BI.27: “GEO accession: GSM772811” AK H3K36me3 BI.27: “GEO accession: GSM621634” AK Input BI.27: “GEO accession: GSM621638.” All the ChIP-seq data were downloaded from the Roadmap database with GEO Series accession number of GSE19465. The Genome assembly is NCBI Build GRCh37/UCSC Build hg19. The nine cell line ChromHMM data were obtained from the UCSC genome browser (28). For adult kidney ChromHMM annotation map was generated using ChromHMM2 (29).
TCGA Data Analysis
Data for RNA-seq for RCC samples was obtained from the TCGA portal. These included 405 CCRCC samples and 68 non-tumor kidney controls (12).
Animals
Mice with TEC-specific overexpression of NOTCH1 intracellular domain and deletion of VHL were achieved by crossing the Kspcre mice (kindly provided by Dr. Igarashi, UTSW) with Gt(ROSA)26Sortm1(NOTCH1)Dam mice (obtained from Jackson Laboratory, stock no. 008159) and VHL floxed mice (Vhltm1Jae, obtained from The Jackson Laboratory, stock no. 004081). The major role of Jag1 and Jag2 is to activate Notch signaling. It has been difficult to express active Jagged1 on cell surface in vivo that would lead to Notch activation. This is mostly because following ligand presentation, Jagged1 is endocytosed with Notch extracellular domain. For these reasons, expression of Notch intracellular domain is the most accepted way to examine the role and effect of Notch signaling in vivo. Animals were identified by tail genomic PCR analysis. Male 8-week-old Kspcre VHLf/f ICNotch1 mice and Kspcre VHLf/f littermate controls were used in this study. Animal studies were approved by the Animal Care Committee of the University of Pennsylvania.
Transcriptome Analysis Overlap
Transcriptome analysis overlap was performed using differentially expressed genes (fold change > 2 and false discovery rate < 0.05) from Notch mouse model and CCRCC expression data. The orthologues were identified using MGI vertebrate homology. For the gene set enrichment analysis, the top 100 significant genes were selected from Notch data, and the enrichment levels were determined for the orthologues in CCRCC expression data.
Immunostaining
Control and RCC human kidney samples were performed using NOTCH1, DLL4, and JAGGED1 specific antibodies as previously described (30).
In Vitro and in Vivo Treatment with LY-3039478
For in vitro studies, LY3039478 was compared with DMSO controls in RCC cell lines. The cell cycle was assessed by FACS using propidium iodide staining. For in vivo studies, LY3039478 was administered via oral gavage formulated in 1% Na-CMC, 0.25% Tween 80, and 0.05% anti-foam. Dosing was done at 8 mg/kg on a Monday, Wednesday, and Friday schedule. Vehicle was used as control. CCRCC xenografts were established in NOD-scid IL2Rγ null mice with subcutaneous implantation using the 769-P cell line. Treatment was started after tumors were established. Tumor measurements were taken at the specified times in triplicate.
Author Contributions
T. D. B., Y. Z., S. H., J. P., M. P., C. H., W. L., N. S., O. G., G. C., Y. Y., Y.-A. K., M. C. I., A. S. D. P., N. V., R. La., R. Lo., M. S., and J. K. did the experiments; J. P., and B. G. contributed samples; A. A. H. analyzed data; B. P. and K. B. contributed reagents; and A. V. and K. S. wrote the manuscript
Supplementary Material
This work was supported by National Institutes of Health Grants R01DK087635, R01076077, and DP3 DK108220 (to K. S.) and a NYSTEM grant and an Albert Einstein Stem Cell Institute grant (to T. D. B.). B. P. and K. B. are employees of Eli Lilly. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Tables S1–S5.
- RCC
- renal cell carcinoma
- CCRCC
- clear cell renal cell carcinoma
- qRT-PCR
- quantitative RT-PCR.
References
- 1. Baldewijns M. M., van Vlodrop I. J., Schouten L. J., Soetekouw P. M., de Bruïne A. P., and van Engeland M. (2008) Genetics and epigenetics of renal cell cancer. Biochim. Biophys. Acta 1785, 133–155 [DOI] [PubMed] [Google Scholar]
- 2. Cohen H. T., and McGovern F. J. (2005) Renal-cell carcinoma. N. Engl. J. Med. 353, 2477–2490 [DOI] [PubMed] [Google Scholar]
- 3. Powles T., Chowdhury S., Jones R., Mantle M., Nathan P., Bex A., Lim L., and Hutson T. (2011) Sunitinib and other targeted therapies for renal cell carcinoma. Br. J. Cancer 104, 741–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Varela I., Tarpey P., Raine K., Huang D., Ong C. K., Stephens P., Davies H., Jones D., Lin M. L., Teague J., Bignell G., Butler A., Cho J., Dalgliesh G. L., Galappaththige D., et al. (2011) Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 469, 539–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Murtaugh L. C., Stanger B. Z., Kwan K. M., and Melton D. A. (2003) Notch signaling controls multiple steps of pancreatic differentiation. Proc. Natl. Acad. Sci. U.S.A. 100, 14920–14925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hakimi A. A., Pham C. G., and Hsieh J. J. (2013) A clear picture of renal cell carcinoma. Nat. Genet. 45, 849–850 [DOI] [PubMed] [Google Scholar]
- 7. Lamprecht B., Bonifer C., and Mathas S. (2010) Repeat-element driven activation of proto-oncogenes in human malignancies. Cell Cycle 9, 4276–4281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu C. Y., Mohtat D., Yu Y., Ko Y. A., Shenoy N., Bhattacharya S., Izquierdo M. C., Park A. S., Giricz O., Vallumsetla N., Gundabolu K., Ware K., Bhagat T. D., Suzuki M., Pullman J., et al. (2014) Kidney cancer is characterized by aberrant methylation of tissue-specific enhancers that are prognostic for overall survival. Clin. Cancer Res. 20, 4349–4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sjölund J., Boström A. K., Lindgren D., Manna S., Moustakas A., Ljungberg B., Johansson M., Fredlund E., and Axelson H. (2011) The notch and TGF-β signaling pathways contribute to the aggressiveness of clear cell renal cell carcinoma. PLoS One 6, e23057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sjölund J., Johansson M., Manna S., Norin C., Pietras A., Beckman S., Nilsson E., Ljungberg B., and Axelson H. (2008) Suppression of renal cell carcinoma growth by inhibition of Notch signaling in vitro and in vivo. J. Clin. Invest. 118, 217–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang Q. B., Ma X., Li H. Z., Ai Q., Liu S. W., Zhang Y., Gao Y., Fan Y., Ni D., Wang B. J., and Zhang X. (2014) Endothelial Delta-like 4 (DLL4) promotes renal cell carcinoma hematogenous metastasis. Oncotarget 5, 3066–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cancer Genome Atlas Research Network (2013) Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alvarez H., Opalinska J., Zhou L., Sohal D., Fazzari M. J., Yu Y., Montagna C., Montgomery E. A., Canto M., Dunbar K. B., Wang J., Roa J. C., Mo Y., Bhagat T., Ramesh K. H., et al. (2011) Widespread hypomethylation occurs early and synergizes with gene amplification during esophageal carcinogenesis. PLoS Genet. 7, e1001356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shao X., Somlo S., and Igarashi P. (2002) Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J. Am. Soc. Nephrol. 13, 1837–1846 [DOI] [PubMed] [Google Scholar]
- 15. Bielesz B., Sirin Y., Si H., Niranjan T., Gruenwald A., Ahn S., Kato H., Pullman J., Gessler M., Haase V. H., and Susztak K. (2010) Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J. Clin. Invest. 120, 4040–4054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Choi K., Ahn Y. H., Gibbons D. L., Tran H. T., Creighton C. J., Girard L., Minna J. D., Qin F. X., and Kurie J. M. (2009) Distinct biological roles for the notch ligands Jagged-1 and Jagged-2. J. Biol. Chem. 284, 17766–17774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weng A. P., Ferrando A. A., Lee W., Morris J. P. 4th, Silverman L. B., Sanchez-Irizarry C., Blacklow S. C., Look A. T., and Aster J. C. (2004) Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271 [DOI] [PubMed] [Google Scholar]
- 18. Sjölund J., Manetopoulos C., Stockhausen M. T., and Axelson H. (2005) The Notch pathway in cancer: differentiation gone awry. Eur. J. Cancer 41, 2620–2629 [DOI] [PubMed] [Google Scholar]
- 19. Wu K., Xu L., Zhang L., Lin Z., and Hou J. (2011) High Jagged1 expression predicts poor outcome in clear cell renal cell carcinoma. Jpn. J. Clin. Oncol. 41, 411–416 [DOI] [PubMed] [Google Scholar]
- 20. Pietras A., von Stedingk K., Lindgren D., Påhlman S., and Axelson H. (2011) JAG2 induction in hypoxic tumor cells alters Notch signaling and enhances endothelial cell tube formation. Mol. Cancer Res. 9, 626–636 [DOI] [PubMed] [Google Scholar]
- 21. Niranjan T., Bielesz B., Gruenwald A., Ponda M. P., Kopp J. B., Thomas D. B., and Susztak K. (2008) The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat. Med. 14, 290–298 [DOI] [PubMed] [Google Scholar]
- 22. Herranz D., Ambesi-Impiombato A., Palomero T., Schnell S. A., Belver L., Wendorff A. A., Xu L., Castillo-Martin M., Llobet-Navás D., Cordon-Cardo C., Clappier E., Soulier J., and Ferrando A. A. (2014) A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 20, 1130–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Massard C., Azaro A., Le Tourneau C., Soria J., Alt M., Smith C., Ohnmacht U., Oakeley G., Patel B., Yuen E., Benhadji K., and Ahnert J. (2015) First-in-human study of LY3039478, a Notch signaling inhibitor in advanced or metastatic cancer. J. Clin. Oncol. 33, [DOI] [PubMed] [Google Scholar]
- 24. Woroniecka K. I., Park A. S., Mohtat D., Thomas D. B., Pullman J. M., and Susztak K. (2011) Transcriptome analysis of human diabetic kidney disease. Diabetes 60, 2354–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oda M., Glass J. L., Thompson R. F., Mo Y., Olivier E. N., Figueroa M. E., Selzer R. R., Richmond T. A., Zhang X., Dannenberg L., Green R. D., Melnick A., Hatchwell E., Bouhassira E. E., Verma A., et al. (2009) High-resolution genome-wide cytosine methylation profiling with simultaneous copy number analysis and optimization for limited cell numbers. Nucleic Acids Res. 37, 3829–3839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Figueroa M. E., Reimers M., Thompson R. F., Ye K., Li Y., Selzer R. R., Fridriksson J., Paietta E., Wiernik P., Green R. D., Greally J. M., and Melnick A. (2008) An integrative genomic and epigenomic approach for the study of transcriptional regulation. PLoS One 3, e1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Figueroa M. E., Wouters B. J., Skrabanek L., Glass J., Li Y., Erpelinck-Verschueren C. A., Langerak A. W., Löwenberg B., Fazzari M., Greally J. M., Valk P. J., Melnick A., and Delwel R. (2009) Genome-wide epigenetic analysis delineates a biologically distinct immature acute leukemia with myeloid/T-lymphoid features. Blood 113, 2795–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ernst J., Kheradpour P., Mikkelsen T. S., Shoresh N., Ward L. D., Epstein C. B., Zhang X., Wang L., Issner R., Coyne M., Ku M., Durham T., Kellis M., and Bernstein B. E. (2011) Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ernst J., and Kellis M. (2012) ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kinney S. R., and Pradhan S. (2013) Ten eleven translocation enzymes and 5-hydroxymethylation in mammalian development and cancer. Adv. Exp. Med. Biol. 754, 57–79 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.