

Abstract
LDL receptor-related protein 1 (LRP1) is a highly modular protein and the largest known mammalian endocytic receptor. LRP1 binds and internalizes many plasma components, playing multiple crucial roles as a scavenger and signaling molecule. One major challenge to studying LRP1 has been that it is difficult to express such a large, highly glycosylated, and cysteine-rich protein, limiting structural studies to LRP1 fragments. Here, we report the first recombinant expression of the complete 61 domains of the full-length LRP1 ectodomain. This advance was achieved with a multistep cloning approach and by using DNA dilutions to improve protein yields. We investigated the binding properties of LRP1 using receptor-associated protein (RAP) as a model ligand due to its tight binding interaction. The LRP1 conformation was studied in its bound and unbound state using mass spectrometry, small-angle X-ray scattering, and negative-stain electron microscopy at neutral and acidic pH. Our findings revealed a pH-dependent release of the ligand associated with a conformational change of the receptor. In summary, this investigation of the complete LRP1 ectodomain significantly advances our understanding of this important receptor and provides the basis for further elucidating the mechanism of action of LRP1 in a whole and integrated system.
Keywords: electron microscopy (EM), mass spectrometry (MS), pH regulation, small-angle X-ray scattering (SAXS), surface plasmon resonance (SPR), LRP1, receptor-associated protein
Introduction
Low-density lipoprotein receptor-related protein-1 (LRP1)2 is the largest member of the mammalian LDL receptor family, further consisting of LDL receptor (LDLR), very low-density lipoprotein receptor (VDLR), apolipoprotein E-receptor-2 (apoER2), LRP1B, megalin, LRP4, LRP5, and LRP6. LRP1 is ubiquitously expressed in brain endothelium, neurons, astrocytes, smooth-muscle cells, macrophages, fibroblasts, and hepatocytes (1). As a scavenger and signaling molecule, it is involved in many biological processes ranging from lipoprotein metabolism, proteinase homeostasis, fibrinolysis, vascular signaling, and development and maintenance of blood-brain barrier integrity (1–3). LRP1 is able to bind and endocytose an array of structurally and functionally different ligands such as apolipoproteins (4), proteinases and proteinase-inhibitor complexes (5), blood coagulation factors (2, 3), growth factors (3, 6), matrix metalloproteinases (7), viruses (8), and bacterial toxins (9). The cellular uptake of ligands, as in the case of other LDL receptor molecules, involves receptor-mediated endocytosis via clathrin-coated pits (10). After uptake, the receptor-ligand complex is delivered to the endosomal compartments, where the ligand dissociates. Although the receptor is recycled to the cell surface, the ligand is further degraded in the lysosomal compartments (11, 12).
Human LRP1, with 4,525 amino acid residues, is one of the largest glycoproteins known. With at least 159 disulfide bonds and 52 predicted N-glycans, LRP1 is a remarkably challenging molecule for expression. It is composed of two non-covalently associated chains, the α-chain (515 kDa) containing the ligand binding regions and the β-chain (85 kDa) composed of the trans-membrane spanning region and intracellular domains (13). The ectodomain of LRP1, which consists of the α-chain and a short extracellular portion of the β-chain, has a highly modular composition resembling that of other LDL receptor molecules. It consists of four clusters, containing in total 31 complement-like cysteine-rich (CR) ligand binding repeats, 22 cysteine-rich epidermal growth factor (EGF)-like repeats, and 8 YWTD six-bladed β-propellers. High-resolution structural information is available for CR repeats only (14–18), and low-resolution electron spectroscopic microscopy images of full-length LRP1 purified from human placenta have previously suggested an elongated “zig-zagged” shape spanning 50–70 nm in length with a globular domain at one end (19). However, detailed structural studies of the full molecule have been hindered by the difficulties in producing recombinant LRP1.
The LRP1 biosynthesis is assisted by the 39-kDa receptor-associated protein (RAP), an endoplasmic reticulum (ER) resident chaperone that contributes to the maturation of different LDL receptors. RAP was initially discovered as a protein that co-eluted with LRP1 when affinity-purified from placenta (5, 20, 21). Later studies showed that RAP binds in the early secretory pathway, preventing premature interaction of LRP1 with other ligands (22–24). RAP dissociates from LRP1 in the Golgi, triggered by the lower pH environment (23). Due to its tight binding to LRP1, recombinant RAP has been widely used in biochemical and cellular assays as a universal antagonist for ligand binding studies (22, 25). RAP is composed of three domains: D1, D2, and D3. Each domain consists of a three-helix bundle connected by flexible linker regions (26). Domain 3 (RAP-D3) has been shown to be the most important for trafficking and folding of LRP1 (27) and exhibits the highest LRP1 binding affinity (28, 29).
RAP and most of the other known LRP1 ligands bind to CR repeats within LRP1 clusters II and IV (30–33), whereas only RAP is able to bind LRP1 cluster III (31, 34). The LRP1 ligand binding model, referred to as an “acidic necklace,” involves calcium-coordinating acidic residues on the CR repeats that interact with positively charged residues of the ligand, preferentially lysines (35–37). This mechanism has been proposed to be widely adopted in ligand-receptor interactions for the LDL receptor family (38). The ligand uncoupling mechanism has been linked to the low-pH environment of the endosomal compartments, which is thought to trigger a structural transition of the LDL receptor from an open active conformation at neutral pH to a closed form at acidic pH (39). However, the mechanism by which LRP1 can recognize and bind such a variety of ligands is still not fully understood. So far, the stoichiometry of LRP1 ligand binding remains unknown. Moreover, binding studies on single clusters have reported somewhat inconsistent results regarding the affinities and specificities of cluster-to-ligand binding events. As suggested previously, these ambiguities may be rationalized by the utilization of non-identical, ligand-specific epitopes and the requirement of multiple LRP1 domains for proper endocytosis (32). Thus, the entire arrangement of domains of LRP1 is needed to fully understand its mode of action. This challenge is addressed in the present study, demonstrating for the first time the recombinant expression of the full-length ectodomain of LRP1. Using RAP as a model ligand, we characterize the affinity and the stoichiometry of the binding to LRP1. Furthermore, we present the conformation of the receptor, in its liganded and unliganded state, at neutral and acidic pH.
Results
Functional Expression of Full-length LRP1
To confirm the proper expression of LRP1 and to investigate its localization, we transfected HEK293T cells with full-length LRP1 (residues 20–4544) containing a C-terminal GFP tag. Fig. 1A illustrates the LRP1 cloning strategy. Expression of the receptor was assessed by taking images with a confocal microscope. LRP1-GFP was expressed by a sub-portion of cells, and it appeared to be present on the cell surface and in subcellular compartments (Fig. 2A), probably endosomes, in agreement with what was reported by Laatsch et al. (12). To quantify how much LRP1 was expressed on the cell surface, we expressed LRP1-GFP in HEK293-Epstein-Barr virus nuclear antigen (EBNA) 1 cells (U-Protein Express) and conducted flow cytometry using a monoclonal antibody against LRP1 ectodomain. We found that ∼50% of the GFP-positive cells are expressing LRP1 on the cell surface (Fig. 2B).
FIGURE 1.

LRP1 cloning strategy and constructs for expression. A, illustration of the multistep strategy adopted to clone full-length LRP1. B and C, schematic representation of the constructs used for expression of full-length LRP1 ectodomain (B) and of full-length RAP (C).
FIGURE 2.

Localization of recombinant full-length LRP1 in HEK293 cells. A, confocal microscopy images of HEK293T cells expressing LRP1-GFP. Nuclei were visualized by DAPI and appear in blue. Bar is 20 μm. B, FACS analysis of LRP1-GFP in HEK293-EBNA1 cells using a monoclonal antibody against LRP1 ectodomain (8G1). Approximately 50% of the GFP-positive cells expressed LRP1 on the cell surface (right panel). Negative controls consisted of cells without any GFP expressed (left panel) and cells expressing LRP1-GFP incubated in the absence of the secondary antibody (middle panel).
Expression of Entire LRP1 Ectodomain with and without RAP
We tested different vectors for optimal expression of the full-length LRP1 ectodomain (residues 20–4409) in HEK293-EBNA1 cells and chose the construct with a C-terminal TEV-StrepII3-His6 tag, which was used for all further experiments. Fig. 1, B and C, illustrates the constructs used for expression. This construct contains the whole α-chain and the extracellular part of the β-chain with an expected molecular mass of 488 kDa without glycans. We did not observe any cleavage of the secreted protein. Expression of LRP1 was performed with and without RAP, demonstrating that RAP is not essential for secretion of LRP1 because yields of about 300 μg of protein per liter of culture were obtained without RAP co-expressed. LRP1 eluted on size-exclusion chromatography (SEC) as a single peak with some aggregation that migrated with the void volume (Fig. 3A). On SDS-PAGE, it appeared as a thick band that migrated higher than the 250-kDa molecular mass marker with less prominent bands at slightly lower molecular masses, most probably due to heterogeneity in the N-linked glycosylation (Fig. 3). Under non-reducing conditions, small amounts of aggregates were observed, which are probably due to improper disulfide bond formation because these higher mass bands were absent under reducing conditions. When LRP1 was co-expressed with RAP, an ∼2-fold increase in LRP1 production was observed, indicating that co-expression of RAP may facilitate LRP1 secretion (Fig. 3B). We observed that RAP was co-secreted and co-purified with LRP1 despite the fact that the RAP construct used for co-expression contained the ER retention signal HNEL at the C terminus. Possibly, the secretion machinery is overwhelmed at high expression levels, as observed before (40). Another band around 70 kDa also co-eluted with LRP1. Bottom-up proteomics analysis identified this band as human Hsp70.
FIGURE 3.

Purification of full-length LRP1 ectodomain and LRP1-RAP complex, and dissociation of RAP at acidic pH. A, SEC profile at pH 7.5 of LRP1 and detection on SDS-PAGE gel. mAU, milliabsorbance units; M, molecular weight markers; βME, β-mercaptoethanol. B, SEC profile at pH 7.5 of LRP1-RAP complex after co-expression and detection on SDS-PAGE gel. C, SEC profile at pH 7.5 of LRP1-RAP complex (green dashed line) superposed to the SEC profile at pH 5.5 of LRP1-RAP (violet line) showing dissociation of LRP1 (peak 1) from RAP (peak 2); detection on SDS-PAGE after SEC at pH 5.5 of LRP1-RAP. D, SEC profile at pH 7.5 of LRP1 (blue line) after SEC at pH 5.5 of LRP1-RAP (violet dashed line) and dissociation of RAP (peak 1 from panel C).
LRP1 Binds RAP with High Affinity at pH 7.4
We used SPR analysis to test the binding of the purified recombinant LRP1 to full-length RAP, RAP-D3, and RAP-D3 K256A/K270A, a variant in which two critical lysine residues were mutated into alanine (41). Initially, we coupled one batch of purified recombinant LRP1 at three different densities (1.3, 3.8, and 8.3 fmol/mm2 respectively) onto a CM5 chip. As expected, RAP-D3 K256A/K270A showed no appreciable interaction, whereas full-length RAP and RAP-D3 bound to the chip with high affinity (Fig. 4A). The binding kinetics, however, did not fit a 1:1 binding model. To rule out mass transport limitations in the binding data as well as protein batch-dependent artifacts, we assessed the binding at three different flow rates (30, 60, and 90 μl/min) for three different batches of purified recombinant LRP1 coupled onto a CM5 chip at 2.5 fmol/mm2. Virtually identical association and dissociation curves were obtained for all analyses, confirming that the LRP1-RAP interaction does not comply with a 1:1 binding model. Therefore, to estimate KD values, we again coupled three different batches of purified recombinant LRP1 at a ligand density of 2.5 fmol/mm2 onto a CM5 chip, repeated the first analysis, and fitted the responses at equilibrium by non-linear regression using a standard hyperbola (GraphPad Prism 4 software) (Fig. 4B). The combined analysis indicated an affinity of 9 ± 5 nm (n = 27) for full-length RAP and 22 ± 2 nm (n = 6) for RAP-D3. The affinity for the RAP-D3 variant containing the K256A/K270A could not be assessed because binding was virtually absent. To estimate the apparent RAP-LRP1 binding stoichiometry, we calculated the ratio of observed Rmax to the theoretical Rmax expected for a 1:1 binding model, assuming a molecular mass of 600 kDa for LRP1, 40 kDa for full-length RAP, and 12.8 kDa for RAP-D3. Both full-length RAP and RAP-D3 bound with a ratio of 1.5 ± 0.1 to LRP1, suggesting that more than one RAP molecule can bind LRP1 simultaneously.
FIGURE 4.

SPR analysis of LRP1-RAP binding interaction. Association and dissociation of RAP, RAP-D3, and RAP-D3 K256A/K270A to recombinant LRP1 was assessed by SPR analysis. Responses at equilibrium were plotted as a function of the analyte concentration. A, one batch of purified recombinant LRP1 was immobilized to a CM5 chip at three different ligand densities (1.3, 3.8, and 8.3 fmol/mm2, respectively). Subsequently, RAP (0–2560 nm), RAP-D3 (0–800 nm), and RAP-D3 K256A/K270A (0–800 nm) were passed over the immobilized LRP1 at a flow rate of 30 μl/min. Binding to LRP1 was corrected for binding in the absence of LRP1. B, three batches of purified recombinant LRP1 were immobilized to a CM5 chip (2.5 fmol/mm2) as described above. Subsequently, RAP (0–2560 nm) was passed over the immobilized LRP1 as described above with a flow rate of 30, 60 or 90 μl/min.
LRP1 Has Two RAP Binding Sites
To further determine the stoichiometry of RAP binding to LRP1, we applied native MS, which preserves non-covalent interactions and the native-like structure of proteins in the gas phase (42). Native MS analysis of LRP1 is particularly challenging because the presence of numerous N-glycans causes a high degree of protein microheterogeneity. To facilitate native MS experiments, LRP1 and RAP were separately produced in N-acetylglucosaminyltransferase I-deficient (GnTI−) HEK293-EBNA1-S cells, which yield shorter and more homogenous N-linked glycan chains (43). The native mass spectrum of LRP1 ectodomain showed a clearly resolved charge-state series, allowing us to calculate its apparent molecular mass to 565.6 ± 0.4 kDa (Fig. 5A).
FIGURE 5.

Native MS analysis of LRP1-RAP interaction. Peak labels indicate the charge state of the respective analyte ion. All LRP1 and LRP1-RAP species are represented by a series of consecutively charged ions (selected charge states are indicated above the peaks), enabling molecular mass determination. These charge state distributions and corresponding molecular masses are color-coded. LRP1 is shown in blue, LRP1-RAP 1:1 complex is shown in orange, and LRP1-RAP 1:2 complex is shown in green. Dissociated LRP1 is shown in violet, when measured at pH 5, and in light blue, when measured at pH 7.5. Peaks shown in black could not be confidently assigned as continuous charge state distributions, suggesting that they represent a mixture of several less abundant species with overlapping charge states. The mass spectra shown as insets have been deconvoluted from m/z to mass domain. A–G, displayed are native mass spectra of LRP1 (A), LRP1-RAP complex formed in vitro after incubating both proteins at different molar ratios (indicated in each mass spectrum) (B), the co-expressed LRP1-RAP complex (C), and pH-shift experiments to study the dissociation and re-association of LRP1 and RAP (D–G). The corresponding experimental conditions are listed in the figure. RSI = relative signal intensity.
Subsequently, native MS was used to monitor in vitro LRP1-RAP complex formation after incubating LRP1 with increasing concentrations of RAP (Fig. 5B). We observed that sub-stoichiometric amounts of RAP (molecular mass ∼39 kDa) are sufficient to form a complex with a molecular mass of 605.6 ± 0.7 kDa, which represents a LRP1-RAP 1:1 complex. More than 2-fold molar excess of RAP is required to form LRP1-RAP 1:2 complex with a molecular mass of 645.3 ± 0.5 kDa. Although the three species exhibit extensively overlapping charge state envelopes, the characteristic spacing between the charge states allows for their unambiguous assignment, enabling us to determine three distinct molecular masses (Fig. 5B). These results suggest the presence of two RAP binding sites on LRP1, which seem to exhibit different binding affinities.
Native MS analysis was also performed after co-expressing LRP1 and RAP in HEK293-EBNA1-S cells (Fig. 5C). Under these conditions, the LRP1-RAP 1:2 complex with a molecular mass of 643.2 ± 0.5 kDa was clearly the most abundant species. Interestingly, charge states corresponding to the LRP1-RAP 1:1 complex were not observed, suggesting that this species is much less abundant or entirely absent when LRP1 and RAP are co-secreted. Thus, during secretion, the LRP1-RAP complex forms preferentially with a 1:2 LRP1:RAP binding stoichiometry. Next, we applied cross-linking MS to assess the regions involved in binding (42). In total, we identified 46 unique lysine-lysine connections (Fig. 6A, supplemental Table 1). Within LRP1, we observed 25 intramolecular cross-links connecting lysines that are up to eight domains apart. Thus, the cross-linking MS data suggest a certain degree of structural compaction but do not provide evidence for a globular conformation. We also verified 15 intramolecular RAP cross-links, mainly representing contacts within RAP-D3 and between RAP-D3 and -D2 (Fig. 6B). Moreover, six intermolecular LRP1-RAP cross-links provide insights into the LRP1-RAP binding interfaces. Three intermolecular cross-links connect RAP-D3 with CR25–27 within LRP1 cluster IV. The other three intermolecular cross-links connect RAP-D2 with the LRP1 β-propeller regions between cluster II and III. Interestingly, the involved RAP-D2 lysines are also internally connected to each other and to RAP-D3 (Fig. 6B). This raises the possibility that RAP exhibits one LRP1 binding region to which all observed intermolecular cross-links can be mapped. By contrast, there is no evidence for intermolecular interactions between the LRP1 regions that are cross-linked to RAP. Therefore, it is more likely that the two groups of LRP1-RAP cross-links correspond to two distinct RAP binding sites on LRP1.
FIGURE 6.
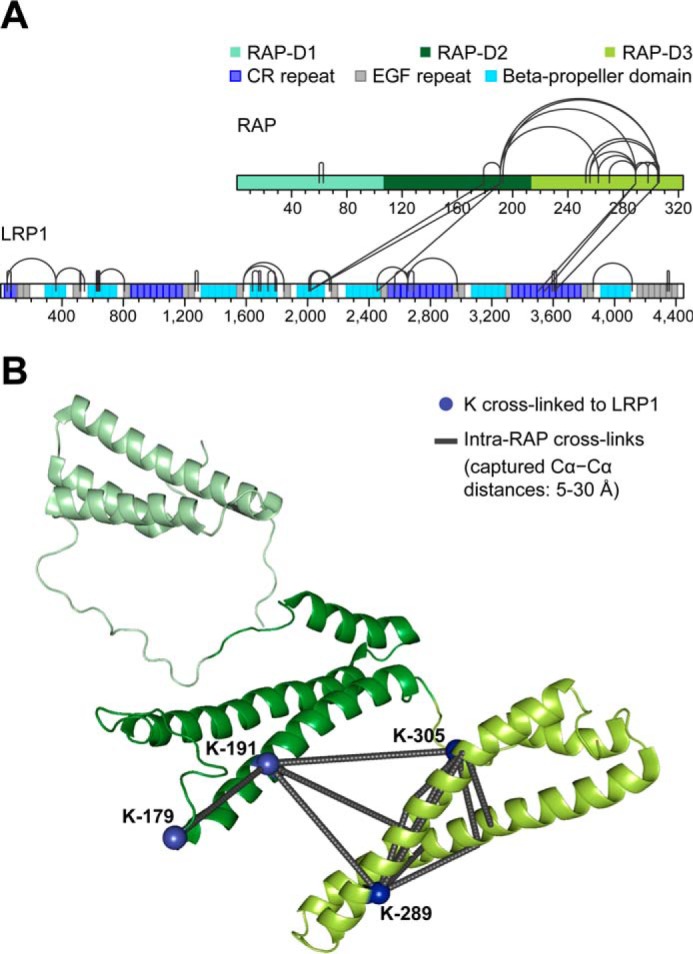
Cross-linking MS analysis of the co-expressed LRP1-RAP complex. A, sequence bar plot of all identified cross-links. The domain boundaries within RAP and LRP1 are taken from Lee et al. (26) and from UniProt entry Q07954, respectively. B, structural representation of intramolecular RAP cross-links. The cross-links were mapped onto all structurally characterized RAP conformers (Protein Data Bank (PDB) entry 2P01). Shown here is the RAP conformer structure that agrees best with the cross-linking distance constraints. RAP lysines involved in intermolecular cross-links to LRP1 are depicted as blue spheres.
Acidic pH Affects RAP Binding and Conformation of LRP1
We investigated whether the expressed soluble LRP1 ectodomain releases RAP at pH <6. SEC analysis at pH 5.5 showed that LRP1-RAP complex dissociates into two peaks corresponding to LRP1 and RAP (Fig. 3C). Complementarily, native MS demonstrated that the in vitro reconstituted LRP1-RAP complex fully dissociates when analyzed at pH 5 (Fig. 5D). The LRP1 charge state distributions at pH 5 and pH 7.5, however, are highly similar (compare Figs. 5A and 5D), suggesting that LRP1 retains a folded conformation at acidic pH.
Next, we probed the reversibility of the pH-induced RAP release. To this end, the co-secreted LRP1-RAP complex was subjected to SEC at pH 5.5, leading to full dissociation of the complex as evidenced by native MS analysis at pH 5 and pH 7.5 (Fig. 5, E and D). Raising the sample pH to 7.5 and incubating it with a molar excess of RAP, however, restored the LRP1-RAP 1:2 complex (Fig. 5F), showing that LRP1-RAP association/dissociation is a reversible, pH-dependent process.
To investigate the effect of the pH on LRP1 conformation, we conducted small-angle X-ray scattering (SAXS) measurements with on-line SEC on soluble LRP1 and on the LRP1-RAP complex at pH 7.5 and pH 5.5. At neutral pH, LRP1 and LRP1-RAP complex behave very similarly as shown by the almost overlapping scattering curves (Fig. 7A). Guinier and distance distribution function analysis showed similar radius of gyration (Rg) values and a maximum particle dimension (Dmax) of ∼39 nm for LRP1 and of ∼38 nm for LRP1-RAP (Fig. 7, B and C, Table 1). Moreover, the Kratky plot of LRP1-RAP shows a small shoulder appearing at higher s ranges, which is also observed for free LRP1, although the curve is noisier in this region (Fig. 7D). This feature hints at a conformation with a globular part and an elongated flexible extremity. A plateau is reached at s = 1.5–2.0 nm−1, indicating partial flexibility for both species. At acidic pH (i.e. conditions where RAP is released), both LRP1 and LRP1-RAP exhibited a different behavior, showing decreased values of Rg and a Dmax of ∼35 nm for LRP1 only and ∼32 nm for LRP1 after RAP dissociation (Table 1). The Kratky plot for LRP1 at pH 5.5 adopts a more bell-like shape, which converges to a plateau around 1.0 nm−1 (Fig. 7D). Also, for LRP1 after RAP release, the Kratky plot indicates a more compact shape when compared with the curves at neutral pH values, although it is noisier. Overall, the SAXS data suggest that, at low pH, LRP1 undergoes a conformational change acquiring a more compact shape. Based on the values of Rg and Dmax, the pH-dependent conformational change appears more enhanced when there is concomitant release of RAP.
FIGURE 7.

SAXS analysis of LRP1 and LRP1-RAP complex at pH 7.5 and pH 5.5. LRP1 at pH 7.5 is shown in blue, LRP1-RAP at pH 7.5 is shown in green, LRP1 at pH 5.5 is shown in red, and LRP1-RAP at pH 5.5 is shown in violet. A, superposition of scattering curves scaled for comparison. rel. units, relative units. B, Guinier plot. C, normalized distance distribution function. D, normalized Kratky plot.
TABLE 1.
SAXS parameters
| LRP1, pH 7.5 | LRP1-RAP, pH 7.5 | LRP1, pH 5.5 | LRP1(-RAP), pH 5.5 | |
|---|---|---|---|---|
| Rg from Guinier (nm) | 11.2 | 10.8 | 10.3 | 9.6 |
| Rg from P(r) (nm) | 11.6 | 11.3 | 10.4 | 9.4 |
| Dmax from P(r) (nm) | 39.1 | 37.9 | 35.3 | 32.3 |
Additional SEC studies showed that, after dissociation from RAP at acidic pH, the LRP1 elution peak shifts back to its usual elution volume at higher apparent molecular masses if the pH is brought back to 7.5 (Fig. 3D). This indicates that the LRP1 conformational change is reversible, which is in good agreement with the observed restorability of the LRP1-RAP interaction (Fig. 5, D–F).
LRP1 Is Structurally Highly Flexible
To obtain structural information complementary to that provided by SAXS, we performed negative-stain EM studies on LRP1 and LRP1-RAP complex after SEC at neutral and at acidic pH. EM of negatively stained particles revealed for all conditions a very heterogeneous population. Such variety in the particle shape points toward a high degree of flexibility, where some particles have a more compact-kinked conformation, whereas others are more elongated. Reference-free alignment and classification yielded multiple orientations of particles with a maximum length of ∼35 nm. In Fig. 8, the most 10 populated 2D-averaged classes with the highest signal-to-noise ratio are shown. Also, when samples were prepared after SEC at pH 5, no significant difference in conformation and size was observed between LRP1 and LRP1-RAP. Possibly, the pH of the negative-stain affected the sample preparation, compromising the analysis of pH-dependent conformational effects and RAP binding.
FIGURE 8.
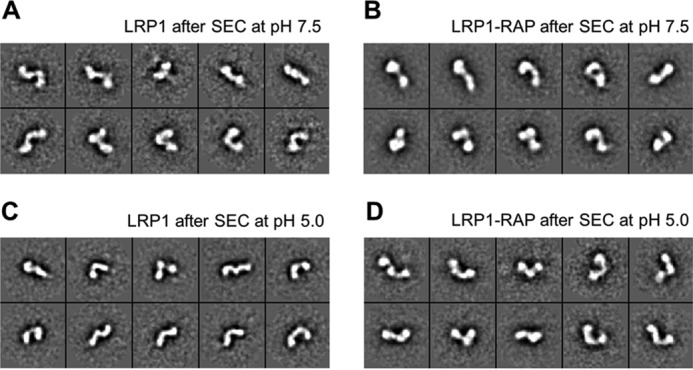
Negative-stain EM 2D class averages of LRP1 and LRP1-RAP complex. The averaged 10 most populated classes with the highest signal-to-noise ratio of a total of ∼2,000 particles manually picked for each sample preparation are shown. The box size is 62 nm2. A, LRP1 after gel filtration at pH 7.5. B, co-expressed LRP1-RAP complex after gel filtration at pH 7.5. C, LRP1 after gel filtration at pH 5.5. D, co-expressed LRP1-RAP after gel filtration at pH 5.5 and dissociation of RAP.
Discussion
LRP1 is a challenging protein to express recombinantly due to its size and high number of glycans and disulfide bridges. We were able for the first time to express the entire ectodomain of LRP1 and characterize its conformation, binding properties, and pH dependence, using RAP as a model ligand. In our experimental setup, RAP was not essential for proper LRP1 secretion, but the presence of co-expressed RAP resulted in higher protein production. This supportive effect is likely caused by stable interactions between RAP and LRP1 because SEC and native MS have shown that they are co-secreted as a stable complex, likely with a 1:2 LRP1:RAP binding stoichiometry (Figs. 3B and 5C). SPR showed apparent affinity constants in the low nanomolar range, which are in agreement with previous studies (35, 41) (Fig. 4B). Native MS-based titration experiments showed that LRP1 harbors two RAP binding sites (Fig. 5B). These sites probably exhibit different binding affinities because sub-stoichiometric amounts of RAP were sufficient to form the LRP1-RAP 1:1 complex, whereas a more than 2-fold molar excess of RAP was needed to observe the LRP1-RAP 1:2 complex. The corresponding LRP1-RAP binding interfaces were probed with cross-linking MS (Fig. 6A). Although cross-linking MS, as such, cannot directly elucidate binding stoichiometries, it is likely that the verified intermolecular connections represent more than one LRP1-RAP interface. In line with previous studies (27, 33), the data show that CR25-CR27 is the most effective RAP binding region within cluster IV and that RAP-D3 is an important LRP1 binding site. A second binding site is probably captured by three intermolecular cross-links connecting the LRP1 β-propeller domains 5 and 6 at the boundary between cluster II and III with RAP-D2.
The presence of two RAP binding sites is consistent with Williams et al. (55) and with the findings that cluster II and IV are the most effective ligand binding regions of LRP1 (31–33, 40, 44). However, it is important to point out that direct cross-links to the CR repeats region of cluster II are absent. A possible explanation is that the cross-linker did not penetrate the immediate cluster II binding site because the cluster II lysines were spatially not accessible, involved in non-covalent interactions, or generally less reactive (e.g. due to the local pKa) (45). This hypothesis is supported by the fact that intramolecular cross-links among the cluster II lysines are also lacking. Instead, the cross-linker may have reacted with lysines that are located in neighboring regions, such as the β-propellers, that might be proximal to the binding interface. Notably, the location of the cross-links between the CR repeats regions of cluster II and III also supports alternative explanations. We cannot exclude that RAP is binding to cluster III, which is known to contain a lower affinity RAP binding site (31, 34). Likewise, the involvement of a yet uncharacterized binding site cannot be ruled out. The previously suggested existence of more than two RAP binding sites, however, is not in agreement with our native MS results.
The overall conformation of LRP1 remains largely unchanged upon RAP binding, as shown by the similarity of the SAXS curves and by the negative-stain EM class averages (Figs. 7A and 8, A and B). Conformational transitions upon RAP binding might be happening at a local level, perhaps influencing the relations of the neighboring domains without producing large structural rearrangements visible at low resolution, as hypothesized by Migliorini et al. (35). SAXS analysis showed a maximum particle diameter of 39 nm for LRP1 at neutral pH (Table 1), whereas from negative-stain EM, we could measure a maximum particle dimension of 35 nm. This difference is probably due to the hydration shell and the flexibility of the system, leading to an apparent bigger dimension for SAXS. Averaged EM classes showed, in fact, that the particles are highly flexible. Unfortunately, the inhomogeneity of the particles hinders any high-resolution 3D structure determination. However, a maximum diameter of less than 40 nm precludes a model where LRP1 is completely elongated because, based on the LDLR X-ray structure (39), it would be spanning almost 100 nm. Therefore, it is reasonable to assume that there is a certain degree of compaction in LRP1 conformation, which is also in agreement with our cross-linking MS data.
According to the proposed ligand internalization mechanism, LRP1 should release its cargo at pH <6 to recycle to the cell surface. SEC studies at pH 5.5 showed complete dissociation of RAP from LRP1 (Fig. 3C). This finding was confirmed by native MS analysis, which moreover indicated that LRP1 remains folded at pH 5 (Fig. 5D). Using negative-stain EM, SAXS, and SEC, we compared the behavior of LRP1 and LRP1-RAP complex at pH <6. Size analysis from SAXS data showed that after RAP release at pH 5.5, LRP1 Dmax decreases from 38 to 32 nm, whereas for unbound LRP1, it shifts from 39 to 35 nm (Fig. 7, Table 1). SEC studies showed a shift of LRP1 elution volume toward lower molecular mass when the pH was acidified (Fig. 3C). Interestingly, when the pH was brought back to neutral values, LRP1 migrated as a higher molecular mass species again, indicating that the conformational change is reversible (Fig. 3D). Correspondingly, the RAP binding capabilities of LRP1 could be restored by shifting the pH from 5 to 7.5 (Fig. 5, E–G). These results suggest that LRP1 switches between a RAP binding-competent conformation at neutral pH and a binding-incompetent conformation at acidic pH. The acidic LRP1 conformation appears to be somewhat more compact, similarly to what has been shown for LDLR (39). Therefore, the release of the LRP1 cargo at acidic pH is probably due to a conformational change of the receptor, possibly with intramolecular interactions competing for the RAP binding sites and promoting its release. Intriguingly, according to the SAXS data, the extent of LRP1 conformational change at acidic pH seems to be influenced by the presence of RAP. This could be due to the existence of two RAP binding sites with different binding kinetics. When the pH is lowered, one RAP molecule could be released firstly, whereas the second, more tightly bound RAP molecule remains bound to LRP1 for a slightly longer time, promoting the conformational change of the receptor. Then, after interacting with both sites and causing compaction of the receptor, the second RAP molecule is also released. This hypothesis also provides an explanation for the previously made observation that RAP can bind more than one LRP1 site at the same time (31). Altogether, this initial structural and functional investigation of the complete LRP1 ectodomain significantly advances our understanding of this complex and important receptor and forms the basis to further characterize its binding properties using the full-length molecule.
Experimental Procedures
Constructs for Recombinant Expression of Full-length LRP1 and Entire Ectodomain
Human LRP1 (isoform 1) cDNA pcDNA 3.1(−) Neo vector with NotI and XhoI restriction sites (kindly provided by Dr. Joachim Herz, University of Texas, Dallas) was used in a multistep cloning approach to clone full-length LRP1 and LRP1 soluble ectodomain into the in-house mammalian expression system. First, the two DNA fragments with nucleotide sequences 57(BglII)-3952 and 3952–6018(XbaI) were obtained in two separate PCR reactions using primers designed to mutate the native BglII site at position 3952. PCR products from both reactions were mixed in an equimolar ratio, and an overlap extension PCR reaction was performed using PfuTurbo Hotstart DNA Polymerase (Agilent Technologies) to obtain the mutated insert 57–6018 (insert 1 in Fig. 1). Insert 1 was further cloned into a gateway recombinational cloning entry vector (pCR8/GW/TOPO, Invitrogen). The DNA sequence of the whole mutated area was confirmed by DNA sequencing. Further, in another two separate PCR reactions, DNA fragments with nucleotide sequences 6018–13635 and 6018–13227 (inserts 2 and 3 in Fig. 1) were obtained using primers that introduced a NotI site at the 3′ end. Inserts 2 and 3 were subsequently cloned into gateway recombinational cloning entry vectors (pCR8/GW/TOPO, Invitrogen), and their DNA sequence was confirmed by DNA sequencing. As a final step, insert 1 containing Xba1 at the 3′ end (obtained by restriction digestion using BglII and XbaI) and insert 2 containing Xba1 at the 5′ end (obtained by restriction digestion with XbaI and NotI) were combined in an equimolar ratio in a ligation reaction to obtain the full-length receptor (nucleotides 57–13635); in another ligation reaction, insert 1 was combined with insert 3 to obtain the complete ectodomain (nucleotides 57–13227). Both constructs had a Bgl2 site at 5′ and a NotI site at 3′ for further cloning into mammalian expression vectors. DNA sequencing revealed that our LRP1 clone is a natural variant with Pro instead of Gln in amino acid position 2900 (UniProt Q07954). Full-length LRP1 was subcloned in expression vector pUPE7.21 with N-terminal cystatin secretion signal and C-terminal eGFP His tag for confocal microscopy experiments. LRP1 ectodomain was subcloned in several mammalian expression vectors including pUPE107.55 (N-terminal cystatin secretion signal and C-terminal TEV-His6), pUPE7.50 (N-terminal cystatin secretion signal and C-terminal TEV-StrepII3-His6), and pUPE3478 (cystatin C-terminal TEV-eGFP-StrepII3), which were used for expression tests. The construct in vector pUPE7.50 (N-terminal cystatin secretion signal and C-terminal TEV-StrepII3-His6) showed the best expression yield.
Constructs for Recombinant Expression of RAP
For co-expression with LRP1, a construct of full-length human RAP (pcDNA kindly provided by Dr. Madelon Maurice, University Medical Center (UMC), Utrecht, The Netherlands) including native signal peptide (residues 1–357) was subcloned using BamHI/NotI sites in pUPE1.02 (tagless vector for intracellular expression). For extracellular expression of human RAP, the construct without native signal peptide and ER retention signal (residues 36–353) was subcloned into pUPE107.03 (N-terminal cystatin secretion signal and C-terminal His6).
Confocal Microscopy
To visualize the cellular localization of LRP1, HEK293T human embryonic kidney cells (kindly provided by W. de Lau, Hubrecht Institute, Utrecht, The Netherlands) were transfected with full-length LRP1 with a C-terminal eGFP-His tag. Cells were grown at 37 °C and 5% CO2 in a 75-cm2 flask in Dulbecco's modified Eagle's medium (ATCC) supplemented with 10% FBS and penicillin/streptomycin. At 3 days after transfection, cells were passaged (TripLE Express) onto poly-d-lysine-precoated Menzel glasses in a 12-well plate. At 6 days after transfection, cells were washed with PBS and fixed with 4% paraformaldehyde (Merck) in PBS. Coverslips were mounted on slides using Hard Set Mounting Medium with DAPI (VECTASHIELD, H-1500) and imaged using a Zeiss LSM700 Confocal Microscope (Carl Zeiss). Images were taken using a Plan Apochromat 63×/1.4 objective lens. Images shown in Fig. 2A are maximum intensity projections generated with ZEN software (Carl Zeiss).
Flow Cytometry
The percentage of LRP1-GFP expressed on the cell surface of HEK293-EBNA 1 cells (U-Protein Express) was determined by flow cytometry. Cells (5 × 106 cells/ml) were incubated with 5 μl/ml anti-LRP1 mouse mAb (8G1, Calbiochem) for 30 min on ice. Samples were washed twice with 1% (w/v) BSA (Merck) in PBS and incubated with an Alexa Fluor 546-conjugated goat anti-mouse secondary antibody (Thermo Fisher) for 30 min on ice. The cells were then fixed with 150 μl of 1% paraformaldehyde (BD Biosciences). Negative controls consisted of cells without any GFP expressed and cells expressing LRP1-GFP incubated in the absence of the secondary antibody. The acquired data were analyzed with FlowJo (TreeStar).
Expression and Purification of LRP1 Ectodomain
Soluble ectodomain of human LRP1 in pUPE7.50 was transiently expressed in HEK293-EBNA1 cells (U-Protein Express). Expression was optimized for protein yield by plasmid titration (46), which indicated that transfections with 10-fold dilutions of expression plasmid in non-expressing dummy plasmid improved LRP1 production approximately 2–3-fold. Protein yield was further improved (∼2-fold) when co-expressed with RAP in ratio LRP1:RAP 10:1. 6 days after transient expression, medium was harvested and 6-fold concentrated using a 10-kDa molecular mass cut-off membrane. LRP1 ectodomain was purified by Strep affinity chromatography followed by SEC on a Superose 6 column (GE Healthcare) equilibrated with 150 mm NaCl, 4 mm CaCl2, and 25 mm HEPES, pH 7.5.
Expression and Purification of RAP
RAP construct for extracellular expression was expressed in HEK293-EBNA1-S cells (43) and purified by nickel-nitrilotriacetic acid affinity chromatography followed by SEC on a Superdex75 column equilibrated with 150 mm NaCl and 25 mm HEPES, pH 7.0. RAP-D3 and RAP-D3 K256A/K270A were produced and purified as described (41).
SDS-PAGE
10 μl of purified protein was diluted with 5 μl of SDS loading dye with or without 6% (v/v) β-mercaptoethanol for reducing and non-reducing SDS-PAGE, respectively. Samples were run on Mini-PROTEAN TGX precast 4–15% gradient gel (Bio-Rad). Gel was stained with Coomassie Blue.
Native MS
LRP1, RAP, and the LRP1-RAP complex were produced recombinantly in HEK293-EBNA1-S cells (43) and subsequently transferred to 150 mm ammonium acetate, pH 7.5, using Vivaspin centrifugal filter units with a 10- or 100-kDa molecular mass cut-off (Sartorius). Prior to native MS analysis, LRP1 and the co-secreted LRP1-RAP complex were diluted to a final concentration of 0.5–1 μm. The RAP concentration was varied according to the molar ratios indicated in Fig. 5B. To assess the protein behavior at pH <6, the protein stock solutions were 6–8-fold diluted with 150 mm ammonium acetate, pH 5. The samples were loaded into gold-coated borosilicate capillaries and analyzed by native nano-electrospray ionization MS on a modified Orbitrap Exactive Plus EMR mass spectrometer (Thermo Fisher Scientific) (47). The instrument was externally calibrated using the sum formula for cesium iodide (CsI) clusters and operated in positive ion mode with the following settings: capillary voltage = 1.4–1.5 kV, source fragmentation voltage = 10 eV, ion injection time = 100 ms, higher-energy collision dissociation energy = 200 eV, N2 gas pressure in higher-energy collision cell = 8–9 × 10−10 bar, mass resolution at m/z 200 = 2,000. The analyte transmission was optimized by manually tuning the ion transfer settings. The mass spectra were analyzed with Xcalibur v2.2 (Thermo Fisher Scientific) and Databridge in combination with MassLynx v4.1 (both from Waters).
Cross-linking MS
The co-expressed LRP1-RAP complex was cross-linked at a concentration of 1 mg/ml using 1 mm bis(sulfosuccinimidyl)suberate (BS3, Thermo Fisher Scientific). The cross-linking reaction proceeded for 45 min at room temperature and was quenched by adding 20 mm Tris-HCl, pH 7.6. The cross-linked samples were denatured with 2 m urea, reduced with 4 mm DTT (30 min at 56 °C), alkylated with 8 mm iodoacetamide (30 min in the dark), and again treated with 4 mm DTT. Next, the samples were deglycosylated by overnight shaking incubation with 0.2 units/μg of N-glycosidase (PNGase) (Roche Applied Science) and subsequently digested by a 6-h incubation with trypsin (Promega) at a 1:60 (w/w) protease:substrate ratio. Both reactions were carried out at 37 °C. The resulting peptide mixture was desalted using Sep-Pak C18 cartridges (Waters), dried under vacuum, dissolved in 10% (v/v) HCOOH, and analyzed by reversed-phase nano-high performance liquid chromatography (column material: Poroshell 120 EC-C18, 2.7 μm (Agilent Technologies))/tandem mass spectrometry. The experiments were conducted using a Proxeon EASY-nLC 1000 coupled to an Orbitrap Elite mass spectrometer (both from Thermo Fisher Scientific) or an ultra-HPLC Agilent 1200 (Agilent Technologies) coupled to an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific). All precursor ions were mass analyzed in the Orbitrap using a mass resolution setting of 60,000 at m/z 200. The most abundant and at least triply charged precursor ions were selected for MS2 experiments, using a top-5 data-dependent acquisition approach (Orbitrap Elite) or the top-speed data dependent mode with 2-s cycle time (Orbitrap Fusion). The precursor ions were fragmented by sequentially applying collision-induced dissociation (CID) and electron transfer dissociation (ETD). The corresponding CID- and ETD-MS2 scans were acquired in the Orbitrap mass analyzer with 15,000 mass resolution at m/z 200.
Cross-linked peptides were identified using the XlinkX software in enumeration mode and validated based on a target-decoy database search strategy, as described previously (48, 49). Cross-link identifications were verified based on the individual XlinkX n-score of the linked peptides. Cross-links were accepted when the more confidently identified linked peptide was verified at a 1% false discovery rate and the less confidently identified linked peptide was verified at a 5% false discovery rate.
SPR
Association and dissociation of RAP, RAP-D3, and RAP-D3 K256A/K270A to recombinant LRP1 was assessed by SPR analysis employing a Biacore T200 biosensor (Biacore AB). Different batches of purified recombinant LRP1 were immobilized at varying ligand densities onto a CM5 sensor chip using the amine-coupling method according to the manufacturer's instructions. Subsequently, RAP (0–2560 nm), RAP-D3 (0–800 nm), and RAP-D3 K256A/K270A (0–800 nm) were passed over the immobilized LRP1 in running buffer containing 150 mm NaCl, 5 mm CaCl2, 0.05% (v/v) Tween 20, and 20 mm HEPES, pH 7.4, at 25 °C with a flow rate of 30, 60, or 90 μl/min. The sensor chip surface was regenerated three times after each concentration of analyte with a buffer containing 150 mm NaCl, 20 mm EDTA, 0.05% (v/v) Tween 20, and 20 mm HEPES, pH 7.4, followed by equilibration using running buffer. Binding to LRP1 was corrected for binding in the absence of LRP1. Responses at equilibrium were plotted as a function of the analyte concentration. To estimate KD values, the responses at equilibrium were fitted by non-linear regression using a standard hyperbola (GraphPad Prism 4 software).
SAXS
SAXS data were collected at the European Synchrotron Radiation Facility (ESRF Grenoble, France) BioSAXS beamline at 12.5 keV (0.9919 Å) with a 2D Pilatus 1M detector (DECTRIS). For online purification, we used the HPLC system consisting of an in-line degasser (DGU-20A5R, Shimadzu), a binary pump (LC-20ADXR, Shimadzu), a valve for buffer selection and gradients, an autosampler (SIL-20ACXR, Shimadzu), a UV-visible array photospectrometer (SPD-M20A, Shimadzu), and a conductimeter (CDD-10AVP, Shimadzu). LRP1 and LRP1-RAP from HEK293-EBNA1 cells were analyzed in different buffer conditions. Protein samples were loaded into vials and automatically injected onto the Superose 6 5/150 analytical column pre-equilibrated with 150 mm NaCl, 4 mm CaCl2, and 25 mm HEPES, pH 7.5, or alternatively with 150 mm NaCl, 4 mm CaCl2, and 25 mm MES, pH 5.5. All data were collected using a sample-to-detector distance of 2.81 m corresponding to a scattering vector s (s = 4π sin θ/λ) range of 0.03–5.0 nm−1. Approximately 1,500 frames (1 frame s−1) were collected per 30-min sample run. Initial data processing was performed automatically using the EDNA pipeline (50), generating azimuthally integrated, calibrated, and normalized one-dimensional profiles for each frame. All frames were compared with the initial frame, and matching frames were merged to create the reference buffer. About 15–30 frames for each run with a consistent Rg, and corresponding to the highest protein concentration based on forward scattering intensity, I(0) values were merged to yield a single averaged frame corresponding to the scattering of an individual SEC purified species. The curves obtained were used for further data processing using PRIMUS of the ATSAS suite (51, 52). Rg values were evaluated within the range of Guinier approximation sRg <1.3 according to the equation: I(s) = I(0) exp(−1/3(sRg)2).
The Rg was also computed from all of the scattering patterns using Porod's law by the calculation of the distance distribution function P(r) using the program GNOM (53), also giving the maximum particle diameter Dmax. SAXS measurements were repeated with different protein batches three times for each sample condition.
Negative-stain EM
5 μl of purified protein (5–10 μg/ml) was applied after SEC to a glow-discharged carbon-coated copper grid (Electron Microscopy Sciences, CF200-Cu) and stained with freshly prepared 0.75% uranyl formate solution. Image acquisition was done using a Tecnai 12 (FEI) operating at 120 kV. Images were recorded with a BM-Eagle CCD camera (FEI) 2,048 × 2,048 at ×30,000 magnification. Approximately 2,000 particles for each sample were manually selected, and projections were subjected to reference-free alignment and classification using the EMAN2 software package (54).
Author Contributions
C. D. N. performed cloning, expression of full-length LRP1, purification, SAXS data collection and analysis, negative-stain EM preparation, and data collection. P. L. performed native and cross-linking MS experiments and analysis. M. v. d. B performed RAP D3 and RAP D3 K256A/K270A expression and purification, SPR experiments, and analysis. P. K. M. designed the cloning strategy and supervised initial expression tests. N. L. purified full-length RAP and helped in expressing LRP1-GFP in HEK293T cells for confocal microscopy. K. M. provided guidance and helped in designing the research. C. D. N., P. L., M. v. d. B., A. J. R. H., and P. G. discussed results and wrote the paper. P. G. supervised the project. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Joachim Herz for providing us with LRP1 pcDNA; Dr. Madelon Maurice for RAP pcDNA; W. de Lau for the provision of HEK293T cells; Joke C. M. Granneman and Dimphna H. Meijer for assistance in expressing LRP1-GFP in HEK293T cells and acquiring images with the confocal microscope; Kok P. M. van Kessel for assistance with FACS experiments; U-Protein Express BV for the provision of protein expression facilities; and Fan Liu for assistance in designing the MS acquisition methods. We also thank The European Synchrotron Radiation Facility (ESRF) for the provision of synchrotron radiation facilities and beamline scientists of the ESRF and the European Molecular Biology Laboratory for assistance, in particular Adam Round and Martha E. Brennich. We are grateful to E. G. Huizinga for the fruitful discussions.
This work was supported by the ManiFold project (Grant 317371) and the BioStruct-X (Grant 283570) embedded in the European Union's Seventh Framework Programme (FP7/2007-2013), the Roadmap Initiative Proteins@Work (Project 184.032.201) funded by The Netherlands Organisation for Scientific Research (NWO), and The Landsteiner Foundation for Blood Transfusion Research (LSBR Fellowship 1517 (to M. v. d. B)). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table 1.
- LRP1
- LDL receptor-related protein 1
- LDLR
- LDL receptor
- RAP
- receptor-associated protein
- CR
- cysteine-rich
- ER
- endoplasmic reticulum
- EBNA
- Epstein-Barr virus nuclear antigen
- SEC
- size-exclusion chromatography
- SAXS
- small-angle X-ray scattering
- TEV
- tobacco etch virus
- CID
- collision-induced dissociation
- ETD
- electron transfer dissociation.
References
- 1. Moestrup S. K., Gliemann J., and Pallesen G. (1992) Distribution of the α2-macroglobulin receptor/low density lipoprotein receptor-related protein in human tissues. Cell Tissue Res. 269, 375–382 [DOI] [PubMed] [Google Scholar]
- 2. Herz J., and Strickland D. K. (2001) LRP: a multifunctional scavenger and signaling receptor. J. Clin. Invest. 108, 779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lillis A. P., Van Duyn L. B., Murphy-Ullrich J. E., and Strickland D. K. (2008) LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 88, 887–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beisiegel U., Weber W., Ihrke G., Herz J., and Stanley K. K. (1989) The LDL-receptor-related protein, LRP, is an apolipoprotein E-binding protein. Nature 341, 162–164 [DOI] [PubMed] [Google Scholar]
- 5. Strickland D. K., Ashcom J. D., Williams S., Burgess W. H., Migliorini M., and Argraves W. S. (1990) Sequence identity between the α2-macroglobulin receptor and low density lipoprotein receptor-related protein suggests that this molecule is a multifunctional receptor. J. Biol. Chem. 265, 17401–17404 [PubMed] [Google Scholar]
- 6. Herz J., Clouthier D. E., and Hammer R. E. (1992) LDL receptor-related protein internalizes and degrades uPA-PAI-1 complexes and is essential for embryo implantation. Cell 71, 411–421 [DOI] [PubMed] [Google Scholar]
- 7. Scilabra S. D., Troeberg L., Yamamoto K., Emonard H., Thøgersen I., Enghild J. J., Strickland D. K., and Nagase H. (2013) Differential regulation of extracellular tissue inhibitor of metalloproteinases-3 levels by cell membrane-bound and shed low density lipoprotein receptor-related protein 1. J. Biol. Chem. 288, 332–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hofer F., Gruenberger M., Kowalski H., Machat H., Huettinger M., Kuechler E., and Blaas D. (1994) Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc. Natl. Acad. Sci. U.S.A. 91, 1839–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kounnas M. Z., Morris R. E., Thompson M. R., FitzGerald D. J., Strickland D. K., and Saelinger C. B. (1992) The α2-macroglobulin receptor/low density lipoprotein receptor-related protein binds and internalizes Pseudomonas exotoxin A. J. Biol. Chem. 267, 12420–12423 [PubMed] [Google Scholar]
- 10. Chen W. J., Goldstein J. L., and Brown M. S. (1990) NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J. Biol. Chem. 265, 3116–3123 [PubMed] [Google Scholar]
- 11. Czekay R. P., Orlando R. A., Woodward L., Lundstrom M., and Farquhar M. G. (1997) Endocytic trafficking of megalin/RAP complexes: dissociation of the complexes in late endosomes. Mol. Biol. Cell 8, 517–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Laatsch A., Panteli M., Sornsakrin M., Hoffzimmer B., Grewal T., and Heeren J. (2012) Low density lipoprotein receptor-related protein 1 dependent endosomal trapping and recycling of apolipoprotein E. PLoS ONE 7, e29385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Herz J., Kowal R. C., Goldstein J. L., and Brown M. S. (1990) Proteolytic processing of the 600 kd low density lipoprotein receptor-related protein (LRP) occurs in a trans-Golgi compartment. EMBO J. 9, 1769–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang W., Dolmer K., and Gettins P. G. (1999) NMR Solution structure of complement-like repeat CR8 from the low density lipoprotein receptor-related protein. J. Biol. Chem. 274, 14130–14136 [DOI] [PubMed] [Google Scholar]
- 15. Dolmer K., Huang W., and Gettins P. G. (2000) NMR solution structure of complement-like repeat CR3 from the low density lipoprotein receptor-related protein: evidence for specific binding to the receptor binding domain of human α2-macroglobulin. J. Biol. Chem. 275, 3264–3269 [DOI] [PubMed] [Google Scholar]
- 16. Simonovic M., Dolmer K., Huang W., Strickland D. K., Volz K., and Gettins P. G. (2001) Calcium coordination and pH dependence of the calcium affinity of ligand-binding repeat CR7 from the LRP: comparison with related domains from the LRP and the LDL receptor. Biochemistry 40, 15127–15134 [DOI] [PubMed] [Google Scholar]
- 17. Jensen G. A., Andersen O. M., Bonvin A. M., Bjerrum-Bohr I., Etzerodt M., Thøgersen H. C., O'Shea C., Poulsen F. M., and Kragelund B. B. (2006) Binding site structure of one LRP-RAP complex: implications for a common ligand-receptor binding motif. J. Mol. Biol. 362, 700–716 [DOI] [PubMed] [Google Scholar]
- 18. Guttman M., Prieto J. H., Handel T. M., Domaille P. J., and Komives E. A. (2010) Structure of the minimal interface between ApoE and LRP. J. Mol. Biol. 398, 306–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Delain E., Barray M., Pochon F., Gliemann J., and Moestrup S. K. (1994) Electron microscopic visualization of the human α2-macroglobulin receptor and its interaction with α2-macroglobulin/chymotrypsin complex. Ann. N.Y. Acad. Sci. 737, 202–211 [DOI] [PubMed] [Google Scholar]
- 20. Ashcom J. D., Tiller S. E., Dickerson K., Cravens J. L., Argraves W. S., and Strickland D. K. (1990) The human α2-macroglobulin receptor: identification of a 420-kD cell surface glycoprotein specific for the activated conformation of α2-macroglobulin. J. Cell Biol. 110, 1041–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kristensen T., Moestrup S. K., Gliemann J., Bendtsen L., Sand O., and Sottrup-Jensen L. (1990) Evidence that the newly cloned low-density-lipoprotein receptor related protein (LRP) is the α2-macroglobulin receptor. FEBS Lett. 276, 151–155 [DOI] [PubMed] [Google Scholar]
- 22. Herz J., Goldstein J. L., Strickland D. K., Ho Y. K., and Brown M. S. (1991) 39-kDa protein modulates binding of ligands to low density lipoprotein receptor-related protein/α2-macroglobulin receptor. J. Biol. Chem. 266, 21232–21238 [PubMed] [Google Scholar]
- 23. Bu G., Geuze H. J., Strous G. J., and Schwartz A. L. (1995) 39 kDa receptor-associated protein is an ER resident protein and molecular chaperone for LDL receptor-related protein. EMBO J. 14, 2269–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Willnow T. E., Rohlmann A., Horton J., Otani H., Braun J. R., Hammer R. E., and Herz J. (1996) RAP, a specialized chaperone, prevents ligand-induced ER retention and degradation of LDL receptor-related endocytic receptors. EMBO J. 15, 2632–2639 [PMC free article] [PubMed] [Google Scholar]
- 25. Iadonato S. P., Bu G., Maksymovitch E. A., and Schwartz A. L. (1993) Interaction of a 39 kDa protein with the low-density-lipoprotein-receptor-related protein (LRP) on rat hepatoma cells. Biochem. J. 296, 867–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee D., Walsh J. D., Migliorini M., Yu P., Cai T., Schwieters C. D., Krueger S., Strickland D. K., and Wang Y. X. (2007) The structure of receptor-associated protein (RAP). Protein Sci. 16, 1628–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Obermoeller L. M., Warshawsky I., Wardell M. R., and Bu G. (1997) Differential functions of triplicated repeats suggest two independent roles for the receptor-associated protein as a molecular chaperone. J. Biol. Chem. 272, 10761–10768 [DOI] [PubMed] [Google Scholar]
- 28. Andersen O. M., Schwarz F. P., Eisenstein E., Jacobsen C., Moestrup S. K., Etzerodt M., and Thøgersen H. C. (2001) Dominant thermodynamic role of the third independent receptor binding site in the receptor-associated protein RAP. Biochemistry 40, 15408–15417 [DOI] [PubMed] [Google Scholar]
- 29. Lazic A., Dolmer K., Strickland D. K., and Gettins P. G. (2006) Dissection of RAP-LRP interactions: binding of RAP and RAP fragments to complement-like repeats 7 and 8 from ligand binding cluster II of LRP. Arch. Biochem. Biophys. 450, 167–175 [DOI] [PubMed] [Google Scholar]
- 30. Horn I. R., van den Berg B. M., van der Meijden P. Z., Pannekoek H., and van Zonneveld A. J. (1997) Molecular analysis of ligand binding to the second cluster of complement-type repeats of the low density lipoprotein receptor-related protein: evidence for an allosteric component in receptor-associated protein-mediated inhibition of ligand binding. J. Biol. Chem. 272, 13608–13613 [DOI] [PubMed] [Google Scholar]
- 31. Neels J. G., van Den Berg B. M., Lookene A., Olivecrona G., Pannekoek H., and van Zonneveld A. J. (1999) The second and fourth cluster of class A cysteine-rich repeats of the low density lipoprotein receptor-related protein share ligand-binding properties. J. Biol. Chem. 274, 31305–31311 [DOI] [PubMed] [Google Scholar]
- 32. Obermoeller-McCormick L. M., Li Y., Osaka H., FitzGerald D. J., Schwartz A. L., and Bu G. (2001) Dissection of receptor folding and ligand-binding property with functional minireceptors of LDL receptor-related protein. J. Cell Sci. 114, 899–908 [DOI] [PubMed] [Google Scholar]
- 33. Meijer A. B., Rohlena J., van der Zwaan C., van Zonneveld A. J., Boertjes R. C., Lenting P. J., and Mertens K. (2007) Functional duplication of ligand-binding domains within low-density lipoprotein receptor-related protein for interaction with receptor associated protein, α2-macroglobulin, factor IXa and factor VIII. Biochim. Biophys. Acta 1774, 714–722 [DOI] [PubMed] [Google Scholar]
- 34. Bu G., and Rennke S. (1996) Receptor-associated protein is a folding chaperone for low density lipoprotein receptor-related protein. J. Biol. Chem. 271, 22218–22224 [DOI] [PubMed] [Google Scholar]
- 35. Migliorini M. M., Behre E. H., Brew S., Ingham K. C., and Strickland D. K. (2003) Allosteric modulation of the ligand binding properties of LRP by the receptor associated protein (RAP) requires critical lysine residues within its carboxyl-terminal domain. J. Biol. Chem. 278, 17986–17992 [DOI] [PubMed] [Google Scholar]
- 36. Fisher C., Beglova N., and Blacklow S. C. (2006) Structure of an LDLR-RAP complex reveals a general mode for ligand recognition by lipoprotein receptors. Mol. Cell 22, 277–283 [DOI] [PubMed] [Google Scholar]
- 37. van den Biggelaar M., Madsen J. J., Faber J. H., Zuurveld M. G., van der Zwaan C., Olsen O. H., Stennicke H. R., Mertens K., and Meijer A. B. (2015) Factor VIII Interacts with the endocytic receptor low-density lipoprotein receptor-related protein 1 via an extended surface comprising “hot-spot” lysine residues. J. Biol. Chem. 290, 16463–16476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Strickland D. K., Au D. T., Cunfer P., and Muratoglu S. C. (2014) Low-density lipoprotein receptor-related protein-1: role in the regulation of vascular integrity. Arterioscler. Thromb. Vasc. Biol. 34, 487–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rudenko G., Henry L., Henderson K., Ichtchenko K., Brown M. S., Goldstein J. L., and Deisenhofer J. (2002) Structure of the LDL receptor extracellular domain at endosomal pH. Science 298, 2353–2358 [DOI] [PubMed] [Google Scholar]
- 40. Willnow T. E., Orth K., and Herz J. (1994) Molecular dissection of ligand binding sites on the low density lipoprotein receptor-related protein. J. Biol. Chem. 269, 15827–15832 [PubMed] [Google Scholar]
- 41. van den Biggelaar M., Sellink E., Klein Gebbinck J. W., Mertens K., and Meijer A. B. (2011) A single lysine of the two-lysine recognition motif of the D3 domain of receptor-associated protein is sufficient to mediate endocytosis by low-density lipoprotein receptor-related protein. Int. J. Biochem. Cell Biol. 43, 431–440 [DOI] [PubMed] [Google Scholar]
- 42. Lössl P., van de Waterbeemd M., and Heck A. J. (2016) The diverse and expanding role of mass spectrometry in structural and molecular biology. EMBO J. 35, 2634–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peng W. C., de Lau W., Madoori P. K., Forneris F., Granneman J. C., Clevers H., and Gros P. (2013) Structures of Wnt-antagonist ZNRF3 and its complex with R-spondin 1 and implications for signaling. PLoS ONE 8, e83110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bloem E., Ebberink E. H., van den Biggelaar M., van der Zwaan C., Mertens K., and Meijer A. B. (2015) A novel chemical footprinting approach identifies critical lysine residues involved in the binding of receptor-associated protein to cluster II of LDL receptor-related protein. Biochem. J. 468, 65–72 [DOI] [PubMed] [Google Scholar]
- 45. Guo X., Bandyopadhyay P., Schilling B., Young M. M., Fujii N., Aynechi T., Guy R. K., Kuntz I. D., and Gibson B. W. (2008) Partial acetylation of lysine residues improves intraprotein cross-linking. Anal. Chem. 80, 951–960 [DOI] [PubMed] [Google Scholar]
- 46. Halff E. F., Versteeg M., Brondijk T. H., and Huizinga E. G. (2014) When less becomes more: optimization of protein expression in HEK293-EBNA1 cells using plasmid titration: a case study for NLRs. Protein Expr. Purif. 99, 27–34 [DOI] [PubMed] [Google Scholar]
- 47. Snijder J., van de Waterbeemd M., Damoc E., Denisov E., Grinfeld D., Bennett A., Agbandje-McKenna M., Makarov A., and Heck A. J. (2014) Defining the stoichiometry and cargo load of viral and bacterial nanoparticles by orbitrap mass spectrometry. J. Am. Chem. Soc. 136, 7295–7299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu F., Rijkers D. T., Post H., and Heck A. J. (2015) Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods 12, 1179–1184 [DOI] [PubMed] [Google Scholar]
- 49. Lössl P., Brunner A. M., Liu F., Leney A. C., Yamashita M., Scheltema R. A., and Heck A. J. (2016) Deciphering the interplay among multisite phosphorylation, interaction dynamics, and conformational transitions in a tripartite protein system. ACS Cent. Sci. 2, 445–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brennich M. E., Kieffer J., Bonamis G., De Maria Antolinos A., Hutin S., Pernot P., and Round A. (2016) Online data analysis at the ESRF BioSAXS beamline. J. Appl. Crystallogr. 49, 203–212 [Google Scholar]
- 51. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., and Svergun D. I. (2003) PRIMUS: a Windows-PC based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 52. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D. T., Konarev P. V., and Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 54. Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., and Ludtke S. J. (2007) EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 [DOI] [PubMed] [Google Scholar]
- 55. Williams S. E., Ashcom J. D., Argraves W. S., and Strickland D. K. (1992) A novel mechanism for controlling the activity of α2-macroglobulin receptor/low density lipoprotein receptor-related protein 1. J. Biol. Chem. 267, 9035–9040 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.