

Abstract
In Escherichia coli, the peptidoglycan cell wall is synthesized by bifunctional penicillin-binding proteins such as PBP1b that have both transpeptidase and transglycosylase activities. The PBP1b transpeptidase domain is a major target of β-lactams, and therefore it is important to attain a detailed understanding of its inhibition. The peptidoglycan glycosyltransferase domain of PBP1b is also considered an excellent antibiotic target yet is not exploited by any clinically approved antibacterials. Herein, we adapt a pyrophosphate sensor assay to monitor PBP1b-catalyzed glycosyltransfer and present an improved crystallographic model for inhibition of the PBP1b glycosyltransferase domain by the potent substrate analog moenomycin. We elucidate the structure of a previously disordered region in the glycosyltransferase active site and discuss its implications with regards to peptidoglycan polymerization. Furthermore, we solve the crystal structures of E. coli PBP1b bound to multiple different β-lactams in the transpeptidase active site and complement these data with gel-based competition assays to provide a detailed structural understanding of its inhibition. Taken together, these biochemical and structural data allow us to propose new insights into inhibition of both enzymatic domains in PBP1b.
Keywords: antibiotics, bacteria, glycosyltransferase, inhibitor, peptidoglycan, β-lactam, moenomycin, penicillin-binding protein 1b, transglycosylation, transpeptidation
Introduction
Most bacteria are encased in a peptide cross-linked glycan net known as the peptidoglycan (PG)2 sacculus. In Gram-negative bacteria such as Escherichia coli, the thin PG layer is sandwiched between the inner and outer membranes and is an essential feature that is inexorably linked to both cell growth and morphogenesis (1). Because of its essential role, accessible periplasmic location, and lack of human orthologs, the PG biosynthetic pathway is targeted by the majority of human antibacterials in clinical use. The final enzymatic steps in PG synthesis are carried out by polysaccharide polymerases called class A penicillin-binding proteins (PBPs). These bifunctional enzymes add new material to the pre-existing PG in two coordinated, yet successive steps. First, the non-reducing end of the C55-PP lipid-activated GlcNAc-MurNAc pentapeptide precursor molecule (lipid II) is attached via a covalent β-1,4-glycosidic bond to the reducing end of a growing nascent (donor) C55-PP activated PG chain by the membrane anchored glycosyltransferase (GTase) activity of PBPs. Second, PBPs cross-link the nascent pentapeptide units to peptides that are already present in the sacculus (transpeptidase, TPase) (2). The latter reaction is the final step in PG biosynthesis and the lethal target of the β-lactam antibiotics, which act as substrate analogs of the terminal d-Ala– on the donor peptide.
In E. coli, two major class A bifunctional enzymes containing both GTase and TPase activity (PBP1a and PBP1b) are anchored to the outer leaflet of the cytoplasmic membrane and are responsible for adding new PG precursor units to the growing sacculus. Either PBP1b or PBP1a is required for cell viability but not both, indicating that the two proteins have overlapping function. However, under normal growth conditions, PBP1b is the predominant synthase during cell division, whereas PBP1a is mainly involved in cell wall elongation (1). Evidence suggests that PBP1a and PBP1b are part of multicomponent synthase complexes in which the spatiotemporal control of the GTase and TPase activities are tightly regulated by accessory proteins (1). E. coli PBP1b consists of five domains: (i) a transmembrane (TM) α-helix at the N terminus (residues 64–87); (ii) a UvrB domain 2 homolog (UB2H) domain (residues 109–200), which is unique to PBP1b and presumably interacts with periplasmic binding partners including the outer membrane-tethered regulatory protein LpoB (1, 3–5); (iii) a membrane-associated GTase domain (residues 203–367); (iv) a linker region connecting the GTase and TPase domains (residues 391–443); and (v) a C-terminal TPase domain (residues 444–736) (Fig. 1).
FIGURE 1.

Overall structure of PBP1b-moenomycin-acyl-β-lactam complexes. A, the crystal structure of PBP1b is shown in surface representation. The TM, UB2H, GTase, linker, and TPase domains are colored cyan, beige, blue, pink, and green, respectively. Moenomycin and acyl-ampicillin bound to the GTase and TPase domain are shown in yellow stick representation. B, topology diagram of PBP1b, color-coded as in A.
The crystal structures of the full-length bifunctional Staphylococcus aureus PBP2 (6) and the isolated Aquifex aeolicus PBP1a GTase domain truncation (7) provided our first glimpse into the molecular details governing PG glycosyltransfer. A large, elongated GTase active site lies at the interface of a predominantly α-helical λ-lysozyme-like “head” subdomain and a unique N-terminal hydrophobic membrane embedded “jaw” subdomain. The head subdomain contains a conserved Glu233, which is the proposed general base required for deprotonation of the acceptor (lipid II) C4 hydroxyl to facilitate a SN2-like nucleophilic attack on the donor MurNAc C1 acyl-phosphate linkage, resulting in departure of the C55-PP leaving group (6). Analysis of the distribution of glycan products in a gel electrophoresis assay using 14C-labeled lipid II revealed that PG GTases catalyze polymerization in a processive manner, meaning that they undergo multiple successive rounds of glycosyltransfer without releasing the growing polymer (7).
The Streptomyces phosphoglycolipid natural product moenomycin is the best characterized PG GTase inhibitor and displays inhibitory constants in the nanomolar range (8). Moenomycin is widely used as a growth promoter in animal feed yet is not effective in humans largely because of poor absorption resulting in suboptimal pharmacokinetic properties (9). Furthermore, although moenomycin potently inhibits purified Gram-negative GTases, its antimicrobial activity is restricted to Gram-positive organisms presumably because of poor outer membrane penetration in the former (10). Therefore, moenomycin analogs that retain potent GTase inhibitory activity and are active against the Gram-negatives are an attractive therapeutic prospect. The crystal structure of moenomycin bound to the donor site of the S. aureus PBP2 GTase revealed that it acts as a lipid IV mimic (C55-PP linked 4 sugar polymer) (6), analogous to the growing PG chain. A detailed knowledge of the moenomycin-PBP1b interactions would aid in the design of analogs that can be used to treat human E. coli infections.
The TPase domain of PBP1b adopts the classical penicilloyl-serine transferase superfamily fold, consisting of two subdomains, a five-stranded antiparallel β-sheet flanked on one face by three α-helices, and an all α-helical domain with the active site sandwiched at the domain interface (11). The TPase active site has three highly conserved sequence motifs: (i) the SXXK motif, which includes the serine nucleophile and general base lysine; (ii) the SXN motif, involved in protonation of the nitrogen leaving group during acylation; and (iii) the KTG(T/S) motif, which lines strand β17 and is involved in oxyanion stabilization and substrate binding (11). Many β-lactams (including piperacillin, cephalexin, and aztreonam) used clinically to treat E. coli and other Enterobacteriaceae infections exclusively target PBP3, which is an essential gene. However, binding to PBP3 triggers the bacterial SOS response that allows the cell to stall at division and thereby escape β-lactam-mediated killing, which requires actively dividing cells (12). An alternative or additional β-lactam target in these bacteria is PBP1b because knock-out strains display hypersensitivity to β-lactam treatment, and binding to PBP1b presumably does not trigger the SOS pathway (13). Currently, the only clinically approved PBP1b-specific β-lactam is cefsulodin, in which clinical utility is restricted to Pseudomonas aeruginosa (14, 15). A structural analysis of β-lactam binding to PBP1b is required to guide further drug design efforts targeted at this important enzyme.
The 2.16 Å resolution crystal structure of E. coli PBP1b in complex with moenomycin revealed important insights into the GTase domain and the global domain architecture (16). However, poorly resolved electron density in the GTase domain led to ambiguities in interpretation of key active site side chains and did not permit modeling of the active site region (residues His240–Thr267). Hence, we revisit the moenomycin-bound PBP1b crystal structure and attain improved GTase active site electron density maps from new crystallization conditions and using feature-enhanced maps (FEM), which are available as part of the Phenix crystallographic software (17). These improved data allow us to unambiguously model key active site side chains and thereby propose new insights into its inhibition and PG glycosyltransfer. Additionally, to understand the effects of β-lactam-mediated inhibition, we solved the crystal structures of multiple acyl-β-lactams bound to the E. coli PBP1b TPase domain by co-crystallization and complemented this with a gel-based competition assay. Taken together, these data provide a detailed structural basis for inhibition of both the GTase and TPase activities of the E. coli PBP1b membrane protein and will aid in the future design of inhibitors targeted at this key bifunctional enzyme.
Results and Discussion
PBP1b Glycosyltransferase Pyrophosphate Sensor Assay
A major advance in peptidoglycan GTase assays came from the development of a continuous kinetic assay using fluorescent dansyl lipid II (i.e. the ϵ-amino group at the position 3 of the stem peptide is covalently modified with a dansyl group) (18). In this assay, polymerization of dansyl-lipid II followed by polymer digestion with a muramidase results in a decrease in fluorescence of the dansyl-muropeptides caused by the removal of the lipid moiety during polymerization (19). Although robust, this assay requires fluorescent derivitization of the limiting lipid II substrate (which is a challenge to attain in large quantities) and a secondary enzyme. Therefore, to test the activity of our purified E. coli PBP1b, we adapted a continuous fluorogenic pyrophosphate sensor assay to monitor glycosyltransfer using the natural lipid II substrate in a low reaction volume. During GTase catalyzed lipid II polymerization, C55-PP is released, and the assay utilizes a fluorogenic anti-pyrophosphate antibody sensor in which fluorescence intensity is directly proportional to the concentration of free pyrophosphate (Fig. 2A) (AAT Bioquest®). Substrate concentration response experiments resulted in a sigmoidal curve with a Hill coefficient of 2.92, indicating that PBP1b-catalyzed glycosyltransfer may display positive cooperativity (Fig. 2B) (20, 21). This observation is in line with a recent surface plasmon resonance study whereby low concentrations of GTase acceptor substrate analogs were found to increase moenomycin donor site affinity in the S. aureus monofunctional GTase. The observed catalytic efficiency (kcat/K0.5) in the pyrophosphate sensor assay for unlabeled lipid II was 1.96 ± 0.10 × 104 m−1 s−1, which is roughly similar to the previously reported kcat/Km of 7.0 × 104 m−1 s−1 for the dansyl-lipid II coupled assay (18). Finally, we performed inhibition assays using the GTase inhibitor moenomycin complex. Moenomycin complex displayed potent inhibition, with an IC50 value of 64.3 ± 3.5 nm (Fig. 2C). Taken together, the pyrophosphate sensor assay provides a convenient alternative assay that has the advantage of utilizing the natural substrate without the need for fluorescent derivatization or coupled enzymes. This assay should be generally transferable to other peptidoglycan GTases and provides a robust system to study the effect of activators and inhibitors in the future. Furthermore, we developed this assay in a low reaction volume 1536-well microplate format that allows the parallel screening of numerous reactions while reducing consumption of the limiting reagent lipid II. Therefore, this assay is readily amenable to high throughput screens to uncover novel inhibitors from large compound libraries.
FIGURE 2.

E. coli PBP1b glycosyltransferase pyrophosphate sensor and thermal aggregation assays. A, schematic representation of pyrophosphate sensor assay. B, substrate concentration response experiment. Various concentrations of lipid II were added to 1 μm of PBP1b. C, PBP1b GTase moenomycin complex inhibition assays. IC50 experiments were performed using 1 μm of enzyme and 45 μm lipid II. D, thermal stabilization of PBP1b in the presence of moenomycin complex. Thermostability of PBP1b was assessed at various moenomycin complex concentrations by differential static light scattering as a measure of the change in thermal aggregation (Tagg) to calculate the aggregation constant (Kagg) for the interaction (see “Experimental Procedures”). All error bars represent standard deviation from triplicate technical replicates.
Structure Solution and Refinement
The moenomycin-boundE. coli PBP1b acyl-ampicillin, cephalexin, CENTA, and aztreonam crystal structures were solved to 2.70, 2.36, 2.31, and 2.42 Å resolution. The PBP1b construct spans amino acids 58–804 (16) encompassing the complete GTase and TPase domains and lacking only the predicted N- and C-terminal disordered regions. The addition of the GTase inhibitor moenomycin is absolutely essential to attain protein crystals and is bound to the GTase donor site in all structures. Thermal aggregation assays revealed that moenomycin has a stabilizing effect on PBP1b thermostability (Kagg = 8.56 ± 0.74 μm, ΔTmax = 3.75 ± 0.12 °C; Fig. 2D), providing a potential reason for its necessity in crystallization. For crystallization of this bitopic membrane protein, it was also necessary to use DM detergent in the final protein buffer. In all structures, the PBP1b protein crystallized in space group P22121 with one protein monomer in the assymetric unit, similar to the previous crystal structure (PDB code 3VMA (16)). PBP1b has been found to form a stable dimer in solution, yet only the monomer has been observed crystallographically. The dimeric form of the enzyme may not facilitate suitable crystal packing, providing a potential reason for the observed crystallographic monomer. The PBP1b-ampicillin, PBP1b-cephalexin, PBP1b-CENTA, and PBP1b-aztreonam structures were built from residues Trp67–Ser797, Trp67–Met799, Ala73–Glu798, and Trp67–Met799 because of missing density at the N and C termini, respectively. In addition, certain flexible loop regions could not be modeled in the structures (Table 1). All structures have favorable stereochemistry and refinement statistics (Table 2). Examination of the ligand omit Fo − Fc and final refined 2Fo–Fc electron density maps reveals that all inhibitors display clear, unambiguous electron density in all structures (Fig. 3). Furthermore, all ligands were refined at full occupancy (Table 2).
TABLE 1.
Unmodeled regions in the E. coli PBP1b crystal structures
| Complex | Unmodeled regions |
|---|---|
| PBP1b-ampicillin | Ser207–Pro208, Glu239–Thr266, Gly406–Val407, Gly427–Lys429 |
| PBP1b-CENTA | Asp234–Leu268, Ser280–Arg286, Lys355–Lys367, Gln381–Leu390, Pro398–Ser349 |
| PBP1b-cephalexin | His240–Ser266, Ser280–Arg286, Gly400–Val401 |
| PBP1b-aztreonam | NAa |
a NA, nonapplicable, no unmodeled regions.
TABLE 2.
X-ray crystallographic data collection and refinement statistics for E. coli PBP1b complexes
| Complex | PBP1b-cephalexin | PBP1b-CENTA | PBP1b-aztreonam | PBP1b-ampicillin |
|---|---|---|---|---|
| Data collection | ||||
| Wavelength (Å) | 1.00 Å | 1.00 Å | 1.00 Å | 1.00 Å |
| Space group | P22121 | P22121 | P22121 | P22121 |
| Cell dimensions | ||||
| a, b, c (Å) | 62.7, 63.8, 297.6 | 62.3, 63.9, 294.6 | 62.5, 64.4, 301.4 | 62.4, 63.8, 299.4 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å)a | 63.76–2.36 (2.42–2.36) | 62.49–2.31 (2.37–2.31) | 64.39–2.42 (2.48–2.42) | 74.8–2.70 (2.85–2.70) |
| Completeness (%)a | 98.7 (99.5) | 92.5 (85.9) | 96.7 (83.6) | 99.9 (100.0) |
| Unique reflectionsa | 49,407 (3638) | 48,263 (3270) | 45,993 (2868) | 33,979 (4857) |
| Redundancya | 4.7 (4.6) | 4.0 (2.6) | 3.5 (2.5) | 6.2 (6.4) |
| I/σ(I) a | 13.2 (2.2) | 13.5 (2.4) | 12.7 (1.9) | 10.4 (2.7) |
| Rmerge (%)a | 0.090 (0.650) | 0.067 (0.410) | 0.058 (0.552) | 0.098 (0.604) |
| Refinement statistics | ||||
| Ligand occupancy β-lactam, moenomycin | 1.00, 1.00 | 1.00, 1.00 | 1.00, 1.00 | 1.00, 1.00 |
| Rwork/Rfree | 0.235/0.250 | 0.253/0.279 | 0.235/0.274 | 0.256/0.285 |
| No. of atoms | ||||
| Protein | 5490 | 5063 | 5765 | 5461 |
| Ligand | 71 | 92 | 77 | 101 |
| Water | 223 | 187 | 233 | 107 |
| r.m.s.d. bonds (Å) | 0.014 | 0.012 | 0.013 | 0.010 |
| r.m.s.d. angles (°) | 1.75 | 1.77 | 1.82 | 1.20 |
| Average B-factors (Å2) | ||||
| Protein | 54.0 | 62.4 | 63.0 | 80.1 |
| Ligand | 98.5 | 103.6 | 106.2 | 85.7 |
| Water | 47.3 | 37.4 | 62.40 | 58.7 |
| Ramachandran statistics | ||||
| Favored (%) | 93.0 | 97.0 | 93.0 | 94.0 |
| Additional (%) | 6.6 | 2.8 | 6.4 | 5.8 |
| Disallowed (%) | 0.4 | 0.2 | 0.6 | 0.2 |
a The values in parentheses represent the highest resolution shell.
FIGURE 3.
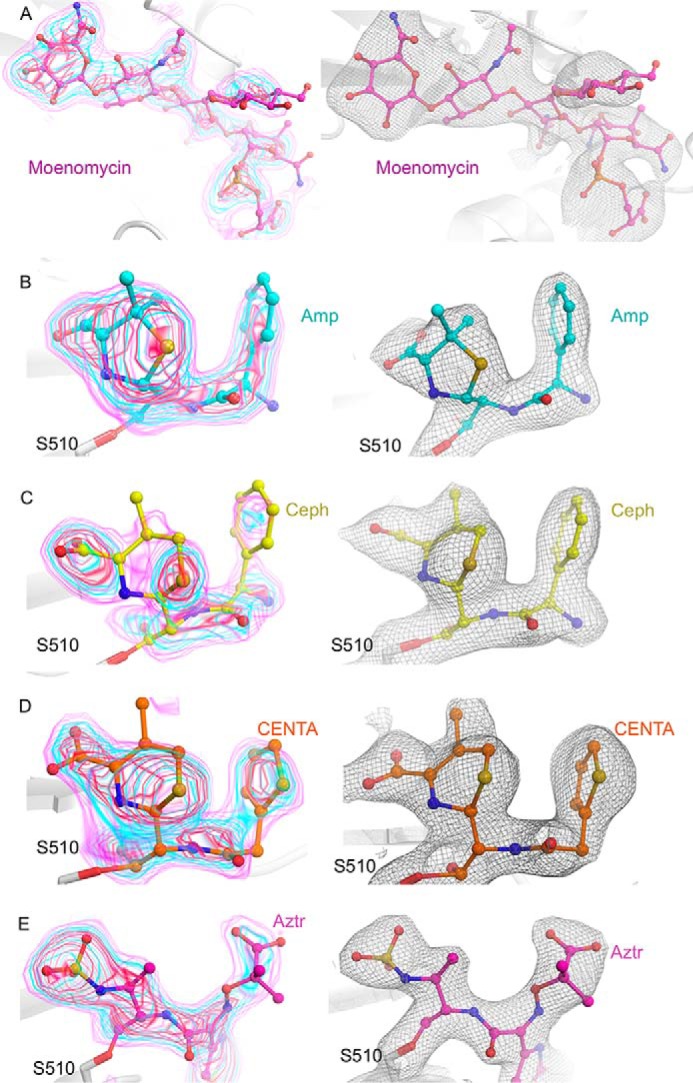
Electron density for E. coli PBP1b-bound ligands. A, moenomycin-bound to the PBP1b GTase domain. Moenomycin is represented as pink sticks with atoms colored by type. B, ampicillin-bound PBP1b TPase domain. Acyl-ampicillin is depicted as teal sticks with atoms colored by type. C, cephalexin-bound PBP1b TPase domain. Acyl-cephalexin is shown as yellow sticks with atoms colored by type. D, CENTA-bound PBP1b transpeptidase domain. Acyl-CENTA is displayed as orange sticks with non-carbon atoms colored by type. E, aztreonam-bound PBP1b TPase domain. Acyl-aztreonam is illustrated as pink sticks with atoms colored by type. In the left panels for A–E, the ligand omit Fo − Fc electron density map is displayed as pink, cyan, and red transparent surface contoured at 2.5, 3.5, and 4.5 σ. In the right panels for A–E, the final refined ligand 2Fo − Fc electron density map is shown as a gray mesh and contoured at 1.0σ.
Improved Electron Density for the E. coli PBP1b GTase Domain
The GTase domain has approximately twice the average atomic temperature factor (B-factors) of the rest of the E. coli PBP1b protein (71 Å2 versus 37 Å2), suggesting that this region has increased conformational variability. This observation is corroborated by a recent small-angle X-ray scattering study on Streptococcus pneumoniae PBP1b, which suggests that the GTase and TPase domains move relative to each other in solution (22). Furthermore, various crystal structures of the bifunctional S. aureus PBP2 display large amplitude conformational differences in relative domain orientations across the different structures (6, 23). An analysis of our P22121 lattice reveals that the E. coli PBP1b crystal contacts are mediated by the TPase, UB2H, and TM domains rather than the GTase domain. This paucity of crystal contacts likely enables conformational freedom in the GTase domain, providing a potential reason for its observed high B-factors and poorly defined electron density. To improve electron density and hence the atomic model in the GTase domain, we turned to the newly developed FEM available as part of the Phenix crystallographic software suite (17). FEM maps utilize a scaling approach to equalize weak and strong signal to improve electron density in areas with moderate signal, such as the PBP1b GTase domain. A combination of unique crystallization conditions including DM detergent (see “Experimental Procedures”), and the use of the FEM 2Fo − Fc maps resulted in a vast improvement when directly compared with the sigma weighted 2Fo − Fc map calculated using Refmac for the previously solved E. coli PBP1b crystal structure in the same space group (PDB code 3VMA (16)), respectively (Fig. 4). Our revised model provides a vastly improved picture of moenomycin-protein interactions within the active site. This new model will prove indispensable for future structure-based drug design efforts and detailed analysis of the PBP1b-catalyzed mechanism of glycosyltransfer. Because of superior data quality, we hereafter limit our analysis to the PBP1b-aztreonam structure when discussing the E. coli PBP1b GTase domain.
FIGURE 4.

Comparison of 3VMA and aztr-PBP1b electron density at the moenomycin binding site. In all figures, the PBP1b protein backbone is shown as a white cartoon with key active site residues depicted as white sticks with non-carbon atoms colored by type. The bound moenomycin ligand is shown as pink sticks. A, electron density at the moenomycin binding site in previously published PBP1b crystal structure (PDB code 3VMA). B, electron density at the moenomycin binding site in the aztr-PBP1b structure. In A and B, the final refined Refmac calculated 2Fo − Fc and FEM 2Fo − Fc electron density maps are displayed as blue and gray mesh around moenomycin and side chains and are contoured at 1.0 σ.
E. coli PBP1b-Moenomycin Interactions
The PBP1b GTase domain FEM-calculated 2Fo − Fc electron density maps allow for a revised and more complete model of moenomycin binding to the donor site because we are able to clearly resolve the position of individual amino acid side chains contacting moenomycin in PBP1b for the first time. Moenomycin is bound in a shallow, elongated groove formed at the interface of the head and jaw subdomains known as the donor binding site (Fig. 5). The moenomycin rings F, E, C, and B form an extended plane across the elongated groove projecting out from the catalytic Glu233 and occupy the traditional −1, −2, −3, and −4 subsites (6). The phosphoric acid diester group projects below the sugar plane directing the C25 lipid tail downward toward the membrane bilayer (Fig. 5). The C25 lipid tail of moenomycin is not resolved in the electron density. Similarly, this lipid tail could not be modeled in either the moenomycin-bound S. aureus PBP2 or A. aeolicus PBP1a crystal structures (6, 7, 10), suggesting that it is not bound in an ordered conformation. More recently, Heaslet et al. were able to model in 15 carbon atoms of the moenomycin polycarbon tail in their S. aureus monofunctional GTase-bound crystal structure, and these data supported earlier structural analysis suggesting a putative channel located between helices α6 and α4 of the jaw subdomain through which the donor lipid tail may pass in the context of the biological membrane (Fig. 5) (24). In a GTase activity assay using fluorescent lipid II, Sung et al. (16) found that the PBP1b TM helix is required for optimal activity. It was later shown that the donor site requires a lipid side chain length of at least 20 carbon units for processive polymerization, suggesting that the hydrophobic tail contributes to donor binding potentially through interaction with the TM helix or hydrophobic residues at the donor binding site (25). Therefore, the design of moenomycin derivatives containing lipid chains that closely mimic the lipid II C55 tail may yield inhibitors with increased affinity and specificity for PG GTases.
FIGURE 5.

Moenomycin binding to the donor site of the E. coli PBP1b GTase domain. A, multiple sequence alignment of PG GTase domains that have been structurally characterized. The sequences are aligned using tree based progressive alignments and displayed according to their secondary structure elements using ClustalW2 (57) and Chimera (58). B, chemical structures of lipid II and moenomycin. C, donor site close-up of moenomycin-bound to PBP1b. The GTase domain protein backbone is shown in blue cartoon representation with key active site residues displayed as blue sticks with non-carbon atoms colored by type. The bound moenomycin is shown as pink sticks with atoms colored by type. Key hydrogen bonding and electrostatic interactions are shown as black dashes.
Previously, the moenomycin EF ring phosphoglycerate moiety was characterized as the minimal pharmacophore required for efficient binding to the S. aureus monofunctional GTase (10). In our revised PBP1b crystal structure, the moenomycin EF ring phosphoglycerate makes extensive contacts with conserved active site residues. In our model, we observe for the first time clear electron density corresponding to the highly conserved Gln271. The Gln271 side chain amide nitrogen forms a hydrogen bond with the moenomycin F ring carboxamide oxygen (Fig. 5). The moenomycin E ring acetyl group nitrogen and oxygen are engaged in hydrogen bonding interactions with the backbone amide oxygen of Val354, and the side chain N-γ of Asn275. Finally, the highly conserved Arg286 and Lys274 side chains interact directly with the moenomycin phosphonate moiety (Fig. 5, A and C). The Lys274 and Arg286 residues are required for optimal lipid II polymerization activity in the S. aureus monofunctional GTase (26) and by analogy to their interaction with the moenomycin phosphonate are likely important for stabilizing the donor lipid pyrophosphate moiety. Taken together, the EF ring phosphonate portion of moenomycin makes an intricate network of hydrogen bonding interactions with conserved residues in and around the E. coli PBP1b catalytic site that are essential for polymerization of the natural substrate. This observation underpins the fact that despite the widespread use of moenomycin as a growth promoter in animal feed, no significant resistance mechanisms have been detected (8). The ability of moenomycin to mimic key interactions of the donor substrate combined with its unprecedented tight binding affinity provides a substantial genetic barrier to the development of resistance by target modification.
The moenomycin CB rings have analogous features to the GlcNAc-MurNAc backbone of the growing PG chain at subsites −3 and −4 and are recognized through hydrogen bonding and Van der Waals contacts by residues located in the extended donor binding cleft. The acetyl group oxygen of the moenomycin C ring is stabilized by hydrogen bonding to the backbone amide nitrogens of Ala357 and Ser358. The C ring OCD hydroxyl is within hydrogen bonding distance 2.7 Å away from the Ser358 side chain O-γ. The side chain O-ϵ of Glu323 is hydrogen bonded to the B ring OCN hydroxyl at 2.9 Å. Additionally, the CB rings are stabilized by Van der Waals contacts with the Tyr315 and Gln318 side chains that form the base of the donor binding cleft in this region (Fig. 5C). Removal of the moenomycin CB rings results in a 10-fold decrease in affinity (10), and we suggest that the above-mentioned weak interactions act to increase moenomycin binding affinity while permitting the plasticity required to facilitate chain extension when considering processive polymerization of the natural substrate.
A combination of NMR-derived distance constraints and molecular dynamics simulations were used to build a three-dimensional model for free moenomycin in solution (27). This work was published before atomic coordinates were made freely available. However, a visual inspection of the model reveals a similar overall conformation to that observed in the E. coli PBP1b-bound structure, lending support to the proposal that the bioactive form of moenomycin is preformed in aqueous solutions. The E. coli PBP1b-bound moenomycin forms three intramolecular hydrogen-bond interactions (ODI-NAU, O3-OCF, and OCP-OCD (Fig. 5)). These interactions likely help stabilize the bound ligand conformation in the moenomycin-PBP1b complex. It is well established that preorganization of the bound form of a ligand in solution can reduce entropy loss upon binding, thereby increasing affinity (28). We suggest that these intramolecular moenomycin hydrogen bonding interactions may serve to minimize entropy loss upon binding by stabilizing the bound conformation in the unbound inhibitor. We note that these intramolecular interactions are generally present in the previously reported GTase structures (6, 10, 16), suggesting that this effect may be a general characteristic of moenomycin.
Modeling of the Membrane-embedded Jaw Subdomain
In our moenomycin-bound PBP1b-aztreonam structure, we resolve the key active site region spanning amino acids His240–Thr267 containing the jaw subdomain (Fig. 6, A and B). In PG GTase domains this region is often highly disordered and typically cannot be modeled in the electron density (6, 7, 16), with a notable exception being the moenomycin-bound S. aureus monofunctional GTase-bound structure (PDB code 3HZS), in which this region is involved in extensive artifactual crystal packing interactions (24). Also, a PBP2 crystal structure was solved in which this region forms a unique π-bulge that was proposed to permit translocation of the growing polymer from the acceptor to the donor site during polymerization (23). In our PBP1b structure, we find that this stretch of amino acids forms an α-4 helix loop that projects out from the active site toward bulk solvent and serves as a steric barrier to physically separate the donor and acceptor binding sites as evidenced by the final refined 2Fo − Fc map and an omit Fo − Fc map computed using a model with His240–Thr267 deleted (Fig. 6A). Acceptor lipid II analogs were shown to bind to a similarly oriented α-4 helix in the 29.5% identical monofunctional S. aureus GTase (26). We therefore suggest that our extended α-4 helix represents an acceptor competent conformational state.
FIGURE 6.

Modeling of the active site region His240–Thr267. A, electron density for previously unresolved α-4 helix loop region of the PBP1b glycosyltransferase domain. The protein backbone is shown as a white cartoon, and individual residues are depicted in stick representation with non-carbon atoms colored by type. Moenomycin is displayed as pink sticks with atoms colored by type. The fully refined 2Fo − Fc and loop omit Fo − Fc electron density maps are displayed as blue and green mesh contoured at 0.6 and 1.5 σ. The density permits modeling of the orientation and secondary structure features of the protein backbone. However, we note that the maps do not allow for high confidence in the exact location of individual amino acid side chains throughout this region, and residues within this span of amino acids contain B-factors that are roughly 1.5 times the GTase domain average (145.0 versus 94.0 Å2). B, GTase active site overlay of E. coli PBP1b-aztreonam and the previous PBP1b moenomycin-bound structure (PDB code 3VMA). The E. coli PBP1b-aztreonam and 3VMA protein backbones are displayed in blue and white cartoon representation, with key active site residues and moenomycin in the E. coli PBP1b-aztr structure shown as sticks with non-carbon atoms colored by type. The structural overlay yielded a r.m.s.d. for all common main chain atoms of 0.8 Å between the two PBP1b structures. C, surface representation of the PBP1b GTase active site cleft. The protein is displayed as a blue surface with moenomycin depicted as in A. Huang et al. (26) solved the crystal structure of a lipid II analog bound to the monofunctional S. aureus GTase acceptor site (31% sequence identity with the E. coli PBP1b GTase domain). The S. aureus monofunctional GTase and bound ligand was overlaid onto the PBP1b-aztr GTase domain (r.m.s.d. for 42 common cα atoms: 1.2 Å) to highlight the expected general location of lipid II binding to the acceptor site. The lipid II analog is displayed as yellow sticks with non-carbon atoms colored by type. D, proposed mechanism for lipid II polymerization by PBP1b. The PBP1b protein backbone is displayed as a blue cartoon with key active site residues shown as blue sticks with non-carbon atoms colored by type.
In light of our structural observations, we can build on previously proposed models for the GTase catalytic cycle. In the free inner membrane-embedded enzyme, to start the catalytic cycle, lipid II binds to the donor site, and a second lipid II molecule binds the extended α4 helix loop at the acceptor site, which is observed unperturbed by crystal packing for the first time in our E. coli PBP1b-aztreonam crystal structure (Fig. 6, A–D) (6, 26, 29). Upon formation of the precatalytic complex with both donor- and acceptor-bound, the general base Glu233, which we now observe directly in the electron density of the PBP1b GTase active site (Figs. 4B and 6D), is appropriately positioned to activate the acceptor GlcNAc C4 hydroxyl for an SN2-like nucleophilic attack on the donor MurNAc C1, resulting in the departure of the negatively charged C55-PP leaving group. The glycosylation reaction results in inversion of the α-linked precursor molecules into a β1,4-linked product. The role of Glu233 as a general base is likely facilitated by electrostatic stabilization of its negative form by the nearby Lys355 and the universally conserved Arg372, which occupy an adjacent hydrophobic pocket, an environment that is thought to enhance this electrostatic effect. Additionally, in our structure we observe clearly resolved electron density for the conserved Gln271 and note that its side chain N-ϵ forms a hydrogen bond with the Glu233 side chain carboxylate (Figs. 4B and 6D), potentially helping to orient the Glu233 carboxylate and further promoting its requisite deprotonated general base form. This proposition is supported by the fact that the E. coli PBP1b Q271A mutant is catalytically inactive (30). There are alternative views regarding stabilization of the leaving group pyrophosphate. In one perspective, the negative charge on pyrophosphate is stabilized by the universally conserved Glu290 either directly or via an intervening magnesium ion (6). In the second view, the conserved Arg286 and Lys274, which are in close proximity to the moenomycin phosphonate moiety, directly protonate the donor pyrophosphate leaving group (26). Following glycosyl bond formation, the product must translocate from the acceptor to the donor site. To accomplish this, the intervening structured α4 helix loop region (which we observed for the first time crystallographically in our E. coli PBP1b-aztreonam structure (Fig. 6A)) must undergo either localized unfolding or structural rearrangement to permit steric passage of the bulky polymer from acceptor to donor site (Fig. 6D). It is possible that this translocation event is permitted by transient formation of a π-bulge in the His240–Thr267 region as is suggested for S. aureus PBP2 (23). A group of conserved basic residues (Arg286, Lys287, and Arg282 in E. coli PBP1b) flank the base of the donor site and are implicated in direct interaction with the pyrophosphate moiety of the donor. It is thought that this region acts as an electropositive sink to attract the translocating polymer during this step (6). Finally, we suggest that the His240–Thr267 region likely refolds into the observed extended α4 helix loop to permit binding to subsequent lipid II acceptors, thus facilitating further rounds of processive chain extension (Fig. 6D).
Inhibition of E. coli PBP1b by β-Lactam Antibiotics
To investigate the molecular details governing inhibition of the E. coli PBP1b TPase domain, we solved acyl-enzyme co-crystal structures of PBP1b in complex with ampicillin, aztreonam, cephalexin, and the chromogenic cephalosporin CENTA (Fig. 7A). To complement these data, we assess the relative PBP1b inhibitory activity of these compounds by a gel-based competition assay using the fluorescent penicillin BOCILLIN FL (31). All crystal structures contain clear, unambiguous Fo − Fc ligand omit electron density for the acyl-β-lactams covalently bound to the O-γ of the catalytic Ser510 in the TPase domain (Fig. 3). The various acyl-β-lactam R1 groups orient away from the active site center toward bulk solvent (Fig. 7, B and C). In all structures, the backbone amide nitrogen atoms of Ser510 and Thr701 constitute a universally conserved oxyanion hole and are within hydrogen bonding distance from the β-lactam acyl carbonyl oxygen. Finally, the C3 carboxylate, C4 carboxylate, and N1 sulfate of the penicillin, cephalosporin, and monobactam antibiotics all project into close proximity to the motif iii threonine residues (Fig. 7, A and B).
FIGURE 7.

Acyl-β-lactam E. coli PBP1b co-crystal complexes and BOCILLIN FL competition assays. A, chemical structure of β-lactams used in this study. B, overlay of acyl-β-lactam complexes. The unbound protein backbone is shown as a white cartoon. The acyl-ampicillin (Amp), cephalexin (Ceph), CENTA, and aztreonam (Aztr) ligands are depicted as teal, yellow, orange, and pink sticks. C, active site overlay of the unbound and acyl-ampicillin-bound E. coli PBP1b TPase active site. The ampicillin-bound and unbound protein backbones are shown as green and white cartoons. Key active site residues are displayed as white and green sticks with non-carbon atoms colored by type for the unbound and acyl-ampicillin-bound structures. The acyl-ampicillin (Amp) is depicted as teal sticks. Putative hydrogen bonding interactions are displayed as black dashes. D, gel-based BOCILLIN FL competition assays to analyze the ability of various unlabeled competitor compounds to bind purified E. coli PBP1b. The error bars indicate standard deviations from three separate technical replicates. NI indicates no inhibition up to 1000 μm compound. IC50 values represent the concentration of unlabeled compound required to reduce the residual binding of BOCILLIN FL by 50%.
Upon binding of the various β-lactams, the overall juxtaposition of catalytic residues within the TPase active site core remains similar to the unbound enzyme (r.m.s.d. for all common cα atoms within the β-lactam bound and unbound TPase domain: 0.3 Å). This observation suggests that the active site is “preorganized” for inhibitor binding and therefore avoids complicated structural rearrangements that can significantly slow acylation rates as is the case for the β-lactam resistant S. aureus PBP2a (32). However, a notable difference between the unbound and β-lactam-bound structures is that the catalytic Ser510 side chain O-γ undergoes a 90° rotation upon acylation (Fig. 7C). This rotation facilitates the necessary positioning of the acyl-β-lactam oxygen in the oxyanion hole.
Upon binding of ampicillin, cephalexin, and CENTA, there is a conformational rearrangement near the N-terminal end of α-25 in proximity to motif iii (illustrated using the ampicillin-bound structure in Fig. 7C). The penicillins and cephalosphorins share an analogous C3 and C4 carboxylic acid, which interacts with the γ-hydroxyl group of Thr701 on motif iii, which undergoes a 90° rotamer shift to facilitate this hydrogen bond. In response to this rotamer shift, the Gly735 backbone amide nitrogen moves 3.0 Å closer to the O-γ of Thr701 upon binding bringing the two groups within hydrogen bonding distance (2.9 Å; Fig. 7C). This hydrogen bonding network likely stabilizes the α-25 helix and as such may contribute substantially to binding affinity.
Acyl-ampicillin-bound E. coli PBP1b
Ampicillin contains the penicillin core consisting of a five-membered thiazolidine heterocycle fused at the 4 and 5 positions to the β-lactam ring (Fig. 7A). Ampicillin binds to all 12 PBPs in E. coli (33) and is used in combination with the β-lactamase inhibitor sulbactam to treat a diversity of Gram-positive and Gram-negative clinical indications (34). Chemically, the only difference between ampicillin and benzyl-penicillin is the presence of an R1 amine group in ampicillin. Interestingly, the benzyl-penicillin IC50 values were roughly half that of ampicillin in BOCILLIN FL competition assays (9.0 ± 0.6 μm versus 22.0 ± 1.8 μm) (Fig. 7D). When overlaying unbound and ampicillin acylated PBP1b crystal structures, we see that the O-δ of Asn703 is 2.6 Å closer to the ampicillin R1 amine upon acylation, resulting in a hydrogen bond between the two groups (Fig. 7C). In the ampicillin-bound structure, it is likely that this hydrogen bond restrains the orientation of the ampicillin α-aminobenzyl R1 group, resulting in an entropic penalty.
Acyl-cephalexin and CENTA-bound E. coli PBP1b
Cephalosporins such as cephalexin and CENTA contain a six-membered dihydrothiazine ring attached to the lactam core. The cephalexin- and CENTA-bound E. coli PBP1b structures provide a molecular basis for cephalosporin-mediated inhibition. A major chemical difference between the CENTA and cephalexin molecules is that cephalexin contains a C3′ methyl group, whereas CENTA has a C3′ 3-carboxyl-4-nitrothiophenol (TNB) moiety (Fig. 7A). Upon acylation, the CENTA dihydrothiazine ring is thought to undergo tautomerization resulting in a shift of the double bond from positions 3–4 to 4–5, with anion formation at position 3 (35). The following reaction depends upon the chemical nature of the R2 functional group. In the case of CENTA, the R2 TNB moiety is an excellent leaving group (36) and is expected to yield a product with elimination of TNB and formation of a 3–3′ double bond (Fig. 8A) (36, 37). This R2 elimination is supported by a lack of TNB electron density in the CENTA-bound PBP1b crystal structure (Figs. 3 and 8B). In contrast, cephalexin contains a C3′ methyl group and thus does not have a good leaving group at position R2 (Fig. 7A and 8C) (38, 39). The CENTA and cephalexin acyl-enzyme crystal structures display remarkably similar overall conformations and ligand-protein hydrogen bonding interactions (Fig. 8, B and D). However, in BOCILLIN FL competition assays, cephalexin displays no inhibition up to 1 mm compound, as compared with the low micromolar IC50 value obtained for CENTA (22.0 ± 1.6 μm; Fig. 7D). These data are corroborated by a previous study whereby E. coli PBP1b-catalyzed transpeptidation of PG was not inhibited by cephalexin or cephradine, which both contain a methyl group at the C3′ position. In contrast, it was found that the third generation cephalosporins cefsulodin and cephaloridine act as inhibitors of E. coli PBP1b (40). Cefsulodin and cephaloridine have a C3′ pyridinium moiety that acts as a leaving group during acylation similar to the CENTA TNB group. It is well established that the C3′ moiety can have a profound influence on acylation rates by delocalizing electrons from the former lactam nitrogen either by acting as an electron withdrawing substituent or leaving group during lactam bond fission (41, 42). Therefore, the presence of a good C3′ leaving group is an important feature to consider when designing cephalosporin-based inhibitors of E. coli PBP1b.
FIGURE 8.

Inhibition of E. coli PBP1b by cephalosporins and aztreonam. A, chemical structure of acyl-CENTA-bound PBP1b. B, active site close-up of CENTA-bound PBP1b. The PBP1b protein backbone is displayed as a green cartoon with key active site residues shown as sticks with atoms colored by type. C, chemical structure of acyl-cephalexin bound PBP1b. D, active site close-up of cephalexin bound PBP1b. The PBP1b protein backbone and active site residues are displayed as in B. In B and D, the CENTA and cephalexin ligands are displayed as orange and yellow sticks with non-carbon atoms colored by atom type. In B and D, putative hydrogen bonding interactions are depicted as black dashes. E, active site close-up of acyl-aztreonam bound to the E. coli PBP1b transpeptidase domain. The PBP1b protein backbone is represented as a green cartoon with key active site residues depicted as green sticks with non-carbon atoms colored by atom type. The acyl-aztreonam (Aztr) is depicted as pink sticks with non-carbon atoms colored by type. F, active site overlay of E. coli PBP1b acyl-aztreonam and P. aeruginosa PBP3 acyl-aztreonam (PDB code 3PBS). The E. coli PBP1b and bound aztreonam are displayed as in A. The P. aeruginosa PBP3 protein backbone is displayed as a white cartoon with key active site residues shown as white sticks with non-carbon atoms colored by type. The PBP3 bound aztreonam ligand is displayed as gray sticks with non-carbon atoms colored by atom type. Key ligand-protein hydrogen bonding interactions are displayed as black and blue dashes for the PBP1b and PBP3 aztreonam-bound structures. In B, residues are labeled according to E. coli PBP1b numbering.
Acyl-aztreonam-bound E. coli PBP1b
The monobactams are predominantly synthetic monocyclic compounds containing variable functional groups at the C3 and C4 positions and a sulfonic acid moiety attached to the N1 nitrogen (Fig. 7A). Aztreonam is currently the only monobactam antibiotic in clinical use and is almost exclusively active against aerobic Gram-negative bacilli (43). In E. coli and other enteric pathogens such as P. aeruginosa, the bactericidal action of aztreonam is attributed predominantly to its potent inhibition of PBP3 (44) rather than PBP1b. Aztreonam displays higher IC50 values in the E. coli PBP1b BOCILLIN FL competition assays than the other β-lactams tested, with the exception of cephalexin (Fig. 7D). A TPase domain overlay of the acyl-aztreonam-bound P. aeruginosa PBP3 (45) with the acyl-aztreonam E. coli PBP1b structure (r.m.s.d. for 70 common cα atoms = 1.3 Å) reveals that the bulky aztreonam aminothiazole and aminopropyl carboxyl moieties occupy roughly analogous positions in the two structures (Fig. 8, E and F). However, in the P. aeruginosa PBP3 structure, the aminopropyl carboxyl moiety of aztreonam forms a bipartite salt bridge with the active site Arg489 guanadino group, and this interaction is absent in the E. coli PBP1b complex. Additionally, in the P. aeruginosa PBP3 acyl-enzyme complex, the aztreonam aminothiazole moiety is stabilized by hydrogen bonding with the Glu291 side chain carboxylate, and this residue is a serine (Ser507) in E. coli PBP1b and is not involved in hydrogen bonding (Fig. 8F). The design of monobactams that replace the aztreonam R1 moiety with side chains analogous to more potent PBP1b inhibitors such as the benzyl side chain of benzyl-penicillin may be an effective design strategy. Development of monobactams targeted at enteric pathogens such as E. coli is an attractive prospect because they represent the only class of β-lactams that avoid hydrolytic inactivation by the rapidly emerging metallo-β-lactamase enzymes (46).
Concluding Remarks
As a major clinical target of the β-lactams for the past half-century, the bifunctional PBPs have gained notoriety as excellent antibacterial targets. Herein, we investigated inhibition of both enzymatic domains in the bifunctional bitopic membrane protein E. coli PBP1b. We provide a pyrophosphate sensor assay that can be used for future in-depth analysis of the kinetic mechanism of glycosyltransfer and to screen for PG GTase inhibitors. A revised analysis of moenomycin binding to the donor site of the GTase domain shows that the EF ring phosphonate portion of moenomycin is involved in an extensive network of hydrogen bonding interactions with highly conserved active site residues that are essential for processive polymerization of the natural substrate. Furthermore, a detailed analysis of various acyl-β-lactams covalently bound to the E. coli PBP1b TPase domain reveals subtle differences in ligand binding that manifest in notable discrepancies in relative inhibitory activity. It is our hope that this study will aid in the informed design of PBP1b inhibitors effective against emerging drug-resistant Enterobacteriacea.
Experimental Procedures
Plasmid Construction, Protein Expression, and Purification
The E. coli PBP1b DNA corresponding to amino acid residues 58–804 was amplified from E. coli K12 genomic DNA. Restriction free cloning was then used to produce a pET-41b expression vector containing PBP1b with a thrombin cleavable C-terminal His8 tag (47).
BL21(DE3) host cells transformed with the E. coli PBP1b expression vector were grown at 37 °C until an A600 of 0.6 was reached, and the samples were cooled to room temperature for 30 min. Protein expression was induced by addition of 1 mm isopropyl β-d-1-thiogalactopyranoside, and the cultures were incubated at 25 °C overnight.
Cell pellets were resuspended in lysis buffer (20 mm Tris, pH 8.0, 300 mm NaCl, 1 EDTA free protease inhibitor tablet from Roche) and lysed by two passes on a French press at a pressure of 1500 p.s.i. The cell lysate was then centrifuged twice at 11,000 rpm for 15 min using a Beckman JA 25.50 rotor to remove unbroken cells and inclusion bodies. The supernatant was then centrifuged at 45,000 rpm for 1 h using a Beckman 60Ti rotor to pellet the membranes. The membranes were homogenized and incubated for 4 h in the presence of extraction buffer (lysis buffer + 20 mm n-dodecyl-β-d-maltopyranoside; Anatrace). The solubilized protein was then purified using nickel chelation chromatography. The column was preincubated in the presence of equilibration buffer (20 mm Tris, pH 8.0, 300 mm NaCl, 1 mm n-dodecyl-β-d-maltopyranoside) and eluted using a linear gradient of imidazole from 0–500 mm. The C-terminal His8 tag was cleaved by the addition of 1 unit of bovine α-thrombin per mg of PBP1b protein and incubation at 4 °C overnight. Following thrombin cleavage, PBP1b was further purified using a Superdex 200 sizing column into crystallization running buffer (20 mm Tris, pH 8.0, 300 mm NaCl, 4.5 mm DM; Anatrace). Peak fractions were pooled and concentrated using a 100-kDa cutoff Amicon centrifugation unit.
Crystallization, Data Collection, and Structure Solution
The moenomycin-bound PBP1b-acyl-β-lactam crystals were grown using the sitting drop vapor diffusion method at 25 °C. Drops contained 1 μl of protein solution (20 mg/ml protein, 100 μm moenomycin, and 2 mm ampicillin, nitrocefin, cephalexin, aztreonam, or CENTA) mixed with an equal volume of precipitant (20% w/v PEG 3350, 0.2 m potassium/sodium tartate, 0.1 m Bis-Tris, pH 8.5). For cryoprotection, the crystals were transferred to mother liquor plus 30% glycerol. We note that our crystallization conditions were different from the previous E. coli PBP1b sitting drop vapor diffusion conditions in which PBP1b (12 mg/ml protein in buffer: 20 mm Tris, pH 8, 300 mm NaCl, 0.28 mm N-dodecyl-N,N-dimethylamine-N-oxide, 1.4 mm moenomycin) was mixed with an equal volume of precipitant (1.2 m sodium formate). The moenomycin was dissolved in double distilled H2O and stored at −20 °C in a 10 mm stock solution and was a kind gift from Aventis Pharma (Frankfurt, Germany).
Diffraction data were collected at Beamline CMCF-2 at the Canadian Light Source in Saskatoon Saskatchewan. The data were collected at a wavelength of 1.0Å and a temperature of 100 K. All data were indexed, integrated, and scaled using Xia2 (48). For cross-validation purposes, a total of 5% of reflections were set aside. The structures were solved by molecular replacement using the program Phaser (49), with chain A of the moenomycin-bound E. coli PBP1b crystal structure as a starting model (PDB code 3VMA (16)). Several cycles of manual building in COOT (50), followed by refinement in REFMAC (CCP4 (51)) were carried out. During late stage refinement, feature enhanced maps (17) were calculated using the Phaser (49) software and were used to guide model building in COOT. The final models are of excellent stereochemical quality and display <0.6% Ramachandran outliers, all of which are consistent with the previous PBP1b model (16). Coordinates and structure factors were deposited in the PDB with accession codes 5HL9, 5HLD, 5HLA, and 5HLB for PBP1b-ampicillin, PBP1b-CENTA, PBP1b-cephalexin, and PBP1b-aztreonam, respectively. The figures representing the PBP1b crystal structures were created using PyMOL (52).
Membrane Purification from E. coli C43 Cells Overexpressing S. aureus MraY and MurG
E. coli C43 host cells were transformed with a pETDuet vector coexpressing MraY and MurG, and the cells were grown at 37 °C until an A600 of 0.6–0.8 was achieved. Protein expression was induced with 1 mm isopropyl β-d-1-thiogalactopyranoside, and the cells were incubated at 37 °C for 4 h with 200 rpm shaking. The cells were harvested by centrifugation and resuspended in lysis buffer (10 mm Tris-HCl, pH 7.5, 20 mm MgCl2, one pulverized complete EDTA free protease inhibitor tablet (Roche), 300 μg/ml lysozyme, and 10 μg/ml DNase I). The cell suspension was homogenized and passed two times through a French press at 13,000 p.s.i. The cells were centrifuged for 30 min at 12,000 rpm at 4 °C using a Thermo FiberLITE F15–6 × 100y rotor. The supernatant was collected and centrifuged for an additional hour at 60,000 × g at 4 °C using a Beckman 70Ti rotor. The membrane pellets were homogenized and resuspended in (20 mm Tris-HCl, pH 7.5, 20 mm MgCl2, 2 mm 2-mercaptoethanol) to a final total protein concentration of 0.6 mg/ml.
Lipid II Synthesis
Undecaprenyl phosphate (C55-P) was prepared from bay leaves as previously described and dissolved in 2:1 chloroform:methanol mixture for long term storage at −80 °C (53). Solvent was evaporated from the C55-P sample using a stream of N2 gas. The final amount of C55-P was determined from the dry weight of the purified compound. Sample integrity and purity was assessed by electrospray ionization (ESI)-TOF mass spectrometry (data not shown). Following evaporation, the sample was dissolved in 2× reaction buffer (100 mm Tris-HCl, pH 8.0, 40 mm MgCl2, 8% v/v glycerol, 2.0% v/v Triton X-100) to a final C55-P concentration of 600 μm with sonication. UDP-MurNAc pentapeptide (with lysine at position 3 on the stem peptide) was prepared as previously described (54), and UDP-N-acetylglucosamine (UDP-GlcNAc) was purchased from Sigma.
For lipid II synthesis, the reaction mixture consisted of (300 μm C55-P, 2 mm UDP-GlcNAc, 2 mm UDP-MurNAc pentapeptide (with lysine at stem peptide position 3), 0.12 mg/ml E. coli C43 membranes harboring S. aureus MraY and MurG, all in reaction buffer in a total volume of 500 μl). The reaction was incubated at 30 °C for 1.5 h with shaking at 200 rpm. The reaction was terminated by addition of 1.5× volume 2:1 n-butanol:6 m pyridinium acetate, pH 4.2. This biphasic mixture was vortexed for 30 s to extract the lipids, and the phases were separated by centrifugation at 6, 400 rpm for 30 s in a microcentrifuge. The organic phase was washed with one equivalent volume of distilled water. The mixture was vortexed and centrifuged as described above, and the organic layer was vacuum-filtered to remove membrane debris. After filtration, the membranes were rinsed several times with butanol, and the organic layer containing lipid II was separated from any residual water. The final purified organic phase was directly injected onto a HPLC Phenomenex Luna C18 reverse phase semipreparative column (5 μm, 100 Å, 250 × 10 mm) with a gradient from 100% buffer A (20 mm ammonium acetate, 65:20:15 methanol:isopropanol:water) to 100% buffer B (20 mm ammonium acetate, 98:2 isopropanol:methanol). Fractions containing lipid II were identified by HPLC analysis with an Agilent Poroshell 120 EC-C18 chromatography column (2.7 μm, 4.6mm × 50 mm) using the same solvent gradient and monitoring the absorbance of the eluate at 210 nm. Peaks corresponding to lipid II were further confirmed by ESI-TOF mass spectrometry to assess molecular weight and purity (data not shown). The final purified lipid II samples were dried down in a 50:50 CHCl3:MeOH mixture, and the final concentration of lipid II was determined by 1H NMR spectroscopy using toluene as an internal standard, and the samples were stored at −80 °C with desiccant. Immediately prior to use in assays, the lipid II stock was resuspended in the following buffer (50 mm HEPES, pH 7.5, 10% DMSO, 10 mm CaCl2, 0.85% C12E8).
E. coli PBP1b Glycosyltransferase Pyrophosphate Sensor Assay
PBP1b-catalyzed glycosyltransfer using purified lipid II (position 3 lysine version) was monitored using the PhosphoWorks fluorometric pyrophosphate assay kit (AAT Bioquest Inc., product no. 21611). The reactions were performed in a 1536-well black assay plate in a total volume of 5 μl at 25 °C according to the manufacturer's protocol. Briefly, various concentrations of purified E. coli PBP1b were incubated with 100 μm lipid II and 1.5× pyrophosphate sensor in final reaction buffer (50 mm HEPES, pH 7.5, 10% DMSO, 10 mm CaCl2, 0.085% C12E8 (Anatrace product no. O330)). Upon polymerization of lipid II by PBP1b, C55-PP is released and binds the pyrophosphate sensor, resulting in a fluorescence signal detected continuously (excitation and emission wavelengths: 360, and 460 nm) using a Synergy H4 multimode plate reader (BioTek). Individual assays were initiated with the addition of lipid II, and initial velocities were collected from the linear portion of the time course (typically from 10–20 min postreaction initiation). Steady-state kinetic values were determined from a substrate dose-response curve using 1 μm of PBP1b and varying concentrations of lipid II (values are taken as averages from triplicate reactions). The kcat value was calculated by titrating increasing concentrations of C55-PP in reaction buffer minus protein to create a standard curve. C55-PP was purchased from Larodan in a 4:1 MeOH:ammonia mixture and stored at −80 °C according to the manufacturer's instructions. Immediately prior to use, the solvent from the C55-PP sample was evaporated off using a N2 stream, and the dried C55-PP was freshly dissolved in assay buffer for immediate use. The concentration of C55-PP was determined using the known mass of the purified dried product provided by the manufacturer. In addition to the manufacturer's quality control analysis, the purity of the C55-PP stock was further verified by ESI-TOF mass spectrometry analysis (data not shown). Inhibition assays were performed using 1 μm E. coli PBP1b and 45.5 μm of lipid II with various concentrations of moenomycin complex (predominantly moenomycin A but also contains other moenomycins; A12, C1, C3, and C4; Santa Cruz Biotechnology). All curve fitting was performed using SigmaPlot. The substrate dose-response curve was fit to a three-parameter sigmoidal Hill equation kinetic model based on equation (Vo = Vm × [s]n/K0.5 + [s]n). The criteria that were used for curve selection were superior fit by visual inspection, sum of squares analysis, and Akaike Information Criterion.
Thermostability Analysis
E. coli PBP1b thermostability was measured as a function of its temperature-dependent aggregation by differential static light scattering (StarGazer-2; Epiphyte Three Inc.) as reported previously (55). Briefly, 0.4 mg/ml purified PBP1b protein was added to various concentrations of moenomycin complex in assay buffer (see above), and the mixture was heated from 25 to 85 °C at a rate of 1 °C/min in a final volume of 9 μl in a clear-bottomed 384-well plate (Nunc). Protein aggregation, as a measure of light scattering, was scanned every 30 s using a CCD camera. Integrated intensities were plotted against temperature, and Boltzmann regression curve fitting was performed using SigmaPlot with the inflection point representing the aggregation temperature (Tagg). The change in Tagg (ΔTagg) from the no-inhibitor control was plotted as a function of moenomycin complex concentration, and the resulting hyperbolic curve was fit to a ligand binding one-site saturation model in SigmaPlot to attain the aggregation constant (Kagg) at ½ ΔTmax.
BOCILLIN FL Gel-based Competition Assays
To assess the relative inhibition of E. coli PBP1b by multiple different small molecules, SDS-PAGE-based concentration response experiments were performed in triplicate using the fluorescent penicillin BOCILLIN FL as a reporter molecule. All reagents were diluted in reaction buffer prior to use (20 mm Tris, pH 8.0, 300 mm NaCl, 4.5 mm DM). To start the reaction, various concentrations of unlabeled compound and 25 μm BOCILLIN FL were simultaneously added to 13 μm of purified E. coli PBP1b protein in a final reaction volume of 35 μl. The reaction was incubated at 25 °C for 30 min prior to addition of 10× SDS-PAGE loading dye. The samples were then boiled for 2 min prior to loading 10 μl on a 10% SDS-PAGE gel. Following electrophoresis, gels were imaged under UV light using a gel imager. Densitometry analysis was performed using ImageJ (56). The individual data points were normalized to the maximum value of the fluorescence intensity, which represents total saturation of protein by BOCILLIN FL in the absence of unlabeled compound. The IC50 values are defined as the compound concentration required to reduce the residual binding of BOCILLIN FL by 50% and were calculated using SigmaPlot.
Author Contributions
D. T. K., G. A. W., and N. C. J. S. designed experiments. D. T. K. cloned, overexpressed, and purified PBP1b; solved all PBP1b co-crystal complexes; performed pyrophosphate sensor and thermostability assays with help from G. A. W.; and performed gel-based BOCILLIN FL competition assays. G. A. W., M. N., and A. F. synthesized lipid II. D. T. K. and N. C. J. S. principally wrote the manuscript with input from all.
Acknowledgments
We thank beamline personnel of CMCF-2 at the Canadian Light Source synchrotron facility (Saskatoon, Canada) for assistance with data collection. We also thank Robert Gale (McMaster University) for instructions regarding the extraction and synthesis of C55-P, Lawrence Amankwa (Centre for Drug Research and Development) for performing mass spectrometry on Lipid II and its precursors and David Grierson (Faculty of Pharmacy, University of British Columbia) for the use of the flash chromatography system to purify C55-P.
This work was supported by the Canadian Institutes of Health Research and Howard Hughes Medical Institute International Scholar Program and infrastructure support from the Canadian Foundation for Innovation, British Columbia Knowledge Development Fund, and the Canada Research Chair Programs to (N. C. J. S.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5HLA, 5HLD, 5HLB, and 5HL9) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PG
- peptidoglycan
- cα
- alpha carbon
- GTase
- glycosyltransferase
- TPase
- transpeptidase
- PBP
- penicillin-binding protein
- PDB
- Protein Data Bank
- r.m.s.d.
- root mean square deviation
- TM
- trans-membrane
- UB2H
- UvrB domain 2 homolog
- DM
- n-decyl-β-d-maltopyranoside
- MurNAc
- N-acetylmuramic acid
- FEM
- feature-enhanced maps
- TNB
- nitrothiophenol
- ESI
- electrospray ionization.
References
- 1. Typas A., Banzhaf M., Gross C. A., and Vollmer W. (2011) From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol. 10, 123–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sobhanifar S., King D. T., and Strynadka N. C. (2013) Fortifying the wall: synthesis, regulation and degradation of bacterial peptidoglycan. Curr. Opin. Struct. Biol. 23, 695–703 [DOI] [PubMed] [Google Scholar]
- 3. Paradis-Bleau C., Markovski M., Uehara T., Lupoli T. J., Walker S., Kahne D. E., and Bernhardt T. G. (2010) Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell 143, 1110–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Egan A. J., Jean N. L., Koumoutsi A., Bougault C. M., Biboy J., Sassine J., Solovyova A. S., Breukink E., Typas A., Vollmer W., and Simorre J. P. (2014) Outer-membrane lipoprotein LpoB spans the periplasm to stimulate the peptidoglycan synthase PBP1B. Proc. Natl. Acad. Sci. U.S.A. 111, 8197–8202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. King D. T., Lameignere E., and Strynadka N. C. (2014) Structural insights into the lipoprotein outer-membrane regulator of penicillin-binding protein 1B. J. Biol. Chem. 289, 19245–19253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lovering A. L., de Castro L. H., Lim D., and Strynadka N. C. (2007) Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis. Science 315, 1402–1405 [DOI] [PubMed] [Google Scholar]
- 7. Yuan Y., Barrett D., Zhang Y., Kahne D., Sliz P., and Walker S. (2007) Crystal structure of a peptidoglycan glycosyltransferase suggests a model for processive glycan chain synthesis. Proc. Natl. Acad. Sci. U.S.A. 104, 5348–5353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ostash B., and Walker S. (2010) Moenomycin family antibiotics: chemical synthesis, biosynthesis, and biological activity. Nat. Prod. Rep. 27, 1594–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goldman R. C., and Gange D. (2000) Inhibition of transglycosylation involved in bacterial peptidoglycan synthesis. Curr. Med. Chem. 7, 801–820 [DOI] [PubMed] [Google Scholar]
- 10. Yuan Y., Fuse S., Ostash B., Sliz P., Kahne D., and Walker S. (2008) Structural analysis of the contacts anchoring moenomycin to peptidoglycan glycosyltransferases and implications for antibiotic design. ACS Chem. Biol. 3, 429–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sauvage E., Kerff F., Terrak M., Ayala J. A., and Charlier P. (2008) The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32, 234–258 [DOI] [PubMed] [Google Scholar]
- 12. Miller C., Thomsen L. E., Gaggero C., Mosseri R., Ingmer H., and Cohen S. N. (2004) SOS response induction by β-lactams and bacterial defense against antibiotic lethality. Science 305, 1629–1631 [DOI] [PubMed] [Google Scholar]
- 13. Pepper E. D., Farrell M. J., and Finkel S. E. (2006) Role of penicillin-binding protein 1b in competitive stationary-phase survival of Escherichia coli. FEMS Microbiol. Lett. 263, 61–67 [DOI] [PubMed] [Google Scholar]
- 14. Wright D. B. (1986) Cefsulodin. Drug Intell. Clin. Pharm. 20, 845–849 [DOI] [PubMed] [Google Scholar]
- 15. Sarkar S. K., Dutta M., Kumar A., Mallik D., and Ghosh A. S. (2012) Sub-inhibitory cefsulodin sensitization of E. coli to β-lactams is mediated by PBP1b inhibition. PLoS One 7, e48598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sung M. T., Lai Y. T., Huang C. Y., Chou L. Y., Shih H. W., Cheng W. C., Wong C. H., and Ma C. (2009) Crystal structure of the membrane-bound bifunctional transglycosylase PBP1b from Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 106, 8824–8829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Afonine P. V., Moriarty N. W., Mustyakimov M., Sobolev O. V., Terwilliger T. C., Turk D., Urzhumtsev A., and Adams P. D. (2015) FEM: feature-enhanced map. Acta Crystallogr. D Biol. Crystallogr. 71, 646–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schwartz B., Markwalder J. A., Seitz S. P., Wang Y., and Stein R. L. (2002) A kinetic characterization of the glycosyltransferase activity of Escherichia coli PBP1b and development of a continuous fluorescence assay. Biochemistry 41, 12552–12561 [DOI] [PubMed] [Google Scholar]
- 19. Offant J., Terrak M., Derouaux A., Breukink E., Nguyen-Distèche M., Zapun A., and Vernet T. (2010) Optimization of conditions for the glycosyltransferase activity of penicillin-binding protein 1a from Thermotoga maritima. FEBS J. 277, 4290–4298 [DOI] [PubMed] [Google Scholar]
- 20. Hutzler J. M., and Tracy T. S. (2002) Atypical kinetic profiles in drug metabolism reactions. Drug Metab. Dispos. 30, 355–362 [DOI] [PubMed] [Google Scholar]
- 21. Tracy T. S., and Hummel M. A. (2004) Modeling kinetic data from in vitro drug metabolism enzyme experiments. Drug Metab. Rev. 36, 231–242 [DOI] [PubMed] [Google Scholar]
- 22. Macheboeuf P., Piuzzi M., Finet S., Bontems F., Pérez J., Dessen A., and Vachette P. (2011) Solution X-ray scattering study of a full-length class A penicillin-binding protein. Biochem. Biophys. Res. Commun. 405, 107–111 [DOI] [PubMed] [Google Scholar]
- 23. Lovering A. L., De Castro L., and Strynadka N. C. (2008) Identification of dynamic structural motifs involved in peptidoglycan glycosyltransfer. J. Mol. Biol. 383, 167–177 [DOI] [PubMed] [Google Scholar]
- 24. Heaslet H., Shaw B., Mistry A., and Miller A. A. (2009) Characterization of the active site of S. aureus monofunctional glycosyltransferase (Mtg) by site-directed mutation and structural analysis of the protein complexed with moenomycin. J. Struct. Biol. 167, 129–135 [DOI] [PubMed] [Google Scholar]
- 25. Perlstein D. L., Wang T. S., Doud E. H., Kahne D., and Walker S. (2010) The role of the substrate lipid in processive glycan polymerization by the peptidoglycan glycosyltransferases. J. Am. Chem. Soc. 132, 48–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang C. Y., Shih H. W., Lin L. Y., Tien Y. W., Cheng T. J., Cheng W. C., Wong C. H., and Ma C. (2012) Crystal structure of Staphylococcus aureus transglycosylase in complex with a lipid II analog and elucidation of peptidoglycan synthesis mechanism. Proc. Natl. Acad. Sci. U.S.A. 109, 6496–6501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kurz M., Guba W., and Vértesy L. (1998) Three-dimensional structure of moenomycin A: a potent inhibitor of penicillin-binding protein 1b. Eur. J. Biochem. 252, 500–507 [DOI] [PubMed] [Google Scholar]
- 28. Martin S. F., and Clements J. H. (2013) Correlating structure and energetics in protein-ligand interactions: paradigms and paradoxes. Annu. Rev. Biochem. 82, 267–293 [DOI] [PubMed] [Google Scholar]
- 29. Bury D., Dahmane I., Derouaux A., Dumbre S., Herdewijn P., Matagne A., Breukink E., Mueller-Seitz E., Petz M., and Terrak M. (2015) Positive cooperativity between acceptor and donor sites of the peptidoglycan glycosyltransferase. Biochem. Pharmacol. 93, 141–150 [DOI] [PubMed] [Google Scholar]
- 30. Terrak M., Sauvage E., Derouaux A., Dehareng D., Bouhss A., Breukink E., Jeanjean S., and Nguyen-Distèche M. (2008) Importance of the conserved residues in the peptidoglycan glycosyltransferase module of the class A penicillin-binding protein 1b of Escherichia coli. J. Biol. Chem. 283, 28464–28470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao G., Meier T. I., Kahl S. D., Gee K. R., and Blaszczak L. C. (1999) BOCILLIN FL, a sensitive and commercially available reagent for detection of penicillin-binding proteins. Antimicrob. Agents Chemother. 43, 1124–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lim D., and Strynadka N. C. (2002) Structural basis for the β lactam resistance of PBP2a from methicillin-resistant Staphylococcus aureus. Nat. Struct. Biol. 9, 870–876 [DOI] [PubMed] [Google Scholar]
- 33. Denome S. A., Elf P. K., Henderson T. A., Nelson D. E., and Young K. D. (1999) Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: viability, characteristics, and implications for peptidoglycan synthesis. J. Bacteriol. 181, 3981–3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rafailidis P. I., Ioannidou E. N., and Falagas M. E. (2007) Ampicillin/sulbactam: current status in severe bacterial infections. Drugs 67, 1829–1849 [DOI] [PubMed] [Google Scholar]
- 35. Feng H., Ding J., Zhu D., Liu X., Xu X., Zhang Y., Zang S., Wang D. C., and Liu W. (2014) Structural and mechanistic insights into NDM-1 catalyzed hydrolysis of cephalosporins. J. Am. Chem. Soc. 136, 14694–14697 [DOI] [PubMed] [Google Scholar]
- 36. Bebrone C., Moali C., Mahy F., Rival S., Docquier J. D., Rossolini G. M., Fastrez J., Pratt R. F., Frère J. M., and Galleni M. (2001) CENTA as a chromogenic substrate for studying β-lactamases. Antimicrob. Agents Chemother. 45, 1868–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mobashery S., and Johnston M. (1986) Reactions of Escherichia coli TEM β-lactamase with cephalothin and with C10-dipeptidyl cephalosporin esters. J. Biol. Chem. 261, 7879–7887 [PubMed] [Google Scholar]
- 38. Vilanova B., Frau J., Donoso J., Muñoz F., and García Blanco F. (1997) β-Lactamase-catalysed hydrolysis of cephalexin: evolution of the cephalosporoate intermediate. J. Chem. Soc. 2, 2439–2444 [Google Scholar]
- 39. Pratt R. F., and Faraci W. S. (1986) Direct observation by 'H NMR of cephalosporoate intermediates in aqueous solution during the hydrazinolysis and β-lactamase-catalyzed hydrolysis of cephalosporins with 3′ leaving groups: kinetics and equilibria of the 3′ elimination reaction. J. Am. Chem. Soc. 108, 5328–5333 [Google Scholar]
- 40. Jha R. K., and de Sousa S. M. (2006) Microplate assay for inhibitors of the transpeptidase activity of PBP1b of Escherichia coli. J. Biomol. Screen. 11, 1005–1014 [DOI] [PubMed] [Google Scholar]
- 41. Boyd D. B. (1984) Elucidating the leaving group effect in the β-lactam ring opening mechanism of cephalosporins. J. Org. Chem. 50, 886–888 [Google Scholar]
- 42. Page M. I., and Proctor P. (1984) Mechanism of β-lactam ring opening in cephalosporins. J. Am. Chem. Soc. 106, 3820–3825 [Google Scholar]
- 43. Johnson D. H., and Cunha B. A. (1995) Aztreonam. Med. Clin. North Am. 79, 733–743 [DOI] [PubMed] [Google Scholar]
- 44. Sykes R. B., and Bonner D. P. (1985) Discovery and development of the monobactams. Rev. Infect. Dis. 7, S579–S593 [DOI] [PubMed] [Google Scholar]
- 45. Han S., Zaniewski R. P., Marr E. S., Lacey B. M., Tomaras A. P., Evdokimov A., Miller J. R., and Shanmugasundaram V. (2010) Structural basis for effectiveness of siderophore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 107, 22002–22007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. King D. T., Worrall L. J., Gruninger R., and Strynadka N. C. (2012) New Delhi metallo-β-lactamase: structural insights into β-lactam recognition and inhibition. J. Am. Chem. Soc. 134, 11362–11365 [DOI] [PubMed] [Google Scholar]
- 47. van den Ent F., and Löwe J. (2006) RF cloning: a restriction-free method for inserting target genes into plasmids. J. Biochem. Biophys. Methods 67, 67–74 [DOI] [PubMed] [Google Scholar]
- 48. Winter G., Lobley C. M., and Prince S. M. (2013) Decision making in xia2. Acta Crystallogr. D Biol. Crystallogr. 69, 1260–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.5.0.4, Schroedinger, LLC, New York [Google Scholar]
- 53. Gale R. T., Sewell E. W., Garrett T. A., and Brown E. D. (2014) Reconstituting poly(glycerol phosphate) wall teichoic acid biosynthesis in vitro using authentic substrates. Chem. Sci. 5, 3823–3830 [Google Scholar]
- 54. Kohlrausch U., and Höltje J. V. (1991) One-step purification procedure for UDP-N-acetylmuramyl-peptide murein precursors from Bacillus cereus. FEMS Microbiol. Lett. 62, 253–257 [DOI] [PubMed] [Google Scholar]
- 55. Vedadi M., Niesen F. H., Allali-Hassani A., Fedorov O. Y., Finerty P. J. Jr., Wasney G. A., Yeung R., Arrowsmith C., Ball L. J., Berglund H., Hui R., Marsden B. D., Nordlund P., Sundstrom M., Weigelt J., et al. (2006) Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. U.S.A. 103, 15835–15840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., and Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 58. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]