

Abstract
Many members of the serine protease inhibitor (serpin) family are activated by glycosaminoglycans (GAGs). Visceral adipose tissue-derived serpin (vaspin), serpin A12 of the serpin family, and its target protease kallikrein 7 (KLK7) are heparin-binding proteins, and inhibition of KLK7 by vaspin is accelerated by heparin. However, the nature of GAG binding to vaspin is not known. Here, we measured vaspin binding of various glycosaminoglycans and low molecular weight heparins by microscale thermophoresis and analyzed acceleration of protease inhibition by these molecules. In addition, basic residues contributing to heparin binding and heparin activation were identified by a selective labeling approach. Together, these data show that vaspin binds heparin with high affinity (KD = 21 ± 2 nm) and that binding takes place at a basic patch on top of β-sheet A and is different from other heparin-binding serpins. Mutation of basic residues decreased heparin binding and activation of vaspin. Similarly, reactive center loop insertion into sheet A decreased heparin binding because it disturbs the basic cluster. Finally, using vaspin-overexpressing keratinocyte cells, we show that a significant part of secreted vaspin is bound in the extracellular matrix on the cell surface. Together, basic residues of central β-sheet A contribute to heparin binding and activation of vaspin. Thus, binding to GAGs in the extracellular matrix can direct and regulate vaspin interaction with target proteases or other proteins and may play an important role in the various beneficial functions of vaspin in different tissues.
Keywords: adipokine, glycosaminoglycan, heparin-binding protein, protease inhibitor, serine protease, serpin, structural biology, kallikrein 7, vaspin
Introduction
Vaspin (serpin A12) was identified in adipose tissue of the OLETF rat type-2 diabetes model and functions as an anti-diabetic and anti-atherogenic adipokine in obesity-related disorders, such as insulin resistance and inflammation (1, 2). Insulin-resistant mouse models exhibit improved glucose tolerance after intraperitoneal vaspin application (3, 4), and transgenic mice overexpressing vaspin are protected from diet-induced obesity and comorbidities, such as glucose intolerance, insulin resistance, and adipose tissue inflammation (5). We found that intraperitoneal and central administration of vaspin leads to reduced food intake and blood glucose in mice (6). A recent study demonstrated that central vaspin regulates hepatic glucose production and insulin signaling via brain-liver communication through the dorsal vagal complex (7). In endothelial and vascular smooth muscle cells, multiple studies demonstrated anti-apoptotic (8), anti-inflammatory (9), and anti-migratory (10) effects of vaspin, suggesting a protective role in the pathogenesis of atherosclerosis. In skin, vaspin expression by keratinocytes suppresses the expression and release of inflammatory cytokines by immune cells (11) and possibly regulates the activation of proinflammatory cytokines, such as chemerin, via KLK7 (12). Whereas vaspin-mediated improvement of glucose tolerance and reduction of food intake were found to be dependent on protease inhibition (4, 13), anti-inflammatory effects in the liver have been linked to interaction with a membrane-associated endoplasmic reticulum chaperone protein (5).
The only protease target of vaspin known so far is human KLK7 (4), a member of a family of 15 serine peptidases with chymotrypsin-like specificity (14). A non-inhibitory vaspin mutant failed to improve glucose tolerance in obese mice, proving that protease inhibition is a prerequisite for vaspin effects on glucose disposal (4). Human insulin is a substrate of KLK7, and vaspin and KLK7 are co-expressed in murine pancreatic islets. We hypothesized that KLK7 inhibition by vaspin may prolong insulin action and increase glucose uptake in insulin-responsive tissues, such as adipose tissue, liver, muscle, and also brain.
We have recently reported that the inhibition of KLK7 by vaspin is critically dependent on an arginine residue in proximity to the reactive center loop (RCL),2 because the native P1′ glutamate residue is essentially repressing inhibition of KLK7 (15). Inhibition of KLK7 by vaspin is furthermore accelerated by heparin (15). Yet, the nature of the interaction between glycosaminoglycans and vaspin has not been explored in much detail.
In this study, we have characterized the binding of unfractionated heparin (UFH), low molecular weight heparins (LMWHs), chondroitin sulfate (CS), and dermatan sulfate (DS) to vaspin and their effects on KLK7 inhibition. Furthermore, we have identified basic residues contributing to heparin binding and localized the putative heparin binding site in vaspin. This binding site is located at a basic cluster on β-sheet A and differs from all known heparin binding sites in other serpins. Third, using a vaspin-overexpressing cell line, we found a major part of secreted vaspin bound in the extracellular matrix, demonstrating the physiological relevance of this interaction.
Results
Binding of Vaspin to Heparin and Defined Heparin Oligomers
We performed microscale thermophoresis (MST) experiments to investigate the interaction of heparins and further GAGs with vaspin. Unfractionated heparin is bound with high affinity and a dissociation constant (KD) of 21 ± 2 nm (Fig. 1A). The low molecular weight heparin enoxaparin (4–24 saccharide units) was bound with a KD of 99 ± 16 nm (Fig. 1B). SDS-denaturation tests ruled out nonspecific binding independent of protein structure (data not shown). No binding was observed for short heparin oligosaccharides of 5 units (fondaparinux) and 8 units (dp8) as well as for the GAGs dermatan sulfate and chondroitin sulfate (Fig. 1C).
FIGURE 1.

GAG binding to fluorescently labeled vaspin analyzed by microscale thermophoresis. Shown is binding of GAG ligands to fluorescently labeled vaspin. Curves are derived from the specific change in the thermophoretic mobility (A and C) or fluorescence change (B) upon titration of ligand to a constant vaspin concentration. A, the fit of binding of UFH to vaspin (10 nm) yielded a KD of 21.6 nm. B, fluorescence change for 50 nm vaspin-RED titrated with enoxaparin. The fit yielded a KD of 99.1 nm. C, normalized fluorescence (F norm) for LMWH oligosaccharides fondaparinux and dp8 as well as dermatan sulfate (DS) and chondroitin sulfate (CS) titrated to vaspin (100 nm). All tested ligands did not bind to vaspin. Results were plotted using GraphPad Prism and KD values calculated using NanoTemper analysis software.
Activation of Vaspin by GAGs
Heparin accelerates KLK7 inhibition by vaspin most likely by bridging both proteins in a ternary complex rather than inducing conformational changes in the inhibitor (15). As also implied by the binding data, neither the 5-mer fondaparinux nor an 8-mer dp8 accelerated complex formation. Notably, also the low molecular weight heparin enoxaparin was not able to accelerate protease inhibition (Fig. 2). Thus, longer saccharide chains of more than 20 units are required to bridge vaspin and KLK7. For the GAGs dermatan and chondroitin sulfate, a slight increase in KLK7 inhibition was observed at ratios above 12.5 (GAG/serpin) (Fig. 2). Because we did not observe binding of DS or CS by vaspin in MST experiments, this slight increase is probably due to moderate activation of KLK7, as reported previously (16).
FIGURE 2.

Effect of heparins and other GAGs on KLK7 inhibition by vaspin. Left, Coomassie-stained SDS gels of a fixed molar ratio (1:3) of vaspin and KLK7 incubated for 1 min in the presence of increasing concentrations of GAGs (0–200-fold inhibitor). The full gel is shown for UFH, whereas for the other GAGs, only the complex bands are presented. The indicated bands are serpin-protease complex (1); active serpin (2); active, tag-cleaved serpin (3); cleaved serpin (4); and protease (5). Right, densitometric quantification of complex band intensities in relation to the molar ratio of GAG to vaspin of the gels on the left.
Basic Residues Surrounding Helix D or Helix H Do Not Contribute to Heparin Binding
To locate the heparin binding site in vaspin, we first investigated potential basic residues in known binding sites of other heparin-binding serpins. For antithrombin (AT) or heparin cofactor II, the heparin binding region is located at helix D, whereas protein C inhibitor (PCI) binds heparin primarily with basic residues in helix H and also helix D (reviewed in Ref. 17). By superposition of the X-ray structures of vaspin and AT bound to heparin, we located Lys40, Arg42 (of helix A), and Lys110 as potential binding residues within helix D. In comparison with AT and heparin cofactor II, vaspin lacks further basic residues in helix D. Superposition with heparin-bound PCI revealed Arg50 (of helix D) and Lys254 and Arg294 within helix H as candidate residues. Mutation of all of these residues did not alter heparin binding analyzed by heparin affinity chromatography (data not shown). Also, the previously reported R302A mutant, corresponding to the binding site for heparin in kallistatin, did not affect heparin binding and activation (Fig. 3, A and B).
FIGURE 3.

Heparin binding and activation of vaspin R302A. A, heparin affinity chromatography of vaspin WT and mutant R302A. Elution via a NaCl gradient (black dotted line) was monitored at 220 nm. B, shown are Coomassie-stained SDS gels of a fixed molar ratio (1:3) of serpin and KLK7 incubated in the presence of increasing concentrations of UFH (0–200-fold inhibitor) for 1 min (vaspin WT, bottom gel) or 30 min (vaspin R302A, top gel). For the R302A mutant, only the complex bands are shown. Although R302A is a dramatically slower inhibitor, complex formation is accelerated, comparable with vaspin WT.
Selective Modification of Lysine Residues
Subsequently, we specifically modified lysine residues by acetylation using a sulfo-NHS-acetate ester. Acetylation of free vaspin in solution resulted in a mass shift of ∼1640 Da, indicating acetylation of all 39 lysines (Fig. 4A), and fully acetylated vaspin was unable to bind heparin (Fig. 5A). CD spectroscopy did not show major changes in protein folding (Fig. 4B). Also, the detection of RCL-cleaved serpin after incubation with KLK7 indicates that the native (RCL-exposed) conformation is still intact after acetylation (Fig. 4C), yet it clearly interferes with the serpin inhibition mechanism, probably by affecting RCL insertion. Native state stability is also decreased with a Tm of 58 °C (versus 70 °C for the unmodified vaspin; Fig. 4D) and results in increased temperature-induced polymerization (Fig. 4E).
FIGURE 4.

Characterization of acetylated vaspin. A, linear mode MALDI-TOF mass spectra of WT and acetylated vaspin. Shown and labeled are the [M + H]+ peaks. The mass difference indicates acetylation of all 39 lysine residues. B, far-UV CD spectra of vaspin (WT or acetylated) at 20 °C are shown (collected in phosphate buffer, pH 7.8, 1.5 μm). C, the reaction of vaspin (WT or acetylated) and KLK7 with and without 12.5-fold UFH was analyzed at the indicated time points for up to 120 min. For WT, heparin accelerates complex formation. For lysine-acetylated vaspin, complex formation is minimal after 120 min, and the majority of serpin is RCL-cleaved. D, thermal unfolding of WT and lysine-acetylated vaspin from single samples observed by far-UV CD at 208 nm at a heating rate of 50 °C/h revealed reduced thermal stability after lysine acetylation. E, SDS-PAGE analysis of polymerization for vaspin (WT or acetylated) after a 60-min incubation at 70 °C.
FIGURE 5.

Specific labeling of vaspin to identify potential heparin binding lysine residues of vaspin. A, heparin affinity chromatography of WT and lysine-acetylated vaspin. Elution via a NaCl gradient (black dotted line) was monitored at 220 nm. Heparin binding was lost after acetylation of lysine residues in vaspin. B, SDS-PAGE analysis of loading (L), flow-through (FT), and elution (E) fractions from heparin affinity chromatography. At different steps of the selective labeling experiment, heparin binding of protein samples was analyzed. Native vaspin bound heparin; no protein was detected in the flow-through. Fully acetylated protein did not bind on the heparin column. After on-column (specific) acetylation, vaspin was still able to bind heparin, confirming specific labeling. Finally, heparin binding was lost after biotinylation of remaining free lysine residues. C, ribbon diagrams of vaspin are presented in the classic view (right) and rotated by 180° (left). Helices are in red, and sheets are in cyan. Candidate lysine residues identified by mass spectrometry are labeled and presented as yellow sticks.
Selective Labeling of Heparin-binding Lysine Residues
To locate the heparin binding site of vaspin and identify important residues, we then used a selective labeling approach, with acetylation of non-binding lysine residues while vaspin was bound to immobilized heparin, followed by biotinylation of the remaining free lysine residues after heparin removal. Selectively acetylated vaspin remained able to bind heparin, when rerun on a heparin column (Fig. 5B). Finally, heparin binding was lost after biotinylation of the remaining free lysine residues (Fig. 5B). Mass spectrometry demonstrated acetylation of ∼31 lysine residues and biotinylation of ∼8 lysine residues (Fig. 6A). Lysine residues were subsequently identified by MS analysis of tryptic peptides after tryptic digestion and isolation of biotinylated fragments (Table 1 and supplemental Fig. 1 and Spectra S1–S8). Three independent selective labeling experiments resulted in the repeated identification of matching peptides and confirmed the selectivity and specificity of the labeling approach. Lys22 and Lys31 are located in the flexible N terminus (not shown in the structure), Lys46 and Lys62 in helix A, Lys296 in helix H, and Lys321 in helix I (Fig. 5C). Lys340 is in the loop region in the bottom of the molecule, and Lys359 is located in strand s5A of central β-sheet A (Fig. 5C).
FIGURE 6.

Heparin binding is not changed after mutation of most candidate lysine residues identified via selective labeling. A, linear mode MALDI-TOF mass spectra of WT, selectively acetylated, and biotinylated vaspin. Shown and labeled are the [M + H]+ peaks. The mass differences indicate acetylation of ∼31 lysine residues and biotinylation of ∼8 lysine residues. B and C, heparin affinity chromatography of T365R in combination with lysine residues identified using the selective labeling experiment. Elution via a NaCl gradient (black dotted line) was monitored at 220 nm.
TABLE 1.
Selectively biotinylated tryptic vaspin peptides identified by mass spectrometry
Peptides are sorted according to the residue number of the lysine residue. Peptides were sequenced by MS/MS, and peak lists were searched against the SwissProt database to identify peptide fragments.
| Labeled residue | m/z observed | m/z theoretical | Error | Peptide sequence | Residues | Supplemental spectrum |
|---|---|---|---|---|---|---|
| ppm | ||||||
| Lys22 | 1312.599 | 1312.602 | 2 | HMK(biotin)PSFSPR | 20–28 | S1 |
| Lys31 | 1974.985 | 1974.945 | −20 | NYK(biotin)ALSEVQGWK(acetyl)QR | 29–42 | S2 |
| Lys46 | 1115.554 | 1115.570 | −15 | MAAK(biotin)ELAR | 43–50 | S3 |
| Lys62 | 1291.639 | 1291.662 | −17 | K(biotin)LAFYNPGR | 62–70 | S4 |
| Lys296 | 1129.601 | 1129.619 | −16 | WK(biotin)TLLSR | 295–301 | S5 |
| Lys321 | 1321.694 | 1321.718 | −19 | K(biotin)TLSYIGVSK | 321–330 | S6 |
| Lys340 | 1988.958 | 1989.001 | −22 | IFEEHGDLTK(biotin)IAPHR | 332–346 | S7 |
| Lys359 | 1217.543 | 1217.565 | −18 | AELK(biotin)MDER | 356–363 | S8 |
Identification of Heparin-binding Residues of Vaspin
Next, we generated and expressed alanine mutants of all identified lysine residues. With the exception of K359A, mutation of candidate lysines identified by the selective labeling approach had no significant effect on heparin binding when analyzed by affinity chromatography (Fig. 6, B and C). We observed a shift of ∼100 mm NaCl needed for elution from immobilized heparin for the K359A mutant compared with the wild type (Fig. 7A and Table 2). Mutation of the neighboring arginine residue Arg211 additionally decreased relative heparin affinity and resulted in a shift of ∼150 mm NaCl compared with the wild type (Fig. 7A and Table 2). We were not able to obtain soluble monomeric protein for any vaspin variant bearing the neighboring R310A mutation (R310A, R310A/K359A, and R211A/R310A/K359A; data not shown).
FIGURE 7.

Mutation of basic residues of central β-sheet A as well as RCL insertion decrease heparin binding of vaspin. Heparin binding vaspin and mutants was assessed by heparin affinity chromatography using a NaCl gradient (black dotted line). A, Elution profiles of WT, K359A, and R211A/K359A vaspin were monitored at 220 nm. Relative heparin affinity is significantly lower for the vaspin mutants. B, elution profiles of WT and A369P compared with cleaved A369P and R211A/K359A/A369P. RCL insertion reduces heparin binding, and this is only slightly more pronounced for the mutant lacking the heparin binding residues of central β-sheet A. C, elution profiles of WT, N-terminally cleaved WT, and R302E as well as cleaved WT vaspin. D, elution profiles of WT, K22A/R28A/K31A, and His tag-cleaved (K22A/R28A/K31A + FXa) and N-terminally cleaved (WT + FXa) vaspin. Loss of N-terminal basic residues as well as loss of His tag sequence do not influence heparin binding of vaspin.
TABLE 2.
Relative heparin affinity of vaspin and mutants analyzed by heparin chromatography
| Vaspin variant | Elution profile | Elution peak |
|---|---|---|
| mm NaCl | mm NaCl | |
| WT | 505–590 | 550 |
| K359A | 425–500 | 460 |
| R211A/K359A | 385–455 | 415 |
| A369P | 490–585 | 540 |
| A369Pcleaved | 415–470 | 445 |
| R211A/K359A/A369Pcleaved | 380–430 | 402 |
| WTN-terminally cleaved (FXa) | 440–510 | 470 |
| WTcleaved | 390–480 | 430 |
| R302EN-terminally cleaved (KLK7) | 445–535 | 488 |
| K22A/R28A/K31A | 524–614 | 566 |
| K22A/R28A/K31AHis tag-cleaved (FXa) | 483–552 | 520 |
Heparin Binding of Cleaved (RCL Inserted) Vaspin
Because Arg211 and Lys359 are located in s3A and s5A, respectively, insertion of the RCL after cleavage will separate the heparin-binding residues. We used the inactive substrate mutant A369P incubated with KLK7 to generate pure cleaved vaspin. Cleaved vaspin A369P showed a reduced heparin affinity, comparable with the R211A/K359A mutant (Fig. 7B). Introduction of R211A and K359A mutations into the A369P mutant (R211A/K359A/A369P) resulted in an additional decrease of heparin affinity after cleavage (Fig. 7B). MST data also clearly reflected the reduced affinity. LMWH enoxaparin was bound with a KD of 1 μm by the R211A/K359A/A369P mutant, a 10-fold reduction compared with wild type vaspin (Fig. 8A).
FIGURE 8.

Mutation of basic residues of central β-sheet A reduces heparin affinity but not stability or general serpin activity. A, fluorescence change for 100 nm R211A/K349A/A369P_vaspin-RED titrated with enoxaparin. The fit yielded a KD of 1 μm, representing a 10-fold decrease in affinity compared with WT vaspin (WT data as in Fig. 1B). B, far-UV CD spectra of vaspin WT, K359A, or R211A/K359A at 20 °C are shown (collected in phosphate buffer, pH 7.8, 1.5 μm). C and D, Coomassie-stained SDS gels of vaspin K359A (B) and R211A/K359A (D) mutants incubated with KLK7 for the indicated times. Complex formation is not affected by these mutations.
Role of N-terminal Basic Residues in Heparin Binding
Two N-terminal lysine residues were identified in the selective labeling experiment, but mutation of both resulted in no marked changes in heparin affinity of vaspin. KLK7 also cleaves recombinant vaspin after Tyr30, resulting in loss of the N-terminal His tag and vaspin residues Lys22–Tyr30 (4). We therefore used the R302E mutant, which due to the negative charge at the exosite for KLK7 is not cleaved within the RCL, but only at the N terminus. We noted a significant decrease of heparin affinity for vaspin (R302E or wild type) devoid of its N-terminal residues (Fig. 7C). Because all experiments were performed using buffers with pH >7.8 and thus above the pKa values for histidine residues (pKa 6.0–7.0), a significant influence of the poly-His tag can be excluded. To determine whether basic N-terminal residues Lys22, Lys31, and Arg28 participate in heparin binding, we generated the triple mutant K22A/R28A/K31A. Heparin affinity of this mutant was as wild type (Fig. 7D). Furthermore, cleavage of the His tag sequence using the intrinsic factor Xa cleavage site and generating vaspin(22–414) also had no apparent effect on heparin affinity (Fig. 7D). These results demonstrate that the N-terminal basic residues of vaspin do not interact with the heparin chain.
Mutation of Basic Residues in Sheet A Decreases Heparin Acceleration of KLK7 Inhibition
Heparin-mediated acceleration of KLK7 inhibition by vaspin was significantly reduced for K359A mutants in comparison with the wild type vaspin (Fig. 9, A and B). Whereas uncatalyzed reaction rates were unchanged (kobs = 7.3 ± 0.6 mm−1·s−1 for WT versus kobs = 7.3 ± 1.5 mm−1·s−1 for R211A/K359A), heparin-induced rate acceleration was reduced by 40% for the R211A/K359A mutant (kobs,UFH = 31.5 ± 1.8 mm−1·s−1 (WT) versus kobs,UFH = 19.3 ± 1.4 mm−1·s−1 (R211A/K359A)). Both mutations had no effects on protein folding, thermal stability, and inhibitory activity (Fig. 8, B–D). Together, the removal of these two basic residues clearly decreased, but did not eliminate, heparin binding or acceleration of the serpin-protease reaction. Importantly, the loss of the N-terminal residues did not affect heparin acceleration of KLK7 inhibition (Fig. 9C).
FIGURE 9.

Mutation of basic residues of central β-sheet A reduces heparin acceleration of KLK7 inhibition by vaspin. A, inhibition of KLK7 by vaspin WT or R211A/K359A with UFH was measured under pseudo-first-order conditions in a discontinuous assay (heparin/serpin ratio of 12.5). Presented is the relative increase in apparent second-order rate constant as x-fold over control (without heparin). B, top panel, complex formation of vaspin WT, K359A, and R211A/K359A with KLK7 was analyzed by SDS-PAGE using a fixed molar ratio of serpin and KLK7 (1:3) incubated in the presence of increasing concentrations of UFH (0–200-fold inhibitor) for 1 min. Only the complex bands from the SDS gels are shown. Bottom panel, densitometric quantification of complex band intensities in relation to the molar ratio of UFH to vaspin of SDS gels in B. The reference band intensity is WT vaspin without heparin. A bell-shaped curve was obtained, but the increase was reduced in comparison with WT for the mutants. C, heparin-induced acceleration of complex formation between vaspin and KLK7 analyzed by SDS-PAGE. A clear and comparable increase in complex band intensity was observed for the serpin-protease reaction with the addition of a 12.5-fold excess of UFH for the full-length vaspin (left gel) and the N-terminally cleaved variant (right gel).
NaCl Solubilization of Vaspin from Extracellular Matrix of Keratinocytes
To address the physiological relevance of the vaspin GAG interaction, we tested whether vaspin is bound and localized on the cell surface or in the extracellular matrix of a vaspin-overexpressing HaCaT cell line. This skin cell line has been generated recently to investigate anti-inflammatory effects of vaspin in skin and obesity-related inflammatory skin diseases, such as psoriasis (11), and expresses and secretes significant and readily detectable amounts of vaspin (Fig. 10A). Cells were treated with heparinase I, 1 m NaCl, or Triton X-100, and vaspin content was determined by ELISA in the cell supernatants. In comparison with untreated HaCaT cells, incubation with 1 m NaCl increased vaspin concentrations in cell supernatants ∼4-fold (Fig. 10B). Incubation with heparinase resulted in a marked increase of vaspin concentration of ∼3-fold (Fig. 10B). To estimate intracellular vaspin, we also permeabilized cells using 1% Triton X-100, which resulted in a dramatic ∼25-fold increase of detectable vaspin in the supernatants (Fig. 10B).
FIGURE 10.

Secreted vaspin by HaCaT cells is retained in the extracellular matrix. A, vaspin concentrations in supernatants of HaCaT cells transfected with an empty control vector or with human vaspin after incubation with Tris buffer for 1 h. B, HaCaT cells were treated with heparinase for 1 h, NaCl for 10 min, or Triton X-100 for 15 min, and vaspin concentration was measured in the cell supernatants via ELISA. Treatment with heparinase and NaCl resulted in significantly increased vaspin supernatant concentrations. Data are expressed as relative to untreated controls. Presented are the mean ± S.E. (error bars) values of at least three experiments performed in triplicates with the exception of the Triton X-100 condition, which was performed twice. Data were analyzed via Kruskal-Wallis test and Dunn's multiple comparisons test; *, p < 0.05.
Discussion
In this study, we have investigated GAG binding of vaspin and identified important residues for heparin binding using a selective labeling approach. We have shown previously that both vaspin and its target protease KLK7 are heparin binding molecules (15). Microscale thermophoresis revealed high affinity binding of heparin by vaspin (KD = 21 nm), which is comparable with that of plasma AT (KD = 10 nm (18)) and protein Z-dependent protease inhibitor (KD = 25 nm (19)). Binding of LMWH enoxaparin by vaspin is strong as well (KD = 99 nm), although 10-fold weaker compared with UFH, and we observed no binding of a defined heparin octasaccharide. Heparin chain lengths of enoxaparin range from 4 to 24, but the majority of chains are of <20 saccharide units (20), and the lower affinity of vaspin for enoxaparin may be simply explained by the longer chain length of UFH (∼50 saccharide units on average).
Enoxaparin is prepared via β-eliminative cleavage of the heparin benzyl ester by alkaline treatment, and the affinity of vaspin for LMWH prepared by other methods (e.g. by heparinase treatment, deaminative cleavage, or oxidative depolymerization) may differ. Also, fractionation of heparin according to vaspin affinity may result in the identification of a vaspin-specific GAG sequence. However, until now, AT remains the only serpin where a specific sequence is known.
The heparin-induced acceleration of KLK7 inhibition by vaspin is dose-dependent with a bell-shaped dose-response curve (15). This indicates a major contribution of the template effect of longer heparin chains to the acceleration of the vaspin-KLK7 reaction, whereby binding of one heparin chain by both the serpin and the protease induces spatial proximity and an increase in reaction rate. Notably, enoxaparin did not accelerate KLK7 inhibition; thus, heparin chain lengths of >20 saccharide units are required for the accelerating bridging mechanism. This is similar to other serpin-protease combinations dependent on longer heparin chains for rate acceleration, such as AT-factor X1, AT-thrombin, or ZPI-factor Xa (>18 units) (21–23).
Any potential conformational changes in vaspin induced by heparin binding seem not to contribute to the heparin-induced acceleration of KLK7 inhibition by vaspin. Also, heparin binding did not obviously increase the serpin substrate pathway.
Glycosaminoglycan binding is primarily mediated by ionic interactions between positively charged side chains of arginine and lysine residues with the negatively charged sulfate groups of the GAGs (24). For many heparin binding serpins, such as AT (25), PAI (26), HC II (27), and protease nexin 1 (28), the heparin binding region is located at helix D and at the beginning of helix A, whereas PCI binds heparin primarily with basic residues in helix H and also helix D (29, 30). In vaspin, helix D does only contain a single lysine (Lys110), with two proximal basic residues Lys40 and Arg42 in helix A. Although the constellation of these residues is somewhat similar to the location of the most important heparin-binding residues in AT (Arg47, Lys125, and Arg129 (25, 31)), mutation of these residues did not affect heparin binding of vaspin. Also, the mutation of basic residues surrounding helix H and D (as for the PCI heparin binding site) did not result in decreased heparin affinity. Together, none of these residues seem to contribute to high affinity heparin binding of vaspin. In kallistatin, the heparin binding site has been located between helix H and β-sheet C (32). This cluster of positively charged residues (K307A/R308A and K312A/K313A, all in s2C) also represents an important exosite for target protease recognition (33). For kallikrein inhibition by kallistatin, the accessible exosite is critical, and obstruction due to heparin binding suppresses kallistatin activity. The corresponding vaspin residue (Arg302) is also an important exosite for KLK7 inhibition (15), although it is not part of a heparin binding site in vaspin, because heparin affinity is not altered for the R302A vaspin mutant, and heparin binding does clearly accelerate protease inhibition (Fig. 3, A and B).
We then used the selective labeling approach to identify candidate lysine residues from among the total of 39 in human vaspin. The only basic residues identified via this approach (Lys359 and Arg211) that clearly affected heparin binding of vaspin are located in the center of a cluster comprising basic residues of the shutter and breach region of central β-sheet A (Fig. 11A). Mutation of these two residues reduced affinity for heparin by 10-fold (Fig. 8A). A further indication for heparin binding at this site is the prominent reduction of heparin affinity after insertion of the RCL into the central β-sheet A. On one hand, the RCL insertion as strand s4A between strands s5A (Lys359) and s3A (Arg211) results in spatial separation of these binding residues. Second, the crystal structure of cleaved vaspin revealed that the RCL hinge region residue Glu366, as part of the newly formed s4A, is placed directly between Arg211 and Lys359 (34) and thus could interfere with heparin binding at this site (Fig. 11B).
FIGURE 11.
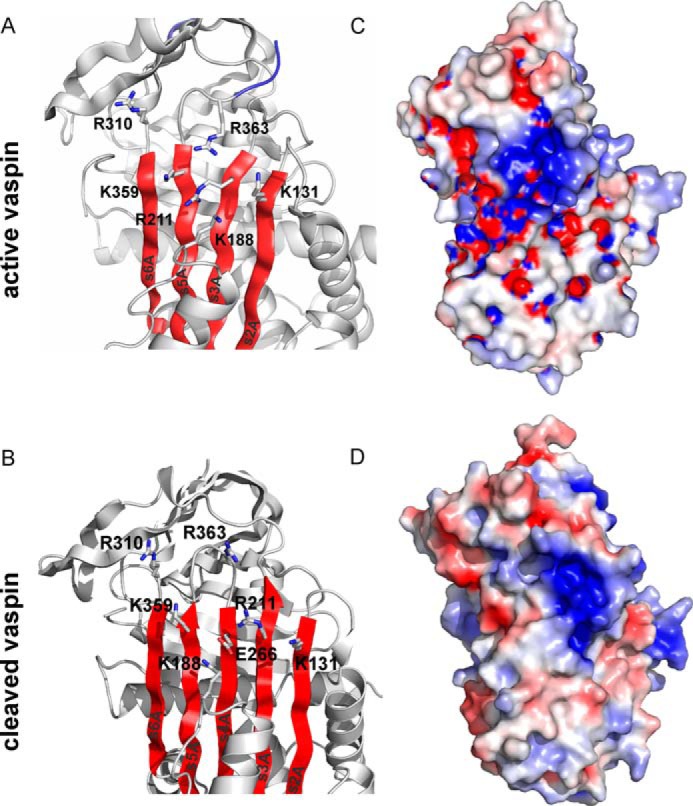
Localization of the putative heparin binding site in vaspin. A and B, a ribbon depiction is shown of the upper part of native vaspin (A, with β-sheet A in red and the RCL in blue; Protein Data Bank code: 4IF8) or cleaved vaspin (B; Protein Data Bank code 5IE0) slightly rotated from the classic view. Side chains of the identified residues contributing to heparin binding (Lys359 and Arg211) and surrounding basic residues are labeled and shown as sticks. C and D, the electrostatic surface and the ribbon diagram of the vaspin (C) and cleaved vaspin (D) molecules are presented in the same orientation as in A. Colors relate to the electrostatic surface potential (blue is positive, and red is negative, −8 to +8 kBT) calculated by APBS (37). The surface charge distribution reveals a prominent basic patch located on top of the β-sheet A.
Vaspin is a highly basic protein (net charge of +10 and a pI of ∼9.3), yet surface charge distribution reveals only one very prominent patch of basic residues (Fig. 11C). Notably, vaspin is the only human serpin with a basic residue at the position of Lys359, whereas an acidic residue (glutamate or aspartate) is highly conserved in the serpin family (34). In the cleaved form of vaspin, this basic patch is considerably narrowed (Fig. 11D). Because mutation of Lys359 and neighboring Arg211 did not fully prevent heparin binding, more basic residues within the cluster (Lys131, Lys188 (next to helix F), Arg310, and Arg363) or elsewhere are contributing to heparin binding. N-terminal basic residues have been reported to contribute to heparin binding of AT (25), and also cleavage of vaspin N-terminal residues resulted in a marked decrease in heparin affinity. But both mutation of all basic N-terminal residues of vaspin in question (Lys22, Arg28, and Lys31) and cleavage of the His tag sequence did not alter heparin binding. Vaspin N-terminal residues are highly flexible (4), and the presence of these additional nine residues may alter heparin affinity by supporting or facilitating heparin binding at the high affinity binding site via interactions with other vaspin residues or domains such as helix F (Fig. 11A).
Importantly, heparin binding at the residues Lys359 and Arg211 is relevant for heparin activation of vaspin. Mutation of these residues reduced heparin-induced rate acceleration by ∼40%, whereas the uncatalyzed inhibition reaction rate is identical to wild type vaspin. In contrast, N-terminal residues contributing to heparin binding are not relevant for rate acceleration via the bridging mechanism. In general, it is remarkable that heparin binding at this region does not hinder but accelerates the serpin inhibition mechanism. Practically, it would be expected that binding of a heparin molecule at β-sheet A should obstruct RCL insertion and thus the serpin inhibition mechanism. The conformational changes after RCL cleavage must be accompanied by release or repositioning of the heparin chain, and this must be fast enough for efficient inhibition. Nevertheless, it very likely explains the moderate activation effect of only ∼5-fold versus up to 1000-fold for serpins binding heparin chains in the D helix.
Previously, studies have reported cell surface localization of vaspin. It has been found enriched on the cell surface of H-4-II-E-C3 cells and in plasma membrane fractions of liver tissue in vaspin transgenic mice (5). There, it is thought to ameliorate hepatic oxidative stress via interaction with the endoplasmic reticulum chaperone GRP78. Furthermore, in liver cells, vaspin was found colocalized with α-2-macroglobulin, a major inhibitor of ECM-degrading matrix metalloproteases.
To investigate the biological relevance of the vaspin-heparin or heparan sulfate (HS) proteoglycan interaction, we used recently established vaspin-overexpressing HaCaT cells as a model system. In HaCaT cells, we found that ∼75% of the secreted vaspin was bound and retained on the cell surface. Heparinase treatment did result in a ∼3-fold increase in vaspin concentration. On one hand, higher vaspin levels measured after the NaCl wash may be a result of heparinase I specificity, which degrades heparin and higher sulfated domains of HS. Vaspin binding to highly sulfated domains could thus hinder enzymatic digestion. On the other hand, a further increase of vaspin release using 1 m NaCl probably indicates that vaspin is also bound to other components of the extracellular matrix or to other proteins on the cell surface, as suggested by the finding of the vaspin-GRP78 interaction (5). Together, these data demonstrate that vaspin, at least in part, is localized in the ECM via interactions with the HS proteoglycan in HaCaT cells. The amount of vaspin retained in the ECM may vary in different cells and tissues.
Together, these results demonstrate that basic residues of vaspin's central β-sheet A contribute to heparin binding and activation of vaspin by the bridging mechanism. Furthermore, via this interaction, vaspin is bound to the extracellular matrix and probably also interacts with further molecules on the cell surface. For many studies reporting vaspin effects on adipocytes, endothelial cells, smooth muscle cells, keratinocytes, and immune cells in the skin, the mechanisms of signal transduction from the extracellular serpin into the cell remain unclear. Binding to glycosaminoglycans on cell surfaces may direct and regulate vaspin interaction with target proteases or other proteins and play an important role in the various beneficial functions of vaspin in different tissues. Thus, our findings give novel insight into vaspin signaling.
Experimental Procedures
Materials
Human recombinant KLK7 and thermolysin were from R&D Systems (Minneapolis, MN). Fluorogenic peptide substrates Abz-KLYSSK-Q-EDDnp and ortho-aminobenzoic acid (Abz)-KLFSSK-glutaminyl-N-[2,4-dinitrophenyl]-ethylenediamine (Q-EDDnp) were a generous gift of Prof. Dr. Maria A. Juliano (Escola Paulista de Medicina, Universidade Federal de São Paulo, Brazil). UFH (from porcine intestinal mucosa with an average molecular mass of 18 kDa), DS (from porcine intestinal mucosa with an average molecular mass of 21 kDa), and CS (from shark cartilage with an estimated average molecular mass of 19 kDa) were all from Sigma-Aldrich; heparin oligosaccharide dp8 was from Iduron (Cheshire, UK); fondaparinux sodium (ArixtraTM) was from Aspen Pharma (Johannesburg, South Africa); and enoxaparin (ClexanTM) was from Sanofi (Paris, France). All calculations were done using Prism version 5.03 (GraphPad Software).
Generation of Recombinant Proteins and Determination of Inhibition Parameters
Mutagenesis, recombinant expression, and purification of vaspin, vaspin mutants, and KLK7 were carried out as described previously (15). Purified proteins were analyzed by reverse phase HPLC, SDS-PAGE, and MALDI-TOF mass spectrometry. Cleaved forms of vaspin mutants were generated as described previously (34). Pseudo-first-order rate constants (kobs) were determined as described previously (15). Briefly, a discontinuous assay was used in the presence of UFH (UFH/vaspin ratio of 12.5) with 10-fold inhibitor concentration to maintain pseudo-first-order conditions. kobs values were normalized to the reaction in the absence of UFH to estimate the heparin-induced rate acceleration. Values presented are the average of at least three experiments from two protein batches.
Microscale Thermophoresis
Vaspin was labeled via lysine side chains using the Monolith NT protein amine-reactive labeling kit RED-NHS (NanoTemper Technologies) according to the manufacturer's instructions. The average number of lysines labeled per vaspin molecule was ∼1, and labeled proteins were stored in MST buffer (50 mm Tris, 150 mm NaCl, 10 mm MgCl2, 0.05% Tween 20, pH 7.6) at −20 °C. Then 10–125 nm vaspin-RED was titrated with serial dilutions of various GAGs (UFH, dp8, fondaparinux, enoxaparin, CS, and DS) in the range of 0.05 nm to 0.63 mm in MST buffer with 0.1% (w/v) BSA. All experiments were performed at room temperature in premium coated capillaries (NanoTemper Technologies), with 12–16 thermophoresis measurements recorded at least in duplicates. For the binding of enoxaparin using 100 nm vaspin-RED or R211A/K359A/A369P_vaspin-RED, ligand-dependent fluorescence enhancement was observed, and absolute fluorescence was used to calculate the KD. Specificity of the binding event was validated by the SDS-denaturation test according to the manufacturer's protocol. Raw data were analyzed using NanoTemper Analysis software and Prism. The principles of microscale thermophoresis to study molecular interactions, such as protein-heparin binding, are described elsewhere (35).
Complex Formation Analysis by SDS-PAGE
Complex formation was analyzed by SDS-PAGE as reported previously (15). Recombinant KLK7 and vaspin or mutants were incubated at a ratio of 3:1 (protease/serpin) using 3.5 μm KLK7 in Tris buffer (50 mm Tris, 150 mm NaCl, pH 8.5). To analyze the effect of GAGs on the vaspin-KLK7 reaction, complex formation assays were performed in the presence of UFH, dp8, fondaparinux, enoxaparin, CS, and DS, and reactions were stopped after 1 min of incubation. Molar ratios of GAG to vaspin ranged from 0.1 to 200. For the R302A mutant, reactions were stopped after 30 min due to the slow reaction rate. After densitometric analysis, complex band intensities were normalized to the reaction without GAG.
Heparin Affinity Chromatography
For each mutant, 200 μg of recombinant protein were run on 1-ml HiTrap heparin HP columns on the ÄKTA protein purification system (both from GE Healthcare, Freiburg, Germany). Elution was performed using an NaCl gradient from 150 mm to 2 m (flow rate, 0.75 ml/min) and monitored at 220 nm.
CD Spectroscopy
CD spectra were recorded on a J-715 spectropolarimeter (Jasco, Tokyo, Japan) as described previously (15).
Lysine Acetylation and Selective Labeling
Primary amines were modified using sulfo-NHS-acetate (Thermo Scientific). For full lysine acetylation, 10 nmol of recombinant vaspin was incubated with a 1000-fold molar excess of sulfo-NHS-acetate in PBS for 90 min at room temperature. Acetylation reactions were stopped by the addition of Tris to a final concentration of ∼200 mm and subsequently dialyzed against Tris buffer.
Selective labeling was achieved as described by Ori et al. (36) with minor changes to the protocol. Vaspin mutant T365R was used in selective labeling experiments because this mutant has a low tendency for aggregate formation. For selective acetylation, 250 μg (∼5 nmol) of vaspin was incubated with 40 μl of heparin-Sepharose bead slurry (Toyopearl AF-Heparin HC-650 M from Tosoh Biosciences GmbH, Stuttgart, Germany) in a total volume of 250 μl of PBS for 10 min at room temperature with gentle agitation. After washing the beads with PBS three times, remaining solvent-accessible lysine residues were acetylated by the addition of 1.5 mg (5.8 μmol) of sulfo-NHS-acetate in 400 μl of PBS. After 10 min, the acetylation reaction was stopped by the addition of Tris to a final concentration of 200 mm. Beads were washed four times with PBS, and vaspin was released in three incubation steps with PB buffer containing 2 m NaCl. Supernatant fractions containing protein were pooled and extensively dialyzed against PBS. Before final biotinylation of lysine residues, the approximate number of acetylated lysine residues was analyzed by MALDI-TOF-MS.
Subsequently, acetylated protein was incubated with a 200-fold molar excess of EZ-Link sulfo-NHS-biotin (Thermo Scientific) for 30 min at room temperature in PBS buffer. Reactions were stopped by the addition of Tris to a final concentration of 200 mm and dialyzed against 10-fold diluted PB. Three selective labeling experiments were analyzed by MALDI-TOF MS after tryptic digestion of selectively labeled vaspin.
Tryptic Digestion and Isolation of Biotinylated Peptides
After dialysis, labeled proteins (100 μg) were digested using Trypsin Gold and ProteaseMAXTM surfactant (according to the manufacturer's protocol; Promega). For isolation of biotinylated peptides, 200 μl of streptavidin-agarose resin (50% slurry; Thermo Scientific) was equilibrated three times with 5 CV of 25 mm NH4HCO3, pH 7.9. Tryptic peptides were incubated with the streptavidin beads for 15 min, and beads were washed four times with 5 CV of 25 mm NH4HCO3, pH 7.9, followed by four times with 5 CV of ultrapure water. Elution of biotinylated peptides was performed in three steps (1.5, 2, and 5 CV) with 75% acetonitrile, 0.1% TFA, 5 mm biotin. Finally, acetonitrile was removed in a SpeedVac vacuum concentrator (Thermo Scientific).
MALDI-TOF Mass Spectrometry
Biotinylated peptides were concentrated and desalted using ZipTip C18-filter tips (Millipore) and analyzed by MALDI-TOF MS and MS/MS using LIFT mode on a Bruker Ultraflex III MALDI TOF/TOF mass spectrometer. Peak lists of combined MS and MS/MS spectra were searched against the SwissProt database using BioTools software (Bruker) and the Mascot search engine (Matrix Science, London, UK) to identify peptide fragments. The following parameters were used for database searches: species, Homo sapiens; enzyme, trypsin; monoisotopic masses; optional modifications, lysine acetylation and lysine biotinylation; mass tolerance MS, 100 ppm; mass tolerance MS/MS, 0.5 Da; maximum missed cleavage sites, 2.
NaCl Extraction of Cell Surface-bound Vaspin
Vaspin-overexpressing HaCaT cells (11), seeded in 12-well plates and grown to confluence, were washed once with 1 ml of Tris buffer (20 mm Tris, 50 mm NaCl, 4 mm CaCl2, pH 7.5) and then incubated with 250 μl of Tris buffer containing either 2 units/ml heparinase I (Sigma-Aldrich) for 1 h, 1 m NaCl for 10 min, or 1% Triton X-100 for 15 min at 37 °C. As controls, cells were incubated in Tris buffer only for 1 h at 37 °C. After incubation, cell culture supernatants were centrifuged (15 min at 37 °C, 15,000 × g). To prevent inefficiency of antibody recognition for vaspin still bound to GAG fragments, NaCl concentrations were adjusted to 500 mm in all cell supernatant samples before vaspin content was determined using the Vaspin (human) ELISA kit.
Author Contributions
D. U. and J. T. H. conceived the study. D. U., K. O., K. A., and J. T. H. conducted experiments. A. S. provided the vaspin-transfected HaCaT cell line. J. P. and N. S. expressed and purified recombinant KLK7. All authors discussed results and commented on the manuscript. J. T. H. supervised the project and wrote the paper.
Supplementary Material
Acknowledgments
The vaspin expression plasmid was kindly provided by Dr. J. Wada (Okayama University Graduate School of Medicine, Okayama, Japan). The substrate peptide used in heparin activation studies was a generous gift of Prof. Dr. Maria A. Juliano (Escola Paulista de Medicina, Universidade Federal de São Paulo, Brazil). We thank Dr. Stephan Schultz for valuable comments on the manuscript.
Note Added in Proof
Jan Pippel and Norbert Sträter were inadvertently omitted as authors from the version of this manuscript that was published as a Paper in Press on December 9, 2016. This error has now been corrected.
This work was supported by the European Union and the Free State of Saxony (to J. T. H.) and by Deutsche Forschungsgemeinschaft SFB1052 “Obesity Mechanisms” Grants C4 (to N. S.), C7 (to J. T. H.), and B5 (to A. S.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Fig. 1 and Spectra S1–S8.
- RCL
- reactive center loop
- UFH
- unfractionated heparin
- LMWH
- low molecular weight heparin
- CS
- chondroitin sulfate
- DS
- dermatan sulfate
- MST
- microscale thermophoresis
- AT
- antithrombin
- PCI
- protein C inhibitor
- ECM
- extracellular matrix
- HS
- heparan sulfate
- CV
- column volumes.
References
- 1. Hida K., Wada J., Zhang H., Hiragushi K., Tsuchiyama Y., Shikata K., and Makino H. (2000) Identification of genes specifically expressed in the accumulated visceral adipose tissue of OLETF rats. J. Lipid Res. 41, 1615–1622 [PubMed] [Google Scholar]
- 2. Heiker J. T. (2014) Vaspin (serpinA12) in obesity, insulin resistance, and inflammation. J. Pept. Sci. 20, 299–306 [DOI] [PubMed] [Google Scholar]
- 3. Hida K., Wada J., Eguchi J., Zhang H., Baba M., Seida A., Hashimoto I., Okada T., Yasuhara A., Nakatsuka A., Shikata K., Hourai S., Futami J., Watanabe E., Matsuki Y., et al. (2005) Visceral adipose tissue-derived serine protease inhibitor: a unique insulin-sensitizing adipocytokine in obesity. Proc. Natl. Acad. Sci. U.S.A. 102, 10610–10615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heiker J. T., Klöting N., Kovacs P., Kuettner E. B., Sträter N., Schultz S., Kern M., Stumvoll M., Blüher M., and Beck-Sickinger A. G. (2013) Vaspin inhibits kallikrein 7 by serpin mechanism. Cell Mol. Life Sci. 70, 2569–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nakatsuka A., Wada J., Iseda I., Teshigawara S., Higashio K., Murakami K., Kanzaki M., Inoue K., Terami T., Katayama A., Hida K., Eguchi J., Horiguchi C. S., Ogawa D., Matsuki Y., Hiramatsu R., Yagita H., Kakuta S., Iwakura Y., and Makino H. (2012) Vaspin is an adipokine ameliorating ER stress in obesity as a ligand for cell-surface GRP78/MTJ-1 complex. Diabetes 61, 2823–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Klöting N., Kovacs P., Kern M., Heiker J. T., Fasshauer M., Schön M. R., Stumvoll M., Beck-Sickinger A. G., and Blüher M. (2011) Central vaspin administration acutely reduces food intake and has sustained blood glucose-lowering effects. Diabetologia 54, 1819–1823 [DOI] [PubMed] [Google Scholar]
- 7. Luo X., Li K., Zhang C., Yang G., Yang M., Jia Y., Zhang L., Ma Z. A., Boden G., and Li L. (2016) Central administration of vaspin inhibits glucose production and augments hepatic insulin signaling in high-fat-diet-fed rat. Int. J. Obes. 40, 947–954 [DOI] [PubMed] [Google Scholar]
- 8. Nakatsuka A., Wada J., Iseda I., Teshigawara S., Higashio K., Murakami K., Kanzaki M., Inoue K., Terami T., Katayama A., Hida K., Eguchi J., Ogawa D., Matsuki Y., Hiramatsu R., et al. (2013) Vaspin inhibits apoptosis of endothelial cells as a ligand for cell-surface GRP78/VDAC complex. Circ. Res. 112, 771–780 [DOI] [PubMed] [Google Scholar]
- 9. Jung C. H., Lee M. J., Kang Y. M., Lee Y. L., Yoon H. K., Kang S. W., Lee W. J., and Park J. Y. (2014) Vaspin inhibits cytokine-induced nuclear factor-κB activation and adhesion molecule expression via AMP-activated protein kinase activation in vascular endothelial cells. Cardiovasc. Diabetol. 13, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Phalitakul S., Okada M., Hara Y., and Yamawaki H. (2012) A novel adipocytokine, vaspin inhibits platelet-derived growth factor-BB-induced migration of vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 423, 844–849 [DOI] [PubMed] [Google Scholar]
- 11. Saalbach A., Tremel J., Herbert D., Schwede K., Wandel E., Schirmer C., Anderegg U., Beck-Sickinger A. G., Heiker J. T., Schultz S., Magin T., and Simon J. C. (2016) Anti-inflammatory action of keratinocyte-derived vaspin: relevance for the pathogenesis of psoriasis. Am. J. Pathol. 186, 639–651 [DOI] [PubMed] [Google Scholar]
- 12. Schultz S., Saalbach A., Heiker J. T., Meier R., Zellmann T., Simon J. C., and Beck-Sickinger A. G. (2013) Proteolytic activation of prochemerin by kallikrein 7 breaks an ionic linkage and results in C-terminal rearrangement. Biochem. J. 452, 271–280 [DOI] [PubMed] [Google Scholar]
- 13. Kim M. S., and Youn B. S. (November 10, 2011) Method of preventing or treating body weight-related disorders by employing vaspin. International Patent WO2011138977 A1 [Google Scholar]
- 14. Debela M., Beaufort N., Magdolen V., Schechter N. M., Craik C. S., Schmitt M., Bode W., and Goettig P. (2008) Structures and specificity of the human kallikrein-related peptidases KLK 4, 5, 6, and 7. Biol. Chem. 389, 623–632 [DOI] [PubMed] [Google Scholar]
- 15. Ulbricht D., Pippel J., Schultz S., Meier R., Sträter N., and Heiker J. T. (2015) A unique serpin P1′ glutamate and a conserved β-sheet C arginine are key residues for activity, protease recognition and stability of serpinA12 (vaspin). Biochem. J. 470, 357–367 [DOI] [PubMed] [Google Scholar]
- 16. Oliveira J. R., Bertolin T. C., Andrade D., Oliveira L. C., Kondo M. Y., Santos J. A., Blaber M., Juliano L., Severino B., Caliendo G., Santagada V., and Juliano M. A. (2015) Specificity studies on kallikrein-related peptidase 7 (KLK7) and effects of osmolytes and glycosaminoglycans on its peptidase activity. Biochim. Biophys. Acta 1854, 73–83 [DOI] [PubMed] [Google Scholar]
- 17. Gettins P. G., and Olson S. T. (2009) Exosite determinants of serpin specificity. J. Biol. Chem. 284, 20441–20445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Olson S. T., Björk I., Sheffer R., Craig P. A., Shore J. D., and Choay J. (1992) Role of the antithrombin-binding pentasaccharide in heparin acceleration of antithrombin-proteinase reactions: resolution of the antithrombin conformational change contribution to heparin rate enhancement. J. Biol. Chem. 267, 12528–12538 [PubMed] [Google Scholar]
- 19. Yang L., Ding Q., Huang X., Olson S. T., and Rezaie A. R. (2012) Characterization of the heparin-binding site of the protein z-dependent protease inhibitor. Biochemistry 51, 4078–4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cosmi B., Fredenburgh J. C., Rischke J., Hirsh J., Young E., and Weitz J. I. (1997) Effect of nonspecific binding to plasma proteins on the antithrombin activities of unfractionated heparin, low-molecular-weight heparin, and dermatan sulfate. Circulation 95, 118–124 [DOI] [PubMed] [Google Scholar]
- 21. Rezaie A. R. (1998) Calcium enhances heparin catalysis of the antithrombin-factor Xa reaction by a template mechanism: evidence that calcium alleviates Gla domain antagonism of heparin binding to factor Xa. J. Biol. Chem. 273, 16824–16827 [DOI] [PubMed] [Google Scholar]
- 22. Huang X., Rezaie A. R., Broze G. J. Jr, and Olson S. T. (2011) Heparin is a major activator of the anticoagulant serpin, protein Z-dependent protease inhibitor. J. Biol. Chem. 286, 8740–8751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bray B., Lane D. A., Freyssinet J. M., Pejler G., and Lindahl U. (1989) Anti-thrombin activities of heparin: effect of saccharide chain length on thrombin inhibition by heparin cofactor II and by antithrombin. Biochem. J. 262, 225–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fromm J. R., Hileman R. E., Caldwell E. E., Weiler J. M., and Linhardt R. J. (1997) Pattern and spacing of basic amino acids in heparin binding sites. Arch. Biochem. Biophys. 343, 92–100 [DOI] [PubMed] [Google Scholar]
- 25. Ersdal-Badju E., Lu A., Zuo Y., Picard V., and Bock S. C. (1997) Identification of the antithrombin III heparin binding site. J. Biol. Chem. 272, 19393–19400 [DOI] [PubMed] [Google Scholar]
- 26. Ehrlich H. J., Gebbink R. K., Keijer J., and Pannekoek H. (1992) Elucidation of structural requirements on plasminogen activator inhibitor 1 for binding to heparin. J. Biol. Chem. 267, 11606–11611 [PubMed] [Google Scholar]
- 27. Whinna H. C., Blinder M. A., Szewczyk M., Tollefsen D. M., and Church F. C. (1991) Role of lysine 173 in heparin binding to heparin cofactor II. J. Biol. Chem. 266, 8129–8135 [PubMed] [Google Scholar]
- 28. Stone S. R., Brown-Luedi M. L., Rovelli G., Guidolin A., McGlynn E., and Monard D. (1994) Localization of the heparin-binding site of glia-derived nexin/protease nexin-1 by site-directed mutagenesis. Biochemistry 33, 7731–7735 [DOI] [PubMed] [Google Scholar]
- 29. Shirk R. A., Elisen M. G., Meijers J. C., and Church F. C. (1994) Role of the H helix in heparin binding to protein C inhibitor. J. Biol. Chem. 269, 28690–28695 [PubMed] [Google Scholar]
- 30. Neese L. L., Wolfe C. A., and Church F. C. (1998) Contribution of basic residues of the D and H helices in heparin binding to protein C inhibitor. Arch. Biochem. Biophys. 355, 101–108 [DOI] [PubMed] [Google Scholar]
- 31. Jin L., Abrahams J. P., Skinner R., Petitou M., Pike R. N., and Carrell R. W. (1997) The anticoagulant activation of antithrombin by heparin. Proc. Natl. Acad. Sci. U.S.A. 94, 14683–14688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen V. C., Chao L., Pimenta D. C., Bledsoe G., Juliano L., and Chao J. (2001) Identification of a major heparin-binding site in kallistatin. J. Biol. Chem. 276, 1276–1284 [DOI] [PubMed] [Google Scholar]
- 33. Chen V. C., Chao L., and Chao J. (2000) A positively charged loop on the surface of kallistatin functions to enhance tissue kallikrein inhibition by acting as a secondary binding site for kallikrein. J. Biol. Chem. 275, 40371–40377 [DOI] [PubMed] [Google Scholar]
- 34. Pippel J., Kuettner E. B., Ulbricht D., Daberger J., Schultz S., Heiker J. T., and Sträter N. (2016) Crystal structure of cleaved vaspin (serpinA12). Biol. Chem. 397, 111–123 [DOI] [PubMed] [Google Scholar]
- 35. Jerabek-Willemsen M., Wienken C. J., Braun D., Baaske P., and Duhr S. (2011) Molecular interaction studies using microscale thermophoresis. Assay Drug Dev. Technol. 9, 342–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ori A., Free P., Courty J., Wilkinson M. C., and Fernig D. G. (2009) Identification of heparin-binding sites in proteins by selective labeling. Mol. Cell. Proteomics 8, 2256–2265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.