

Abstract
On the basis of diet induced obesity and KO mice models, nitric oxide is implied to play an important role in the initiation of dyslipidemia induced insulin resistance. However, outcomes using iNOS KO mice have so far remained inconclusive. The present study aimed to assess IR in iNOS KO mice after 5 weeks of LFD feeding by monitoring body composition, energy homeostasis, insulin sensitivity/signaling, nitrite content and gene expressions changes in the tissues. We found that body weight and fat content in KO mice were significantly higher while the respiratory exchange ratio (RER), volume of carbon dioxide (VCO2), and heat production were lower as compared to WT mice. Furthermore, altered systemic glucose tolerance, tissue insulin signaling, hepatic gluconeogenesis, augmented hepatic lipids, adiposity, as well as gene expression regulating lipid synthesis, catabolism and efflux were evident in iNOS KO mice. Significant reduction in eNOS and nNOS gene expression, hepatic and adipose tissue nitrite content, circulatory nitrite was also observed. Oxygen consumption rate of mitochondrial respiration has remained unaltered in KO mice as measured using extracellular flux analyzer. Our findings establish a link between the NO status with systemic and tissue specific IR in iNOS KO mice at 5 weeks.
Nitric oxide (NO), an important gaseous signaling molecule, regulates numerous physiological processes having its synthesis being regulated by Ca+2 dependent nitric oxide synthases (eNOS and nNOS) and/or Ca+2 independent inducible nitric oxide synthase (iNOS)1,2. Intracellular compartmentalization of iNOS is observed in metabolic organs such as primarily kupffer cells, periportal hepatocytes, cytoplasm and plasma membrane of white adipose tissue (WAT) adipocytes, cell membrane (sarcolemma) and cytoplasm (sarcoplasm) of skeletal muscle myocytes3,4. However, under normal conditions constitutive expression of iNOS is predominant in kupffer cells and minor in hepatocytes4. Studies reported so far suggest a prominent role of NO/NOS in the pathogenesis of metabolic syndrome, insulin resistance (IR), obesity and diabetes type I/II5,6,7. Augmented visceral fat deposition is an independent risk factor for IR8 and clinical studies have shown that higher body mass index in obese patients correlates positively with IR, hyperinsulinemia and glucose intolerance8,9.
In vitro studies on C2C12 myotubes using NO donor (GSNO) or ectopic iNOS expression demonstrated IRS-1 degradation10. High fat diet (HFD) fed obese (DIO) and ob/ob mice exhibited elevated iNOS expression in liver, skeletal muscle and adipose tissue5,6 and also the inactivation of insulin receptor β/insulin receptor substrate-1 (IRS-1) and AKT11,12. Treatment of ob/ob mice with iNOS selective inhibitor reversed fasting hyperglycemia5. Moreover, iNOS KO mice fed with 30% fructose for 8 weeks were reported to be protected against hepatic steatosis and inflammation13. However, selective over expression of iNOS in liver increased obesity/IR, hepatic IR, and mild hyperglycemia7. On the contrary, rats fed on chow diet and NOS inhibitors showed increased fat deposition, and enhanced level of circulating and hepatic lipids14,15,16. Interestingly, HFD fed iNOS KO mice were protected from diet induced systemic IR even though they exhibited elevated body weight, fat deposition and fasting hyperglycemia17. Hyper-insulinemic-euglycemic (HE) clamp study demonstrated a significantly reduced glucose uptake by the skeletal muscle in aged mice, but not in the young adult iNOS KO mice18. Studies published so far thus found iNOS, to be both protective and detrimental in IR and insulin signaling. It is evident that most of the studies conducted on iNOS KO mice did not examine the systemic IR and insulin signaling in all the metabolic organs but monitored insulin signaling/IR in only one or two organs after feeding standard low fat/chow diet or HFD (55–60%) with diverse regimens (16–18 weeks)17,19,20. However, parameters related to energy homeostasis such as food intake, energy utilization and physical activity, and genes responsible for glucose and lipid metabolism were not profiled in these studies17,20.
These contradictory outcomes associated with iNOS studies and IR could also be due to the use of variable protocols such as amount of glucose (1–2 g) and insulin (0.35 to 1.5 IU) administered for monitoring the tolerance, duration of fasting (4–12 h), glucose/insulin levels and the types of diets (10–60% kcal in energy)17,20. The present study was undertaken to address the anomalies in the role of iNOS in the incidence of IR by using WT and iNOS KO mice fed on low fat diet (LFD) (often used as isocaloric diet to assess high fat diet induced dyslipidemia) for 5 weeks. Circulating glucose, insulin and lipids were measured in the both on ad libitum fed and on fasted (6 h & 12 h time points) mice. To systematically assess incidence of IR in the iNOS KO mice, ITT, GTT, PTT, as well as insulin signaling proteins and expression of selective genes involved in the glucose and lipid metabolism were measured by Western blotting and RT-PCR respectively. Further, WT and iNOS KO mice were assessed for total circulatory nitrite, tissue specific nitrite (liver, adipose and skeletal muscle) and NOS (eNOS, nNOS) isoforms gene expressions followed by hepatic and adipose tissue isolated mitochondrial OCR and UCP2 gene expressions. Our results demonstrate the incidence of tissue specific (liver and adipose tissue but not skeletal muscle) and systemic IR as a resultant of reduced NO availability in iNOS KO mice fed with LFD for 5 weeks.
Results
Weight and body composition of WT and iNOS KO mice
Significant gain in body weight was noticed in iNOS KO mice from third week of LFD feeding and it continued until five weeks (Fig. 1a,b). Whole body fat was enhanced while lean mass was reduced as measured by ECHO-MRI (Fig. 1c). Liver weight was markedly increased, however no significant change in heart, spleen and kidney weight was observed in iNOS KO mice as compared to WT (Fig. 1d).
Figure 1. Gross parameters in wild type (WT) and iNOS knockout (KO) mice.

(a) Body weight, (b) % increase in body weight, (c) % change in whole body fat/lean mass and (d) tissue weights (n = 6–9). Comparison between groups was done by Student’s t test. *p < 0.05 and **p < 0.01 vs WT.
Food intake, energy utilization and physical activity
Food intake and physical activity in iNOS KO mice was not affected during 5 weeks of LFD compared to WT (Fig. 2a,e). However, VCO2, RER and heat production were reduced significantly as compared to WT controls (Fig. 2b–d). Furthermore, metabolic rates as indicated by BMR and RMR were almost similar in KO and WT mice (Fig. 2f).
Figure 2. Food intake, energy utilization and physical activity in WT and iNOS KO mice.

(a) Food consumption, (b) VCO2, (c) RER, (d) heat production, (e) X-axis movement and (f) metabolic rates (n = 6–8). L, D represents Light and Dark phases of a single day while 1, 2 and 3 indicates days of experiment. Comparison between groups was done by two way ANOVA followed by Bonferroni post hoc test (WT vs KO). *p < 0.05 and **p < 0.01 vs WT.
Glycemic control and insulin sensitivity
A significant increase in serum insulin and glucose levels in KO mice indicated a compensatory insulin action to maintain euglycemia for 6 h fasting (Table 1). Subsequent studies in overnight fasted group showed no significant change in glucose and insulin levels. Marked difference was observed in HOMA-IR and QUICKI but not HOMA-B between WT and KO mice, indicated systemic IR with functional β-cells in iNOS KO mice21,22 (Table 1). Subsequently, tissue insulin sensitivity and glucose disposal to metabolic organs were measured using glucose and insulin tolerance tests. A significant increase in the blood glucose level in iNOS KO mice was found untill 2 h after intra-peritoneal (i.p.) glucose or pyruvate administration (GTT & ITT) (Fig. 3a,b). Similarly, PTT analysis for insulin sensitivity was also adversely affected in WT and KO mice, suggesting a persistent hepatic gluconeogenesis (Fig. 3c). Real time PCR data indicated that hepatic and adipose tissue gluconeogenetic enzymes (PEPCK, G6PC) and their transcriptional regulators (FOXOA1, PGC-1α and PGC-1β) were found to be augmented by 2–3 folds (Fig. 4a,b). Insulin stimulated hepatic glycogen level (Fig. 3d) was significantly reduced, but skeletal muscle glycogen content was not altered (Fig. 3d). qPCR analysis of glucose transporters in WT and KO mice showed a significant decrease in hepatic GLUT-2 with unaltered adipose tissue GLUT-2, 4 (Fig. 4c).
Table 1. Biochemical parameters in 5 weeks LFD fed WT and KO mice.
| S. No. | Parameter | 5 weeks |
|||||
|---|---|---|---|---|---|---|---|
| 6 h fasted |
Overnight fasted |
Fed (ad libitum) |
|||||
| WT | KO | WT | KO | WT | KO | ||
| 1 | TG (mg/dl) | 81.55 ± 3.35 | 178.37 ± 6.96*** | 79.42 ± 7.07 | 238.29 ± 32.25*** | 104.26 ± 8.10 | 151.09 ± 9.03** |
| 2 | TC (mg/dl) | 99.22 ± 3.59 | 175.87 ± 6.38*** | 82.51 ± 6.69 | 256.09 ± 17.55*** | 133.92 ± 7.67 | 178.08 ± 15.55* |
| 3 | LDL (mg/dl) | 22.71 ± 1.70 | 25.36 ± 2.21 | 44.31 ± 3.25 | 56.19 ± 7.38 | 17.54 ± 1.43 | 18.95 ± 1.11 |
| 4 | HDL (mg/dl) | 34.45 ± 2.93 | 32.66 ± 2.34 | 43.72 ± 3.11 | 51.27 ± 6.43 | 23.14 ± 1.37 | 26.94 ± 1.70 |
| 5 | NEFA (mmol/L) | 0.92 ± 0.04 | 1.69 ± 0.07*** | 1.45 ± 0.14 | 3.25 ± 0.11*** | 0.74 ± 0.04 | 1.00 ± 0.09* |
| 6 | Glucose (mg/dl) | 142.13 ± 2.41 | 166.38 ± 5.61** | 112.75 ± 5.36 | 126.63 ± 5.07 | 182.88 ± 4.82 | 197.38 ± 7.01 |
| 7 | Insulin (ng/ml) | 0. 69 ± 0.03 | 1.08 ± 0.13* | 0.43 ± 0.03 | 0.54 ± 0.05 | 1.49 ± 0.06 | 3.40 ± 0.48** |
| 8 | HOMA-IR | — | — | 2.62 ± 0.22 | 3.79 ± 0.49* | — | — |
| 9 | HOMA-B | — | — | 75.96 ± 10.34 | 68.35 ± 5.53 | — | — |
| 10 | QUICKI | — | — | 0.33 ± 0.0038 | 0.32 ± 0.0054* | — | — |
| 11 | ALT (IU/L) | 31.55 ± 2.11 | 27.20 ± 0.97 | 31.78 ± 3.19 | 27.21 ± 1.88 | 36.53 ± 4.65 | 27.09 ± 2.29 |
| 12 | T. Bilirubin (mg/dl) | 0.13 ± 0.02 | 0.11 ± 0.02 | 0.15 ± 0.01 | 0.14 ± 0.02 | 0.16 ± 0.01 | 0.17 ± 0.01 |
| 13 | Creatinine (mg/dl) | 0.29 ± 0.01 | 0.24 ± 0.03 | 0.32 ± 0.02 | 0.30 ± 0.02 | 0.28 ± 0.01 | 0.26 ± 0.01 |
Results are expressed as mean ± SEM (n = 8–10).
Figure 3. Systemic glycemic control and tissue biochemistry.

(a) GTT (n = 8), (b) ITT (n = 10), (c) PTT (n = 9), (d) hepatic and skeletal muscle glycogen content (n = 5), (e) hepatic, adipose tissue and skeletal muscle Oil red O staining (n = 5), (f,g) hepatic triglycerides and cholesterol (n = 5). Comparison between groups was done by Student’s t test. *p < 0.05, **p < 0.01 and ***p < 0.001 vs WT.
Figure 4.
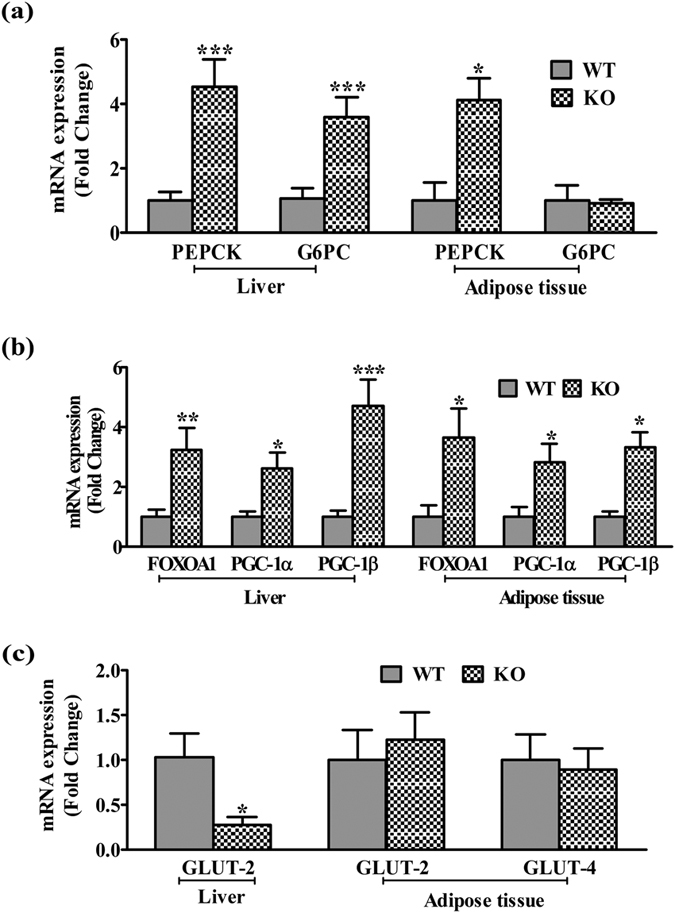
qPCR expressions of (a) Enzymes and (b) Transcriptional regulators involved in hepatic (n = 9–11) and adipose tissue (n = 4) gluconeogenesis, and (c) Glucose transporters (GLUTs, n = 4). Comparison between groups was done by Mann-Whitney U test. *p < 0.05, **p < 0.01 and ***p < 0.001 vs WT.
Insulin signaling proteins
To gain further insight, phosphorylation status of IRS-1 (Ser 307) and AKT-1/2/3 (Ser 473) was monitored during insulin stimulated conditions in liver, skeletal muscle and adipose tissue23. IRS-1 phosphorylation on Ser 307 was enhanced in liver and adipose tissues but not in skeletal muscle (Fig. 5a–c). AKT-1/2/3 phosphorylation at Ser 473 being prerequisite for normal insulin signaling was reducedin liver and adipose tissues, but not in skeletal muscle of KO mice (Fig. 5d–f).
Figure 5. Effect of 5 weeks LFD feeding on insulin signaling proteins in liver, adipose and skeletal muscle tissues.

Western blots analyses of (a–c) pIRS-1 (Ser 307) and (d–f) pAKT-1/2/3 (Ser 473) in liver, adipose and skeletal muscle tissues. Bar diagrams represent mean ± SEM of the change in image density (n = 3). Comparison between groups was done by Student’s t test. *p < 0.05 vs WT Basal and ##p < 0.01 vs WT Insulin.
Systemic cytokine status
Previous studies have shown that sub-acute inflammation is crucial for metabolic disorders. We thus measured the circulating inflammatory and anti-inflammatory cytokines in WT and iNOS KO mice. Serum cytokine profile analyses indicated only a trend of increase in IL-1β, which was statistically not significant (p > 0.05). However, the levels of other pro-inflammatory cytokines (IFN-γ, GM-CSF, IL-6, TNF-α) were remain unchanged. On the contrary, anti-inflammatory cytokines, IL-2 and IL-4 were significantly increased in iNOS KO mice (Fig. 6a).
Figure 6.
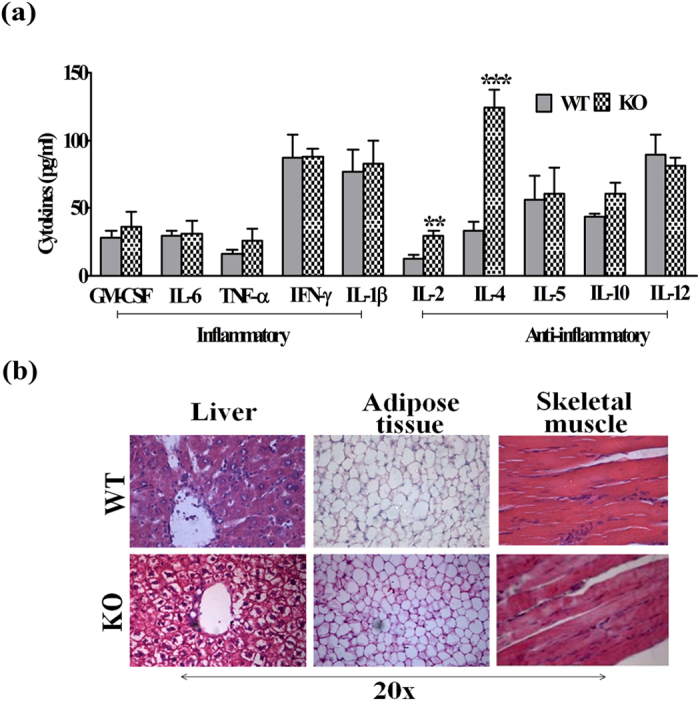
(a) Serum cytokine status (n = 6–8) and (b) HE staining in WT and iNOS KO mice. Comparison between groups was done by Student’s t test. **p < 0.01 and ***p < 0.001 vs WT.
Systemic and hepatic lipid content
iNOS KO mice fed ad libitum on LFD exhibited a profound increase in the serum TG, TC, and NEFA during fasted as well as in fed conditions (Table 1). The increase in serum lipids was more pronounced in overnight fasted iNOS KO mice as compared to 6 h fasted or ad libitum fed mice. However, no significant change was observed in the serum LDL and HDL in fasted or ad libitum fed mice. Pathophysiology of IR is often associated with excessive lipid accumulation in adipose tissue, lipolysis and hepatic de novo lipogenesis24. Morphological assessment by HE staining detected the presence of white spots around bluish nucleus on pink cytoplasmic background in iNOS KO mice liver (Fig. 6b). Oil red O staining of three metabolic organs indicated a significant lipid accumulation in liver and adipose tissue but not in skeletal muscle of iNOS KO mice (Fig. 3e). Estimation of hepatic triglyceride and total cholesterol levels confirmed increased lipid accumulation in iNOS KO mice (Fig. 3f,g). Further, the qPCR analysis of genes involved in hepatic lipogenesis (FAS, ACC1 & 2, HMGCR, SREBP-1C and SREBP-2) and metabolism (PPAR-α, γ, LXR-α, β, LPL, ApoE, LDLR, ABCG5 and ABCG8) indicated 2–4 fold augmentation in the expression of genes coding for triglyceride (FAS, ACC1 & 2, SREBP-1C), total cholesterol (HMGCR & SREBP-2) synthesis and β-oxidation (PPAR-α, γ, LXR-α, β) suggesting that iNOS KO mice are prone to hepatic steatosis (Fig. 7a,b). Surprisingly, augmentation in lipid efflux genes (ApoE, LDLR, ABCG5 & ABCG8) in iNOS KO mice liver suggests simultaneous increase in the lipid efflux (Fig. 7d). Subsequently, qPCR analysis of adipose tissue and skeletal muscle lipid metabolism associated genes revealed a significant increase in genes coding for lipogenesis and lipolysis in former but not in latter tissue (Fig. 7c).
Figure 7. Hepatic and adipose tissue lipid modulating genes.
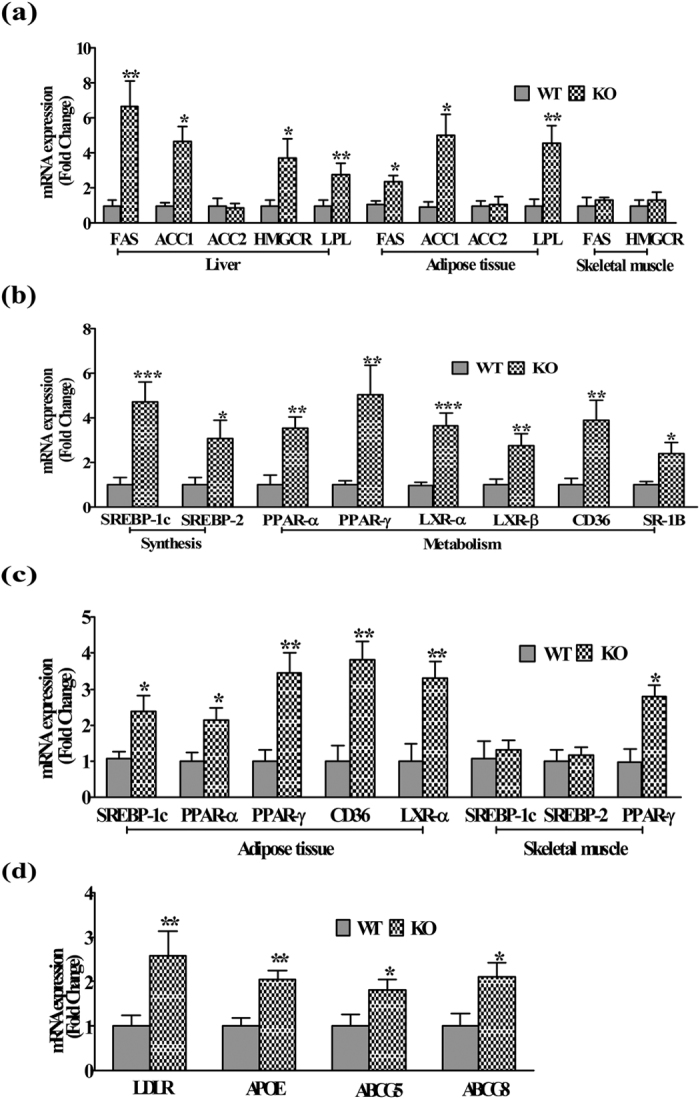
(a) qPCR expressions of enzymes and (b,c) key regulators of hepatic (n = 8–12), adipose tissue (n = 6–10), skeletal muscle (n = 4) lipid metabolism, and (d) hepatic lipid excretion (n = 10). Comparison between groups was done by Mann-Whitney U test. *p < 0.05, **p < 0.01 and ***p < 0.001 vs WT.
NO levels in plasma, liver, adipose and skeletal tissue
We checked circulatory and tissue NO content in insulin resistant iNOSKO mice after 5 weeks of LFD feeding. Nitrite content in plasma (Fig. 8a), liver, and adipose (Fig. 8b) were significantly reduced except in the skeletal muscle (Fig. 8b). Further, iNOS deletion led to attenuated eNOS and nNOS gene expressions in liver and adipose tissues but not in skeletal muscle. These results signify that inhibition of iNOS is the major regulator of NO, eNOS and nNOS in tissue insulin resistance (IR) resulting in systemic IR (Fig. 8c). Previous reports suggest that alterations in tissue NO affects mitochondrial function and biogenesis3. Mitochondrial basal oxygen consumption rate (OCR) in isolated hepatic (Fig. 8d,e) and adipose tissue (Fig. S1a,b) showed a decreased trend. However, a significant difference was observed in the adipose tissue of KO mice. Nonetheless, the OCR and ECAR (data not shown) were not affected in both the liver and adipose tissue. Further, 5 weeks LFD fed WT and KO mice demonstrated similar hepatic and adipose tissue ATP levels (Fig. 8f) and UCP2 (Fig. 8g) gene expression.
Figure 8. NO dependent systemic IR in iNOS KO mice.

Total nitrite levels in (a) Plasma and (b) metabolic tissues (liver, adipose tissue and skeletal muscle) (n = 5), qPCR expressions of (c) eNOS, nNOS and (g) UCP2 (n = 6), (d,e) hepatic mitochondrial OCR (n = 3), and (f) tissue ATP (n = 5) in 5 weeks LFD fed WT and iNOS KO mice. Comparison between groups was done by Student’s t test except qPCR studies where Mann-Whitney U test was applied. *p < 0.05 an **p < 0.01 vs WT.
Discussion
Obesity linked IR is a multi-factorial pathology. Enhanced oxidative (ROS)/nitrosative (RNS) stress and inflammatory cytokinesis is well documented during the initial stages of IR25,26,27. The present study was undertaken to extensively evaluate incidences of IR by monitoring weight & fat gain, food intake, physical activity, energy utilization, circulating lipids, systemic insulin resistance, insulin signaling in the metabolic organs and the level of circulating inflammatory cytokines in LFD fed iNOS KO mice. Low fat diet as an isocaloric diet is often used in studies conducted on HF induced dyslipidemia, and was also used for mice feeding in this study28. Moreover, an optimum duration of five weeks feeding was considered appropriate in the present study, as alterations in redox status are expected to be evident during initiation of IR. Weight gain in iNOS KO mice correlated with body fat and lean mass as measured by ECHO-MRI29. Perreault and Marette, like us observed significant body weight gain after 16 weeks of 55% HFD feeding to iNOS KO mice17. Epididymal and retroperitoneal white adipose tissue (WAT) in iNOS KO mice were significantly increased in 18 weeks fed 55% HFD diet17,19. Surprisingly, Becerril et al. had observed a significant reduction in body weight and total WAT after 12 weeks of chow diet feeding in iNOS KO mice30. On the contrary, other studies reported similar level of weight gain in WT and iNOS KO mice fed on chow diet up to 22 months or HFD (55–60% kcal) for 18 weeks18,19,20. Our study is strengthened by monitoring other tissue weights apart from enhanced liver weight in line with other published data17,19. In the present study, significant reduction in RER, VCO2, and heat production was noticed with no change in food intake and physical activity in iNOS KO mice, indicating altered energy utilization. A single study on iNOS KO mice measured similar parameters, but failed to find alterations in VO2 and RER following HF feeding for 18 weeks19. Nevertheless, parameters measured by us following LFD feeding in WT mice were comparable to the reported values6,19,28,30,31,32. HF feeding to WT mice exhibited augmented serum lipids, altered insulin signaling, and expression of metabolically important enzymes/transcription factors31. iNOS KO mice exhibited altered expression of metabolically important genes (Figs 4 and 7), implying a regulatory role of iNOS/NO in glucose and lipid homeostasis, as reported for eNOS and nNOS KO mice33,34,35,36,37,38,39.
Treatment of rodents with non-specific pharmacological NOS inhibitors like L-NNA or L-NAME for 2–8 weeks resulted in enhanced body fat, serum lipids and IR as observed by us in iNOS KO mice14,15,16. Genetic knockout of single (eNOS/nNOS)33 or triple (eNOS, nNOS and iNOS)40 null mice are pre-disposed to IR; on the contrary, iNOSKO have so far remain inconsistent17,18,19,20. Diet induced obesity and IR studies in iNOS KO were conducted mostly after 16–18 weeks of HF diet feeding17,19,20. However, in the present study iNOS KO mice exhibited a significant change in lipids, glucose and insulin even after 5 weeks of LFD feeding (Fig. 3 and Table 1). Long term (16–18 weeks) feeding with 55% HFD led to fasting hyperglycemia (5 h) with low insulin (Perreault and Marette) which was inconsistent with Dallaire et al., though similar dietary regimens were used17,19. In the present study we measured most of the parameters (lipids, insulin, glucose and insulin signaling proteins), by using three different feeding time points (6 h, 12 h fasting and food ad libitum). Significant changes in glucose and insulin were evident only after 6 h fasting and in fed condition but not after overnight fasting, while serum lipids remain altered. Normal blood glucose and serum insulin in overnight but not in 6 h fasted KO mice signify their pre-diabetic insulin resistant state. This might be attributed to some secondary systemic effects rather than primary iNOS absence which needs to be studied7. None of the studies on iNOS KO mice found systemic IR and β-cell functionality, as observed by us using HOMA indices. Gene profile of glucose and lipid metabolism as seen in this study was also not examined by others. We found both hepatic gluconeogenesis and altered glycogen content in iNOS KO mice as compared to their WT counterparts. Further, data from tolerance tests (GTT, PTT and ITT), skeletal muscle glycogen content and Western blots of insulin signaling confirmed that mice lacking iNOS had altered body glucose homeostasis and insulin sensitivity in hepatic and adipose tissue but not in skeletal muscle. Most of the previous studies on iNOS KO mice performed GTT and ITT and never assessed for PTT17,19,20. All the reported studies involving whole body iNOS deletion but not myeloid specific KO, failed to observe systemic IR in iNOS KO mice17,19,20. The difference in the present study and other reports is of the duration of feeding (16–18 weeks), composition of the diet (55 to 60% kcal) and the protocols (Dose and fasting time for biochemical estimations) used to assess IR17,19,20. There are two studies in the literature, which assessed IR signaling proteins [p-IRS (AKT 473) and/or PI3K activity] at 16 or 18 weeks of 55% HFD feeding17,19. Our results highlights the presence of systemic IR which the other studies neglected, nonetheless our results concur with published reports on hepatic and adipose tissue IR and the absence of skeletal muscle IR in iNOS KO mice17,19. A significant decrease in plasma total nitrite in insulin resistant iNOS KO mice, which is in line with the earlier published report18. Perreault and Marette, observed a marked decrease in eNOS gene expression in chow diet fed iNOS KO mice WAT but not in skeletal muscle17. On the contrary, nNOS expression was reduced in muscle and was not expressed in WAT17. Variations in the expression of the respective genes might be related to the sensitivity of the technique used for gene assessments. The present study highlights the requirement of optimal iNOS levels in the maintenance of normal glucose/lipid metabolism and insulin sensitivity.
Hyperinsulinemic euglycemic (HE) clamp study in the adult and old mice showed a significant decrease in the whole body and skeletal muscle glucose uptake in old (22 months) but not in adult (7 months) iNOS KO mice18. Conversely, improved muscle glucose uptake in the isolated soleus muscles of young and old iNOS KO mice has also been published recently41. These studies highlight that skeletal muscle cell glucose utilization and metabolism is differently regulated in iNOS KO mice. Though we did not carry out glucose uptake studies, it was however evident that the change in skeletal muscle glycogen, IR marker proteins and qPCR gene expressions was statistically not significant. Glutathione and NO are imperative for insulin sensitivity, its modulation in iNOS KO mice is however an unexplored area so far and needs attention42. Reactive oxygen species (ROS) and H2O2 also act as a secondary messengers in insulin signaling and mimic insulin effects43, their excessive production has been implicated in IR pathophysiology43. The significance of ROS and associated redox modulators in insulin resistant iNOS KO mice remains to be explored. Obesity linked IR is described as a sub-acute inflammatory disorder and inflammatory cytokines are the crucial players44. Nevertheless, protective role of iNOS against tissue degradation has recently been reported45. iNOS KO macrophages exhibited significantly higher basal generation of H2O2 as well as LPS induced TNF-α and MIP-2 production, suggesting dual role of iNOS in inflammation46. Status of inflammatory cytokines in the current study, dissociates inflammation with IR in iNOS KO mice as we failed to observe any significant change in these cytokines. This is also in agreement with a previous study where standard chow fed adult iNOS KO mice showed no significant variation in muscular and adipose pro-inflammatory cytokine gene expressions18. In old age mice (22 months), iNOS KO mice showed significant increase in pro-inflammatory and a decrease in anti-inflammatory cytokine gene expressions18. The altered cytokine levels in old but not in adult iNOS KO mice coincided with reduced glucose uptake, suggesting that mice lacking NOS2 were pre-disposed to IR with the advancement of age18.
As evident from hepatic insulin signaling data, we and others17,19 have not found protection in the hepatic tissue against IR in the insulin signaling. We extended this observation by measuring hepatic lipids which was profoundly increased in KO mice. Serum lipid levels in KO mice were also enhanced with fasting, possibly due to high rates of lipid oxidation and systemic IR47. Moreover, IR at liver and adipose tissue level was substantiated by measuring genes responsible for lipid metabolism (synthesis, breakdown and/or efflux) in iNOS KO mice. Further, data obtained from Oil red O staining and lipogenic gene expressions rule out the role of skeletal muscle in pre-disposing iNOS KO mice to systemic IR. In the present study, we report for the first time that functional markers of liver or kidney injury (ALT, T. Bilirubin and Creatinine) were not altered at 5 weeks of LFD between both WT and iNOSKO mice. It is clearly evident that LFD fed iNOS KO mice exhibited impairment in glucose and lipid metabolism correlating well with altered body composition and energy homeostasis. Excessive iNOS was often linked with IR5,6,7, on the contrary, even its absence has been associated with systemic IR.
It is well documented that NO at physiological concentrations acts as an endogenous messenger in maintaining systemic glucose and lipid homeostasis by promoting their utilization but not synthesis3. Gene manipulation of endothelial NOS (eNOS) in mice has highlighted its importance in maintaining systemic glucose and lipids by regulating metabolism in three major metabolic organs (liver, skeletal muscle and adipose tissue)33,34,35,36,37,38. On the other hand, studies on nNOS KO mice have demonstrated its crucial role in regulating metabolism of only peripheral organs excluding liver33,39. Previous literature and data from hepatic, adipose tissue nitrite and eNOS, nNOS gene expressions associate tissue specific as well as systemic IR in KO mice, while no change in skeletal muscle nitrite and NOS expression correlated with their normal insulin signaling. Reduction in hepatocyte, adipocyte NO synthesis due to attenuated eNOS and nNOS expression in the absence of iNOS, seems to be responsible for decreased NO and systemic IR in iNOS KO mice3. On the other hand, our previous experiments demonstrated, the crucial role of hepatic and adipose tissue metabolic deregulation as a predisposing factor for IR in mice lacking iNOS. Kleemann et al. demonstrated that HFD induced IR is a tissue dependent process which starts with liver and culminates in white adipose tissue (WAT)48. They observed that IR development is paralleled by tissue-specific gene expression changes, metabolic adjustments, changes in lipid composition, and inflammatory responses in liver and WAT but not in skeletal muscle48. Further, they demonstrated that alterations in skeletal muscle are largely opposite to those in liver and WAT, which signifies that pathology of IR involve compromised liver and adipose tissue in the initial stages followed by skeletal muscle in the later stages48. Hepatic and adipose tissue IR in iNOS KO mice in the present study goes in line with the statement that they are primarily compromised in IR pathology48. Altered NO content and tissue metabolism in liver and adipose tissue of insulin resistant iNOS KO mice signify that reduction in their tissue NO pool, affected the normal utilization of glucose and lipids, thereby pre-disposing iNOS KO mice to systemic and tissue IR.
It is well known that NO is important for mitochondrial biogenesis via peroxisome proliferator activated receptor-γ coactivator 1α (PGC-1α)3,49. PGC-1α acts as a master regulator of mitochondrial oxidative phosphorylation via upregulation of transcription factors PPAR-α and NRFs, and uncoupling proteins (UCPs) respectively3. Augmented mitochondrial UCP2 levels in insulin resistant metabolic organs indicate ATP depletion and mitochondrial dysfunction50. Almost similar mitochondrial OCR, ATP levels and UCP2 gene expression (Fig. 8f,g) were observed in WT and iNOS KO mice indicating a normal mitochondrial function. This might be probably due to non-stimulation of PGC-1α expression and its downstream PPAR-α is not regulated within 5 weeks of LFD feeding leading to mitochondrial dysfunction50,51. It could be that PGC-1α up-regulation might not necessarily be dependent only on NO signaling pathways39,52 or requires more time to affect the downstream pathways leading to change in the mitochondrial function. Mitochondrial OCR levels in KO mice were unaffected after feeding with 5 weeks LFD, thus suggesting mitochondrial dysfunction is not initiated, during the initial stages of IR. We demonstrated that systemic and tissue IR in iNOS KO mice is a resultant of perturbed glucose and lipid availability affecting insulin signaling along with altered tissue NO availability, but not due to mitochondrial dysfunction. Studies involving fat enriched obesogenic diets in WT and iNOS KO mice on mitochondrial functionality and incidence of IR at later time points might further help to understand the role of NO on mitochondrial performance. Therapeutic efficacy of dietary supplements containing NO donors (NaNO2) or its substrates (L-Arginine) were extensively studied53,54,55,56,57,58. Administration of NaNO2 (50–150 mg/L) in drinking water for 10 days/4 weeks have shown to improve insulin resistance parameters in db/db and KKAy KO mice models of type 2 diabetes54,55. Similarly, dietary supplementation of L-Arginine to rodents or patients with metabolic syndrome and obesity improved systemic IR and energy homeostasis53,56,57,58. Systemic IR due to reduced NO levels in iNOS KO mice suggests usefulness of NO donors/L-arginine to treat IR, obesity and metabolic syndrome. Studies involving the nitrite pharmacotherapy for treating insulin resistant iNOS KO mice are out the scope of the present study which needs future investigations. In summary, the present study thus suggests importance of optimal iNOS along with eNOS and nNOS in the maintenance of glucose/lipid metabolism and insulin sensitivity.
Materials
RevertAid H Minus first strand kit for cDNA synthesis and (2X) PCR master mix were obtained from Fermentas Life Sciences (Vilnius, Lithuania). Maxima SYBR Green (2X) RT-PCR Master Mix was purchased from Roche Applied Science (Lewes, UK). IRS-1(C-20, sc-559), pIRS-1 (Ser 307) (sc-33956) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, USA) while AKT-1/2/3 (11E7, 4685), pAKT-1/2/3 (Ser 473) (D9E, 4060) and GAPDH (14C10, 2118) antibodies were procured from Cell Signaling Technology (Danvers, MA). All the primers used in the study were from Integrated DNA technology (India). Human insulin (Humulin, 5000 IU/ml) was from El Lilly, USA and rest of the chemicals including antibody against β-Actin (AC-15, A3854) were from Sigma Aldrich Co. (St. Louis, USA).
Methods
Animals
Eight week old male C57BL/6 WT and iNOS KO mice (Jackson Labs; 002609) were housed in IVC cages (Tecniplast, Italy) at 24 ± 2 °C with free access to food and water. Experiments were approved by the Institutional Animal Ethics Committee, Council for Scientific and Industrial Research-Central Drug Research Institute (CSIR-CDRI) (IAEC/2014/43) and conducted in accordance with the Guidelines of Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA). Mice used in the present study were randomly genotyped as described earlier59,60. Animals were kept for five weeks on LFD [Research diets, D12450H], which contains 10% kcal fat which is 35% less compared to HFD (4057 kcal% energy). Body weight in each group was measured weekly from day zero to the completion of five weeks.
Body composition analysis
Mice were analyzed by echo MRI for body composition (E26-226-RM Echo MRI LLC, USA), by applying radio frequency pulses at a distinct static magnetic field as described earlier61. Each mouse during the measurement period was placed into a thin wall plastic cylinder (4.5 cm diameter) allowing only limited vertical and horizontal movement.
Indirect calorimetry
Metabolic measurements were performed in conscious and unrestrained WT, KO mice using six station oxymax CLAMS [CLAMS-CIS-6 with CLAMS-C-6MR, Columbus Instruments, USA] as described earlier62. The system was calibrated against a standard gas mixture to measure O2 consumed (VO2, ml/kg/h) and CO2 generated (VCO2, ml/kg/h). Metabolic rate (VO2) and respiratory exchange ratio (RER) (VCO2/VO2, mL/kg/min) were evaluated over a 3-day period. Energy expenditure (Kcal/h; heat production) was calculated using a rearrangement of the Weir equation as per the instruction of the manufacturer [(3.815 + 1.232*RER)*VO2 (A mean of 50 values per mouse)].
Serum parameters
Mice were categorized in fasted (overnight, 6 h) and ad libitum fed groups to measure their circulating lipids [total cholesterol (TC), triglycerides (TG), low and high density lipoprotein (LDL, HDL)] using commercial kits from Randox, UK. Insulin levels were measured by using ELISA kits (Crystal Chem, USA); while ALT, total bilirubin and creatinine were monitored using kits from Merck, Germany. Effect of fasting was assessed at two time points (6 h, overnight) as defined earlier7.
Tolerance tests
Mice fasted for 6 h were administered 2 g/kg D-Glucose, 2 g/kg sodium pyruvate and 0.6 IU/kg insulin by the intraperitoneal (i.p.) route to assess glucose (GTT), pyruvate (PTT) and insulin tolerance tests (ITT) respectively, as per the protocols used earlier63. Blood glucose was monitored using glucometer (Roche Diagnostics, India) at 0, 15, 30, 60 and 120 min after administration of either glucose, pyruvate or insulin injection. Indices of insulin sensitivity/IR, beta cell (HOMA-IR, HOMA-B) functionality and the quantitative insulin check index of insulin sensitivity (QUICKI) were calculated as per the formulae used by other investigators21,22.
Tissue biochemistry
The fasted mice (6 h) were sacrificed after anesthesia with ether to retrieve the organs. Liver triglyceride was measured using Abcam Triglyceride quantification kit (Abcam, ab65336) and cholesterol using Amplex Red Assay kit (Invitrogen, A12216). Tissue glycogen was analysed using Biovision (K 646–100) kit. ATP levels in hepatic and adipose tissue homogenates were estimated calorimetrically using ATP assay kit (Abcam, ab83355) as per manufacturer’s instructions.
Hematoxylin and Eosin (HE) staining
Formalin fixed tissues were processed in graded concentrations of ethanol followed by xylene prior to liquid paraffin infiltration64. The paraffin embedded tissue blocks were sectioned (4–5 μm slices), HE stained images were captured in the bright field mode using an up-right microscope (DM5000, Leica Microsystems, Germany).
RNA isolation and Real time PCR
Quantitative gene expression analysis was performed using SYBR Green as described previously65. Briefly, total RNA was extracted using TRIZOL reagent, and cDNA was synthesized using RevertAid H Minus first strand cDNA synthesis kit. Real-Time PCR (qPCR) for various genes was performed using LightCycler 480II Real-Time PCR system (Roche Applied Science, Indianapolis, IN). 18S rRNA was used as reference gene for normalization to calculate the expression of candidate genes. List of various primers used for qPCR analysis has been shown in Table S1.
Immunoblotting
Protein samples (40 μg) were run on 8% SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes (Amersham), blocked with 5% BSA in TBST (for 2 h, at RT)and then probed with primary antibodies [diluted in 1:1 ratio of TBST (pH 7.4) and 5% BSA, incubated overnight at 4 °C]. Membranes were later incubated with horse radish peroxidase-linked secondary IgGs (1:10,000) for 2 h at RT, washed and visualized using ECL detection kit (Amersham Biosciences, UK). The signals were captured and normalized with GAPDH for liver and skeletal muscle proteins, or β-actin for adipose tissue proteins, to assess the fold change with respect to WT controls66.
Total nitrite estimation
Plasma and tissue total nitrite (nitrate and nitrite) were estimated using Griess reagent method67. Pre-activated cadmium pellets were used for reduction of 100 μl of plasma or 100 mg tissue (liver, adipose and skeletal muscle) homogenates for 4 h at RT. Equal volumes (1:1) ratio of supernatant and Griess reagent were incubated for 30 min (37 °C in dark), followed by deprotinization with 3% trichloroaceticacid (TCA) and reading at 545 nm. Concentration of test samples total nitrite was calculated using sodium nitrite as standard.
Mitochondrial respiratory measurements
Tissue mitochondria isolation followed by oxygen consumption rates measurements (OCR, a reliable indicator of mitochondrial function) were performed as recommended by Seahorse Biosciences68. Briefly, hepatic and adipose tissues were minced in ~10 volumes of mitochondrial isolation buffer (MIB, pH 7.2) followed by homogenization in dounce homogenizer. Tissue homogenates were initially centrifuged at 800 g to remove the cell debris followed by its supernatant at 8000 g for 10 min at 4 °C. The pellets obtained were washed twice in MIB and finally resuspended in 1X mitochondrial assay buffer (MAS, pH 7.2) buffer with 10 mM (pH 7.2) succinate. Equal quantity of mitochondria was normalized with the equal mitochondrial protein content using BCA reagent. After incubation at 37 °C for 8–10 min, plates were placed in XFp Bioanalyzer, and mitochondrial OCR was measured before and after sequential addition of respiratory reagents like 4 mM ADP, 3.16 μM Oligomycin, 4 μM FCCP, and 4 μM Antimycin A68.
Statistical Analysis
Data has been summarized as mean ± SEM (standard error of the mean). Groups were compared by independent Student’s t test or Mann-Whitney U test wherever applicable. Groups were also compared by two way analysis of variance (ANOVA) and the significance of mean difference between the groups (WT and KO) was done by Bonferroni post hoc test after adjusting for multiple contrasts. Prior to analysis, data was ascertained for normality and homogeneity of variance by Shapiro-Wilk’s test/Kolmogorov-Smirnov test and Levene’s test respectively. A two-tailed (α = 2) p < 0.05 was considered statistically significant. Analyses were performed on STATISTICA software (Version 7.1 StatSoft, Inc. USA) and graphs were made on GraphPad Prism (Trail version 5.01).
Conclusion
Previous studies on iNOS KO mice, using HFD with prolonged regimens demonstrated protection against systemic IR. However, the present study even after using LFD only for 5 weeks, observed glucose intolerance, enhanced hepatic glucose production, lipid accumulation, reduced circulatory and tissue nitrite, IR and altered expression of the genes involved in the NO synthesis, lipid and glucose metabolism in iNOS KO mice. Interestingly iNOS mice did not exhibit any change in physical activity but VCO2, RER, and heat production were significantly less, further suggesting altered metabolism. We for the first time report systemic IR without alteration in the serum cytokines, ruling out the pivotal role of inflammation in IR. We also did not observe skeletal muscle IR but found hepatic and adipose tissue IR in iNOS KO mice. Taken together, IR in iNOS KO, imply crucial role of NO in the regulation of energy and metabolic homeostasis.
Additional Information
How to cite this article: Kanuri, B. N. et al. Altered glucose and lipid homeostasis in liver and adipose tissue pre-dispose inducible NOS knockout mice to insulin resistance. Sci. Rep. 7, 41009; doi: 10.1038/srep41009 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Acknowledgments
The study was supported by a financial grant to Madhu Dikshit from Council of Scientific and Industrial Research (CSIR) network project BSC0102. We gratefully acknowledge the award of research fellowships by the Council of Scientific and Industrial Research (CSIR), New Delhi, India to K.B.N and Indian Council of Medical Research, New Delhi, India to J.S.K. We are grateful to Mr. M.P.S Negi from biometry and statistics division, CSIR-CDRI for helping in the statistical analysis of the data. The authors are sincerely thankful to Ms. Dipika Awasthi and Ms. Sheela Nagarkoti for assistance in conducting experiments. We thank Dr. Naibedya Chattopadhyay for providing ECHO-MRI instrument for body composition analysis and Mr. Subhashis Pal for excellent help during data acquisition and analysis. We acknowledge the scientific and technical inputs during this work given by Prof. Parimal Misra (Dr. Reddy’s Institute of Life Sciences), Dr. S. Sanyal, Dr. A.K. Tamrakar and Dr. Sachin Kumar (CSIR-CDRI, Lucknow).
Footnotes
Author Contributions B.N.K., J.S.K., S.C.R., P.P., and A.P.G., performed the experimentations. B.N.K. analyzed data and wrote the manuscript; J.R.G. helped in analysis of calorimetry results and gave critical inputs relating to experimental design; K.J. provided genotyped WT and KO mice used in the studies and helped in editing manuscript; M.D. conceptualized and supervised the study, planned and designed experiments, and wrote the manuscript.
References
- Förstermann U. & Sessa W. C. Nitric oxide synthases: regulation and function. European heart journal 33, 829–837 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan C. Nitric oxide synthase in innate and adaptive immunity: an update. Trends in immunology 36, 161–178 (2015). [DOI] [PubMed] [Google Scholar]
- Jobgen W. S., Fried S. K., Fu W. J., Meininger C. J. & Wu G. Regulatory role for the arginine–nitric oxide pathway in metabolism of energy substrates. The Journal of nutritional biochemistry 17, 571–588 (2006). [DOI] [PubMed] [Google Scholar]
- Leifeld L. et al. Inducible nitric oxide synthase (iNOS) and endothelial nitric oxide synthase (eNOS) expression in fulminant hepatic failure. Journal of hepatology 37, 613–619 (2002). [DOI] [PubMed] [Google Scholar]
- Fujimoto M. et al. A role for iNOS in fasting hyperglycemia and impaired insulin signaling in the liver of obese diabetic mice. Diabetes 54, 1340–1348 (2005). [DOI] [PubMed] [Google Scholar]
- Ha S.-K. & Chae C. Inducible nitric oxide distribution in the fatty liver of a mouse with high fat diet-induced obesity. Experimental Animals 59, 595–604 (2010). [DOI] [PubMed] [Google Scholar]
- Shinozaki S. et al. Liver-specific inducible nitric-oxide synthase expression is sufficient to cause hepatic insulin resistance and mild hyperglycemia in mice. Journal of Biological Chemistry 286, 34959–34975 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchernof A. & Després J.-P. Pathophysiology of human visceral obesity: an update. Physiological reviews 93, 359–404 (2013). [DOI] [PubMed] [Google Scholar]
- Reaven G. M. Insulin resistance: the link between obesity and cardiovascular disease. Medical Clinics of North America 95, 875–892 (2011). [DOI] [PubMed] [Google Scholar]
- Sugita H. et al. Inducible nitric-oxide synthase and NO donor induce insulin receptor substrate-1 degradation in skeletal muscle cells. Journal of Biological Chemistry 280, 14203–14211 (2005). [DOI] [PubMed] [Google Scholar]
- Yasukawa T. et al. S-nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. Journal of Biological Chemistry 280, 7511–7518 (2005). [DOI] [PubMed] [Google Scholar]
- Carvalho-Filho M. A. et al. S-Nitrosation of the Insulin Receptor, Insulin Receptor Substrate 1, and Protein Kinase B/Akt A Novel Mechanism of Insulin Resistance. Diabetes 54, 959–967 (2005). [DOI] [PubMed] [Google Scholar]
- Spruss A., Kanuri G., Uebel K., Bischoff S. C. & Bergheim I. Role of the inducible nitric oxide synthase in the onset of fructose-induced steatosis in mice. Antioxidants & redox signaling 14, 2121–2135 (2011). [DOI] [PubMed] [Google Scholar]
- Khedara A., Goto T., Morishima M., KAYAsHiTA J. & KATO N. Elevated Body Fat in Rats by the Dietary Nitric Oxide Synthase Inhibitor, l-Nω Nitroarginine. Bioscience, biotechnology, and biochemistry 63, 698–702 (1999). [DOI] [PubMed] [Google Scholar]
- Goto T. et al. Feeding the nitric oxide synthase inhibitor LN ω nitroarginine elevates serum very low density lipoprotein and hepatic triglyceride synthesis in rats. The Journal of nutritional biochemistry 10, 274–278 (1999). [DOI] [PubMed] [Google Scholar]
- Sadri P. & Lautt W. W. Blockade of hepatic nitric oxide synthase causes insulin resistance. American Journal of Physiology-Gastrointestinal and Liver Physiology 277, G101–G108 (1999). [DOI] [PubMed] [Google Scholar]
- Perreault M. & Marette A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nature medicine 7, 1138–1143 (2001). [DOI] [PubMed] [Google Scholar]
- Cha H.-N. et al. Lack of inducible nitric oxide synthase does not prevent aging-associated insulin resistance. Experimental gerontology 45, 711–718 (2010). [DOI] [PubMed] [Google Scholar]
- Dallaire P. et al. Obese mice lacking inducible nitric oxide synthase are sensitized to the metabolic actions of peroxisome proliferator–activated receptor-γ agonism. Diabetes 57, 1999–2011 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M. et al. Inducible nitric oxide synthase deficiency in myeloid cells does not prevent diet-induced insulin resistance. Molecular Endocrinology 24, 1413–1422 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama H. et al. Quantitative insulin sensitivity check index and the reciprocal index of homeostasis model assessment in normal range weight and moderately obese type 2 diabetic patients. Diabetes Care 26, 2426–2432 (2003). [DOI] [PubMed] [Google Scholar]
- Matthews D. et al. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28, 412–419 (1985). [DOI] [PubMed] [Google Scholar]
- Kain V. et al. Co-activator binding protein PIMT mediates TNF-α induced insulin resistance in skeletal muscle via the transcriptional down-regulation of MEF2A and GLUT4. Scientific reports 5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshkani R. & Adeli K. Hepatic insulin resistance, metabolic syndrome and cardiovascular disease. Clinical biochemistry 42, 1331–1346 (2009). [DOI] [PubMed] [Google Scholar]
- Matsuzawa-Nagata N. et al. Increased oxidative stress precedes the onset of high-fat diet–induced insulin resistance and obesity. Metabolism 57, 1071–1077 (2008). [DOI] [PubMed] [Google Scholar]
- Galili O. et al. Early experimental obesity is associated with coronary endothelial dysfunction and oxidative stress. American Journal of Physiology-Heart and Circulatory Physiology 292, H904–H911 (2007). [DOI] [PubMed] [Google Scholar]
- Esposito K. et al. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans role of oxidative stress. Circulation 106, 2067–2072 (2002). [DOI] [PubMed] [Google Scholar]
- Maessen D. E. et al. Delayed intervention with pyridoxamine improves metabolic function and prevents adipose tissue inflammation and insulin resistance in high-fat diet-induced obese mice. Diabetes db151390 (2015). [DOI] [PubMed] [Google Scholar]
- Smemo S. et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507, 371–375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerril S. et al. Deletion of inducible nitric-oxide synthase in leptin-deficient mice improves brown adipose tissue function (2010). [DOI] [PMC free article] [PubMed]
- Boudina S. et al. Early mitochondrial adaptations in skeletal muscle to diet-induced obesity are strain dependent and determine oxidative stress and energy expenditure but not insulin sensitivity. Endocrinology 153, 2677–2688 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatori M. et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell metabolism 15, 848–860 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar R. R., Wu Y., Shen H.-Q., Zhu J.-S. & Baron A. D. Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes 49, 684–687 (2000). [DOI] [PubMed] [Google Scholar]
- Cook S. et al. Clustering of cardiovascular risk factors mimicking the human metabolic syndrome X in eNOS null mice. Swiss medical weekly 133, 360–363 (2003). [DOI] [PubMed] [Google Scholar]
- Lee-Young R. S. et al. Endothelial nitric oxide synthase is central to skeletal muscle metabolic regulation and enzymatic signaling during exercise in vivo. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 298, R1399–R1408 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansbury B. E. et al. Overexpression of endothelial nitric oxide synthase prevents diet-induced obesity and regulates adipocyte phenotype. Circulation research 111, 1176–1189 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abudukadier A. et al. Tetrahydrobiopterin Has a Glucose-Lowering Effect by Suppressing Hepatic Gluconeogenesis in an Endothelial Nitric Oxide Synthase–Dependent Manner in Diabetic Mice. Diabetes 62, 3033–3043 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansbury B. E. & Hill B. G. Regulation of obesity and insulin resistance by nitric oxide. Free radical biology and medicine 73, 383–399 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldelli S., Barbato D. L., Tatulli G., Aquilano K. & Ciriolo M. R. The role of nNOS and PGC-1α in skeletal muscle cells. J Cell Sci 127, 4813–4820 (2014). [DOI] [PubMed] [Google Scholar]
- Tsutsui M. et al. Significance of nitric oxide synthases: Lessons from triple nitric oxide synthases null mice. Journal of pharmacological sciences 127, 42–52 (2015). [DOI] [PubMed] [Google Scholar]
- Ropelle E. R. et al. Targeted disruption of inducible nitric oxide synthase protects against aging, S-nitrosation, and insulin resistance in muscle of male mice. Diabetes 62, 466–470 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarino M. P., Afonso R. A., Raimundo N., Raposo J. F. & Macedo M. P. Hepatic glutathione and nitric oxide are critical for hepatic insulin-sensitizing substance action. American Journal of Physiology-Gastrointestinal and Liver Physiology 284, G588–G594 (2003). [DOI] [PubMed] [Google Scholar]
- Bashan N., Kovsan J., Kachko I., Ovadia H. & Rudich A. Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiological reviews 89, 27–71 (2009). [DOI] [PubMed] [Google Scholar]
- Glass C. K. & Olefsky J. M. Inflammation and lipid signaling in the etiology of insulin resistance. Cell metabolism 15, 635–645 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E. et al. Toll-like receptor 4 signaling confers cardiac protection against ischemic injury via inducible nitric oxide synthase-and soluble guanylate cyclase-dependent mechanisms. The Journal of the American Society of Anesthesiologists 114, 603–613 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler P. et al. Response of alveolar macrophages from inducible nitric oxide synthase knockout or wild-type mice to an in vitro lipopolysaccharide or silica exposure. Journal of Toxicology and Environmental Health Part A 66, 995–1013 (2003). [DOI] [PubMed] [Google Scholar]
- Hoeks J. et al. Prolonged fasting identifies skeletal muscle mitochondrial dysfunction as consequence rather than cause of human insulin resistance. Diabetes 59, 2117–2125 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleemann R. et al. Time-resolved and tissue-specific systems analysis of the pathogenesis of insulin resistance. PloS one 5, e8817 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisoli E. et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299, 896–899 (2003). [DOI] [PubMed] [Google Scholar]
- Chavin K. D. et al. Obesity induces expression of uncoupling protein-2 in hepatocytes and promotes liver ATP depletion. Journal of Biological Chemistry 274, 5692–5700 (1999). [DOI] [PubMed] [Google Scholar]
- Brand M. D. & Nicholls D. G. Assessing mitochondrial dysfunction in cells. Biochemical Journal 435, 297–312 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leary S. C. & Shoubridge E. Mitochondrial biogenesis: which part of “NO” do we understand? Bioessays 25, 538–541 (2003). [DOI] [PubMed] [Google Scholar]
- Monti L. D. et al. L-arginine enriched biscuits improve endothelial function and glucose metabolism: a pilot study in healthy subjects and a cross-over study in subjects with impaired glucose tolerance and metabolic syndrome. Metabolism 62, 255–264 (2013). [DOI] [PubMed] [Google Scholar]
- Jiang H. et al. Dietary nitrite improves insulin signaling through GLUT4 translocation. Free radical biology and medicine 67, 51–57 (2014). [DOI] [PubMed] [Google Scholar]
- Ohtake K. et al. Dietary nitrite supplementation improves insulin resistance in type 2 diabetic KKA y mice. Nitric Oxide 44, 31–38 (2015). [DOI] [PubMed] [Google Scholar]
- Jobgen W. et al. Dietary L-arginine supplementation reduces white fat gain and enhances skeletal muscle and brown fat masses in diet-induced obese rats. The Journal of nutrition jn. 108.096362 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemmensen C., Madsen A. N., Smajilovic S., Holst B. & Bräuner-Osborne H. L-Arginine improves multiple physiological parameters in mice exposed to diet-induced metabolic disturbances. Amino acids 43, 1265–1275 (2012). [DOI] [PubMed] [Google Scholar]
- McKnight J. R. et al. Beneficial effects of L-arginine on reducing obesity: potential mechanisms and important implications for human health. Amino acids 39, 349–357 (2010). [DOI] [PubMed] [Google Scholar]
- Jyoti A. et al. Interaction of inducible nitric oxide synthase with Rac2 regulates reactive oxygen and nitrogen species generation in the human neutrophil phagosomes: Implication in microbial killing. Antioxidants & redox signaling 20, 417–431 (2014). [DOI] [PubMed] [Google Scholar]
- Dubey M. et al. L-plastin S-glutathionylation promotes reduced binding to β-actin and affects neutrophil functions. Free radical biology and medicine (2015). [DOI] [PubMed] [Google Scholar]
- Taicher G. Z., Tinsley F. C., Reiderman A. & Heiman M. L. Quantitative magnetic resonance (QMR) method for bone and whole-body-composition analysis. Analytical and bioanalytical chemistry 377, 990–1002 (2003). [DOI] [PubMed] [Google Scholar]
- Wang Y., Zheng Y., Nishina P. M. & Naggert J. K. A new mouse model of metabolic syndrome and associated complications. Journal of Endocrinology 202, 17–28 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowe J. E. et al. Metabolic phenotyping guidelines: assessing glucose homeostasis in rodent models. Journal of Endocrinology 222, G13–G25 (2014). [DOI] [PubMed] [Google Scholar]
- Baker F. J. & Silverton R. E. Introduction to medical laboratory technology. (Butterworth-Heinemann, 2014). [Google Scholar]
- Khanna V. et al. Cholesterol diet withdrawal leads to an initial plaque instability and subsequent regression of accelerated iliac artery atherosclerosis in rabbits. PloS one 8, e77037 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash P. et al. Silymarin ameliorates fructose induced insulin resistance syndrome by reducing de novo hepatic lipogenesis in the rat. European journal of pharmacology 727, 15–28 (2014). [DOI] [PubMed] [Google Scholar]
- Dubey M. et al. Nitric oxide-mediated apoptosis of neutrophils through caspase-8 and caspase-3-dependent mechanism. Cell Death & Disease 7, e2348 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers G. W. et al. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PloS one 6, e21746 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]