

Abstract
Statistical similarity analysis has been instrumental in elucidation of the voluminous microarray data. Genes with correlated expression profiles tend to be functionally associated. However, the majority of functionally associated genes turn out to be uncorrelated. One conceivable reason is that the expression of a gene can be sensitively dependent on the often-varying cellular state. The intrinsic state change has to be plastically accommodated by gene-regulatory mechanisms. To capture such dynamic coexpression between genes, a concept termed “liquid association” (LA) has been introduced recently. LA offers a scoring system to guide a genome-wide search for critical cellular players that may interfere with the coexpression of a pair of genes, thereby weakening their overall correlation. Although the LA method works in many cases, a direct extension to more than two genes is hindered by the “curse of dimensionality.” Here we introduce a strategy of finding an informative 2D projection to generalize LA for multiple genes. A web site is constructed that performs on-line LA computation for any user-specified group of genes. We apply this scoring system to study yeast protein complexes by using the Saccharomyces cerevisiae protein complexes database of the Munich Information Center for Protein Sequences. Human genes are also investigated by profiling of 60 cancer cell lines of the National Cancer Institute. In particular, our system links the expression of the Alzheimer's disease hallmark gene APP (amyloid-β precursor protein) to the β-site-cleaving enzymes BACE and BACE2, the γ-site-cleaving enzymes presenilin 1 and 2, apolipoprotein E, and other Alzheimer's disease-related genes.
Keywords: microarray, gene expression, protein complex, Saccharomyces cerevisiae, Alzheimer's disease
Microarray technologies enable simultaneous measurement of transcript abundance at the full-genome scale. The voluminous data generated under various conditions contain numerous messages about gene regulation and protein function. They are invaluable for deciphering the complex cellular circuitry. But the type of information distillable from a given expression database can vary substantially, depending on the computational/statistical/mathematical method applied (1–5). In this paper, we focus on a subtle pattern of coexpression that has become relatively easy to detect by means of the recently developed concept of “liquid association” (LA) (6).
Profile similarity is a concept underlying many microarray elucidation procedures. Consider a matrix with each row representing one gene and each column representing one condition. The jth value in the ith row shows the level of expression for gene i under condition j. The expression profile for a gene refers to the corresponding row in the matrix. Profile similarity can be measured by the correlation between two rows. It has been thought that genes with similar expression profiles are likely to be functionally associated. The encoded proteins may participate in the same pathway, form a common structural complex, or be regulated by the same mechanism.
Although coexpressed genes are likely to be functionally associated, the profiles of most functionally associated genes turn out to be uncorrelated. One reason is the high noise level of microarray data. Another explanation is that not all genes are regulated at the mRNA level. Yet a third possibility can be described in terms of LA. This more advanced concept of statistical association originates from the need to describe a situation as schematized in Fig. 1 Left, wherein two opposing trends between X and Y are displayed. The positive and negative correlations cancel each other out, rendering the overall correlation insignificant. It would be valuable to learn why and how the change of trend occurs. But for real data, such hidden trends are not easy to detect directly from the scatterplot of X and Y. To alleviate the difficulty, we look for additional variables that may be associated with the change of the trend. LA quantifies how well a candidate variable Z can be used for this purpose. There are two types of change. A positive value of LA indicates the change from a negative to positive correlation as the value of Z increases. A negative value of LA indicates just the opposite way of changing, from a positive to negative correlation. The adjective “liquid,” as opposed to “solid” or “steady,” depicts this subtle pattern of association between X and Y.
Fig. 1.

LA and PLA. (Left) Illustration of the concept of LA. X and Y are uncorrelated because the two opposing trends nullify each other. Low values of Z (green) are associated with the positive trend, whereas high values of Z (red) are associated with the negative trend. Z plays a mediator role. The LA score is negative in this case. For the gene expression application, each dot represents one condition under which the expression levels of genes X, Y, and Z are measured. (Right) Illustration of projection-based LA. The expression profiles for a group of p genes with n conditions can be viewed as n points in p-dimensional space; p = 4 is shown here with each blue axis representing one gene. High-dimensional data are difficult to visualize directly. Two projections (pink and yellow arrows) are sought so that the LA pattern can be revealed.
Why is the LA suitable for describing subtle coexpression patterns? First, many genes have multiple functions and their biological roles may be dependent on the often-varying cellular state. Second, two proteins engaged in a common process under some conditions may disengage and embark on activities of their own under other conditions. This fact implies that both the strength and the pattern of association between the expression profiles of X and Y may vary as the intrinsic cellular state changes. Third, if the cellular state change is correlated with the expression of a third gene Z, then the correlation change may be detected by conditioning on Z. Fourth, because the relevant state variable is often unknown, to find out which genes can act as candidate for a mediator Z, a genome-wide search is appropriate. However, this search is an insurmountable challenge if inspection of the gene expression activity were to be done by direct examination of the scatterplots by eye. Yeast, for example, has >6,000 genes. The total number of triplets would be >36 billion. The situation is even worse for studying human genes. Consequently, an easy-to-compute index of how likely one is to find a LA pattern is desirable. After some mathematical derivation, a formula of LA was given by Li (6), which turns out to be simple enough to serve the purpose.
The Stanford cell-cycle database (http://genome-www.stanford.edu/cellcycle) was used to show how some subtle gene regulation patterns in yeast could be found only by the LA method. One example reveals how the enzymes associated with the urea cycle/arginine biosynthesis are expressed to ensure proper metabolite flow along this metabolic pathway. In particular, the expression profiles of X = ARG2 (acetylglutamate synthase) and Y = CAR2 (ornithine aminotransferase) are uncorrelated. But after a genome-wide search for the LA score leaders had been conducted for this pair, an enzyme adjacent in the pathway, CPA2 (arginine-specific carbamoyl-phosphate synthase, large subunit), was found at the 8th place from the negative score end. The correlation between ARG2 and CAR2 changes from positive to negative as the expression of CPA2 increases. This change of correlation reflects well a remarkable cellular control on the influx and efflux of ornithine in response to the arginine demand. The high level of CPA2 indicates a cellular state ready for arginine biosynthesis. Under this state, we observe a negative correlation between ARG2 and CAR2. Up-regulation of ARG2 is concomitant with down-regulation of CAR2, thereby preventing the newly synthesized ornithine from leaving the urea cycle.
LA was further applied in a functional genomic study of the National Cancer Institute's anticancer drug screen (7). In all, 60 representative human cell lines from seven cancer types (lung, colon, melanoma, kidney, ovary, brain, and leukemia) were selected, and their responsiveness over a broad range of concentration for tens of thousands of anticancer compounds was tested. More recently, molecular characterization of these cell lines was made available by profiling thousands of genes with microarrays (http://dtp.nci.nih.gov/docs/cancer/cancer_data.html) (8–10). In correlating drug sensitivity profiles with gene expression, most drugs of known molecular mechanism turn out to be uncorrelated with their molecular-target genes. In ref. 7, LA is used to find candidate genes that intervene, confound, and weaken the drug–gene correlation.
The LA measure deals with only two genes. How to bypass this limitation in studies that involve multiple genes at one time? In this article, we take an informative low-dimension projection approach as schematized in Fig. 1 Right. First, the expressions of a group of p genes under n conditions are viewed as n points in a p-dimensional space. While we wish to visualize how the points are distributed, this is hard to do for p > 3. To sidestep the obstacle, a promising strategy is to project the data to a lower-dimensional space. The popular principal component analysis (PCA) uses the directions with the largest variance for projection. But because our goal is to reveal the LA pattern as clearly as possible after projection, we next develop a different formulation of informative projection.
Theory
Our theory is presented in terms of continuous random variables. Suppose all variables are standardized to have mean 0 and variance 1 so that the correlation between variables X and Y is equal to E(XY). With the presence of a third variable Z, we denote the conditional expectation E(XY|Z = z) by g(z). The overall correlation between X and Y, E(XY), is equal to Eg(Z). We regard g(z) as the coexpression measure between gene X and gene Y when gene Z is expressed at level z. The derivative g′(z) quantifies how g(z) varies as z increases. LA of X and Y with respect to Z is defined by LA(X, Y|Z) = Eg′(Z). A simple estimate of LA is available under the normal assumption for Z: LA(X, Y|Z) = E(XYZ). A normal score transformation on each gene profile is performed before analysis.
To extend LA for a group of p genes, let X denote a vector of p variables, X1,..., Xp, where each variable measures the expression level of one gene. A one-dimensional projection of X is a linear combination a′X = a1X1 + · · · + apXp with norm ∥a∥ = 1. For a 2D projection, we require that the two projection directions a, b be orthogonal to each other: a′b = a1b1 + · · · + apbp = 0. After projection, the liquid association between a′X and b′X mediated by Z becomes
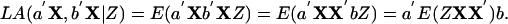 |
The most informative 2D projection for revealing the LA pattern can be found by maximizing |a′E(ZXX′)b| over any pair of orthogonal projection directions a, b.
This maximization can be done by eigenvalue decomposition on E(ZXX′):
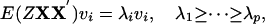 |
where vi are eigenvectors and λi are eigenvalues. The following theorem conveys the final result. The proof is given later.
Theorem. Assume that Z is normal with mean 0 and standard deviation 1. Subject to ∥a∥ = ∥b∥ = 1 and the orthogonal condition a′b = 0, the maximum for the absolute value of LA(a′X, b′X|Z) is equal to (λ1 - λp)/2. The optimal 2D projection directions are given by a = (v1 + vp)/√2 (or -a), b = (v1 - vp)/√2 (or -b).
The proper signs of a and b are determined in the following way. Let 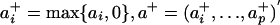 and let
and let 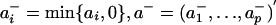 . If var(a+′X) ≥ var(a-′X), set sign(a) = 1 and keep a as the first projection direction; otherwise set sign(a) = -1 and use -a as the first projection direction. The sign of the second projection vector b is determined in the same way. The resulting liquid association is called the projection-based liquid association (PLA) for X mediated by Z and is denoted by PLA(X|Z).
. If var(a+′X) ≥ var(a-′X), set sign(a) = 1 and keep a as the first projection direction; otherwise set sign(a) = -1 and use -a as the first projection direction. The sign of the second projection vector b is determined in the same way. The resulting liquid association is called the projection-based liquid association (PLA) for X mediated by Z and is denoted by PLA(X|Z).
Proof: By eigenvalue decomposition, we put 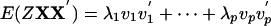 . Represent two candidate projection directions a, b as a = c1v1 + · · · + cpvp, b = d1v1 + · · · + dpvp under the constraints
. Represent two candidate projection directions a, b as a = c1v1 + · · · + cpvp, b = d1v1 + · · · + dpvp under the constraints
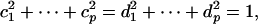 |
[1] |
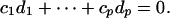 |
[2] |
Because LA(a′X, b′X|Z) = a′ E(ZXX′)b = c1d1λ1 + · · · + cpdpλp, we need to find the maximum of |c1d1λ1 + · · · + cpdpλp|. Denote 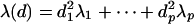 . Now, using constraint 2, we see that
. Now, using constraint 2, we see that
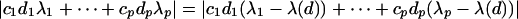 |
[3] |
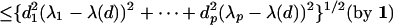 |
The last expression can be viewed as the standard deviation of a discrete random variable U with 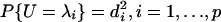 . To maximize the standard deviation, the probability mass has to be placed only on the endpoints λ1, λp. Thus we have
. To maximize the standard deviation, the probability mass has to be placed only on the endpoints λ1, λp. Thus we have 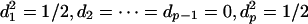 . Without lost of generality, we take d1 = 1/√2 = dp and return to inequality 3. We need only to maximize |c1 - cp|(λ1 - λp)/√2. Using Eq. 1, this can be achieved at c1 =√2, cp = -√2, c2 = · · · = cp-1 = 0. This proves the theorem.
. Without lost of generality, we take d1 = 1/√2 = dp and return to inequality 3. We need only to maximize |c1 - cp|(λ1 - λp)/√2. Using Eq. 1, this can be achieved at c1 =√2, cp = -√2, c2 = · · · = cp-1 = 0. This proves the theorem.
Method
To summarize what we have developed, for a group of p genes X = (X1,..., Xp)′ and a candidate mediator Z, the procedure of PLA comprises the following steps.
(i) Apply the normal score transformation to each gene profile.
(ii) For any two genes Xi, Xj in the group, compute the original LA score L(Xi, Xj|Z) = (Xi1Xj1Z1 + · · · + XimXjmZm)/m, where m denotes the total number of conditions and Xik denotes the expression of gene i under condition k.
(iii) Put the LA scores in a p by p matrix and use it to estimate E(ZXX′).
(iv) Conduct an eigenvalue decomposition on the matrix obtained in step iii to find the eigenvectors v1,..., vp and eigenvalues λ1 ≥ · · · ≥ λp.
(v) Let a = [v1 + vp]/√2 and b = [v1 - vp]/√2 and determine sign(a), sign(b) as in the discussion following the statement of the Theorem.
(vi) Set PLA(X|Z) = sign(a) sign(b)(λ1 - λp)/2.
We assess the statistical significance of the score PLA(X|Z) by comparing it to a reference distribution obtained by permutation. This reference distribution is given for each PLA output table in the supporting information. More precisely, for each group of genes X of interest, we generate a large number of artificial profiles Z* by randomly permuting (Z1,..., Zm). Then we evaluate each PLA(X|Z*) and pool the results to form the reference distribution. As usual, the p value can be determined by counting how often PLA(X|Z*) exceeds PLA(X|Z).
Results
Both yeast and human genes are studied. For yeast, we use protein complexes from the Saccharomyces cerevisiae-Protein Complexes database of the Munich Information Center for Protein Sequences (MIPS; http://mips.gsf.de/proj/yeast/catalogues/complexes/index.html). Two gene expression datasets are considered: the Stanford cell-cycle database (11) and a yeast segregation database generated by Brem et al. (12). For the human gene study, we use the cDNA gene expression database for the 60 cancer cell lines of the National Cancer Institute (8, 9).
Cytoplasmic Translation Initiation Complex eIF2. The Stanford cell-cycle data are used here. MIPS assigns three genes to this complex: SUI2, SUI3, and GCD11, encoding the α, β, and γ subunits of eukaryotic translation initiation factor eIF2. eIF2 acts by binding and delivering the initiator 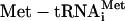 to the 40S ribosomal subunit in a GTP-dependent manner (13). The correlations between these three gene profiles are low: 0.37, 0.35, and 0.15, respectively, for (GCD11, SUI3), (GCD11, SUI2), and (SUI2, SUI3). Take them as X and apply PLA for a genome-wide search; the output is given in Table 1, which is published as supporting information on the PNAS web site. Among the top 20 genes with the best positive PLA scores, we find ribosome small subunits (RPS26A, and RPS23A), ribosome large subunit assembly and maintenance (RPL11B, RPL10, and DBP10), and rRNA processing genes (IFH1, and DBP10).
to the 40S ribosomal subunit in a GTP-dependent manner (13). The correlations between these three gene profiles are low: 0.37, 0.35, and 0.15, respectively, for (GCD11, SUI3), (GCD11, SUI2), and (SUI2, SUI3). Take them as X and apply PLA for a genome-wide search; the output is given in Table 1, which is published as supporting information on the PNAS web site. Among the top 20 genes with the best positive PLA scores, we find ribosome small subunits (RPS26A, and RPS23A), ribosome large subunit assembly and maintenance (RPL11B, RPL10, and DBP10), and rRNA processing genes (IFH1, and DBP10).
Fig. 3 Upper, which is published as supporting information on the PNAS web site, shows the optimal LA-projection as mediated by Z = RPS26A, whereas Fig. 3 Lower gives scatterplots between individual genes. A subtle coherent pattern of activation is revealed. When RPS26A is up-regulated (points coded in red triangles), we find high expression of SUI3 from Fig. 3 Lower Right and a positive correlation between GCD11 and SUI2 from Fig. 3 Lower Left. When RPS26A is down-regulated (points coded in blue diamonds), the coherence dissolves.
TIF4631, TIF4632, and CAF20. Stanford cell-cycle data are used here. These three genes participate in translation initiation complex eIF4F. TIF4631 and TIF4632 encode two similar proteins, which play a positive pole in translation (14). They bind to the mRNA cap-binding protein CDC33p. In contrast, CAF20, which encodes a small protein, p20, is a negative regulator of translation. It represses cap-dependent translation initiation through the competitive binding to CDC33p (15). We applied PLA to this group. The result is given in Table 2, which is published as supporting information on the PNAS web site. The gene with best negative PLA score is TIF5, which encodes the translation initiation factor eIF5.
Antagonistic Pattern in CAF20 and TIF4631 Expression. A closer examination of the expression pattern between TIF5, CAF20, and TIF4631 was undertaken. In Fig. 2 Lower Left, down-regulation of TIF5 (points coded in blue diamonds) is concomitant with low expression of CAF20. The TIF5-encoded protein eIF5 is required for the joining of the 60S ribosome subunit with the preinitiation complex to begin the translation (16). When the expression of TIF5 is low, cytoplasmic translation is likely to be less active. Cells do not express much CAF20 because of nothing to repress. In contrast, when the TIF5 expression is up (points coded in red color), a negative trend is visible between TIF4631 and CAF20 (Fig. 2 Lower Right). This result shows well the competitive roles between CAF20 and TIF4631 in translation initiation.
Fig. 2.

Translation initiation complex eIF4F. (Upper) The optimal LA projection as mediated by TIF5.(Lower Left) Down-regulation of TIF5 indicates weak CAF20 activity. (Lower Right) A negative trend between CAF20 and TIF4621 is observed when TIF5 is up-regulated, reflecting the antagonistic roles of CAF20 and TIF4621 in translation regulation.
In addition to TIF5, the list of PLA score leaders includes RPL30 (structural constituent of the ribosome), YTM1 (microtubule-associated protein, ribosomal large subunit biogenesis), PNO1, and RAP1. PNO1 encodes a component of the 90S preribosome, which is required for pre-18S rRNA processing (17). RAP1 (repressor activator protein) is involved in diverse processes. In its role as a transcription activator, the largest group of target genes is those that encode ribosomal proteins (18).
Kinetochore Protein Complex. The yeast segregation data generated by Brem et al. (12) are used in this example. MIPS puts nine genes in this group (CBF2, SKP1, CTF13, CEP3, CBF1, CSE4, MIF2, BIR1, and CBF5). Accurate chromosome segregation depends on a specialized chromosomal structure, the kinetochore/centromere. CBF2 is essential for chromosome segregation and movement of centromeres along microtubules. The SKP1, CBF2, CTF13, and CEP3 products form a multisubunit complex, which binds to the CDEIII domain of the centromere. CBF1 interacts with the CDEI domain of the centromere. CSE4, MIF2, and BIR1 are also involved in chromosome segregation. CBF5 encodes a major low-affinity 55-kDa centromere/microtubule-binding protein. CTF13 is excluded from analysis because its expression profile has >20% missing values. The correlations between genes in this group are mostly quite low (see Table 3, which is published as supporting information on the PNAS web site).
Table 4, which is published as supporting information on the PNAS web site, gives the PLA score leaders after the genome-wide search. Three leading genes, SFI1, SPC72, and FIN1 are localized in spindle pole body. SFI1 protein has conserved centrin-binding sites and an essential function in budding yeast spindle pole body duplication (19). FIN1 protein forms cell cycle-specific filaments between spindle pole bodies (20). SPC72 controls proper migration of the nucleus. It interacts with the microtubule-binding protein STU2 and participates in mitotic chromosome segregation (21).
We turn to the gene with the best negative PLA score, YFR044C (CNN1). This gene encodes a kinetochore protein copurified with NNF1p [Saccharomyces Genome Database (SGD) annotation], but its molecular function is still unknown. In contrast, NNF1p is better understood. It is a spindle pole protein required for accurate chromosome segregation (22). The correlation between the expression profiles of YFR044C and NNF1 is weak (0.34). This finding prompted us to apply the regular LA analysis to this pair. The gene with the best positive LA score turns out to be MSH3, a gene with the second-best positive score in the earlier PLA result given in Table 4. MSH3 protein forms a complex with MSH2 protein, which is active in mismatch repair and recombination (23). Interestingly, the gene that has the highest correlation (0.77) with MSH2 is STU2, and the binding partner of SPC72 is discussed earlier.
AP-1 Complex. The yeast segregation data are used. The AP-1 complex is a heterotetramer composed of two large subunits (APL2 and APL4), one medium subunit (APM1), and one small subunit (APS1). Clathrin-coated vesicles budding from the trans-Golgi network interact with AP-1 complex. The correlations among these four genes are low (see Table 5, which is published as supporting information on the PNAS web site). The output of applying PLA is given in Table 6, which is published as supporting information on the PNAS web site. Leading on the positive-score side is ENT4. The ENT4 protein contains the epsin N-terminal homology (ENTH) domain, which is essential in clathrin-dependent endocytosis (24). To help the study of other genes in the output, we submitted them to the Gene Ontology Term Finder in SGD for an automatic analysis. From the enriched terms in branch of the biological process, we found END3 (endocytosis, polar budding), SSO1 (Golgi to plasma membrane transport; nonselective vesicle fusion), CHS5 (Golgi to plasma membrane transport, spore-wall assembly), BUD7 (bud site selection; clathrin-coated vesicle); SEC61 (protein–endoplasmic reticulum targeting), SRP54 (protein–endoplasmic reticulum targeting), APL2, and ENT4.
Other Complexes. We have constructed a web site (http://kiefer.stat.ucla.edu/LAP) that gives the standard correlation and PLA results for each complex listed in the Saccharomyces cerevisiae-Protein Complexes database of MIPS. Researchers can quickly browse the results for the complex of interest to them. A gene ontology (GO) term summary table is provided for the output genes along with clickable buttons for submitting to GO term Finder of SGD. For instance, the SPB (spindle pole body) component complex_205 consists of 16 genes. Using Stanford cell cycle data, we remove two genes because of abundant missing values. The rest of them are used as a group for PLA application. The GO analysis on the 20 genes with the best negative PLA scores shows 7 genes in “cell proliferation” (P = 0.00065). Among them, five genes are in “cell cycle”: SWI5, CDC39, SET3, SNT1, and CLB2.
As another example, GO term Finder shows that the PLA output for the translation elongation eEF1 complex_225 (consisting of six genes) has nine genes in the cellular component “ribosome” (P value 0.00002). They are RPL9B, RPL21A, RPS9B, CDC19, RPL23A, RPS23B, RPS11B, RPS6B, and RPL14B.
Genetic Markers. In the yeast segregation experiment by Brem et al. (12), the genetic marker profiles were also available. There are in total 3,312 markers, which cover >99% of the genome. A marker profile keeps track of the parental strains from which each of the 40 offspring inherits the marker. Our web site allows for treating them as Z and applying the same procedure as described in Method to compute the PLA scores. For instance, use the translation elongation eEF1 complex as X and specify the genetic marker files as Z. From the PLA output given in Table 7, which is published as supporting information on the PNAS web site, we find two markers located at genes RPL28 and RPL24B that function as ribosome structural constituents and another two markers located at two genes participating in ribosome biogenesis: NSA1 (constituent of 66S preribosomal particle, involved in 60S ribosomal unit biogenesis) and MTR3 (35S primary transcript processing). Because marker profiles are highly correlated with the chromosome positions of the markers, many neighboring markers appear in Table 7. RPL28 and NSA1 fall within a block of four markers, YGL101W, YGL103W (RPL28), YGL110C, and YGL111W (NSA1), whereas RPL24B and MTR3 embrace a block of five markers: YGR148C (RPL24B), YGR149W, YGR150C, YGR157W, YGR158C (MTR3). According to both MIPS and SGD, a MTR3 mutant strain has defects in rRNA synthesis/processing. For our data, MTR3 is the gene having the third-strongest negative correlation with the MTR3 marker profile; see Table 8, which is published as supporting information on the PNAS web site.
Tumor Suppressor p53. The well known human tumor suppressor p53 is encoded by the gene TP53. A keyword query on TP53 produces four hits in our database: TP53, TP53INP1 (P53-inducible nuclear protein), TPBP1 (p53-binding protein 1), and TPBP2 (p53-binding protein 2). The correlations between these genes are low, falling between -0.0805 and 0.1904. We applied PLA to this group; see the output Table 9, which is published as supporting information on the PNAS web site. We found SMARCA4 (SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a, member 4) at fourth place on the negative score side. The p value is far below 0.1%. The coefficients for the two projection directions are 0.97, 0.045, -0.015, and 0.236, and -0.090, 0.722, 0.629, and 0.2716, respectively. Because the weights on TPBP2 (0.236 and 0.2716) are the lowest, we drop it from the group and apply PLA again to the three remaining genes. We find SMARCA4 at the first place. By using genetic and biochemical approaches, SMARCA4 was shown to interact with tumor suppressor p53; SWI/SNF complex is necessary for the activation of p53-mediated transcription (25).
Alzheimer's Disease (AD) Genes. Amyloid-β peptide is the predominant component of senile plaques in the brains of patients with AD. It is derived from the amyloid-β precursor protein (APP) by the consecutive proteolytic cleavage by β-secretase at the N terminus and by γ-secretase at the C terminus. APP is a widely expressed cell surface protein. Its normal role was linked to the control of gene expression in ref. 26, where the C-terminal intracellular fragment of APP was found to interact with the nuclear adaptor protein Fe65 (encoded by APBB1) and the histone acetyltransferase Tip60 (encoded by HTATIP). We compared the profiles of APBB1 and HTATIP with the profile of APP and found that the correlations (-0.06 and -0.27, respectively) are quite low. In search of genes that may play a role in weakening the correlation, we first applied the ordinary LA to the pair APP and PBB1. We found a β-site APP-cleaving enzyme BACE2 from the best 20 genes with negative LA scores; see Table 10, which is published as supporting information on the PNAS web site. We then applied LA again to the pair APP and HTATIP. This time we found a major component of γ-secretase PSEN1 (presenilin 1) to be in second place in the best positive LA scores; see Table 11, which is published as supporting information on the PNAS web site.
Then we took APP, PSEN1, PSEN2, APBB1, and APBB2 as a group and applied PLA; see Table 12, which is published as supporting information on the PNAS web site. In addition to recovering BACE2, we found CTSB (cathepsin B) and APOD (apolipoprotein D) from our short list of PLA-score leaders. Cathepsin B, a lysosomal cysteine proteinase also known as amyloid precursor protein secretase, is found elevated in the amyloid plaques of AD brains. On the other hand, apolipoprotein D (ApoD), a component of high-density lipoprotein, is elevated in association with several central nervous system disorders, including AD. ApoD has been proposed to be an especially robust marker for brain regions specifically affected by particular neuropathologies (27).
This connection brought our attention to apolipoprotein E (ApoE), which has been implicated in the pathology of AD ever since inheritance of the ε4 allele was shown to be an important risk factor for the development of AD. The expression profiles of APOE and APOD were again found to be uncorrelated. So we applied the ordinary LA to this pair and found APOC1 in our list of LA-score leaders; see Table 13, which is published as supporting information on the PNAS web site. APOC1 is located adjacent to APOE on chromosome 19. Applying the ordinary LA again to the pair APOE and APOC1, we found FE65L2 (amyloid-β precursor protein binding family B, member 3); see Table 14, which is published as supporting information on the PNAS web site. We also used ordinary LA for the pair APBB1 and APOE and found BACE (β-site APP-cleaving enzyme 1) and APOB (apolipoprotein B); see Table 15, which is published as supporting information on the PNAS web site.
Discussion
Coexpression of functionally associated genes can be dependent on the often-varying cellular state. To survive, living organisms have developed a plastic gene-regulatory mechanism to accommodate/facilitate the inherent state change. This mechanism results in subtle gene expression patterns hard to recognize by standard similarity analysis based on correlation. LA and its higher-dimension generalization emerge as analytic tools for investigating the dynamic nature of coexpression at the genome-wide scale. The method bypasses the need to specify the cellular state in the first place.
Historically, Fisher and Yates (see ref. 28) had advocated the use of normal score transformation before applying Pearson correlation to gain robustness in data with a nonnormal distribution. Motivated from a different consideration and fueled by Stein's lemma (48), LA also uses the normal score transformation. Algebraically, LA appears to be a natural three-variable extension of the Fisher–Yates modification of Pearson correlation. However, a further extension of LA by considering the average product of four (or more) gene profiles is not pursued here because a lot more samples will be needed to ensure the stability of higher-moment statistics. Such difficulty of extending a statistical procedure from the low- to the high-dimensional situation is generally referred to as “the curse of dimensionality” in the statistical literature (29).
We took the approach of informative projection to bypass the hurdle. Given a group X of p gene expression profiles and a candidate mediator gene Z, we looked for an optimal 2D projection for revealing the LA pattern as clearly as possible. Through a theorem we provide, the optimal projection is easy to find. It involves an eigenvalue decomposition of a p by p matrix E(ZXX′) consisting of the original pair-wise LA scores. As in the original LA, the simplicity of PLA allows for a speedy full-genome evaluation to find a short list of PLA-score-leading genes Z.
We demonstrated how the PLA can be applied to study protein complexes in yeast provided by MIPS. Our examples include complexes for translation initiation, elongation, protein transportation, and chromosome segregation. We are able to find functionally associated genes that mediate changes in the coexpression pattern of the complex. We discuss the biological relevance of some PLA-score leaders, including gene YFR046C (CNN1) for the kinetochore complex, gene ENT4 for the AP-1 complex, and gene TIF5 for revealing an antagonistic pattern in TIF4631 and CAF20 expression. The functional relevance of TIF5 is confirmed by a recent report (30) showing that eIF5 (the TIF5-encoded protein) interacts with a distinct HEAT domain of yeast eIF4G (product of TIF4631 and TIF4632).
We have constructed a web site that offers on-line analysis of gene-expression data. The system integrates standard correlation, LA, and PLA analyses under a common forum. We gave an example to show how our system can shed light on the expression networks of important genetic diseases. In our study of the AD gene APP, our system reveals the involvement of the β- and the γ-secretases as well as other AD-related genes at the gene-expression level. As reviewed in ref. 31, the four genes APP, PSEN1, PSEN2, and APOE, which are definitively linked to inherited forms of AD, have been shown to increase the production and/or deposition of amyloid-β in the brain. They are important biochemical targets for drug screening and therapeutic development.
Our method contributes to the understanding about the biological roles of human genes, of which the vast majority are still poorly understood. Genes appearing together as the LA or PLA score leaders are likely to be functional associated. For example, consider the gene RAB1A (a member of the RAS oncogene family) that appears in our Table 12 given earlier for PLA results in the AD study. RAB proteins are thought to regulate the targeting and fusion of membranous vesicles during organelle assembly and transport. From the same table, we find TC10 (also known as ARHQ), a member of the RAS superfamily of small GTP-binding proteins involved in insulin-stimulated glucose uptake; MGC46235, tubulin tyrosine ligase (TTL) involved in the posttranslational modification of tubulin; and MYO10, myosin X. Using a recently developed functional screening system, Komano et al. (32) found that RAB1A protein can act as a regulator of amyloid-β generation.
Most recently, Phiel et al. (33) showed that GSK3A (glycogen synthase kinase 3 α) regulates production of AD amyloid-β peptides. They noted that GSK3A also phosphorylates the tau protein (MAPT), the principal component of neurofibrillary tangles in AD, and suggested that inhibition of GSK3A may offer a new therapeutic approach to AD. We conducted LA analysis for the pair APP and GSK3A. From the output Table 16, which is published as supporting information on the PNAS web site, we find several genes that have already appeared in Table 12: BACE2, LARGE, TCF3, APC4, SPS, CXCL14, DCLRE1A, NIP30, and MGC40414.
Our system also offers a few variants in computation that might be used for other exploratory purpose. One option is to replace the constraints |a| = |b| = 1 and a′b = 0byvar(a′X) = var(b′X) = 1 and cov(a′X, b′X) = 0. Another option is useful for dealing with two distinct groups X, Y of genes. The objective is to find one projection a for X and one projection b for Y that maximizes the absolute value of the projected LA score. This can be done by using the singular value decomposition of E(ZXY′) to maximize a′E(ZXY′) b, subject to |a| = |b| = 1. The first pair of singular vectors will serve for this purpose. We also offer an option that uses the constraint var(a′X) = var(b′X) = 1.
In our web site, all datasets are maintained by the relational database software mysql (34). The intercommunication between the server and the database is powered by php (35). Users can specify a set of X, Y, and Z profiles by keywords, chromosome locations, and gene or drug names. High-score genes are returned to the user's browser for immediate connection to Locus Link or SGD. Our system is located at http://stat.ucla.edu/kiefer/LAP. The current server is a Mac G4 (1-GHz dual processor), which takes about 2 sec to return results for a query with two gene profiles as X, two gene profiles as Y, and 9,706 gene profiles (cell-line data) as Z. For the eigenvalue decomposition, we use C functions from Numerical Recipe (36) and integrate them into mysql and php. For a query on a yeast complex with 25 genes, it takes less than 1 min to evaluate all PLA scores and find the PLA-score-leading genes.
One issue that merits further investigation is the biological relevance of other genes with equally high PLA scores, which we did not discuss. Recall that the assumption behind our method is the existence of a hidden cellular state that governs the coexpression of a group of genes under study. In each example, we reported a selective set of genes whose activities best represent the hidden cellular state, given the current limited knowledge on gene functions. In Tables 17–22, which are published as supporting information on the PNAS web site, we show that the majority of other unreported genes are likely to be coregulated in response to the same cellular state change characterized by the reported genes. In the case of cytoplasmic translation initiation complex eIF2, we have reported six genes (RPS26A, RPS23A, RPL11B, RPL10, DBP10, and IFH1) involved in the making of cytoplasmic ribosome. Two unreported genes, TGS1 and ARC1, also have apparent roles associated with the translation mechanism. ARC1 participates in tRNA aminoacylation for protein translation and in exporting tRNA from nucleus to cytoplasm. TSG1 (small nuclear RNA/small nucleolar RNA cap hypermethylase) plays a role in the maturation of pre-mRNAs and pre-rRNA (37). Thus collectively, up-or-down regulation of these eight genes may point to a cellular state change about the rate of translation activity. The rest of the genes in Table 1 are either unknown or functionally diverse. However, we find a broadly consistent pattern of correlation between them and the eight representative genes; see Table 17. This correlation suggests that many of these genes are likely to be coregulated in response to the cellular state change. For example, COG2 (endoplasmic reticulum-to-Golgi transport, intra-Golgi transport, retrograde transport; protein binding), and ARL1 (protein-vacuolar targeting) may be indicative of the supportive mechanism required immediately after translation. Indeed, genetic interaction studies have shown that continued functioning of the secretory pathway is essential for ribosome synthesis (38, 39). Tables 18–20 provide similar results for the other three yeast complexes.
For the p53 group, in addition to SMARCA4, we find four other genes, CAV1, NRG1, PDCD2, and CITED2, from Table 9. The correlation between these genes and others shows a consistent pattern of coregulation (Table 21). CAV1 (caveolin-1) mediates cell cycle arrest through a p53/p21-dependent pathway (40). NRG1 (neuregulin 1) activates a p53-dependent pathway in cancer cells (41). The human programmed cell death-2 (PDCD2) gene is a target of BCL6 repression (42), whereas the disruption of the p53 pathway affects the development of BCL6-expressing B cell lymphomas (43). CITED2 encodes a CBP-p300-interacting transactivator, and CBP-p300 is involved in mediating p53 degradation (44).
For the AD gene group, Table 22 shows a consistent pattern of coregulation for BACE2, CTSB, APOD, RAB1A, and GAB2 with other genes. GAB2, encoding a GRB2-associated binding protein, is included because Russo et al. (45) showed that in human brain, tyrosine-phosphorylated C-terminal fragments of APP form stable complexes with the adaptor proteins ShcA and GRB2. Three genes, SNX9, ADAM9, and ADAM15, from Tables 13, 14, and 16, respectively, are also noteworthy. Sorting nexin-9 (SNX9) interacts with the cytoplasmic domains of metalloprotease disintegrins ADAM9 and ADAM15 (46). Recently, Asai et al. (47) determined that ADAM9 displays the APP α-secretase activity.
Supplementary Material
Acknowledgments
This work was funded in part by National Science Foundation Grants 0104038 and 0201005.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: LA, liquid association; PLA, projection-based LA; MIPS, Munich Information Center for Protein Sequences; SGD, Saccharomyces Genome Database; AD, Alzheimer's disease; APP, amyloid-β precursor protein.
References
- 1.Eisen, M. B., Spellman, P. T., Brown, P. O. & Botstein, D. (1998) Proc. Natl. Acad. Sci. USA 95, 14863-14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tamayo, P., Slonim, D., Mesirov, J., Zhu, Q., Kitareewan, S., Dmitrovsky, E., Lander, E. S. & Golub, T. R. (1999) Proc. Natl. Acad. Sci. USA 96, 2907-2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alter, O., Brown, P. O. & Botstein, D. (2000) Proc. Natl. Acad. Sci. USA 97, 10101-10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marcotte, E. M., Pellegrini, M., Thompson, M. J., Yeates, T. O. & Eisenberg, D. (1999) Nature 402, 83-86. [DOI] [PubMed] [Google Scholar]
- 5.Zhou, X., Kao, M. C. & Wong, W. H. (2002) Proc. Natl. Acad. Sci. USA 99, 12783-12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li, K.-C. (2002) Proc. Natl. Acad. Sci. USA 99, 16875-16880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li, K.-C. & Yuan, S. (2004) Pharmacogenomics J. 4, 127-135. [DOI] [PubMed] [Google Scholar]
- 8.Ross, D. T., Scherf, U., Eisen, M. B., Perou, C. M., Rees, C. R., Spellman, P., Iyer, V., Jeffrey, S. S., Van de Rijn, M., Waltham, M., et al. (2000) Nat. Genet. 24, 227-235. [DOI] [PubMed] [Google Scholar]
- 9.Scherf, U., Ross, D. T., Waltham, M., Smith, L. H., Lee, J. K., Tanabe, L., Kohn, K. W., Reinhold, W. C., Myers, T. G., Andrews, M. D., et al. (2000) Nat. Genet. 24, 236-244. [DOI] [PubMed] [Google Scholar]
- 10.Staunton, J. E., Slonim, D. K., Coller, H. A., Tamayo, P., Angelo, M. J., Park, J., Scherf, U., Lee, J. K., Reinhold, W. O., Weinstein, J. N., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 10787-10792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spellman, T. P., Sherlock, G., Zhang, Q. M., Iyer, R. V., Anders, K., Eisen, B. M., Brown, O. P., Botstein, D. & Futcher, B. (1998) Mol. Biol. Cell 9, 3273-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brem, R., Yvert, G., Clinton, R. & Kruglyak, L. (2002) Science 296, 752-755. [DOI] [PubMed] [Google Scholar]
- 13.Nika, J., Rippel, S. & Hannig, E. M. (2001) J. Biol. Chem. 276, 1051-1056. [DOI] [PubMed] [Google Scholar]
- 14.Ramirez, C. V., Vilela, C., Berthelot, K. & McCarthy, J. E. (2002) J. Mol. Biol. 318, 951-962. [DOI] [PubMed] [Google Scholar]
- 15.Ptushkina, M., von der Haar, T., Vasilescu, S., Frank, R., Birkenhäger, R. & McCarthy, E. G. J. (1998) EMBO J. 17, 4798-4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Das, S. & Maitra, U. (2001) Prog. Nucleic Acid Res. Mol. Biol. 70, 207-231. [DOI] [PubMed] [Google Scholar]
- 17.Senapin, S., Clark-Walker, G. D., Chen, X. J., Seraphin, B. & Daugeron, M. C. (2003) Nucleic Acids Res. 31, 2524-2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lieb, J. D., Liu, X., Botstein, D. & Brown, P. O. (2001) Nat. Genet. 28, 327-334, and erratum (2001) 29, 100. [DOI] [PubMed] [Google Scholar]
- 19.Kilmartin, J. V. (2003) J. Cell Biol. 162, 1211-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Hemert, M. J., Deelder, A. M., Molenaar, C., Steensma, H. Y. & van Heusden, G. P. (2003) J. Biol. Chem. 278, 15049-15055. [DOI] [PubMed] [Google Scholar]
- 21.Usui, T., Maekawa, H., Pereira, G. & Schiebel, E. (2003) EMBO J. 22, 4779-4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Euskirchen, G. M. (2002) Eukaryot. Cell 1, 229-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saparbaev, M., Prakash, L. & Prakash, S. (1996) Genetics 142, 727-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wendland, B., Steece, K. E. & Emr, S. D. (1999) EMBO J. 18, 4383-4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee, D., Kim, J. W., Seo, T., Hwang, S. G., Choi, E. J. & Choe, J. (2002) J. Biol. Chem. 277, 22330-22337. [DOI] [PubMed] [Google Scholar]
- 26.Cao, X. & Südhof, T. C. (2001) Science 293, 115-120, and erratum (2001) 293, 1436. [DOI] [PubMed] [Google Scholar]
- 27.Thomas, E. A., Laws, S. M., Sutcliffe, J. G., Harper, C., Dean, B., McClean, C., Masters, C., Lautenschlager, N., Gandy, S. E. & Martins, R. N. (2003) Biol. Psychiatry 54, 136-141. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez, R. N. (1982) in Encyclopedia of Statistical Sciences, eds. Kotz, S. & Johnson, N. L. (Wiley, New York), Vol 2, pp. 193-204. [Google Scholar]
- 29.Hubert, P. (1985) Ann. Stat. 13, 435-526. [Google Scholar]
- 30.He, H., von der Haar, T., Singh, C. R., Ii, M., Li, B., Hinnebusch, A. G., McCarthy, J. E. & Asano, K. (2003) Mol. Cell. Biol. 23, 5431-5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Selkoe, D. J. (2001) Physiol. Rev. 81, 741-766. [DOI] [PubMed] [Google Scholar]
- 32.Komano, H., Shiraishi, H., Kawamura, Y., Sai, X., Suzuki, R., Serneels, L., Kawaichi, M., Kitamura, T. & Yanagisawa, K. (2002) J. Biol. Chem. 277, 39627-39633. [DOI] [PubMed] [Google Scholar]
- 33.Phiel, C. J., Wilson, C. A., Lee, V. M. & Klein, P. S. (2003) Nature 423, 435-439. [DOI] [PubMed] [Google Scholar]
- 34.Axmark, D., Widenius, M. M., Cole, J. & DuBois, P. (1997) MySQL Reference Manual (MySQL, Uppsala).
- 35.Sklar, D. & Trachtenberg, A. (2003) PHP Cookbook (O'Reilly, Sebastopol, CA)
- 36.Press, H. W., Teukolsky, A. S., Vetterling, T. W. & Flannery, P. B. (2002) Numerical Recipes in C (Cambridge Univ. Press, New York), 2nd Ed.
- 37.Mouaikel, J., Bujnicki, J., Tazi, J. & Bordonne, R. (2003) Nucleic Acids Res. 31, 4899-4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mizuta, K. & Warner, J. R. (1994) Mol. Cell. Biol. 14, 2493-2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyoshi, K., Miyakawa. T. & Mizuta, K. (2001) Nucleic Acids Res. 29, 3297-3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galbiati, F., Volonte, D., Liu, J., Capozza, F., Frank, P. G., Zhu, L., Pestell, R. G. & Lisanti, M. P. (2001) Mol. Biol. Cell 12, 2229-2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bacus, S. S., Yarden, Y., Oren, M., Chin, D. M., Lyass, L., Zelnick, C. R., Kazarov, A., Toyofuku, W., Gray-Bablin, J., Beerli, R. R., et al. (1996) Oncogene 12, 2535-2547. [PubMed] [Google Scholar]
- 42.Baron, B. W., Anastasi, J., Thirman, M. J., Furukawa, Y., Fears, S., Kim, D. C., Simone, F., Birkenbach, M., Montag, A., Sadhu, A., Zeleznik-Le, N. & McKeithan, T. W. (2002) Proc. Natl. Acad. Sci. USA 99, 2860-2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kusam, S., Vasanwala, F. H. & Dent, A. L. (2004) Oncogene 23, 839-844. [DOI] [PubMed] [Google Scholar]
- 44.Matt, T., Martinez-Yamout, M. A., Dyson, H. J. & Wright, P. E. (2004) Biochem. J. 381, 685-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Russo, C., Dolcini, V., Salis, S., Venezia, V., Zambrano, N., Russo, T. & Schettini, G., (2002) J. Biol. Chem. 277, 35282-35288. [DOI] [PubMed] [Google Scholar]
- 46.Howard, L., Nelson, K. K., Maciewicz, R. A. & Blobel, C. P. (1999) J. Biol. Chem. 274, 31693-31699. [DOI] [PubMed] [Google Scholar]
- 47.Asai, M., Hattori, C., Szabo, B., Sasagawa, N., Maruyama, K., Tanuma, S. & Ishiura, S. (2003) Biochem. Biophys. Res. Commun. 301, 231-235. [DOI] [PubMed] [Google Scholar]
- 48.Stein, C. (1981) Ann. Stat. 9, 1135-1151. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}