

Abstract
The novel immune-type receptor (NITR) genes encode a unique multigene family of leukocyte regulatory receptors, which possess an extracellular Ig variable (V) domain and may function in innate immunity. Artificial chromosomes that encode zebrafish NITRs have been assembled into a contig spanning ≈350 kb. Resolution of the complete NITR gene cluster has led to the identification of eight previously undescribed families of NITRs and has revealed the presence of C-type lectins within the locus. A maximum haplotype of 36 NITR genes (138 gene sequences in total) can be grouped into 12 distinct families, including inhibitory and activating receptors. An extreme level of interindividual heterozygosity is reflected in allelic polymorphisms, haplotype variation, and family-specific isoform complexity. In addition, the exceptional diversity of NITR sequences among species suggests divergent evolution of this multigene family with a birth-and-death process of member genes. High-confidence modeling of Nitr V-domain structures reveals a significant shift in the spatial orientation of the Ig fold, in the region of highest interfamily variation, compared with Ig V domains. These studies resolve a complete immune gene cluster in zebrafish and indicate that the NITRs represent the most complex family of activating/inhibitory surface receptors thus far described.
Large families of activating and inhibitory leukocyte regulatory receptors have been characterized in human and mouse (1–5) and effect a number of functions relating to innate immunity as well as the regulation of cell–cell interactions. The mammalian Fc and natural killer (NK) receptors are the best characterized in terms of function of the activating/inhibitory receptor multigene families (1); additional families of these receptors also have been described (5). The NK receptors are the most genetically diverse and include the killer Ig-type receptors (KIRs), which are encoded in a continuous gene complex on chromosome 19 in human, and the C-type lectin Ly49 receptor genes, which are encoded on chromosome 6 in mouse (6). NK receptors effect self/nonself recognition through interactions with MHC I or MHC I-related products (2–4). Although KIRs and Ly49s are encoded by unrelated gene families and possess structurally distinct ectodomains, they both effect similar functions and use shared mechanisms of intracellular signaling.
Both Ig-(7) and C-type lectin (8, 9) receptors, which share some structural features with mammalian NK receptors, have been identified in several different genera of bony fish (10). The Ig-type receptors are encoded by novel immune-type receptor (NITR) genes and typically possess Ig variable (V) and intermediate (I) ectodomains (11), transmembrane regions, and cytoplasmic immunoreceptor tyrosine-based inhibition motifs (ITIMs) (12–14). Putative activating forms are structurally similar but contain a positively charged amino acid residue within the transmembrane region (7, 14). Several families of NITRs are expressed abundantly in NK lines in bony fish that exhibit alloreactive specificity (14, 15).
At this point, there is no comprehensive understanding of the actual numbers of NITR genes in a single species, the composition of the chromosomal locus/loci at which they are encoded, their overall organization, patterns of diversity, and the relationship of variation in NITR structure to that of the other Ig receptors that mediate immune recognition. The developmental genetics and other unique features of the zebrafish suggest that it is an ideal model for understanding the function of novel genes such as the NITRs. The enormous level of complexity, as well as the genetic, signaling, and other structural findings described here, are entirely consistent with a role for these genes in the recognition of diverse determinants.
Materials and Methods
Sequencing, Mapping, and Analysis of Gene Transcripts. P1 artificial chromosome (PAC) screening, RACE, cDNA sequencing, identification of sequence-tagged site (STS) markers, and radiation hybrid mapping were carried out as described (13, 14). Partial mapping and sequencing of PACs (AB strain) and bacterial artificial chromosomes (BACs) (Tübingen strain) were conducted through the Zebrafish Genome Project at the Sanger Institute, Cambridge, United Kingdom (www.sanger.ac.uk/Projects/D_rerio). The Otter system was used to manually annotate sequences based on publicly available protein, cDNA, and EST resources by using blast (www.ncbi.nlm.nih.gov/blast) (16). acedb (www.acedb.org) was used to visualize the gathered information and to manually create gene structures based on this supporting evidence (17).
Analysis of Activating Function. A nitr9-L cDNA was subcloned into pLF, which is derived from pcDNA3 (Invitrogen) and introduces an amino-terminal peptide signal sequence (from zebrafish Nitr3a) and a FLAG epitope tag (13). Transfection through recombination of the viral vector SP11 with the WR strain of vaccinia virus (18), fluorescence-activated cell sorter (FACS) analysis of FLAG-tagged NITRs (13), and redirected cytolysis of the P815 (19) target have been described previously. A human DAP12 cDNA (a gift from E. Vivier, Université de la Méditerranée, Marseille-Luminy, France) was subcloned into pLF and shuttled into pSP11 to generate a wild-type (wtDAP12) viral vector. A mutant form of DAP12 (mutDAP12) was generated by mutating (boldface) the amino-terminal tyrosine of the immunoreceptor tyrosine-based activating motif (ITAM) (ESPYQELQGQRSDVYSDL) to an alanine (ESPAQELQGQRSDVYSDL) with a site-directed mutagenesis kit (QuikChange, Stratagene).
Comparative Modeling of NITR V Domains. Theoretical models of NITR V domains were generated by using the programs implemented by the automated protein homology-modeling server SWISS-MODEL (http://swissmodel.expasy.org) (20). The rms deviations and distances were calculated with lsqkab by using Collaborative Computational Project Number 4 (ccp4) (21). Figures were generated with grasp (21).
Results and Discussion
The NITR Gene Cluster Is Composed of Multiple Families. Four families of NITR genes were described previously in zebrafish: nitr1, nitr2, nitr3, and nitr4 (13). Probes complementing these individual families were used to screen a high-density filter array of a zebrafish genomic PAC library that was generated from pooled blood of ≈200 AB strain zebrafish (22). Positively hybridizing PAC insert regions were fingerprinted, and those PACs judged to be informative were submitted to high-throughput sequencing as part of the Zebrafish Genome Project. A single BAC (DKEY-7N10), from the Tübingen strain of zebrafish, was identified through sequence overlap within the Zebrafish Genome Project database. By using this approach, eight additional families of NITRs as well as genes and other sequences that are associated with the NITR gene cluster were identified through blast analysis, exon/intron boundary and transmembrane prediction, directed searches for short NITR sequence motifs and, in some cases, specific cDNA recovery/isolation using RACE. In total, 138 gene sequences define 36 individual genes that, based on V-region sequences, distribute into 12 families. The previously described nitr3 and nitr3-related (nitr3r) genes (13) group together and collectively define the nitr3 family. Table 1, which is published as supporting information on the PNAS web site, presents the revised nomenclature for the previously published zebrafish NITR cDNAs. To date, the genome sequencing project has produced whole-genome shot-gun assemblies with an estimated coverage of 97% as well as BAC and PAC sequences totaling >1 Gb. Within these available sequences, it has not been possible to define additional NITRs belonging to known families or to identify additional NITR families. If additional NITR genes are present, their sequences vary significantly from known NITRs and/or they are not linked closely with known NITRs.
The NITR Gene Cluster Is Encoded in ≈0.35 Mb. Of the 46 PACs that hybridize to NITR-specific probes, 16 have been sequenced fully. Standard sequence assembly algorithms fail to consolidate the relationships between all of the various NITR-encoding PACs; however, visual inspection of the relative position, order (inter-spersion), and transcriptional orientation of various NITR genes define a series of contigs that project a relatively compact locus, in which extensive interindividual differences are evident (Fig. 1A and see below). This projected map is validated by a solved sequence contig encompassing three PACs (BUSM1-173M20, BUSM1-105L16, and BUSM1-150L22) and one BAC (DKEY-7N10). Table 2, which is published as supporting information on the PNAS web site, describes the characterization of 11 additional AB-line PACs that are predicted to overlap the BAC (DKEY-7N10). The intergenic distances between nitr12, nitr7a, nitr3d, nitr1d, nitr1j, nitr5, and nitr6c predict a minimum distance between nitr12 and nitr6c of 329 kb and a maximum distance of 357 kb. A hypothetical maximum (complexity) haplotype consists of 36 NITR genes and multiple NITR pseudogenes (Fig. 1B). The NITR gene cluster is bounded by an uncharacterized glycine transporter gene and the map4k2 and men1 genes (Fig. 1C).
Fig. 1.

Contig map of the NITR gene cluster on zebrafish chromosome 7. (A) Coordinates for AB-strain PAC clones from the BUSM1 library and a single Tübingen BAC clone (DKEY-7N10) are indicated on the right or left. Genes encoding NITRs and their transcriptional orientation are indicated by arrowheads, which are color-coded to correspond to 12 different families. Vertical lines between NITR genes of individual PAC/BAC clones define allelic correspondence. Full-length pseudogenes are indicated by truncated arrowheads. Numerous smaller-length pseudogenes also are present in this genomic region and are not shown. A length reference in kilobase pairs (kb) is shown. PAC/BAC lengths, but not gene lengths, are shown to scale. (B) A predicted maximum haplotype of the NITR gene cluster based on the contig map in A. Different genes within a NITR family are distinguished by adding a letter (a–o) after the gene symbol (e.g., nitr1a, nitr1b, nitr1c, etc.) Gene symbols for full-length pseudogenes include a P (e.g., nitr1eP, nitr2cP, and nitr4bP). (C) The locus is flanked on the right by map4k2 and men1 genes and on the left by an uncharacterized glycine transporter gene (elongated arrowheads). One or more putative C-type lectin genes are present between nitr8 and nitr7a.(D) Relative positions of zebrafish STSs, including one EST, fl04c06, which is a nitr1o transcript, are indicated. STSs were used to map the NITR gene cluster to chromosome 7 by using two radiation hybrid panels (13). Distance (cR) to the nearest framework markers is based on the current T51 radiation hybrid panel map.
Multiple STSs within and flanking the NITR gene cluster have been mapped to a single region of zebrafish chromosome 7 by using the T51 and LN54 radiation hybrid panels (Fig. 1D and ref. 13). The span between the flanking STS markers (CRI0006 and CRI0009) is 13 cRs on the T51 radiation hybrid panel (January 2, 2004, update). There is no indication from mapping data, PAC screening, and whole-genome sequencing that more than one cluster of NITR genes (of the nitr1–12 types) is present in the zebrafish genome.
C-Type Lectins Are Encoded Within the NITR Gene Cluster. Between one and three putative C-type lectins, which share 87–99% identity at the amino acid level (data not shown), were identified in the region between nitr8 and nitr7a (Fig. 1B). These C-type lectin sequences lack transmembrane domains and introns but possess a polyadenylation signal sequence; their transcriptional status is unknown. A single cDNA encoding a killer cell C-type lectin receptor (cKLR), which possesses a positively charged residue within the transmembrane region and appears to be phylogenetically related to the mammalian NKG2/CD94 family of NK receptors (9) was identified recently in Paralabidochromis chilotes, another species of bony fish. Although no clear orthologs of cKLR can be identified currently in the zebrafish genome, blast searches indicate that cKLR shares low similarity with multiple zebrafish C-type lectin genes, including those encoded in the NITR gene cluster on chromosome 7 as well as genes on chromosomes 1, 11, and 16.
The NITR Cluster Demonstrates Genetic Variation in the AB Line of Zebrafish. Certain NITRs that are present in some PACs are absent from overlapping PACs (Fig. 6, which is published as supporting information on the PNAS web site). This allelic complexity is well exemplified by the nitr1g/nitr1h and nitr2d/nitr2e genes in Fig. 1 A. It was considered that nitr2d and nitr2e may represent different alleles of the same gene; however they are classified as different genes because their V domains share only 58–63% identity at the peptide level. In addition, individual PACs possess either the nitr1g and nitr2d genes (BUSM1-139E19, BUSM1-173M20, and BUSM1-221H6) or the nitr1h and nitr2e genes (BUSM1-186F8, BUSM1-175P12, and BUSM1-219H16), suggesting different haplotypes (Fig. 6).
Heterozygosity in both coding and noncoding regions of the NITR gene cluster was investigated by using PCR amplification of genomic DNA of haploid AB-line embryos, which are viable to ≈48 h. Four representative polymorphic regions were selected and could be distinguished on the basis of product size. When applied to haploid genomic DNA, single PCR bands were identified from each haploid genome; however, for certain polymorphisms, either form is detected from sibling haploid embryos, suggesting parental heterozygosity at that sequence (data not shown). Both interstrain polymorphism and intrastrain heterozygosity have been maintained in the AB line after ≈5 years (the period between library construction and analyses reported here) (23–25). Zebrafish are recalcitrant to inbreeding, despite efforts to maintain highly homozygous strains (23, 26). The interindividual variation seen in this library, which derives from >200 animals (22), reflects heterozygous advantage, which likely influences overall fecundity. These extensive polymorphisms may reflect interindividual variation in NITR function.
NITR Families Differ in Overall Organization and Function. Genomic analysis led to the discovery of NITR families 5–12. These families differ from the other NITR gene families in terms of the number of ectodomains as well as the presence or absence of joining (J) or J-like sequences (Table 3, which is published as supporting information on the PNAS web site), ITIM, an ITAM-related sequence, a transmembrane region, and a charged residue within the transmembrane region (Fig. 2).
Fig. 2.

Schematic representation of diverse NITRs in zebrafish. Predicted protein structures of multiple zebrafish NITRs are shown. Protein domains include the following: a variable Ig domain (V); an intermediate Ig domain (I) (11); a transmembrane region (TM) denoted by a helical structure; a positively charged residue midmembrane ( ); a prototypic joining-like region including the GXGTXLX(V/I/L) peptide sequences (J); a J-like variant (see Table 3) is indicated by an orange oval; a conventional ITIM including the (I/V/L)XYXX(I/V/L) peptide sequences; a variant of the conventional ITIM (itim); and variant of the conventional ITAM sequence (itam). An asterisk (*) indicates that full-length cDNA clones have been identified and sequenced confirming these structures; other structures are predicted from genomic sequence. SP, alternative RNA splicing variants; PM, polymorphic variants of the same gene.
); a prototypic joining-like region including the GXGTXLX(V/I/L) peptide sequences (J); a J-like variant (see Table 3) is indicated by an orange oval; a conventional ITIM including the (I/V/L)XYXX(I/V/L) peptide sequences; a variant of the conventional ITIM (itim); and variant of the conventional ITAM sequence (itam). An asterisk (*) indicates that full-length cDNA clones have been identified and sequenced confirming these structures; other structures are predicted from genomic sequence. SP, alternative RNA splicing variants; PM, polymorphic variants of the same gene.
Phylogenetic Analyses Suggest Divergent Evolution of the NITR Cluster. A phylogenetic comparison of all of the zebrafish NITR V domains (Fig. 3) is consistent with the vertical alignments and contig relationships shown in Fig. 1A. The predicted NITR families group together with 98–100% confidence. However, an examination of two NITR families, nitr1 and nitr2 (Fig. 7, which is published as supporting information on the PNAS web site), reveals that some V domains, even for those of the same family, show moderate to extensive synonymous nucleotide substitutions per synonymous site, e.g., 0.304 between nitr1d/g/h/i subfamilies and nitr1a/b/c/f/k subfamilies (Table 4, which is published as supporting information on the PNAS web site), indicating that single or relatively small-scale gene duplication events have occurred independently at various times during the evolution of this cluster. Taking the phylogenetic relationships, the numbers of synonymous nucleotide substitutions per synonymous site among NITR V families, and the location of genes into consideration, it is not possible to assign any regions within the NITR cluster as having arisen via a large-scale duplication event.
Fig. 3.

Phylogenetic analysis of zebrafish NITR V domains. V domains encoded by all zebrafish NITR genes defined in the contig map shown in Fig. 1 were aligned with clustalw and phylogenetic and molecular evolutionary analyses were conducted by using mega 2.1 (30) as described (31). Colors are used to distinguish the NITR families. The confidence for all of the branch points defining each NITR family is 98–100%. Branch lengths are measured in terms of the number of amino acid substitutions, with the scale indicated at the bottom of the figure.
When the zebrafish Nitr V domains were compared in phylogenetic analyses with 55 NITR V-domain sequences available from pufferfish, catfish, and trout, it was difficult to identify potential orthologs, partially because of occasional duplication of genes for a given evolutionary lineage (Fig. 8, which is published as supporting information on the PNAS web site). However, it is clear that different species possess distinct sets of NITR V families while maintaining divergent repertoires. Considering the fact that most, if not all, zebrafish NITR V-domain sequences are revealed here and that a substantial portion of the pufferfish NITR V repertoire was identified by searching Fugu rubripes genomic database (fugu.hgmp.mrc.ac.uk), these results reflect a general pattern of Nitr V-domain evolution rather than a sampling bias of the genes identified. Because an occasional duplication of genes is observed and NITR pseudogenes are present in the zebrafish genome, it can be concluded that the NITR multigene families have been subject to the birth-and-death process of evolution, as in the case of major histocompatibility complex, Ig, T cell antigen receptor (TCR), and NK receptor genes.
Alternative mRNA Splicing and Allelic Variation Contribute to NITR Complexity. We previously characterized five different nitr4 transcripts (13), which now can be assigned as products of different alleles of a single functional nitr4 gene that exhibits alternative mRNA splicing between the V and I exons. Two full-copy length splice variants of nitr9 have been identified: nitr9-long (nitr9-L) encodes an intact V domain, and nitr9-short (nitr9-S) eliminates 31 V-domain residues and the Ig fold (see Figs. 2 and Fig. 9A, which is published as supporting information on the PNAS web site). Alternative splicing also occurs between the leader and V-domain exons of nitr7b that eliminates 11 residues at the amino terminus of the V domain (GenBank accession no. AY606016). A single allele of nitr5 (BUSM1-178A3) lacks the coding sequence for 11 residues within the I-domain exon (see Figs. 2 and 9B). Although most alleles of nitr2d appear functional, a single allele (BUSM1-139E19) encodes a pseudogene with a premature stop codon in the I domain and no consensus alternative mRNA splice donor sites.
Nitr9 Is an Activating NITR. The nitr9 gene was identified initially in the Tübingen strain BAC DKEY-7N10 and represents the only zebrafish NITR gene that encodes a positively charged residue in the transmembrane region, a characteristic feature of activating forms of NK, Fc (1), and related receptors (5). Analyses of genomic and partial cDNA sequence of nitr9 from the AB strain indicate that this is a conserved feature of this gene (data not shown). We previously demonstrated the predicted function of inhibitory NITRs (13). To address the function of Nitr9, a putative activating receptor, a FLAG-tagged nitr9-long (nitr9-L) cDNA was cotransfected with wild-type DAP12 (wt-DAP12) into NKL cells. DAP12 is known to associate with mammalian NK-activating receptors and is required to signal downstream, resulting in tumorolytic function. After Nitr9-L was cross-linked with an anti-FLAG antibody, the treated NKL cells were exposed to P815 target cells in a redirected cytolysis assay. Nitr9-L cross-linking resulted in 14.7% cytotoxicity (Fig. 4). In contrast, 2.9% cytotoxicity was observed when the same epitope-tagged NITR was cotransfected with a mutant DAP12. Likewise, infection and cross-linking of either Nitr9-L or wtDAP12 alone results in minimal cytotoxicity (data not shown). The redirected killing of P815 by using an antibody specific for endogenous NKG2D is >47% (data not shown). These studies establish that zebrafish Nitr9 (i) has the potential for functioning as an activating receptor and (ii) can interact with mammalian DAP12, although under the experimental conditions used, this association may not be specific to that particular adaptor.
Fig. 4.

Functional analysis of Nitr9. (A) Fluorescence-activated cell sorter analysis of epitope (FLAG)-tagged Nitr9-L and wild-type DAP12 (wtDAP12) cotransfected (vaccinia) NKL cells indicating an 80.4% infection rate. (B) Redirected cytolysis of mastocytoma P815 by NKL cells cotransfected with FLAG-tagged Nitr9-L and wtDAP12 or a mutant DAP12 (mutDAP12). Killing is approximately 5-fold greater with wtDAP12 than with mutDAP12 or other controls.
nitr5 encodes an unusual cytoplasmic sequence. Although the prototypic sequence of an ITAM is YXX(V/L/I)X7–8YXX(V/L/I), the presence of a unique Tyr (YATVNTSSKYSRV) in the cytoplasmic region of nitr5 (see Fig. 9C) could correspond to an ITAM, assuming that a two-residue-shorter spacing difference is consistent with activating function. However, two of the six nitr5 sequences from the AB strain reported here (BUSM1-105L16 and BUSM1-150L22) encode a His instead of a Tyr in the second position (HSRV) of the putative ITAM and partial sequencing of one nitr5 allele from the Tübingen strain identified a His in this same position (data not shown). These allelic variations in the putative ITAM sequence make it unclear whether Nitr5 functions in direct activation, inhibition, or possibly in both pathways.
Comparative Models Suggest Structural Differences Between NITRs and V-Type Ig Domains. Alignment of the V domains from representative Nitr1 sequences defines three general stretches of hypervariable (HV) regions (see Fig. 10, which is published as supporting information on the PNAS web site). High variation is observed between Cys-23 and Trp-41, Gly-47 and Leu-89, and between Cys-104 and the J-like sequence, which correspond to HV region 1 (HV1), HV2, and HV3, respectively. The positioning of the HV1 and HV3 regions within the NITR V domains corresponds to the positions of complementarity-determining region 1 (CDR1) and CDR3 within Ig/TCR V domains, respectively; however, the positioning of the HV2 region overlaps CDR2 positioning. The intrafamily pattern variation evident in HV1 and HV2 is generally reminiscent of germ-line variation seen in Ig/TCR genes.
Homology models representing V domains of NITRs were generated by comparative protein structure analysis (27). For the Nitr1g V domain, pairwise alignments with 112 residues identified solved structures of Ig domains that range in sequence identity, e.g., 1qnzh, VH region = 35.2% (28); 1k4c, VL region = 33.6%; and 1qsed, TCR Vα = 27.8% (29).
Overall, the predicted Nitr1g V-domain structure is most similar to a VH region as indicated by average rms deviations of 0.8 Å for Cα atoms and high model confidence factors in the framework residues of the Ig fold (Fig. 5A). However, there are notable differences in the stem and loop region encoded by the HV2 region. The stem of the HV2 region deviates from the VH region at Ser-49 of the Nitr1g V domain in the central portion of the C′ strand, causing the conformation of the loop region to be altered. The apex of the HV2 loop in Nitr1g is shifted 15 Å, compared with the CDR2 (C′C″ loop) of the most similar VH region. Comparative analysis and superposition of models representative of the other Nitrs and Nitr1g show a range of deviations (5–15 Å) at the CDR2 analogous region, compared with solved V-type Ig domains. These observations suggest that NITR V domains may differ significantly from Ig and TCR V regions in the CDR2 analogous region (HV2) and in the C′ strand.
Fig. 5.
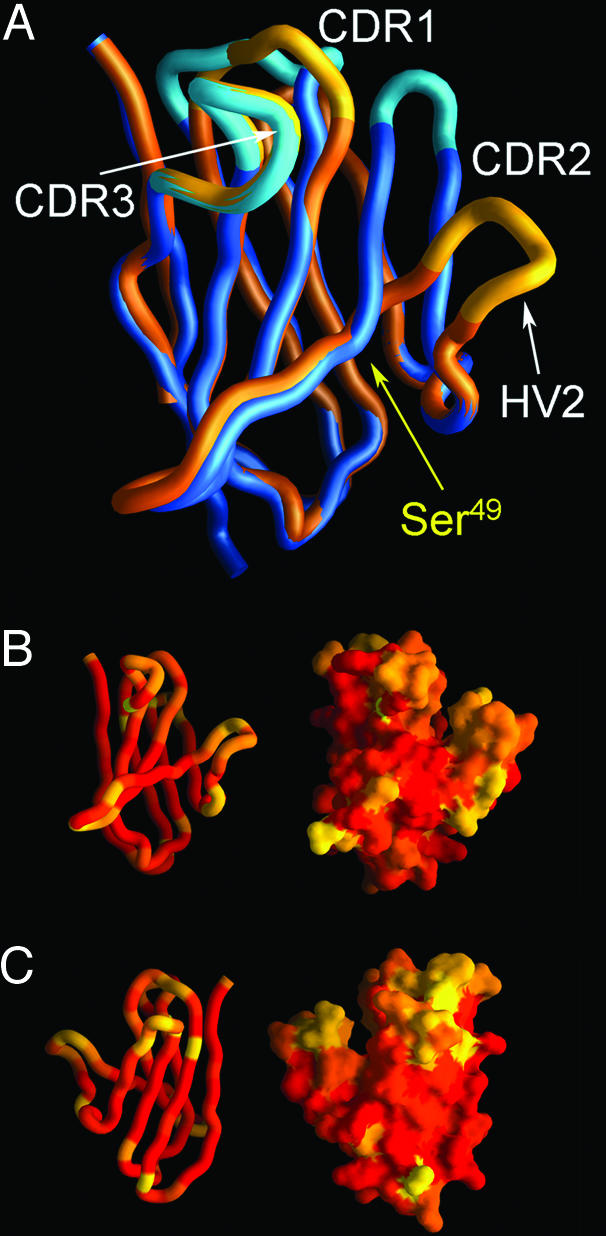
Homology models of NITR V domains show structural variability in CDR analogous loops. (A) A structural model of the Nitr1g V domain (in gold) is shown superimposed on a homologous Ig domain (in blue) (Protein Data Bank ID code 1QNZ). The distance between equivalent Cα atoms at the apex of the HV2 and CDR2 loop is 15 Å. The position of Ser-49 of the Nitr1g V domain is indicated. In B and C, variability between the Nitr1 family V domains is displayed on a structural homology model of Nitr1g. Variability is color-coded based on sequence identity and graded from red (60–100% identical) to orange (40–60% identity) and yellow (<40% identity). The NITR V domain is oriented to show the top sheet (A′GFCC′) of the Ig domains in A and B and the back sheet (ABED) in C.
To interpret the potential significance of intrafamily variation within NITRs, the sequence variability of zebrafish Nitr1 V domains (see Fig. 10) was mapped on the comparative model of Nitr1g to determine which structural elements exhibit the highest degree of sequence diversity. A representation of the backbone and molecular surface of the Nitr1g V domain is shown in Fig. 5 B and C, in which Nitr1 sequence identity is color-coded. These data demonstrate that within the Nitr1 family, the HV1 and HV2 regions are more diverse (sequence-wise and structurally) than HV3. Overall, these data indicate that NITR V domains are similar to Ig and TCR domains, with the striking exception of the HV2 region, which exhibits a structural shift that places the loop further from the other CDRs. The functional role of the distinctive arrangement of HV regions in NITR V regions is unknown but likely creates ligand-binding specificities that differ from those exhibited by Ig or TCR.
Insight from Comprehensive Analysis of the Zebrafish NITR Locus. Although the precise function(s) of NITRs is not yet known, it is likely that their membrane disposition and signaling are equivalent to other activating/inhibitory leukocyte regulatory receptors. Of the various gene clusters encoding activating/inhibitory leukocyte regulatory receptors, the NITR cluster reported here is the most complex yet described. The patterns of sequence variation in the V region are related to those typifying interfamily variation in Ig/TCR genes, suggesting that certain features of receptor function may be shared among NITRs, Igs, and TCRs. However, a significant deviation occurs in the orientation of the region of highest variation in the V region of NITRs, compared with Igs/TCRs, suggesting that the NITRs, which do not undergo somatic rearrangement, may recognize very different ligands or recognize ligands in a manner that is different from the rearranging adaptive receptors. The large number of NITR alleles defined here permits comparison with NITRs that have been identified in other species. The minimal degree of interspecies relatedness of NITR sequences is consistent with rapid evolutionary change, suggesting that NITRs may be recognizing a self-determinant in a manner equivalent to that seen with NK receptors. The mechanisms underlying the transition from a V region that potentially recognizes a self-determinant to one recognizing a peptide in the context of a self-determinant (as with TCRs) is significant in understanding the divergence of immune specificity. The comprehensive definition of the NITR gene cluster in zebrafish reported here represents a significant step toward approaching this basic question.
Supplementary Material
Acknowledgments
We thank Diana Skapura for DNA sequencing, Dr. E. Vivier for the DAP12 cDNA, Dr. M. Ekker for the LN54 radiation hybrid mapping panel, and Barbara Pryor for editorial assistance. G.W.L. is supported by grants from the National Institutes of Health and The Pediatric Cancer Foundation. J.A.Y. is supported by grants from the National Science Foundation and the American Cancer Society and by funding from the H. Lee Moffitt Cancer Center and Research Institute. The Zebrafish Genome Project is supported by the Wellcome Trust.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: NITR, novel immune-type receptor; V, variable; PAC, P1 artificial chromosome; BAC, bacterial artificial chromosome; ITAM, immunoreceptor tyrosine-based activating motif; NK, natural killer; I, intermediate; ITIM, immunoreceptor tyrosine-based inhibition motif; TCR, T cell antigen receptor; HV, hypervariable region; CDR, complementarity-determining region; STS, sequence-tagged site.
Data deposition: The PAC, BAC, STS, and cDNA sequences have been deposited in the GenBank and ZFIN databases [accession nos. AL591382 (BUSM1-105L16), AL591482 (BUSM1-116C14), AL591405 (BUSM1-125J23), AL591418 (BUSM1-139E19), AL592105 (BUSM1-150L22), AL591497 (BUSM1-173M20), AL591401 (BUSM1-175P12), AL591372 (BUSM1-178A3), AL591391 (BUSM1-186F8), AL691521 (BUSM1-186K12), AL591427 (BUSM1-219H16), AL591476 (BUSM1-263J20), AL732544 (BUSM1-6I1), AL591406 (BUSM1-213M14), AL591420 (BUSM1-221H6), AL844870 (BUSM1-169I8), AL954849 (DKEY-7N10), G68151 (STS-CRI0002), G68152 (STS-CRI0003), G68153 (STS-CRI0004), G68154 (STS-CRI0005), G68155 (STS-CRI0006), G68156 (STS-CRI0007), BV096859 (STS-CRI0009), AY570232 (nitr1o cDNA), AY570233 (nitr5 cDNA), AY570234 (nitr6b cDNA), AY570235 (nitr7b cDNA), AY606016 (nitr7b splicing variant, partial cDNA), AY570236 (nitr8 cDNA), AY570237 (nitr9-long cDNA), and AY570238 (nitr9-short cDNA)].
References
- 1.Ravetch, J. V. & Lanier, L. L. (2000) Science 290, 84-89. [DOI] [PubMed] [Google Scholar]
- 2.Vilches, C. & Parham, P. (2002) Ann. Rev. Immunol. 20, 217-251. [DOI] [PubMed] [Google Scholar]
- 3.Yokoyama, W. M. & Plougastel, B. F. M. (2003) Nat. Rev. Immunol. 3, 304-316. [DOI] [PubMed] [Google Scholar]
- 4.Cerwenka, A. & Lanier, L. L. (2000) Nat. Rev. Immunol. 1, 41-49. [DOI] [PubMed] [Google Scholar]
- 5.Davis, R. S., Wang, Y.-H., Kubagawa, H. & Cooper, M. D. (2001) Proc. Natl. Acad. Sci. USA 98, 9772-9777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trowsdale, J., Barten, R., Haude, A., Stewart, C. A., Beck, S. & Wilson, M. J. (2001) Immunol. Rev. 181, 20-38. [DOI] [PubMed] [Google Scholar]
- 7.Litman, G. W., Hawke, N. A. & Yoder, J. A. (2001) Immunol. Rev. 181, 250-259. [DOI] [PubMed] [Google Scholar]
- 8.Zhang, H., Nichols, K., Thorgaard, G. H. & Ristow, S. S. (2001) Immunogenetics 53, 751-759. [DOI] [PubMed] [Google Scholar]
- 9.Sato, A., Mayer, W. E., Overath, P. & Klein, J. (2003) Proc. Natl. Acad. Sci. USA 100, 7779-7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoder, J. A. (2004) Comp. Biochem. Physiol. C 138, 271-280. [DOI] [PubMed] [Google Scholar]
- 11.Harpaz, Y. & Chothia, C. (1994) J. Mol. Biol. 238, 528-539. [DOI] [PubMed] [Google Scholar]
- 12.Strong, S. J., Mueller, M. G., Litman, R. T., Hawke, N. A., Haire, R. N., Miracle, A. L., Rast, J. P., Amemiya, C. T. & Litman, G. W. (1999) Proc. Natl. Acad. Sci. USA 96, 15080-15085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoder, J. A., Mueller, M. G., Wei, S., Corliss, B. C., Prather, D. M., Willis, T., Litman, R. T., Djeu, J. Y. & Litman, G. W. (2001) Proc. Natl. Acad. Sci. USA 98, 6771-6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hawke, N. A., Yoder, J. A., Haire, R. N., Mueller, M. G., Litman, R. T., Miracle, A. L., Stuge, T., Shen, L., Miller, N. & Litman, G. W. (2001) Proc. Natl. Acad. Sci. USA 98, 13832-13837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen, L., Stuge, T. B., Bengten, E., Wilson, M., Chinchar, V. G., Naftel, J. P., Bernanke, J. M., Clem, L. W. & Miller, N. W. (2004) Dev. Comp. Immunol. 28, 139-152. [DOI] [PubMed] [Google Scholar]
- 16.Searle, S. M., Gilbert, J., Iyer, V. & Clamp, M. (2004) Genome Res. 14, 963-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jekosch, K. (2004) Methods Cell Biol. 77, in press. [DOI] [PubMed]
- 18.Jiang, K., Zhong, B., Gilvary, D. L., Corliss, B. C., Hong-Geller, E., Wei, S. & Djeu, J. Y. (2000) Nat. Immunol. 1, 419-425. [DOI] [PubMed] [Google Scholar]
- 19.Wei, S., Gilvary, D. L., Corliss, S., Sebti, S., Sun, J., Straus, D. B., Leibson, P. J., Trapani, J. A., Hamilton, A. D., Weber, M. J., et al. (2000) J. Immunol. 165, 3811-3819. [DOI] [PubMed] [Google Scholar]
- 20.Schwede, T., Kopp, J., Guex, N. & Peitsch, M. C. (2003) Nucleic Acids Res. 31, 3381-3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicholls, A., Sharp, K. A. & Honig, B. (1991) Proteins 11, 281-296. [DOI] [PubMed] [Google Scholar]
- 22.Amemiya, C. T. & Zon, L. I. (1999) Genomics 58, 211-213. [DOI] [PubMed] [Google Scholar]
- 23.Johnson, S. L. & Zon, L. I. (1999) Methods Cell Biol. 60, 357-359. [DOI] [PubMed] [Google Scholar]
- 24.Stickney, H. L., Schmutz, J., Woods, I. G., Holtzer, C. C., Dickson, M. C., Kelly, P. D., Myers, R. M. & Talbot, W. S. (2002) Genome Res. 12, 1929-1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rawls, J. F., Frieda, M. R., McAdow, A. R., Gross, J. P., Clayton, C. M., Heyer, C. K. & Johnson, S. L. (2003) Genetics 163, 997-1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nechiporuk, A., Finney, J. E., Keating, M. T. & Johnson, S. L. (1999) Genome Res. 9, 1231-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fiser, A., Feig, M., Brooks, C. L. & Sali, A. (2002) Acc. Chem. Res. 35, 413-421. [DOI] [PubMed] [Google Scholar]
- 28.Tugarinov, V., Zvi, A., Levy, R., Hayek, Y., Matsushita, S. & Anglister, J. (2000) Structure Fold. Des. 8, 385-395. [DOI] [PubMed] [Google Scholar]
- 29.Ding, Y. H., Baker, B. M., Garboczi, D. N., Biddison, W. E. & Wiley, D. C. (2000) Immunity 11, 45-56. [DOI] [PubMed] [Google Scholar]
- 30.Kumar, S., Tamura, K., Jakobsen, I. B. & Nei, M. (2001) Bioinformatics 17, 1244-1245. [DOI] [PubMed] [Google Scholar]
- 31.Yoder, J. A., Mueller, M. G., Nichols, K. M., Ristow, S. S., Thorgaard, G. H., Ota, T. & Litman, G. W. (2002) Immunogenetics 54, 662-670. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}