

Abstract
Background
The transient global cerebral hypoperfusion/reperfusion achieved by induction of Bilateral Common Carotid Artery Occlusion followed by Reperfusion (BCCAO/R) may trigger a physiological response in an attempt to preserve tissue and function integrity. There are several candidate molecules among which the endocannabinoid system (ECS) and/or peroxisome-proliferator activated receptor-alpha (PPAR-alpha) may play a role in modulating oxidative stress and inflammation. The aims of the present study are to evaluate whether the ECS, the enzyme cyclooxygenase-2 (COX-2) and PPAR-alpha are involved during BCCAO/R in rat brain, and to identify possible markers of the ongoing BCCAO/R-induced challenge in plasma.
Methods
Adult Wistar rats underwent BCCAO/R with 30 min hypoperfusion followed by 60 min reperfusion. The frontal and temporal-occipital cortices and plasma were analyzed by high performance liquid chromatography-mass spectrometry (HPLC-MS) to determine concentrations of endocannabinoids (eCBs) and related molecules behaving as ligands of PPAR-alpha, and of oxidative-stress markers such as lipoperoxides, while Western Blot and immunohistochemistry were used to study protein expression of cannabinoid receptors, COX-2 and PPAR-alpha. Unpaired Student’s t-test was used to evaluate statistical differences between groups.
Results
The acute BCCAO/R procedure is followed by increased brain tissue levels of the eCBs 2-arachidonoylglycerol and anandamide, palmitoylethanolamide, an avid ligand of PPAR-alpha, lipoperoxides, type 1 (CB1) and type 2 (CB2) cannabinoid receptors, and COX-2, and decreased brain tissue concentrations of docosahexaenoic acid (DHA), one of the major targets of lipid peroxidation. In plasma, increased levels of anandamide and lipoperoxides were observed.
Conclusions
The BCCAO/R stimulated early molecular changes that can be easily traced in brain tissue and plasma, and that are indicative of the tissue physiological response to the reperfusion-induced oxidative stress and inflammation. The observed variations suggest that the positive modulation of the ECS and the increase of proinflammatory substances are directly correlated events. Increase of plasmatic levels of anandamide and lipoperoxides further suggests that dysregulation of these molecules may be taken as an indicator of an ongoing hypoperfusion/reperfusion challenge.
Keywords: Bilateral common carotid artery occlusion, Reperfusion, Endocannabinoid system, Lipoperoxides, COX-2, Cerebral cortex
Background
The transient Bilateral Common Carotid Artery Occlusion followed by Reperfusion (BCCAO/R) is a model of transient global hypoperfusion [1]. This model does not lead to an authentic ischemic insult in the rodent brain due to the presence of efficient collateral systems, which allow for a cerebral blood flow compensation within a few minutes [2]. However, it has been recently shown that, in the rat, the cerebral transient hypoperfusion followed by reperfusion, such as that induced by the BCCAO/R, causes detectable and consistent tissue changes such as a decrease of docosahexaenoic acid (DHA), one of the most abundant polyunsaturated fatty acid in neuronal membrane phospholipids, an increase of the cyclooxygenase-2 (COX-2) [3] and significant microvascular alterations [4].
The endocannabinoids (eCBs) are endogenous lipid mediators involved in a wide variety of biological processes. The most frequently studied eCBs are N-arachidonoyl-ethanolamide (AEA, anandamide), belonging to the superfamily of N-acylethanolamides (NAEs), and 2-arachidonoyl-glycerol (2-AG). AEA and 2-AG are not stored in vesicles but they are synthesized ‘on demand’ and released from plasmatic membranes immediately after their production when cells are challenged with potentially harmful stimuli [5–7]. Accumulating data show that eCBs, with their signaling-mediating receptors, and enzymes involved in their synthesis and degradation, constitute an extensive system with relative basic levels of each component that is modulated by different and concurring molecular mechanisms [8, 9]. Growing evidence supports a general role for eCBs and congeners in the preservation of metabolic homeostasis and responsivity of the brain to stress, and indicate that they are involved in inflammation and act as endogenous neuroprotectants in cerebral ischemia [5, 9–18]. As for the likely involvement of eCB in neuroinflammation, it has been shown that 2-AG and AEA are substrates for COX-2 [19] and it is relevant that the eCB neuroprotective activity may be mediated by preventing excessive expression of COX-2 [11, 20, 21]. Conversely, COX-2 is a key regulator of eCB signalling [22]. Moreover, it has been shown that the peroxisome-proliferator activated receptor (PPAR)-alpha mediates the anti-inflammatory effects of the non-cannabinoid palmitoylethanolamide (PEA) that behaves as one of its endogenous ligands [23]. Though the role of eCB system in neuroprotection is somewhat controversial, data on focal cerebral ischemia followed by reperfusion show that i) intraperitoneally administered PEA and AEA reduce the size of infarcted tissue [7], ii) both pharmacological blockade [24] and genetic deletion of CB1 receptor reduce the infarct volume and improve neurological function [25], and iii) CB2 activation attenuates the cerebral ischemia/reperfusion-induced microcirculatory dysfunction [13].
In this study, we investigated whether the eCB system may be directly correlated to the oxidative events triggered by the BCCAO/R-induced transient global hypoperfusion in rat brain, and we chose to examine the forebrain areas that are directly and selectively reached by the internal carotid artery branches. With this aim, we examined, prior to and after induction of BCCAO/R, cerebral tissue and plasmatic concentrations of eCBs and of oxidative stress markers such as polyunsaturated fatty acid (PUFA) hydroperoxides. Parallel immunochemical analyses, carried out by means of Western Blot and immunohistochemistry, investigated possible BCCAO/R-induced changes in the occurrence and tissue distribution of CB1 and CB2 receptors, COX-2, and the transcription factor peroxisome-proliferator activated receptor (PPAR)-alpha. Results are discussed in view of the possible use of eCBs and lipoperoxides as markers of an ongoing transient cerebral global hypoperfusion.
Methods
Experimental procedure
Animals and keeping
For 1 week before the start of the experiment, fifty six adult male Wistar rats (Harlan-Italy, Udine, Italy), weighing 210 ± 20 g (mean ± SD) were housed under controlled temperature (21 ± 2 °C), relative humidity (60 ± 5%) and artificial 12 h light/dark cycle, avoiding all stressful stimuli. Animal handling and care throughout the experimental procedures met with national (Legislative Decree n. 26, 04/04/2014) and international (Directive 2010/63/EU in Europe) laws and policies. The experimental protocols were carried out in compliance with the guidelines of the Animal Ethics Committee of the University of Cagliari. Standard laboratory food (A04, Safe, Augy, France) and water were freely available ad libitum.
According to the optimum standard for the evaluation of lipids in tissue and plasma [6, 26], animals received no food for 12 h before surgery. Rats were randomly assigned to two groups (n = 12/group): one group was submitted to BCCAO and reperfused (BCCAO/R) and one group was sham-operated. Surgery was performed in all cases between 13:00 and 16:30 p.m..
Surgery
Surgical procedure for induction of BCCAO/R was adapted from the method of Iwasaki et al. [27] and performed in all cases between 13:00 and 16:30 p.m.. Rats were anesthetized with intraperitoneal administration of Equitesin (4.2% w/v chloral hydrate, 2.12% w/v MgSO4, 16.2% w/w pentobarbital, 39.6% w/w propylene glycol, and 10% w/w ethanol in sterile distilled H2O) (0.5 ml/100 g bodyweight). After a midline cervical incision and blunt dissection of muscles, the common carotid arteries (CCA) were exposed while leaving the vagus nerve intact. Cerebral blood flow reduction was produced by placement of two atraumatic microvascular clips for 30 min on CCA. The reperfusion period was achieved by removing the clips and restoring blood flow through the stenosed vessels for 60 min. The control animals, used to determine the effects of anaesthesia and surgical manipulation of the results, were represented by sham-operated rats that underwent surgery without CCA occlusion.
Sampling
At the end of the procedure, brain samples were collected either as fresh tissue for lipid analysis and Western Blot or after transcardial perfusion fixation with ice cold 4% formaldehyde in 0.1 M phosphate buffer (PB), pH 7.4 for immunohistochemistry. The frontal cortex was rapidly dissected out by a transverse cut made at the level of the optic chiasm, at the approximate bregma level of −1.0 mm [28], and frozen at −80 °C until HPLC or Western Blot analysis. Temporal-occipital cortex, dissected out by a transverse cut at the approximate bregma level of −4.5 mm, was also sampled as a control cortical area not irrorated by the internal carotid artery branches. Blood was quickly collected from the trunk of killed animals into heparinised tubes and centrifuged at 1500 g for 10 min at −8 °C. The resulting plasma was frozen at −20 °C until assayed for lipids. Immunohistochemistry was used to determine the tissue distribution and cellular localization of the same markers used for the Western Blot analysis. Perfused brains were dissected out and then rinsed overnight in 0.1 M PB, pH 7.3, containing 20% sucrose. After sucrose infiltration, samples were embedded in Optimal Cutting Temperature (OCT) medium for cryostat sectioning. For each assay, the investigator was blind with respect to the experimental condition of the rats.
Endocannabinoid and congener quantifications
Frozen tissues were homogenized and extracted with 50 mM chloroform/methanol/Tris–HCl, pH 7.5 (2:1:1, v/v), containing internal deuterated standards for anandamide (AEA), 2-arachidonoyl-monoacylglycerol (2-AG), palmitoylethanolamide (PEA) and oleoylethanolamide (OEA) quantification by isotope dilution ([2H]8 AEA, [2H]5 2-AG, [2H]4 PEA, [2H]4 OEA; Cayman Chemical, Ann Arbor, MI, USA). AEA, 2-AG, PEA, and OEA were quantified by liquid chromatography–atmospheric pressure chemical ionization–mass spectrometry [1100 HPLC system (Agilent Technologies, Santa Clara, CA, USA) equipped with MS Detector 6110 single quadrupole] and using selected ion monitoring at M1 values for the four compounds and their deuterated homologs, as described previously [26]. Concentrations (nmoles/g; nmoles/ml) are shown as histograms in Fig. 1.
Fig. 1.
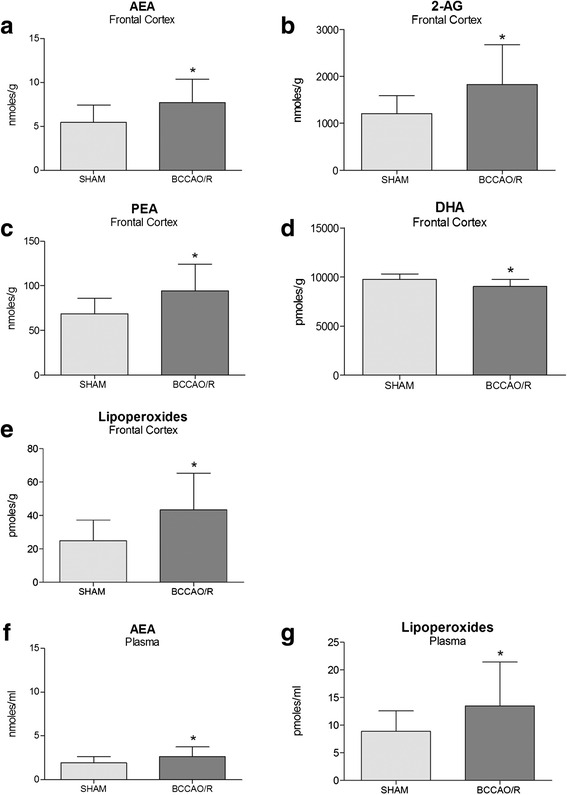
Endocannabinoids and fatty acid profile in frontal cortex and plasma. High performance liquid chromatography (HPLC) analysis of frontal cortex (a−f) and plasma (g, h) of sham-operated and bilateral common carotid artery occlusion followed by reperfusion (BCCAO/R) rats. N-arachidonoyl-ethanolamide (AEA) (a, e), 2-arachidonoyl-glycerol (2-AG) (b), palmitoylethanolamide (PEA) (c), docosahexaenoic acid (DHA) (e) and lipoperoxides (f, h) concentrations are reported as mean values of 12 sham-operated and 12 BCCAO/R rats. Error bars depict S.D.. Unpaired Student’s t test: *p < 0.05
Tissue fatty acid quantifications
Total lipids were extracted from different brain areas using chloroform/methanol 2:1 (v/v). Aliquots were mildly saponified as previously described [29] in order to obtain free fatty acids for high-performance liquid chromatography (HPLC) analysis. Separation of fatty acids was carried out with an Agilent 1100 HPLC system (Agilent Technologies) equipped with a diode array detector as previously reported [6]. Concentrations (nmoles/g; nmoles/ml) are shown as histograms in Fig. 1.
Western blot
Tissue homogenates were prepared in a 2% solution of sodium dodecyl sulfate (SDS) containing a cocktail of protease inhibitors (cOmplete, Mini Protease Inhibitor Cocktail Tablets, Roche, Basel, Switzerland). Protein concentrations were determined using the Lowry method of protein assay [30] with bovine serum albumin as standard. Proteins for each tissue homogenate (40 μg), diluted 3:1 in 4× loading buffer (NuPAGE LDS Sample Buffer 4×, Novex by Life Technologies, Carlsbad, CA, USA), were heated to 95 °C for 10 min and separated by SDS-polyacrilamide gel electrophoresis (SDS-PAGE) using precast polyacrylamide gradient gel (NuPAGE 4–12% Bis-Tris Gel Midi, Novex by Life Technologies) in the XCell4 Sure Lock™ Midi-Cell chamber (Life Technologies). Internal mw standards (Precision Plus Protein™ WesternC™ Standards, Bio-Rad, Hercules, CA, USA) were run in parallel. Two gels at a time were run for Coomassie staining and immunoblotting, respectively. Proteins for immunoblotting were electrophoretically transferred on a polyvinylidene fluoride membrane (Amersham Hybond™-P, GE Healthcare, Little Chalfont, United Kingdom) using the Criterion™ Blotter (Bio-Rad). Blots were blocked by immersion in 20 mM Tris base and 137 mM sodium chloride (TBS) containing 5% milk powder and 0.1% Tween 20 (TBS-T), for 60 min at room temperature and incubated overnight at 4 °C with rabbit polyclonal antisera directed against CB1 receptor (Synaptic System, Göttingen, Germany), diluted 1:500, CB2 receptor (MyBioSource, San Diego, CA, USA), diluted 1:1000, COX-2 (residues 570–598) (Cayman Chem.), diluted 1:200, and PPAR-α (Thermo Scientific, Waltham, MA, USA), diluted 1:1000, in TBS containing 5% milk powder and 0.02% sodium azide, were used as primary antisera. After TBS-T rinse, blots were incubated for 60 min, at room temperature, with peroxidase-conjugated goat anti-rabbit serum (Sigma Aldrich, Saint Louis, MO, USA), diluted 1:10,000 in TBS/T. Loading controls were obtained by stripping and immunostaining the membranes with a mouse monoclonal antibody against the housekeeping protein glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (EMD Millipore, Darmstadt, Germany), diluted 1:1000, as primary antiserum, and a peroxidase-conjugated goat anti-mouse serum (EMD Millipore), diluted 1:5000, as secondary antiserum. In order to control for non specific staining, blots were stripped and incubated with the relevant secondary antiserum. After TBS-T rinse, protein bands were visualized using the ECL chemiluminescent system according to the protocol provided by the company (GE Healthcare), under ImageQuant LAS 4000. Approximate molecular weight (mw) and relative optical density (O.D.) of immunolabelled protein bands were evaluated by a “blind” examiner, and were quantified by comparing the position of relevant bands on the digital images with those of the GAPDH bands, respectively. The ratio of the intensity of COX-2- and PPAR-α-positive bands to the intensity of GAPDH-positive ones was used to compare relative expression levels of these proteins following BCCAO/R procedure. O.D. was quantified by ImageJ (http://imagej.nih.gov/ij/). Concentrations (nmoles/g; nmoles/ml) are shown as histograms in Fig. 2.
Fig. 2.
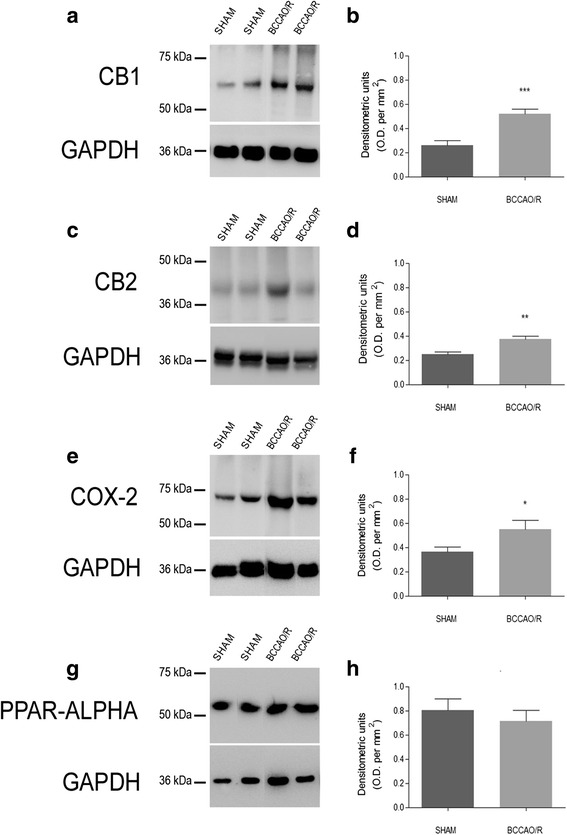
Western Blot analysis. Cannabinoid receptors CB1 (a, b) and CB2 (c, d), cyclooxygenase-2 (COX-2) (e, f) and peroxisome-proliferator activated receptor-alpha (PPAR-alpha) (g, h) in the frontal cortex of sham and bilateral common carotid artery occlusion followed by reperfusion (BCCAO/R) rats. b, d, f, h: densitometric analysis of the band gray levels expressed as a percentage of the optical density (O.D.) ratio of immunostained bands to those of GAPDH. Data are reported as mean values of 12 sham-operated and 12 BCCAO/R rats. Error bars depict S.D. Unpaired Student’s t test: *p < 0.05
Immunohistochemistry
The avidin–biotin–peroxidase complex (ABC) and the indirect immunofluorescence techniques were used to process cryostat semiconsecutive sections (16 μm thick), collected on chrome alum-gelatin coated slides. Coronal serial sections at + 4.70 to - 1.70 mm and at - 3.5 to - 8.0 mm Bregma levels, respectively, were used to focus the observations on the frontal and temporal-occipital cortex [28]. For the ABC, the endogenous peroxidase activity was blocked with 0.1% phenylhydrazine in phosphate buffered saline (PBS) containing 0.2% Triton X-100 (PBS/T), followed by incubation with 20% of either normal goat or normal horse serum (Vector, Burlingame, CA, USA) for 1 h at RT. For ABC, the primary antibodies were rabbit polyclonal antibody against CB1 (Synaptic System), diluted 1:1000 and COX-2 (Cayman Chem.), diluted 1:300. For immunofluorescence, the primary antibodies were goat polyclonal antiserum against COX-2 (SantaCruz Biotechnology), diluted 1:100, mouse monoclonal antibody against Iba1 (WAKO), diluted 1:1000, rabbit polyclonal antiserum against Glial Fibrillary Acidic Protein (GFAP) (DAKO), diluted 1:1000. Incubations with primary antiserum were carried out overnight at 4 °C. Biotin-conjugated goat anti-rabbit serum (Vector, Burlingame, CA, USA), diluted 1:400, was used as secondary antiserum in the ABC method; Alexa Fluor 488 or 594 donkey anti-goat, anti-mouse and anti-rabbit sera (Invitrogen, Eugene, OR, USA), diluted 1:500, were used as secondary antiserum in the immunofluorescence technique. The ABC reaction product was revealed with a biotin-conjugated goat anti-rabbit serum (Vector, Burlingame, CA, USA), diluted 1:400, as secondary antiserum. The ABC (BioSpa Div. Milan, Italy), diluted 1:250, followed by a solution of 0.1 M PB, pH 7.3, containing 0.05% 3,3’-diaminobenzidine (Sigma Aldrich), 0.01% hydrogen peroxide and 0.04% nickel ammonium sulfate were used to reveal the reaction product. Incubations with secondary antiserum and ABC lasted 60 min and were performed at RT. Negative control preparations were obtained by incubating tissue sections in parallel with either PBS-T alone or with the relevant primary antiserum preabsorbed with an excess of the corresponding peptide antigen. Slides were observed by the same examiner, who was “blind” with respect to the animals’ treatment, with an Olympus BX61 microscope, equipped with epifluorescence illumination, and digital images (Figs. 4, 5) were captured with a Leica DF 450C camera.
Fig. 4.

Cyclooxygenase-2 (COX-2)-like immunoreactivity in the frontal cortex. Brain sections of sham-operated and BCCAO/R rats are shown in (a, b) and (c, d), respectively. b, d represent a higher magnification of the microscopic fields squared in (a) and (c), respectively. Panels (a–d) are representative of observations carried out in 6 sham-operated and 6 BCCAO/R rats. Positive cell bodies are mostly distributed in the superficial cortical layers. m, molecular layer; wm, white matter. Scale bars: 50 μm
Fig. 5.
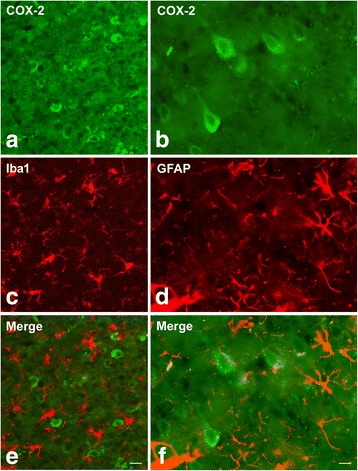
Double immunofluorescence for cyclooxygenase-2 (COX-2) (a, b, e, f) and either Iba1 (c, e) or glial fibrillary acidic protein (GFAP) (d, f) in the frontal cortex of BCCAO/R rats. Scale bars E = A, C: 25 μm; F = B, D: 10 μm
Statistical analysis
Data from the two experimental groups, sham-operated animals and the BCCAO/R ones, are depicted in the figures as mean ± standard deviation (S.D.) and statistical differences were determined by unpaired Student’s t-test.
Results
eCB and fatty acid profiles in brain tissue
In the frontal cortex AEA, 2-AG, and PEA were found significantly increased in BCCAO/R rats as compared to the sham-operated ones (p < 0.05) by 41, 51 and 37%, respectively (Fig. 1a–c), while DHA decreased by about 7% (p < 0.001) (Fig. 1d), and lipoperoxides, considered as a molecular marker of oxidative stress, increased by 57% (p < 0.05) (Fig. 1e). No statistically significant changes were observed in the temporal-occipital cortex (data not shown).
eCB and fatty acid profiles in plasma
In BCCAO/R rats, analysis of eCBs and congeners revealed that levels of AEA increased significantly by about 38% compared to sham rats (p < 0.05) (Fig. 1f), whereas no change was observed in 2-AG, PEA, and OEA levels. A strong increase of lipoperoxides could also be detected (52%; p < 0.05) (Fig. 1g). No statistically significant changes were observed in the temporal-occipital cortex (data not shown).
Western blot
After the BCCAO/R, protein changes were detected only in the frontal cortex homogenates, whereas the temporal-occipital cortex appeared unaffected. Thus, the relative levels of the CB1 receptor protein increased by 101% (p < 0.0001) (Fig. 2a, b), that of CB2 by 51% (p < 0.05) (Fig. 2c, d) and that of COX-2 by 50% (Fig. 2e, f) (p < 0.05). No changes were detectable for the relative levels of PPAR-α (Fig. 2g, h). The antibodies against CB1 and COX-2 were also the only ones to produce a reliable immunostaining in tissue sections of rat cerebral cortex. For this reason, the following immunohistochemical data are based exclusively on the immunoreactivity that they provided.
Immunohistochemistry
In order to find a possible association between the molecular changes observed by HPLC and Western Blot analyses and the tissue morphology, immunoreactivities to CB1 and COX-2 were also examined in the cerebral cortex. All markers labelled neuronal structures distributed throughout the rostro-caudal extension of the frontal cortex (Figs 3 and 4) and the temporal-occipital cortex (data not shown).
Fig. 3.

CB1-like immunoreactivity in the frontal cortex. Brain sections of sham-operated and BCCAO/R rats are shown in (a) and (b), respectively. Panels (a) and (b) are representative of observations carried out in 6 sham-operated and 6 BCCAO/R rats. Positive nervous structures are distributed throughout the cortical layers. m, molecular layer; wm, white matter. Scale bars: 50 μm
CB1 receptor-antibody labelled a dense to moderate plexus of beaded fibers and a discrete number of neuronal cell bodies distributed throughout the cortical layers (Fig. 3). As a general rule the density of labelled nerve terminals and fibers appeared higher in BCCAO/R animals (Fig. 3b) than in the sham-operated ones (Fig. 3a). By contrast, the COX-2-like immunoreactivity appeared as intracytoplasmic granules of different density in the perikaryon and proximal processes of neuronal cells (Fig. 4), being intensely stained and easily detectable in layers II/III and V. As a general rule, the staining intensity and density of labelled structures were higher in BCCAO/R (Fig. 4b) than in sham-operated rat brains (Fig. 4a). Representative double immunostainings for COX-2 and either glial marker Iba1 (for microglia) or GFAP (for astrocytes), carried out in the BCCAO/R rats, showed the absence of colocalization and demonstrated that COX-2 immunolabelling was localized to neuronal cell bodies (Fig. 5).
Discussion
The primary findings of this study are that BCCAO for 30 min followed by 60 min reperfusion of the brain is sufficient to cause molecular changes including: i) a significant positive modulation of the eCB system, involving the cerebral tissue and plasmatic levels of eCBs and congeners and tissue expression of the CB1 receptor; ii) a decrease of DHA tissue levels; iii) an increase of lipoperoxide levels; iv) a parallel increase of COX-2 expression in frontal cortex suggesting that they are part of a physiological response to the hypoperfusion/reperfusion insult.
Modulation of the eCB system by hypoperfusion/reperfusion
According to previous studies [12, 31, 32], our data point out that in cerebral cortex there are basal differences in the occurrence and relative concentrations of eCBs, the tissue content of which is increased after BCCAO/R. This last observation is in line with studies showing that brain focal ischemia and reperfusion activate the eCB system by driving a combination of biochemical adaptations of the NAE degrading and synthesizing enzymes that, collectively, lead to the accumulation of eCBs [12, 33]. The eCB congener PEA is an important player in the endogenous defense against neuroinflammation [33] and has beneficial pharmacological effects in several animal models of inflammation [11, 33–35]. In particular, PEA may exert neuroprotective effects by modulating the immune cells [36], activating PPAR-alpha [23, 37] and blunting COX-2 activity [11]. Exogenous administration of PEA in an acute stroke model is effective in reducing the infarct size [7]. Moreover, in human stroke eCBs and congeners have been shown to become detectable during the acute phase of an ischemic stroke where they may play a role through multiple potential mechanisms [17]. Research into this suggested that the inhibition or modulation of the enzymatic breakdown of PEA may represent a complementary therapeutic approach to counteract neuroinflammation [33].
Our data also show that after BCCAO/R a concomitant 38% increase of AEA could be detected in plasma. It is difficult to speculate whether the eCB modifications in brain tissue are directly correlated to the observed increase in AEA plasma levels. Clinical studies support the notion that neurological and neuropsychiatric disorders are characterized by detectable changes in eCB plasma levels [17 and refs therein]. Raise in the eCB levels in acute stroke patients have been detected by in vivo microdialysis [5] and plasma analysis [17]. Interestingly, the increases of AEA and PEA plasma level have been reported to show a positive correlation with infarct volume and/or neurological disability, so that patients with higher AEA and PEA levels had greater neurological impairment [17]. It has been proposed that a contribution to eCB and congener plasma levels might be due to a brain damage “spill over” effect to the peripheral circulation, the extent of which may reflect the severity of central nervous system pathology [14, 17]. On the other hand, it has been reported that the BCCAO modifies the pial microvasculature and that restoration of blood circulation in reperfusion causes a reactive hyperaemia [4]. Whether similar changes in vascular tone are responsible for setting off or modulating the brain “spill over” of eCBs into systemic circulation is an important issue to be taken into consideration. As for the functional meaning of the increase in eCB levels following the BCCAO/R, it can be inferred that it contributes to neuroprotection by different pathways. Whether in the BCCAO/R model used in this study the apparently concomitant raise in eCB concentrations and COX-2 protein levels are mutually dependent events is an issue that remains to be clarified [38].
In this study, we have also demonstrated that, after BCCAO/R, higher concentration of eCBs are associated with an increase of the relative levels of CB1 and CB2 receptor proteins. We may speculate that CB1 may act synergistically to increase eCB tone, which may be critical for protection against transient hypoperfusion/reperfusion cerebral insult [25]. Moreover, in agreement with previous observations [39], the extensive CB1-positive innervation that we observed in the cerebral cortex may suggest that intrinsic local circuits may contribute to the modulation of the reperfusion-induced tissue challenge. It is tempting to hypothesize that the BCCAO/R-induced increase of relative CB2 protein levels adds to this framework, probably regulating the production of pro-inflammatory molecules by glial cells, through which CB2 may either prevent the detrimental effects of neuroinflammatory reaction or participate in adaptive changes to the brain insult [40, 41]. As already proposed [42, 43], a parallel increase of the levels of eCBs and CB1 receptors indicates a sensitization of the cannabinoidergic system that may contribute to the regulation of cellular functions that depend upon CB1 receptor activation. It can be suggested that, after BCCAO/R, this sensitization modulates events such as neurotransmitter release, calcium cellular influx, oxidative stress damage, and vascular tone that appear to be crucial in the response of cerebral tissue to the ischemia/reperfusion insult. Further, in light of the evidence that eCBs may act as ligands for receptors other than CBs [44], it is interesting that the endocannabinoid and endovanilloid pathways have been found to antagonistically interact to adjust synaptic strength of inhibitory synapses [45].
Evidences for hypoperfusion/reperfusion-induced oxidative stress
In line with our previous findings [3], the present data indicate that the BCCAO/R causes a significant decrease of DHA, a polyunsaturated fatty acid that is richly endowed and avidly retained in the brain [46, 47] and confirm that the BCCAO/R model is sufficient to perturb tissue homeostasis by disrupting the normal DHA levels and thus by potentially affecting the efficiency of membrane-depending molecular mechanisms [48]. DHA is recognized to be particularly liable to lipid peroxidation [49] and, therefore, to be potentially apt to contribute to the hypoperfusion/reperfusion-induced oxidative stress. However, several lines of evidence have associated increased levels of DHA with tissue protection in neuroinflammation [49], suggesting that DHA per se does not increase susceptibility to oxidative stress. Data in rodents and healthy humans support this inference, as dietary supplementation with n-3 PUFAs does not affect lipid peroxidation [50, 51]. By contrast, since DHA has been shown to have a role in neuroprotection after brain hypoxia and ischemia, it is relevant that recent prophylactic and therapeutic approaches for cerebrovascular disease take into account the pathways of brain accretion and delivery of DHA [48, 52].
In our study, eCB changes were further characterized by a marked increase of tissue and plasmatic concentrations of lipoperoxides that is one of the hallmarks of the reperfusion-induced oxidative stress [53, 54]. Lipoperoxides are quite unstable compounds that are capable of extending the free radical oxidative damage, and forming proinflammatory substances [53, 54]. The increase in lipoperoxide levels without any evident histological alteration of cerebral tissue is in line with previous experimental findings on a rat model of BCCAO/R similar to ours [55].
Clinical implications
Increase of levels of lipoperoxides and a concomitant increase of their catabolism in peroxisomes have been shown to be directly correlated with the hypoperfusion/reperfusion-induced oxidative challenge in patients undergoing carotid endoarterectomy [55]. In particular, it is interesting that peroxisomal beta-oxidation increased during the first 30 min of reperfusion only in patients having contralateral carotid stenosis higher than 50% to decrease thereafter within 2 h from reperfusion [55].
Several physiopathological conditions, not necessarily associated with early obvious neurological signs [56], share the occurrence of cerebral hypoperfusion episodes for which the detection of molecular indicators in the early hours may be useful in clinical settings to prevent irreversible cerebral damage. The question of whether plasmatic changes of AEA and lipoperoxides could represent additional specific markers in humans should be further investigated.
Conclusions
The present study showed that the 30/60 min BCCAO/R procedure activates the ECS in rat and induces parallel changes in the fatty acid tissue profile (namely decreased levels of DHA and increased the lipoperoxides) and COX-2 levels in the rat frontal cortex. In addition, we found that BCCAO/R increased plasmatic levels of anandamide and lipoperoxides. The molecular changes induced by the BCCAO/R are evaluated on the basis of a single time point of reperfusion and, so far, this aspect represents an intrinsic limitation. Additional studies are warranted to evaluate both the time course of these changes during longer time points of reperfusion (e.g., at 6, 12 and 24 h after BCCAO/R) and the possible effects of dietary compounds in preventing BCCAO/R-induced oxidative stress.
In conclusion this study shows that BCCAO/R-induced positive modulation of the ECS. As far as we aware, this is the first study that has investigated early changes that can be easily traced in brain tissue as well as in plasma, and may be interpreted as indicative of the tissue physiological response to the oxidative stress induced by the BCCAO/R. The variations observed suggest that the activation of the ECS and the increase of pro-inflammatory substances are events that may be directly correlated.
Aknowledgements
The study was partially supported by Fondazione Banco di Sardegna. LP was funded with a research fellowship by the Regione Autonoma della Sardegna.
Funding
This work was supported by Grant from University of Cagliari (Progetti di Ricerca di Interesse Dipartimentale, PRID 2015) and Regione Autonoma della Sardegna (LP).
Availability of data and materials
The datasets obtained during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors’ contributions
MQ and SB conceived the study, participated in its design and coordination and drafting, and wrote the manuscript; LP conceived and participated in the design of the study, carried out the protein analysis, elaborated the data; TM performed the surgery, carried out protein analysis and elaborated the data; MPS and MB carried out the tissue sampling and performed the statistical analysis of western blot data; SL, GC and EM, participated in the design of the study, performed the eCB analysis and elaborated the data; LM performed the fatty acid analysis and elaborated the data; MC gave a critical contribution to the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- AEA, anandamide
N-arachidonoyl-ethanolamide
- 2-AG
2-arachidonoyl-glycerol
- BCCAO/R
Bilateral common carotid artery occlusion/reperfusion
- COX-2
Cyclooxygenase-2
- DHA
Docosahexaenoic acid
- eCBs
Endocannabinoids
- ECS
Endocannabinoid system
- GAPDH
Glyceraldehyde phosphate dehydrogenase
- NAE
N-acylethanolamides
- OEA
Oleoylethanolamide
- PB
Phosphate buffer
- PBS
Phosphate buffered saline
- PEA
Palmitoylethanolamide
- PPAR
Peroxisome-proliferator activated receptor
- PUFA
Polyunsaturated fatty acids
- SDS
Sodium dodecyl sulfate
- TBS-T
Tris-buffered saline-Tween 20
Contributor Information
Marina Quartu, Phone: 0039 070 6754084, Email: quartu@unica.it.
Laura Poddighe, Email: laura.poddighe@unica.it.
Tiziana Melis, Email: tiziana.melis@unica.it.
Maria Pina Serra, Email: mpserra@unica.it.
Marianna Boi, Email: marianna.boi@unica.it.
Sara Lisai, Email: saralisy@alice.it.
Gianfranca Carta, Email: Giacarta28@hotmail.com.
Elisabetta Murru, Email: m.elisabetta.murru@tiscali.it.
Laura Muredda, Email: lau.muredda1@studenti.unica.it.
Maria Collu, Email: mcollu@unica.it.
Sebastiano Banni, Email: banni@unica.it.
References
- 1.Traystman RJ, Kirsch JR, Koehler RC. Oxygen radical mechanisms of brain injury following ischemia and reperfusion. J Appl Physiol. 1991;71:1185–95. doi: 10.1152/jappl.1991.71.4.1185. [DOI] [PubMed] [Google Scholar]
- 2.Coyle P, Panzenbeck MJ. Collateral development after carotid artery occlusion in Fischer 344 rats. Stroke. 1990;21:316–21. doi: 10.1161/01.STR.21.2.316. [DOI] [PubMed] [Google Scholar]
- 3.Quartu M, Serra MP, Boi M, Pillolla G, Melis T, Poddighe L, et al. Effect of acute administration of Pistacia lentiscus L. essential oil on rat cerebral cortex following transient bilateral common carotid artery occlusion. Lipids Health Dis. 2012;11:8. doi: 10.1186/1476-511X-11-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lapi D, Vagnani S, Pignataro G, Esposito E, Paterni M, Colantuoni A. Rat pial microvascular responses to transient bilateral common carotid artery occlusion and reperfusion: quercetin’s mechanism of action. Front Physiol. 2012;3:99. doi: 10.3389/fphys.2012.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schäbitz WR, Giuffrida A, Berger C, Aschoff A, Schwaninger M, Schwab S, et al. Release of fatty acid amides in a patient with hemispheric stroke: a microdialysis study. Stroke. 2002;33:2112–4. doi: 10.1161/01.STR.0000023491.63693.18. [DOI] [PubMed] [Google Scholar]
- 6.Melis MP, Angioni E, Carta G, Murru E, Scanu P, Spada S, et al. Characterization of conjugated linoleic acid and its metabolites by RPHPLC with diode array detector. Eur J Lipid Sci Technol. 2001;103:617–21. doi: 10.1002/1438-9312(200109)103:9<617::AID-EJLT6170>3.0.CO;2-C. [DOI] [Google Scholar]
- 7.Schomacher M, Müller HD, Sommer C, Schwab S, Schäbitz WR. Endocannabinoids mediate neuroprotection after transient focal cerebral ischemia. Brain Res. 2008;1240:213–20. doi: 10.1016/j.brainres.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 8.Di Marzo V, Petrosino S. Endocannabinoids and the regulation of their levels in health and disease. Curr Opin Lipidol. 2008;18:129–40. doi: 10.1097/MOL.0b013e32803dbdec. [DOI] [PubMed] [Google Scholar]
- 9.Di Marzo V. The endocannabinoid system: its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol Res. 2009;60:77–84. doi: 10.1016/j.phrs.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 10.Conti S, Costa B, Colleoni M, Parolaro D, Giagnoni G. Antiinflammatory action of endocannabinoid palmitoylethanolamide and the synthetic cannabinoid nabilone in a model of acute inflammation in the rat. Br J Pharmacol. 2002;135:181–7. doi: 10.1038/sj.bjp.0704466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costa B, Conti S, Giagnoni G, Colleoni M. Therapeutic effect of the endogenous fatty acid amide, palmitoylethanolamide, in rat acute inflammation: inhibition of nitric oxide and cyclo-oxygenase systems. Br J Pharmacol. 2002;137:413–20. doi: 10.1038/sj.bjp.0704900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hillard CJ. Role of cannabinoids and endocannabinoids in cerebral ischemia. Curr Pharm Des. 2008;14:2347–61. doi: 10.2174/138161208785740054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang M, Adler MW, Abood ME, Ganea D, Jallo J, Tuma RF. CB2 receptor activation attenuates microcirculatory dysfunction during cerebral ischemic/reperfusion injury. Microvasc Res. 2009;78:86–94. doi: 10.1016/j.mvr.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Centonze D, Battistini L, Maccarrone M. The endocannabinoid system in peripheral lymphocytes as a mirror of neuroinflammatory diseases. Curr Pharm Res. 2008;14:2370–42. doi: 10.2174/138161208785740018. [DOI] [PubMed] [Google Scholar]
- 15.Pacher P, Haskó G. Endocannabinoids and cannabinoid receptors in ischaemia-reperfusion injury and preconditioning. Br J Pharmacol. 2008;153:252–62. doi: 10.1038/sj.bjp.0707582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pellegrini-Giampietro DE, Mannaioni G, Bagetta G. Post-ischemic brain damage: the endocannabinoid system in the mechanisms of neuronal death. FEBS J. 2009;276:2–12. doi: 10.1111/j.1742-4658.2008.06765.x. [DOI] [PubMed] [Google Scholar]
- 17.Naccarato M, Pizzuti D, Petrosino S, Simonetto M, Ferigo L, Grandi FC, et al. Possible anandamide and palmitoylethanolamide involvement in human stroke. Lipids Health Dis. 2010;9:47. doi: 10.1186/1476-511X-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shohami E, Cohen-Yeshurun A, Magid L, Algali M, Mechoulam R. Endocannabinoids and traumatic brain injury. Br J Pharmacol. 2011;163:1402–10. doi: 10.1111/j.1476-5381.2011.01343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozak KR, Crews BC, Morrow JD, Wang LH, Ma YH, Weinander R, et al. Metabolism of the endocannabinoids, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. J Biol Chem. 2002;277:44877–85. doi: 10.1074/jbc.M206788200. [DOI] [PubMed] [Google Scholar]
- 20.Jhaveri MD, Richardson D, Robinson I, Garle MJ, Patel A, Sun Y, et al. Inhibition of fatty acid amide hydrolase and cyclooxygenase-2 increases levels of endocannabinoid related molecules and produces analgesia via peroxisome proliferator-activated receptor-alpha in a model of inflammatory pain. Neuropharmacol. 2008;55:85–93. doi: 10.1016/j.neuropharm.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, Chen C. Endocannabinoid 2-arachidonoylglycerol protects neurons by limiting COX-2 elevation. J Biol Chem. 2008;283:22601–11. doi: 10.1074/jbc.M800524200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hermanson DJ, Gamble-George JC, Marnett LJ, Patel S. Substrate-selective COX-2 inhibition as a novel strategy for therapeutic endocannabinoid augmentation. Trends Pharmacol Sci. 2014;35:358–67. doi: 10.1016/j.tips.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, et al. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol. 2005;67:15–9. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- 24.Muthian S, Rademacher DJ, Roelke CT, Gross GJ, Hillard CJ. Anandamide content is increased and CB1 cannabinoid receptor blockade is protective during transient, focal cerebral ischemia. Neuroscience. 2004;129:743–50. doi: 10.1016/j.neuroscience.2004.08.044. [DOI] [PubMed] [Google Scholar]
- 25.Parmentier-Batteur S, Jin K, Mao XO, Xie L, Greenberg DA. Increased severity of stroke in CB1 cannabinoid receptor knock-out mice. J Neurosci. 2002;22:9771–5. doi: 10.1523/JNEUROSCI.22-22-09771.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Marzo V, Goparaju SK, Wang L, Liu J, Bàtkai S, Jàrai Z, et al. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature. 2001;410:822–5. doi: 10.1038/35071088. [DOI] [PubMed] [Google Scholar]
- 27.Iwasaki Y, Ito S, Suzuki M, Nagahori T, Yamamoto T, Konno H. Forebrain ischemia induced by temporary bilateral common carotid occlusion in normotensive rats. J Neurol Sci. 1989;90:155–65. doi: 10.1016/0022-510X(89)90098-1. [DOI] [PubMed] [Google Scholar]
- 28.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 6. Amsterdam-Boston: Elsevier Academic Press; 2007. [Google Scholar]
- 29.Banni S, Carta G, Contini MS, Angioni E, Deiana M, Dessi MA, et al. Characterization of conjugated diene fatty acids in milk, dairy products, and lamb tissues. J Nutr Biochem. 1996;7:150–5. doi: 10.1016/0955-2863(95)00193-X. [DOI] [Google Scholar]
- 30.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurements with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 31.Bisogno T, Berrendero F, Ambrosino G, Cebeira M, Ramos JA, Fernandez-Ruiz JJ, et al. Brain regional distribution of endocannabinoids: implications for their biosynthesis and biological function. Biochem Biophys Res Commun. 1999;256:377–80. doi: 10.1006/bbrc.1999.0254. [DOI] [PubMed] [Google Scholar]
- 32.Liedhegner ES, Sasman A, Hillard CJ. Brain region-specific changes in N-acylethanolamine contents with time of day. J Neurochem. 2014;128:491–506. doi: 10.1111/jnc.12495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skaper SD, Facci L, Barbierato M, Zusso M, Bruschetta G, Impellizzeri D, et al. N-Palmitoylethanolamine and neuroinflammation: a novel therapeutic strategy of resolution. Mol Neurobiol. 2015;52:1034–42. doi: 10.1007/s12035-015-9253-8. [DOI] [PubMed] [Google Scholar]
- 34.De Filippis D, D’Amico A, Cipriano M, Petrosino S, Orlando P, Di Marzo V, et al. Levels of endocannabinoids and palmitoylethanolamide and their pharmacological manipulation in chronic granulomatous inflammation in rats. Pharmacol Res. 2010;61:321–8. doi: 10.1016/j.phrs.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 35.Balvers MG, Verhoeckx KC, Meijerink J, Wortelboer HM, Witkamp RF. Measurement of palmitoylethanolamide and other N-acylethanolamines during physiological and pathological conditions. CNS Neurol Disord Drug Targets. 2013;12:23–33. doi: 10.2174/1871527311312010007. [DOI] [PubMed] [Google Scholar]
- 36.Skaper SD, Facci L, Giusti P. Glia and mast cells as targets for palmitoylethanolamide, an anti-inflammatory and neuroprotective lipid mediator. Mol Neurobiol. 2013;48:340–52. doi: 10.1007/s12035-013-8487-6. [DOI] [PubMed] [Google Scholar]
- 37.Hansen HS. Palmitoylethanolamide and other anandamide congeners. Proposed role in the diseased brain. Exp Neurol. 2010;224:48–55. doi: 10.1016/j.expneurol.2010.03.022. [DOI] [PubMed] [Google Scholar]
- 38.Davies BL, Arpin DJ, Volkman KG, Corr B, Reelfs H, Harbourne RT, et al. Neurorehabilitation strategies focusing on ankle control improve mobility and posture in persons with multiple sclerosis. J Neurol Phys Ther. 2015;39:225–32. doi: 10.1097/NPT.0000000000000100. [DOI] [PubMed] [Google Scholar]
- 39.Egertová M, Elphick MR. Localisation of cannabinoid receptors in the rat brain using antibodies to the intracellular C-terminal tail of CB. J Comp Neurol. 2000;422:159–71. doi: 10.1002/(SICI)1096-9861(20000626)422:2<159::AID-CNE1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 40.Benito C, Tolón RM, Pazos MR, Núñez E, Castillo AI, Romero J. Cannabinoid CB2 receptors in human brain inflammation. Br J Pharmacol. 2008;153:277–85. doi: 10.1038/sj.bjp.0707505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernández-Ruiz J, Moro MA, Martínez-Orgado J. Cannabinoids in neurodegenerative disorders and stroke/brain trauma: from preclinical models to clinical applications. Neurotherapeutics. 2015;12:793–806. doi: 10.1007/s13311-015-0381-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Svízenská I, Dubový P, Sulcová A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures-a short review. Pharmacol Biochem Behav. 2008;90:501–11. doi: 10.1016/j.pbb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 43.Vinod KY, Arango V, Xie S, Kassir SA, Mann JJ, Cooper TB, et al. Elevated levels of endocannabinoids and CB1 receptor-mediated G-protein signalling in the prefrontal cortex of alcoholic suicide victims. Biol Psychiatry. 2005;57:480–6. doi: 10.1016/j.biopsych.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 44.Di Marzo V, Breivogel CS, Tao Q, Bridgen DT, Razdan RK, Zimmer AM, et al. Levels, metabolism, and pharmacological activity of anandamide in CB(1) cannabinoid receptor knockout mice: evidence for non-CB(1), non-CB(2) receptor-mediated actions of anandamide in mouse brain. J Neurochem. 2000;75:2434–44. doi: 10.1046/j.1471-4159.2000.0752434.x. [DOI] [PubMed] [Google Scholar]
- 45.Lee SH, Ledri M, Tóth B, Marchionni I, Henstridge CM, Dudok B, et al. Multiple forms of endocannabinoid and endovanilloid signaling regulate the tonic control of GABA release. J Neurosci. 2015;35:10039–57. doi: 10.1523/JNEUROSCI.4112-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia MC, Ward G, Ma YC, Salem N, Jr, Kim HY. Effect of docosahexaenoic acid on the synthesis of phosphatidylserine in rat brain in microsomes and C6 glioma cells. J Neurochem. 1998;70:24–30. doi: 10.1046/j.1471-4159.1998.70010024.x. [DOI] [PubMed] [Google Scholar]
- 47.Niemoller DT, Bazan NG. Docosahexaenoic acid neurolipidomics. Prostaglandins Other Lipid Mediat. 2010;91:85–9. doi: 10.1016/j.prostaglandins.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mayurasakorn K, Williams JJ, Ten VS, Deckelbaum RJ. Docosahexaenoic acid: brain accretion and roles in neuroprotection after brain hypoxia and ischemia. Curr Opin Clin Nutr Metab Care. 2011;14:158–67. doi: 10.1097/MCO.0b013e328342cba5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strokin M, Sergeeva M, Reiser G. Role of Ca2 + −independent phospholipaseA2 and n-3 polyunsaturated fatty acid docosahexaenoic acid in prostanoid production in brain: perspectives for protection in neuroinflammation. Int J Devl Neurosci. 2004;22:551–7. doi: 10.1016/j.ijdevneu.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 50.Ando K, Nagata K, Yoshida R, Kikugawa K, Suzuki M. Effect of n-3 polyunsaturated fatty acid supplementation on lipid peroxidation of rat organs. Lipids. 2000;35:401–7. doi: 10.1007/s11745-000-538-6. [DOI] [PubMed] [Google Scholar]
- 51.Nälsén C, Vessby B, Berglund L, Uusitupa M, Hermansen K, Riccardi G, et al. Dietary (n-3) fatty acids reduce plasma F2 isoprostanes but not prostaglandin F2alpha in healthy humans. J Nutr. 2006;36:1222–8. doi: 10.1093/jn/136.5.1222. [DOI] [PubMed] [Google Scholar]
- 52.Williams JJ, Mayurasakorn K, Vannucci SJ, Mastropietro C, Bazan NG, Ten VS, et al. N-3 fatty acid rich triglyceride emulsions are neuroprotective after cerebral hypoxic-ischemic injury in neonatal mice. PLoS One. 2013;8:2. doi: 10.1371/journal.pone.0056233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adibhatla RM, Hatcher JF. Altered lipid metabolism in brain injury and disorders. Subcell Biochem. 2008;49:241–68. doi: 10.1007/978-1-4020-8831-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Niki E. Lipid peroxidation: physiological levels and dual biological effects. Free Radic Biol Med. 2009;47:469–84. doi: 10.1016/j.freeradbiomed.2009.05.032. [DOI] [PubMed] [Google Scholar]
- 55.Yanpallewar SU, Hota D, Rai S, Kumar M, Acharya SB. Nimopidine attenuates biochemical, behavioral and histopathological alterations induced by acute transient and long-term bilateral common carotid occlusion in rats. Pharmacol Res. 2004;49:143–50. doi: 10.1016/j.phrs.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 56.Banni S, Montisci R, Sanfilippo R, Finco G, Sanna D, Giordano E, et al. Physiological response to lipid peroxidation in ischemia and reperfusion during carotid endarterectomy. Lipids Health Dis. 2010;9:41. doi: 10.1186/1476-511X-9-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets obtained during and/or analyzed during the current study are available from the corresponding author on reasonable request.