

Abstract
The spleen tyrosine kinase (SYK) regulates immune cell activation in response to engagement of a variety of receptors, making it an intriguing target for the treatment of inflammatory and autoimmune disorders as well as certain B‐cell malignancies. We have previously reported on the discovery and preclinical characterization of PRT062607, a potent and highly selective inhibitor of SYK that exhibits robust anti‐inflammatory activity in a variety of animal models. Here we present data from our first human studies aimed at characterizing the pharmacokinetics (PK), pharmacodynamics (PD), and safety of PRT062607 in healthy volunteers following single and multiple oral administrations. PRT062607 demonstrated a favorable PK profile and the ability to completely inhibit SYK activity in multiple whole‐blood assays. The PD half‐life in the more sensitive assays was approximately 24 hours and returned to predose levels by 72 hours. Selectivity for SYK was observed at all dose levels tested. Analysis of the PK/PD relationship indicated an IC50 of 324 nM for inhibition of B‐cell antigen receptor‐mediated B‐cell activation and 205 nM for inhibition of FcεRI‐mediated basophil degranulation. PRT062607 was safe and well tolerated across the entire range of doses. Clinical PK/PD was related to in vivo anti‐inflammatory activity of PRT062607 in the rat collagen‐induced arthritis model, which predicts that therapeutic concentrations may be safely achieved in humans for the treatment of autoimmune disease. PRT062607 has a desirable PK profile and is capable of safely, potently, and selectively suppressing SYK kinase function in humans following once‐daily oral dosing.
Keywords: SYK, PRT062607, autoimmune, allergy, lymphoma, immunopharmacology
Spleen tyrosine kinase (SYK) is a cytosolic protein tyrosine kinase that, upon receptor engagement, binds to diphosphorylated immune tyrosine activation motif (ITAM), resulting in tyrosine phosphorylation of multiple downstream substrates that ultimately regulate the cell's survival, functional activation, differentiation, and clonal expansion. In B cells, signaling through the B‐cell antigen receptor (BCR) is transduced following antigen ligation by its association with immunoglobulins Igα and Igβ, each bearing an ITAM, leading to cellular activation, clonal expansion, and differentiation into Ig‐secreting plasma cells.1 In addition to its role in B‐cell development and the generation of antibody‐producing plasma cells,2, 3 SYK is also required for BCR‐mediated uptake and presentation of antigen on MHC.4 Fc receptors also associate with ITAM‐containing proteins and utilize SYK for the initiation of Ig‐mediated inflammatory responses by mast cells and basophils (via FcεR)5 and for the promotion of antigen presentation, oxidative burst, phagocytosis, and cytokine release by eosinophils, neutrophils, dendritic cells, and macrophages (via FcγRs).6 FcγR‐dependent antigen/antibody immune complex uptake and presentation by antigen‐presenting cells are also dependent on SYK.7 SYK inhibition may therefore indirectly suppress T‐cell activation. Several integrins similarly rely on ITAM‐mediated SYK activation,8 which is critical for neutrophil oxidative burst and leukocyte recruitment to sites of inflammation. Hence, SYK inhibition has the potential to intervene in several autoimmune mechanisms that act in concert to promote tissue damage and warrants further exploration as a therapeutic target in both B‐cell malignancies and autoimmune/allergicy disorders.
PRT062607 (also referred to as P505‐15) is a potent and highly selective small‐molecule inhibitor of SYK discovered by Portola Pharmaceuticals Inc (South San Francisco, California). The specificity and potency of SYK inhibition by PRT062607 was previously evaluated in purified kinase assays and cellular assays as well as in rodent models to determine its pharmacokinetics (PK) and pharmacodynamics (PD) and impact on inflammatory disease. In a specificity screen of 270 kinases, PRT062607 exhibited greater than 80‐fold selectivity over the next most potently inhibited kinase and did not cross‐react with the structural homologue ZAP70.9 Other published reports demonstrated the ability of PRT062607 to selectively and completely suppress BCR‐ and FcR‐mediated immune functional responses in whole blood from healthy volunteers and in whole blood from patients with active rheumatoid arthritis.10 The antitumor activity of PRT062607 in diffuse large B‐cell lymphoma cell lines and primary CLL was also demonstrated.11, 12, 13
Described herein, a single ascending dose (SAD) escalation study in healthy volunteers was initiated at 3 mg and escalated to 400 mg, at which complete inhibition of SYK was achieved in multiple relevant assays performed in whole blood collected over a 72‐hour period following dosing. A multiple ascending dose (MAD) escalation study was also conducted in healthy volunteers, who were dosed once daily for 10 consecutive days, starting at 11 mg and escalating to 110 mg. The aim of the first in‐human studies was to evaluate the safety, tolerability, PK, and PD of PRT062607 following oral dosing. The PD variables measured in whole blood of dosed subjects included both SYK‐dependent (BCR‐induced pERK and up‐regulation of cell surface CD69 in B cells, and FcεRI‐induced up‐regulation of basophil cell surface CD63) and SYK‐independent (BCR‐induced pSYK and PMA‐induced pERK in B cells, fMLP‐induced up‐regulation of basophil cell surface CD63, and CD3/CD28‐induced up‐regulation of T‐cell surface CD69) measures of immune cell functional responses. Complete inhibition of SYK in peripheral whole‐blood assays was achieved at steady state at the 110‐mg dose. No inhibition of SYK‐independent immune functional responses was observed. PK/PD evaluations enabled dose projections for efficacy in human autoimmune disease based on exposures required for efficacy in animal models.
Methods
Clinical Trial Design
Study protocols were approved by the local site IRB. Written informed consent was obtained from all subjects prior to study participation.
The SAD study was a single‐center, blinded, randomized, placebo‐controlled, ascending, single‐dose study of an oral PRT062607 suspension or its matching placebo, administered to healthy subjects. Doses ranged from 3 to 400 mg. Within the first 2 groups (n = 4 subjects), 1 subject received placebo and 3 received PRT062607. Within the next groups (n = 8 subjects), 2 subjects received placebo and 6 received PRT062607. At each dose level, 2 sentinel subjects (1 active, 1 placebo) were dosed at least 48 hours in advance of the other subjects in their respective group. There was at least a 1‐week observation period after dosing at each of the dose levels. The primary objectives were to (1) assess the safety and tolerability of single ascending oral doses of PRT062607 in healthy subjects, and to (2) assess the PK and PD properties of single ascending oral doses of PRT062607.
The MAD study was a single‐center, double‐blind, randomized, placebo‐controlled, ascending, multiple‐dose study of a solid formulation (11 mg gelatin capsules) of PRT062607 or its matching placebo, administered to healthy subjects every 24 hours for 10 consecutive days. A total of 32 subjects were enrolled, divided into 4 sequential groups of 8 subjects at each dose level (6:2 active:placebo). For each subject, the total duration of the study was up to 17 weeks (4 weeks predose, 15 days in the clinical study unit, and 9‐11 weeks after the last dose of study drug, until the final outpatient follow‐up visit). The primary objectives were to (1) assess the safety and tolerability of multiple ascending oral doses of PRT062607 in healthy subjects and (2) determine the PK and PD properties of multiple ascending oral doses of a solid formulation of PRT062607 at steady state. Regular safety examinations were conducted throughout both studies.
Sample Collection
PK Collection
In the SAD study, blood samples for determination of PRT062607 concentrations were collected at predose and 0.5, 1, 2, 3, 4, 6, 9, 12, 24, 48, 72, 96, and 144 hours postdose. Blood sample collection intervals were shorter in the 2 lowest dose groups: up to 24 hours postdose for the 3‐mg group and up to 48 hours postdose for the 10‐mg group. Urine was collected prior to drug administration for analysis of baseline drug concentration. Batched urine was collected after oral administration at the following intervals: 0 to 12, 12 to 24, 24 to 48, 48 to 72, and 72 to 96 hours postdose. In the MAD study, blood samples were collected on day 1 (first dose) and 10 (steady state) at predose and 0.5, 1, 2, 3, 4, 6, 9, 12, and 24 hours postdose, and additional 48‐, 72‐, 96‐, and 144‐hour postdose samples were collected for day 10. For the 110‐mg group, additional 120‐ and 192‐hour postdose samples were collected for day 10. Peak (2 and/or 4 hours postdose) and trough (predose; to assess time to steady state) samples were collected during the course of the study on days 4, 7, 8, and 9. Urine was collected prior to drug administration for analysis of baseline drug concentration. Batched urine was collected after oral administration on days 1 and 10 at 0‐24 hours postdose.
PD Collection
In the SAD study PD samples were collected at predose and 1, 2, 4, 6, 9, 12, 24, 48, and 72 hours postdose for all dose groups. In the MAD study PD samples were collected on day 1 at predose and 1, 2, 4, 6, 9, 12, and 24 hours postdose, on day 4 at predose and 2 and 4 hours postdose, and on day 10 at predose and 0, 1, 2, 4, 6, 9, 12, 24, 48, and 72 hours postdose.
Analytical Methods
A sensitive and specific liquid chromatography‐tandem mass spectrometry (LC‐MS/MS) method was developed and validated by Worldwide Clinical Trials (Austin, Texas) and was used to determine PRT062607 concentrations in K2EDTA human plasma and urine. PRT062607 and stable labeled internal standard were extracted from human plasma and urine by solid‐phase extraction. Urine samples were fortified with 3‐[(3‐cholamidopropyl) dimethylammonio]‐1‐propanesulfonate (CHAPS) prior to extraction in order to prevent adsorption to polypropylene. The plasma method was validated for a range of 0.500 to 500 ng/mL, based on the analysis of 0.100 mL of human plasma. The urine method was validated for a range of 50.0 to 1000 ng/mL and 10.0 to 1000 ng/mL for the SAD and MAD study, respectively, based on the analysis of 0.100 mL of human urine. Chromatographic separations were performed over a Phenomenex Synergi Polar‐RP column (50 × 2.0 mm, 4 μm) (Phenomenex, Torrance, California). MS/MS analysis was performed using a Sciex API‐4000 triple quadrupole mass spectrometer with a TurboSpray ion source (Applied Biosystems, Foster City, California). The peak area of the m/z 394→360 PRT062607 product ion was measured against the peak area of the m/z 397→363 internal standard product ion. For the plasma assay, intra‐assay precision (%CV) and accuracy (% bias) were within 0.8% to 4.5% and –10.1% to 7.8%, respectively, and interassay precision (%CV) and accuracy (% bias) were 2.0% to 3.8% and −7.3% to 6.0%, respectively. For the urine assay, intra‐assay precision and accuracy were within 1.4% to 10.6% and −12.6% to 0.0%, respectively, and interassay precision and accuracy were 2.5% to 7.8% and −11.0% to −3.7%, respectively.
PK Analysis
SAD Study
Pharmacokinetic calculations based on PRT062607 plasma concentrations were performed by Quintiles Inc (Durham, North Carolina) using noncompartmental analysis and appropriate models (Model 200 extravascular administration) in WinNonlin Professional Version 5.2 (Pharsight Corporation, Mountain View, California). Individual plasma concentration data from each subject and the exact time points for blood sampling were used throughout the analysis. Pharmacokinetic parameters calculated from plasma PRT062607 concentrations included, but were not limited to, maximum plasma concentration (Cmax), time of maximum concentration (tmax), area under the plasma concentration‐time curve from time 0 to time of last measurable concentration (AUClast), area under the plasma concentration‐time curve from time 0 extrapolated to infinity (AUC0‐∞), apparent terminal rate constant (λz), and apparent terminal half‐life (t1/2). Dose‐normalized values for Cmax and AUClast were calculated and captured as Cmax/D and AUC/D, respectively. From urine data, cumulative amount excreted from time 0 to 96 hours postdose (Ae0‐96) and cumulative fraction of dose excreted unchanged in the urine from time 0 to 96 hours postdose (fe0‐96) were also determined.
MAD Study
Pharmacokinetic calculations based on PRT062607 plasma concentrations were performed by Covance Laboratories (Princeton, New Jersey) using noncompartmental analysis in WinNonlin Enterprise Version 5.2 (Pharsight Corporation, Mountain View, California). Individual plasma concentration data from each subject and the exact time points for blood sampling were used throughout the analysis. Pharmacokinetic parameters calculated from plasma PRT062607 concentrations included, but were not limited to, Cmax, tmax, AUC0‐24, λz, and t1/2. Dose‐normalized values for Cmax and AUC0‐24 were calculated and captured as Cmax/D and AUC/D, respectively. Accumulation ratio based on Cmax was calculated as Cmax,day 10/Cmax,day 1 and for AUC as AUC0‐24,day 10/AUC0‐24,day 1. Peak‐to‐trough ratio was computed as the ratio of Cmax on day 10 to the 24‐hour concentration postdose on day 10. Fluctuation on day 10 was calculated as (Cmax ‐ Cmin)/Cavg where Cmin is the minimum concentration and Cavg is the average concentration. From urine data, cumulative amount excreted from time 0 to 24 hours postdose (Ae0‐24) and cumulative fraction of dose excreted unchanged in the urine from time 0 to 24 hours postdose (fe0‐24) were also determined. Renal clearance (CLr) was calculated as the ratio of Ae0‐24 to AUC0‐24 on day 10.
Statistical Analysis for PK
Quantitative variables were summarized using descriptive statistics including N, arithmetic mean, standard deviation (SD), geometric mean, coefficient of variation (CV in %), median, and minimum and maximum values.
Reagents
The chemical structure of PRT062607 4‐((3‐(2H‐1,2,3‐triazol‐2‐yl)phenyl)amino)‐2‐(((1R,2S)‐2‐aminocyclohexyl)amino)pyrimidine‐5‐carboxamide acetate, also referred to as P505‐15, is published.9
Antibodies obtained from BD Biosciences (San Jose, California) include mouse anti–human SYK tyrosine (Y) 352/ZAP70Y319 phycoerythrin, CD3 allophycocyanin, CD19 allophycocyanin, CD14 alexafluor 488 and allophycocyanin conjugates, CD80 and CD86 phycoerythrin, CD69 phycoerythrin, MHCII peridinin‐chlorophyll protein, and CD1a allophycocyanin. The following reagents were procured from other sources: ERK threonine (T) 202/Y204 alexafluor 488 (Cell Signaling Technologies, Danvers, Massachusetts), goat anti–human IgE and anti–human IgD (Bethyl Laboratories, Montgomery, Texas), CD3/CD28 Dynabeads (Life Technologies Corporation, Grand Island, New York), CD14 microbeads and MS columns for monocyte purification from whole blood (Miltenyi Biotech, Auburn, California), recombinant human IL4 and GM‐CSF (R&D Systems, Minneapolis, Minnesota), BasoTest and Phagoburst kits for measuring basophil degranulation and neutrophil oxidative burst, respectively (Orpegen Pharma, Heidelberg, Germany), lymphoprep solution for peripheral blood mononuclear cell isolation (Stem Cell Technologies, Vancouver, Canada), phosphate buffered saline (PBS), bovine serum albumin (BSA), phorbol 12‐myristate 13‐acetate (PMA), and lipopolysaccharide (LPS) were all obtained from Sigma‐Aldrich (St. Louis, Missouri). Sheep red blood cells were purchased from MP Biomedicals (Burlingame, California), and sheep red blood cell opsonizing antibody was obtained from Cell Biolabs, Inc (San Diego, California).
Human Whole‐Blood Assays
Whole blood collected into lithium heparin vacutainer tubes was used for all assays. Basophil degranulation was measured using the BasoTest kit as previously described.9 Blood was stimulated with fMLP (supplied in the BasoTest kit) as an SYK‐independent specificity control or with anti‐IgE to cross‐link FcεRI leading to SYK‐dependent basophil degranulation. Basophil degranulation was measured by up‐regulation of surface‐expressed CD63. B‐cell activation was measured by CD69 up‐regulation on B cells following stimulation overnight (16‐18 hours) with anti‐IgD antibody or with PMA (80 nM). BCR signaling responses were measured by intracellular phospho‐flow cytometry detecting LYN‐mediated SYK Y352 phosphorylation as a specificity control, and ERK Y204 phosphorylation as an event downstream of SYK. PMA was used at a final concentration of 80 nM to induce ERK Y204 phosphorylation via protein kinase C as a specificity control. The B‐cell assays were also previously described in detail.9 In the MAD study, T‐cell function following different stimulations was also assessed. Blood (100 μL) was stimulated overnight with 10 μL CD3/CD28 Dynabeads or with 80 nM PMA. Blood was then stained with antibodies to detect surface CD3 and CD69. For each assay, percentage inhibition was calculated by normalizing to the signaling or functional response obtained with the predose sample.
Dendritic Cell Activation
An aliquot of 30 mL human whole blood was layered over 15 mL of lymphoprep density medium and centrifuged at 400g for 20 minutes to obtain PBMCs. Recovered cells were washed once in PBS containing 1% BSA and 2 mM EDTA (isolation buffer) and resuspended in 360 μL of ice‐cold isolation buffer. Then, 40 μL of CD14 microbeads was added to the cells and incubated 30 minutes on ice. Cells were washed once in isolation buffer and purified over an MS column per the manufacturer's recommendations. Isolated monocytes were suspended in tissue culture medium and verified for purity (>95%) by FACS analysis staining with CD14‐specific antibody. Monocytes were then aliquotted into 24‐well plates and cultured with 12.5 ng/mL IL4 and 20 ng/mL GM‐CSF for 5 days, after which they were stained for CD14 (monocyte marker) and CD1a (immature dendritic cell marker) and assessed for differentiation to immature dendritic cells by flow cytometry. Immature dendritic cells were then aliquotted 0.5 × 106 cells per well in a 6‐well plate and preincubated for 1 hour with various concentrations of PRT062607, then stimulated overnight with 1 μg/mL LPS as an SYK‐independent stimulation control, or with 50 μL antibody‐opsonized sheep red blood cells (opRBC) to elicit SYK‐dependent FcγR‐induced cellular activation. opRBC were prepared by washing 200 μL RBCs with PBS; they were then suspended in 1 mL PBS containing 2 μL opsonization solution and incubated at 37°C for 30 minutes. The RBCs were then washed twice in PBS and suspended in 1 mL PBS. Dendritic cell activation was measured by flow cytometry the next day by surface staining for CD80/86 and MHCII.
Neutrophil Oxidative Burst
Heparinized blood, 100 μL, was aliquotted into FACS tubes and preincubated with various concentrations of PRT062607 or vehicle control for 1 hour at 37°C in a tissue culture incubator prior to stimulation. Cells were stimulated with 50 μL opRBC as described before or with 20 μL of anE coli suspension used as an SYK‐independent stimulation control (supplied in the PhagoBurst kit). Blood was incubated with stimulations (or 50 μL of the supplied washing buffer as a nonstimulation control) for 10 minutes in a 37°C water bath. Detection of oxidative burst was performed as described in the protocols supplied with the PhagoBurst test kit.
Rat Collagen‐Induced Arthritis Model and Whole Blood Phospho‐Flow
The rat collagen‐induced arthritis (CIA) model was previously described in detail.9 Briefly, male Sprague‐Dawley rats were immunized with bovine collagen and randomized into treatment groups on development of hind‐paw inflammation with clinical scores of 1 to 2. Whole blood was drawn from immunized rats with inflammation scores of 1 to 2 into lithium‐heparin tubes, and 100‐μL aliquots were preincubated for 1 hour in a 37°C tissue culture incubator with various concentrations of PRT062607. Blood was then stimulated for 5 minutes with biotinylated anti–rat IgD, followed by the addition of 1 μg streptavidin for an additional 10 minutes. Whole blood was then fixed and lysed by the addition of 3 mL prewarmed (37°C) BD FACS Lyse Buffer (BD Biosciences, San Jose, California). Cells were washed twice in PBS, and cell membranes were permeablized in 50% methanol/PBS (prechilled to −20°C) for 1 hour at 4°C. Cells were then washed in PBS containing 1% bovine serum albumin and stained for 1 hour at room temperature with mouse anti–rat IgM phycoerythrin conjugate and rabbit anti–human/mouse ERK Y204 Alexafluor 488. B cells were gated based on IgM‐positive staining for evaluation of BCR‐induced signaling to ERK.
PD Analysis
The data for PD assays were analyzed using the software R together with the drc package.14 The equation was used to perform robust regression on the dose‐response data, which were transformed to percentage of inhibition.
The parameters c and d are the lower and upper limits with d fixed to 100% inhibition. The parameter e represents the dose with a response halfway between c and d (ED50), and b the relative slope around e. The approximate errors were calculated using the delta method.15 The data were visualized using the ggplot2 package. Doses required for specific response levels were calculated by inverse regression using the fitted model. Excel, Prism (GraphPad Software Inc, San Diego, California), and FlowJo (Ashland, Oregon) were used to analyze flow cytometry data.
Results
Pharmacokinetics of PRT062607
The PK and PD of PRT062607 were evaluated in 2 consecutive clinical studies following oral administration in healthy volunteers. In both studies escalation was terminated when complete target inhibition was achieved based on a range of SYK‐specific PD assays. Figure 1 represents the plasma concentration‐time profiles following single doses in the SAD study and at steady‐state (day 10) following 10 consecutive doses in the MAD study. Mean PK parameters for all cohorts in the SAD and MAD (at steady state) studies are summarized in Tables 1 and 2. In the SAD study, exposure parameters (AUC0‐24 and Cmax) increased with increasing dose of PRT062607 within the entire dose range of 3 mg to 400 mg with the exception of the 200‐mg dose, which exhibited slightly higher exposure than the 300‐mg dose. The increase in exposure parameters were more than dose proportional from 10 mg to 200 mg. Between the 200‐mg and 400‐mg doses, the exposure continued to increase in an approximately dose‐proportional manner. Mean Cmax and AUC as they relate to dose are presented graphically in Supplemental Figures S1A and B. Mean apparent terminal half‐life (range 57 to 94 hours) as well as median time to peak plasma concentration (range 1.0 to 4.0 hours) were similar across the selected dose range and appeared to be independent of the administered dose. Intersubject variability (%CV) for exposure parameters (Cmax and AUClast) were generally low to moderate (<40% for 6 out of 7 dose cohorts). Mean fraction of PRT062607 dose excreted as unchanged drug in urine ranged from 1.47% to 5.07%, indicating that urinary excretion is not the primary pathway for elimination of PRT062607.
Figure 1.
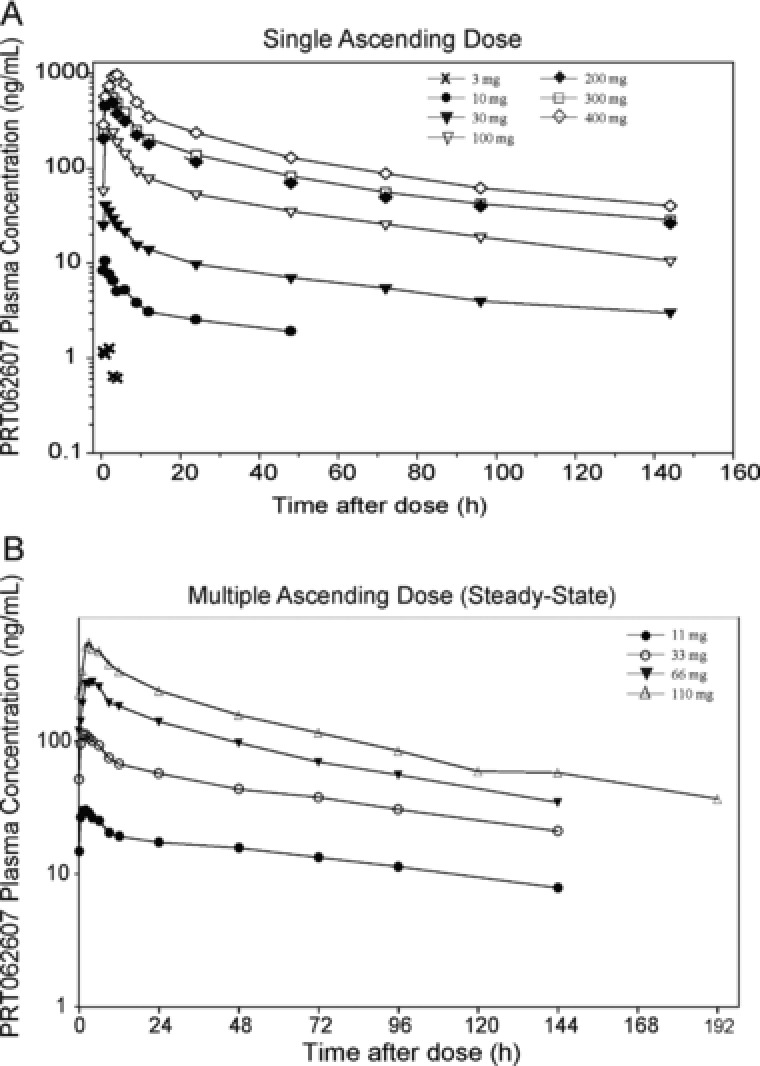
Plasma concentration‐time profiles following oral administration of PRT062607. Plasma concentrations (y‐axis, ng/mL) over time (x‐axis, hours) are shown for the SAD (A) and MAD (B) studies. Dose groups are represented by symbols, as shown. Results for the MAD study represent steady‐state concentrations (day 10).
Table 1.
Mean (SD) Plasma and Urine Pharmacokinetic Parameters of PRT062607 from SAD Study
| Dose (mg) | N | Cmax (ng/mL) | AUClast (ng·h/mL) | Cmax/D (ng/mL)/(mg) | AUC/D (ng·h/mL)/(mg) | t1/2 (hours) | tmax a (hours) | Ae0‐96 (mg) | fe0‐96 (%) |
|---|---|---|---|---|---|---|---|---|---|
| 3 | 3 | 1.5 (0.4) | b | 0.5 (0.1) | b | b | 1.0 (0.5, 2.0) | NDc | NDc |
| 10 | 3 | 10.5 (2.6) | 150 (60.1) | 1.1 (0.3) | 15.0 (6.0) | 57.1 (9.0) | 1.0 (1.0, 1.0) | NCd | NCd |
| 30 | 6 | 42.9 (20.1) | 878 (341) | 1.4 (0.7) | 29.3 (11.4) | 56.7 (6.3) | 1.0 (0.5, 3.0) | 0.4 (0.3) | 1.5 (0.9) |
| 100 | 6 | 263 (48.1) | 5400 (1100) | 2.6 (0.5) | 52.6 (9.8) | 69.8 (30.9) | 2.0 (1.0, 3.0) | 2.4 (0.5) | 2.4 (0.5) |
| 200 | 6 | 629 (189) | 13,700 (3770) | 3.2 (0.9) | 68.3 (18.8) | 77.2 (31.6) | 2.0 (2.0, 3.0) | 4.3 (0.8) | 2.2 (0.4) |
| 300 | 6 | 558 (160) | 11,800 (2370) | 1.9 (0.5) | 39.3 (8.0) | 93.8 (46.3) | 2.1 (1.0, 3.0) | 7.0 (2.0) | 2.3 (0.7) |
| 400 | 6 | 996 (261) | 22,300 (4770) | 2.5 (0.7) | 55.7 (11.9) | 65.1 (13.3) | 4.0 (3.0, 4.0) | 20.2 (14.3) | 5.1 (3.6) |
Median (Min, Max) presented for tmax.
Values were not reported due to incomplete concentration‐time profile and insufficient detectable concentrations in terminal phase.
ND: Not determined due to urine collection issues.
NC: Not calculated as all samples were BLQ (< 50 ng/mL).
Table 2.
Mean (SD) Plasma and Urine Pharmacokinetic Parameters of PRT062607 from MAD Study (Day 10)
| PRT062607 Dose | |||||
|---|---|---|---|---|---|
| PK Parameter | Units | 11 mg | 33 mg | 66 mg | 110 mg |
| N | 6 | 6 | 6 | 6 | |
| Cmax | ng/mL | 32.6 (11.6) | 118 (42.7) | 317 (164) | 557 (200) |
| AUC0‐24 | ng·h/mL | 507 (206) | 1810 (619) | 4630 (1847) | 8487 (2357) |
| Cmax/D | (ng/mL)/mg | 3.0 (1.1) | 3.6 (1.3) | 4.8 (2.5) | 5.1 (1.8) |
| AUC/D | (ng·h/mL)/mg | 46.1 (18.7) | 54.9 (18.7) | 70.1 (28.0) | 77.2 (21.4) |
| t1/2 | Hours | b | b | 61.3 (10.2) | 75.3 (11.2) |
| tmax a | Hours | 1.5 (0.5, 2.0) | 1.5 (1.0, 2.1) | 4.0 (2.0, 4.0) | 3.0 (3.0, 6.0) |
| Accumulation Ratio | |||||
| Cmax | 2.9 (2.7) | 2.1 (0.7) | 3.2 (1.4) | 3.3 (2.5) | |
| AUC | 5.4 (2.6) | 3.2 (1.1) | 4.3 (2.1) | 5.4 (5.1) | |
| Peak/Trough Ratio | 2.0 (0.6) | 2.1 (0.2) | 2.2 (0.3) | 2.3 (0.5) | |
| Fluctuation | % | 90.7 (43.9) | 88.0 (15.9) | 98.8 (24.1) | 92.8 (30.7) |
| Ae0‐24 | mg | 0.4 (0.1) | 1.3 (0.0) | 3.7 (0.6) | 6.96 (1.7) |
| fe0‐24 | % | 3.3 (1.3) | 4.0 (0.1) | 5.6 (1.0) | 6.3 (1.5) |
| CLr | mL/(min·kg) | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.0) | 0.2 (0.1) |
Median (Min, Max) presented for tmax.
Values were not reported due to incomplete concentration‐time profile and insufficient detectable concentrations in the terminal phase.
In the MAD study, PRT062607 was readily absorbed (median tmax between 1.5 to 4.0 hours postdose) and slowly eliminated. Mean apparent terminal t1/2 values at dose levels of 66 and 110 mg were 61.3 and 75.3 hours, respectively. Mean Cmax and AUC0‐24 values generally increased in a greater than dose‐proportional manner from 11 to 110 mg. Mean Cmax and AUC as they relate to dose are presented in Supplemental Figures S1C and D. Following dose increases of 3‐, 6‐ and 10‐fold, Cmax increased 3.6‐, 9.7‐, and 17‐fold, and AUC0‐24 increased 3.6‐, 9.1‐, and 17‐fold, respectively. There was approximately a 3‐ to 5‐fold accumulation of PRT062607 (based on AUC) after once‐daily dosing for 10 days. Comparison of the trough (predose) levels collected on days 4, 7, 8, 9, and10 of the study (data not shown) indicated that steady state appeared to have been achieved by day 7. Mean peak‐to‐trough ratio on day 10 ranged from 2.00 to 2.30 across all the dose cohorts. At steady state, intersubject variability (CV%) for exposure parameters was generally low to moderate (≤45% for all dose cohorts). Additional plasma concentration‐time profiles and mean PK parameters for all cohorts following administration of the first dose of PRT062607 in the MAD study (day 1) are presented in Supplemental Figure S2.
Renal excretion was a minor route of elimination for PRT062607. The mean percentage of unchanged drug recovered in urine over 24 hours ranged from 3.29% to 6.32% on day 10. Mean renal clearance observed on day 10 at the different dose levels appeared to be dose independent and represented a small fraction of the total apparent oral clearance (data not shown).
PK/PD Relationship in Healthy Normal Volunteers
Previous publications have shown the specificity of SYK inhibition on in vitro addition of PRT062607 to human whole blood and in animal models of various diseases.9, 10 To confirm the PK/PD relationship in healthy volunteers following oral dosing, we initiated SAD and MAD studies in which time‐matched blood samples were collected for PK and PD analyses to determine the PK/PD relationship in humans. Percentage inhibition of BCR‐mediated B‐cell activation (Figure 2A) and FcεR1‐mediated basophil degranulation (Figure 2B) were plotted against the plasma concentration for each sampling time‐point. Data from the SAD (filled circles) and from the MAD (open circles) are overlaid. The overall IC50 for SYK in the B‐cell activation assay was 324 nM, and in the basophil degranulation assay it was 205 nM. IC90 for Syk in the B‐cell activation assay was 1.46 μM, and in the basophil degranulation assay it was 0.71 μM. Table 3 depicts the percentage inhibition of SYK in various assays extrapolated from the PK/PD plots. The drug effect on BCR signaling was evaluated only in the SAD study. The concentration‐response curve is shown in Figure 2C. The assay is more technically challenging and therefore variable, but as in the activation assay, specific inhibition of the signaling pathway was observed. SYK‐mediated pERK Y204 was inhibited with an IC50 of 660 nM. No concentration‐dependent inhibition of LYN‐mediated pSYK Y392 was observed (Figure 2D), demonstrating specificity of action on the BCR signaling pathway.
Figure 2.
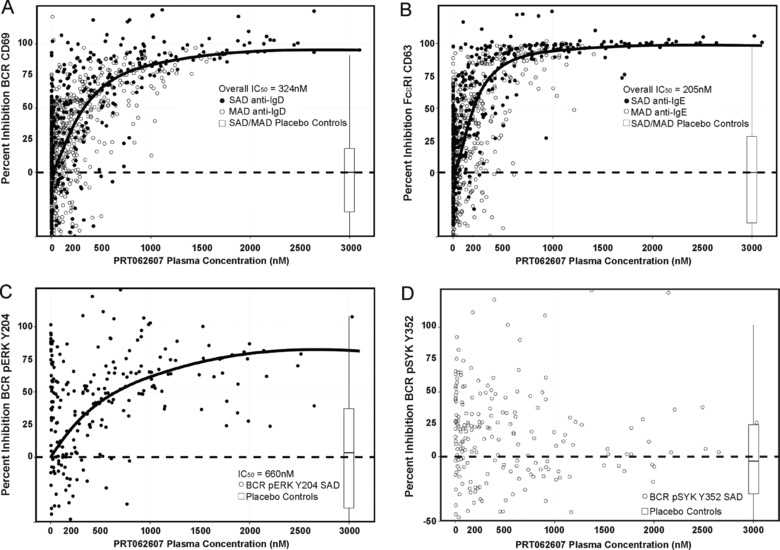
Oral administration of PRT062607 results in concentration‐dependent and selective inhibition of SYK. PRT062607 PK/PD relationships are shown. (A and B) Data from the SAD (filled circles) and MAD (open circles) studies are overlaid. Percentage inhibition of BCR/SYK‐mediated B‐cell activation (A), FcεR1/SYK‐mediated basophil degranulation (B), BCR/SYK‐mediated pERK Y204 (C), and BCR/LYN‐mediated pSYK Y352 (D) are shown on the y‐axes, relative to PRT062607 plasma concentration (nM) on the x‐axes. The inset box and whisker plot depict the median percentage inhibition (horizontal black line), the range in which 95% of the data fell (defined by box dimensions), and the 2.5% outliers in either direction (error bars) observed for each assay in healthy volunteers receiving placebo control. IC50s generated from nonlinear fits of all data combined are shown.
Table 3.
Inhibitory Concentrationsa
| SAD & MAD Merged (All Cohorts) | ||||||||
|---|---|---|---|---|---|---|---|---|
| B‐Cell Activation (CD69) | Basophil Degranulation (CD63) | |||||||
| 95% Confidence | 95% Confidence | |||||||
| IC | Estimate (nM) | Standard Error | Lower | Upper | Estimate (nM) | Standard Error | Lower | Upper |
| 10 | 72 | 4 | 63 | 80 | 59 | 3 | 53 | 65 |
| 20 | 125 | 5 | 115 | 135 | 93 | 4 | 86 | 101 |
| 25 | 152 | 6 | 141 | 164 | 110 | 4 | 103 | 117 |
| 30 | 181 | 6 | 169 | 194 | 127 | 4 | 119 | 134 |
| 40 | 245 | 9 | 228 | 262 | 163 | 4 | 155 | 171 |
| 50 | 324 | 13 | 297 | 350 | 205 | 4 | 196 | 213 |
| 60 | 427 | 21 | 386 | 469 | 257 | 5 | 248 | 266 |
| 70 | 578 | 35 | 509 | 647 | 330 | 6 | 319 | 342 |
| 75 | 687 | 46 | 596 | 778 | 381 | 7 | 367 | 395 |
| 80 | 836 | 63 | 712 | 961 | 448 | 10 | 429 | 467 |
| 90 | 1458 | 146 | 1172 | 1744 | 709 | 22 | 665 | 753 |
| 100 | 2432 | 299 | 1845 | 3019 | 1082 | 46 | 991 | 1173 |
Inhibitory concentrations (IC) in BCR‐induced B‐cell activation and FcεR1‐induced basophil degranulation assays corresponding to PRT062607 exposure (nM) were estimated using all PK/PD data from the SAD and MAD cohorts, fit using the software R together with the drc package. Standard error and upper and lower 95% confidence intervals are shown.
Figure 3 demonstrates the specificity and reversible nature of SYK inhibition following a single dose of PRT062607, consistent with its mechanism of action (noncovalent ATP‐competitive binding to SYK). Whole blood isolated from volunteers at baseline and at several time points postdosing were evaluated in 3 assays. B‐cell activation (as measured by up‐regulation of cell surface CD69) was induced by ligating the BCR (SYK‐dependent) or by inducing protein kinase C using PMA (SYK‐independent). Basophil degranulation (as measured by up‐regulation of cell surface CD63) was induced by cross‐linking the FcεR1 (SYK‐dependent) or by ligating formyl peptide receptor 1 (fMLP: SYK‐independent degranulation). The BCR signaling pathway was monitored by intracellular phospho‐flow cytometry following BCR ligation. SYK‐mediated pERK Y204 was measured, as was LYN‐mediated pSYK Y352 (as a specificity control). PMA was additionally used as an SYK‐independent stimulation to monitor PRT062607 effect on protein kinase C–induced pERK Y204. The data shown in Figure 3 are for the 200‐ and 300‐mg dose cohorts combined (SAD study), as the plasma exposure to drug was not different in the 2 cohorts. BCR‐mediated B‐cell activation was inhibited by >75% between 1 and 4 hours and returned to predose levels after 24 hours following study drug administration. As expected, no inhibition of PMA‐induced B‐cell activation was observed (Figure 3A). FcεR1‐mediated basophil degranulation was >80% suppressed on average between 1 and 6 hours postdose, which was also reversible and returned to baseline by approximately 48 to 72 hours after study drug administration. No inhibition of SYK‐independent fMLP‐mediated basophil degranulation was observed (Figure 3B). In the B‐signaling assay, SYK‐mediated pERK Y204 was inhibited by approximately 75% at 2 hours following study drug administration and returned to baseline by 48 hours. No consistent inhibition of BCR‐mediated pSYK Y352 or PMA‐mediated pERK Y204 was observed (Figure 3C). On each plot, the average plasma exposure to PRT062607 in time‐matched blood samples is shown by the black tracing.
Figure 3.
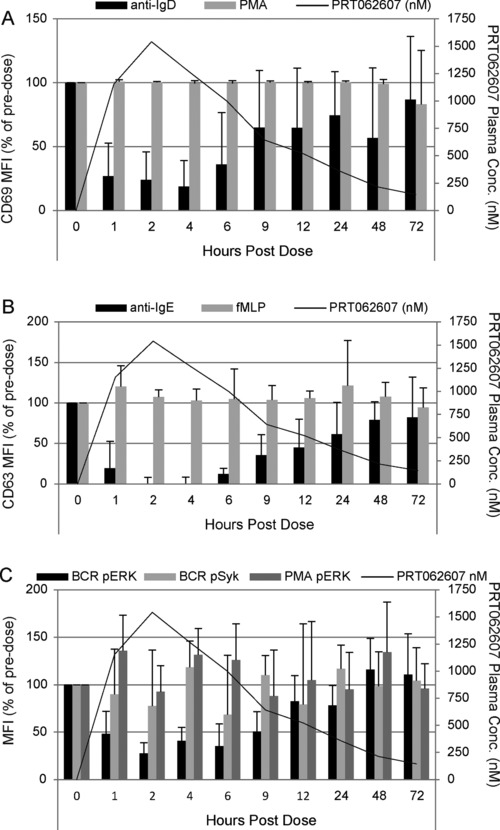
Kinetics of SYK inhibition following a single oral dose of PRT062607. The percentage of predose levels of BCR/SYK‐ (anti‐IgD; dark gray bars) or PMA/PKC‐ (light gray bars) induced B‐cell activation (A), FcεR1/SYK‐ (anti‐IgE; dark gray bars) or fMLP‐ (light gray bars) induced basophil degranulation (B), and BCR/SYK‐mediated pERK Y204 (black bars), BCR/LYN‐mediated pSYK Y352 (light gray bars), and PMA/PKC‐mediated pERK Y204 (dark gray bars) (C) are shown on the first (left) y‐axis. The average PRT062607 plasma concentration (nM) is presented as a tracing and defined on the second (right) y‐axis. Hours postdose are depicted on the x‐axis. Error bars represent the standard error of the mean.
Similar results were observed in the MAD study. Only modest inhibition of SYK signaling in the basophil degranulation assay was observed at the 33‐mg dose. A more robust inhibition of SYK‐mediated signaling was achieved after 10 days of dosing of 66 mg PRT062607, with data indicating approximately 80% (or greater) inhibition in the basophil degranulation assay and approximately 60% inhibition in the B‐cell activation assay. The highest dose tested (110 mg) resulted in complete inhibition of SYK in the basophil degranulation assay and a mean of approximately 80% inhibition on day 10 in the B‐cell activation assay. Because the plasma concentration of PRT062607 diminished with time following dosing, the effect on SYK inhibition in both assays was also diminished, again revealing the reversible nature of the inhibition. Further, at all dose levels studied, there was no effect of PRT062607 plasma concentration on the SYK‐independent stimulations (fMLP and PMA; data not shown).
The closest structural homologue to SYK is ZAP70, responsible for signaling and functional responses downstream of the T‐cell antigen receptor (TCR).16 We previously reported that PRT062607 does not inhibit ZAP70 in a purified kinase assay (SYK IC50 = 1 nM, ZAP70 IC50 = 1050 nM).9 To further confirm this observation, we implemented in the MAD study measures of T‐cell function following CD3/CD28 cross‐linking (ZAP70‐dependent) or PMA stimulation (PKC‐dependent) to determine if PRT062607 affected T‐cell functional responses following oral dosing. As shown in Figure 4, no effect on CD3/CD28‐ (filled circles) or PMA‐induced (open circles) T‐cell activation was observed. These data support the specificity of action of PRT062607 against SYK and demonstrate that it does not cross‐react with ZAP70 to elicit an inhibitory T‐cell response. Data are also shown in Figure 4B at the 110‐mg dose level over time.
Figure 4.
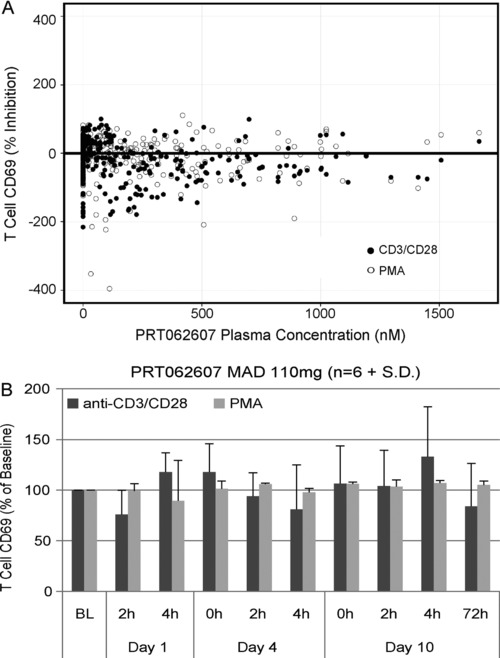
PRT062607 does not inhibit T‐cell functional responses following 10 days of consecutive dosing in healthy volunteers. (A) Percentage inhibition of T‐cell activation following CD3/CD28 cross‐linking (black‐filled circles) or PMA stimulation (gray‐filled circles) is shown across the plasma concentrations achieved (x‐axis; nM) at steady state following 10 consecutive once‐daily doses from the MAD study. (B) Data from the highest‐dose cohort (110 mg once daily) are presented as percentage of predose baseline (BL) over time, shown in days and hours (h).
Tolerability
The evaluation of safety included adverse events (AEs), safety laboratory tests (hematology, serum chemistry, immune function test panel, urinalysis), vital signs (sitting), 12‐lead ECGs (supine), cardiac monitoring, and physical examination.
In the SAD study there were no serious adverse events (SAEs), and no subject was withdrawn as a result of treatment‐emergent adverse events (TEAEs). A total of 40 AEs were reported, including 1 predose AE of mild headache. Overall, 39 TEAEs were reported by 19 subjects; 37 TEAEs were reported by 17/42 (40%) subjects receiving PRT062607, and 2 TEAEs were reported by 2/12 (17%) subjects receiving placebo. All of the TEAEs had resolved by the time of the last subject follow‐up. The TEAEs with the highest subject incidence were headache (6 PRT062607 subjects and 1 placebo subject), dizziness, abdominal pain, nausea, and diarrhea (3 PRT062607 subjects each) and rhinitis (2 PRT062607 subjects). There was no consistent relationship between increasing dose of PRT062607 and number and incidence of TEAEs. The greatest subject incidence of events was in the 200‐mg and 300‐mg PRT062607 dose groups (both 67%); however, the highest (400‐mg) dose group had a similar subject incidence of TEAEs to placebo (17% for both groups). The 200‐mg PRT062607 dose group also had the greatest number of reported TEAEs (15), which was higher than any of the other dose groups. Subjects in the 300‐mg PRT062607 dose group reported 7 TEAEs, and those in the 400‐mg dose group reported 3 TEAEs, which was similar to the number reported by the placebo group (2 TEAEs). Most (36/39) TEAEs were mild intensity; 3 moderate intensity TEAEs (dizziness, headache, and musculoskeletal pain) were reported by 3 subjects, all of whom received PRT062607.
In the MAD study there were no SAEs, and no subjects were discontinued due to AEs. The AEs reported for all treatments were predominantly mild in severity. Overall, 71 of 96 AEs (74%) reported following PRT062607 were considered by the investigator to be related to the study drug. The frequency of AEs was highest at the 110‐mg dose of PRT062607 compared to the lower doses, and also compared to the placebo treatment, with the frequency for the 11‐, 33‐, and 66‐mg doses of PRT062607 being similar to that for placebo; however, the incidence of AEs was similar across the treatments. Overall, 96 AEs were reported by 13 (54.2%) subjects following administration of PRT062607 compared with 19 AEs reported by 7 (87.5%) subjects following placebo. The majority (93%) of AEs for the 110‐mg dose (55% of AEs in the study) were reported by 2 subjects, both of whom completed the study and remained on the study drug. TEAEs reported at the 110‐mg dose were all mild in nature and included gastrointestinal symptoms (nausea, diarrhea, abdominal discomfort), hyperhidrosis, fatigue, and loss of appetite.
In neither study were there any consistent trends in safety laboratory test results, vital signs data or 12‐lead ECG findings following treatment with PRT062607 when compared to placebo.
PRT062607 Inhibits FcR Function in Healthy Normal Leukocytes
SYK is also critical for FcγR signaling,6, 17, 18 which was not measured as part of the clinical trials due to limitations in accessible blood volume. We therefore evaluated the ability of PRT062607 to inhibit immune‐complex–driven FcγR‐mediated DC activation and neutrophil oxidative burst using in vitro assays. Immature dendritic cells were generated from IL4/GMCSF stimulated peripheral blood monocytes isolated from healthy donors (Figure 5A) and then stimulated with antibody‐opsonized sheep red blood cells (opRBC) in the presence or absence of a concentration range of PRT062607 (Figure 5B). Cellular activation was partially inhibited by an FcγRII‐specific antibody (4.3). PRT062607 inhibited FcγR‐mediated DC activation, as measured by up‐regulation of surface CD80/86 and MHCII on the cell surface (Figure 5B and C, top). No inhibition of LPS‐induced DC activation (SYK‐independent) was observed (Figure 5C, bottom). In whole blood from healthy donors, neutrophils were induced to release reactive oxygen species (ROS) by stimulation with opRBC to cross‐link FcγR, or by E coli to induce ROS via SYK‐independent toll‐like receptor mechanisms. As shown in Figure 6A, opRBC‐induced ROS generation was completely abrogated by pretreating blood samples with 4 μM PRT062607. In replicate experiments from different donors, PRT062607 inhibited opRBC‐ but not E coli‐induced ROS formation in a concentration‐dependent manner (Figure 6B). Importantly, the concentrations safely achieved in the human SAD and MAD studies are sufficient to inhibit FcR‐mediated cellular responses in whole blood in vitro.
Figure 5.
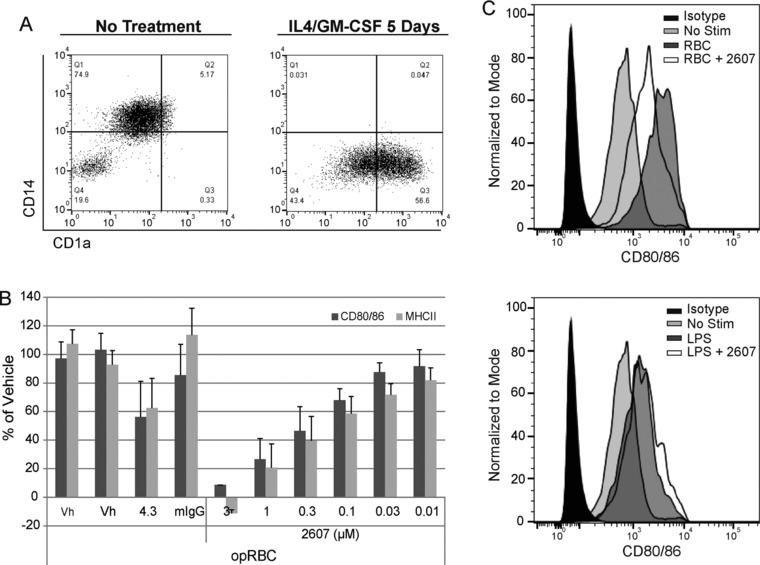
Inhibition of immune‐complex–mediated DC activation in vitro. (A) Differentiation of purified peripheral blood monocytes into imDCs via IL4/GM‐CSF is shown based on CD14 and CD1a expression. (B) Data represent DC activation as measured by surface expression of CD80/86 and MCHII from replicate (n = 5) experiments. Pretreatment of cells with vehicle (Vh), anti‐FcRIIa antibody (4.3) or an isotype control (mIgG), or various concentrations of PRT062607 (μM) prior to stimulation with opsonized sheep red blood cells (opRBC) is indicated. Bars represent mean percentage of vehicle plus standard deviation. (C) Representative CD80/86 expression data from opRBC (SYK‐dependent)‐stimulated cells (top histogram overlay) and LPS (SYK‐independent)‐stimulated cells (bottom histogram overlay). Unstimulated cells are shown (No Stim); 1 μM PRT062607 was used as indicated (+2607).
Figure 6.
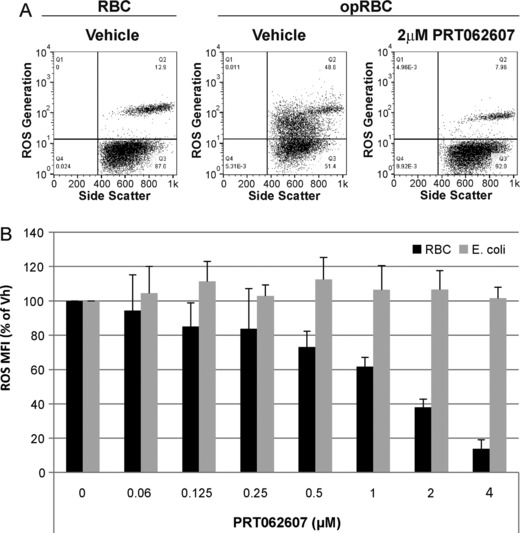
Inhibition of immune‐complex–mediated NE oxidative burst in vitro. (A) Representative FACS plots demonstrating the effect of 4 μM PRT062607 on immune complex (opRBC)‐mediated neutrophil oxidative burst (y‐axis). (B) Replicate experiments from 5 independent healthy blood donors. Data represent mean neutrophil oxidative burst (ROS) as a percentage of vehicle control, plus standard deviation on the y‐axis. PRT062607 concentration (μM) is presented on the x‐axis. Black bars represent cells stimulated with opRBC (SYK‐dependent), and gray bars represent cells stimulated with E coli (non‐SYK dependent).
The percentage inhibition of SYK required for efficacy in an in vivo model of inflammatory autoimmune disease was then estimated. We previously demonstrated the exposure‐efficacy relationship of PRT062607 in the rat CIA model of inflammatory arthritis.9 Supplemental Table S1A details the doses tested, exposures achieved, and corresponding percentage inhibition of hind‐paw inflammation. The minimally effective exposure observed in this study was at the 10 mg/kg dose, resulting in a Cav,ss of 0.72 μM. To estimate the percentage of SYK inhibition in rats at this exposure, we stimulated rat whole blood (immunized with bovine collagen and presenting with hind‐paw inflammation scores of 1 to 2) with anti‐BCR antibody and measured phosphorylation of ERK Y204. Blood from rats was tested independently and in triplicate on 3 separate days for BCR‐mediated induction of pERK Y204 and for responsiveness to increasing concentrations of PRT062607 (Figure 7A, bar graph). Data from 20 rats with inflammation scores of 1 to 2 were pooled, and percentage inhibition was calculated to generate a concentration‐response profile (Figure 7A, curve‐fit). Average plasma concentrations achieved at the 10 and 15 mg/kg dose levels in the rat CIA study were extrapolated back to the percentage inhibition of the BCR signaling pathway by PRT062607 and related to the percentage inhibition of joint swelling in the model. As indicated in the associated table, a concentration of 0.72 μM corresponds to 40% inhibition of the BCR/SYK signaling pathway and 55% inhibition of hind‐paw inflammation. Similarly, 1.34 μM corresponds to a 57% inhibition of the BCR/SYK pathway and to a 90% inhibition of hind‐paw inflammation. Figure 7B demonstrates the inability of PRT062607 to inhibit PMA‐induced pERK Y204 in rat blood, indicating that the assay behavior compares well with the human system. Percentage inhibition of the pathway at Cmin,ss, Cmax,ss, and Cav,ss at the 10 mg/kg dose is detailed in Supplemental Table S1B and ranges from 21% to 47% inhibition. To estimate the therapeutic exposures of PRT062607 that may be required for efficacy in patients with autoimmune diseases, we used the SAD and MAD PK/PD relationships to extrapolate what doses would be required to maintain SYK inhibition levels above IC25, IC50, IC75, and IC90 (Table 4). Efficacy in the rat CIA model appears to be achievable at approximately 50% inhibition of SYK. This level of inhibition is maintained at Cmin,ss in humans with oral doses ranging between 51 and 75 mg.
Figure 7.
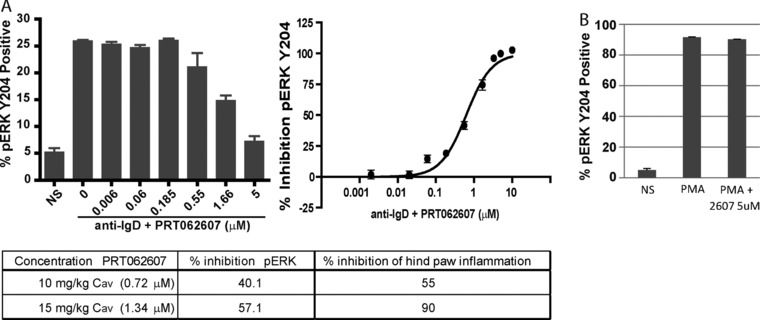
Inhibitory potency of PRT062607 against SYK activity in rat whole blood. (A) The bar graph represents replicate experiments (n = 3) from a single rat whole‐blood sample, showing the percentage of B cells that are positive for pERK Y204 following no stimulation (NS) or BCR stimulation (anti‐IgD) in the presence of the indicated concentrations of PRT062607. Data from 20 rats included as part of a CIA study were normalized to generate percentage inhibition relative to vehicle control and plotted using a 3‐parameter nonlinear curve fit to estimate the PRT062607 concentration‐effect relationship for SYK inhibition in rat whole blood. The table depicts the dose groups (10 and 15 mg/kg) that demonstrated statistically significant efficacy in the rat CIA model, the Cav,ss achieved at each dose, the percentage inhibition of pERK Y204, and the percentage inhibition of hind‐paw inflammation. (B) The percentage of pERK Y204‐positive B cells in rat whole blood following no stimulation (NS) or stimulation with PMA in the presence of vehicle or 5 μM PRT062607 (n = 3).
Table 4.
Concentrations Required to Maintain Various Levels of Target Inhibitiona
| B‐Cell Activation (CD69) | Basophil Degranulation (CD63) | |||||||
|---|---|---|---|---|---|---|---|---|
| Conc (nM) | ||||||||
| Dose | AUC | Cmax | Dose | AUC | Cmax | |||
| Target | CD63 | CD69 | (mg) | (ng·h/mL) | (ng/mL; nM) | (mg) | (ng·h/mL) | (ng/mL; nM) |
| Maintain above IC25 | 110 | 152 | 41 | 1888 | 123; 313 | 33 | 1366 | 89; 226 |
| Maintain above IC25 (upper 95% CI) | 117 | 164 | 43 | 2049 | 134; 341 | 35 | 1486 | 97; 247 |
| Maintain above IC50 | 205 | 324 | 71 | 4057 | 265; 674 | 51 | 2571 | 168; 427 |
| Maintain above IC50 (upper 95% CI) | 213 | 350 | 75 | 4378 | 286; 727 | 52 | 2651 | 173; 440 |
| Maintain above IC75 | 381 | 687 | 121 | 8556 | 559; 1421 | 80 | 4780 | 312; 793 |
| Maintain above IC75 (upper 95% CI) | 395 | 778 | 133 | 9761 | 638; 1622 | 82 | 4941 | 323; 821 |
| Maintain above IC90 | 709 | 1458 | 215 | 18,196 | 1188; 3019 | 124 | 8837 | 577; 1467 |
| Maintain above IC90 (upper 95% CI) | 753 | 1744 | 251 | 21,771 | 1422; 3614 | 129 | 9399 | 614; 1561 |
Concentrations were extrapolated from PK/PD curve fit. Data were then used to estimate the dose (mg) required to maintain the calculated target concentration. Cmax is shown in ng/mL and nM.
Discussion
Therapeutic targeting of the BCR signaling pathway has been validated clinically for the treatment of B‐cell malignancies by 2 small‐molecule kinase inhibitors. Bruton's tyrosine kinase (BTK) inhibitor ibrutinib is indicated for the treatment of CLL, MCL, and Waldenström's macroglobulinemia.19, 20, 21, 22 Similarly, the PI3Kδ inhibitor idelalisib is approved for the treatment of relapsed/refractory CLL in combination with B‐cell depletion by rituximab23 and as a single agent for the treatment of relapsed/refractory follicular lymphoma.24 Although SYK is upstream of BTK and PI3K on the BCR signaling pathway, the tissue expressions of these 3 kinases are divergent, indicating that each target does reflect its distinct biology. In spite of the overall efficacy of the 2 agents (ibrutinib and idelisib), recurring adverse events that appear to be drug related have been identified for both. Neutropenia, thrombocytopenia, diarrhea, and fatigue have emerged as common safety concerns in treated patients. In addition, increased rates of adverse events have been observed in combination studies with idelisib and may limit its potential to be used in conjunction with other therapies. Unique kinase selectivity patterns of small‐molecule inhibitors may also offer the opportunity to achieve distinct efficacy and safety profiles as a consequence of targeting SYK, BTK, or PI3K.
Thus far, SYK as a therapeutic target has not been explored as thoroughly as BTK and PI3Kδ. Fostamatinib (a nonspecific SYK inhibitor) was originally evaluated in patients with relapsed/refractory B‐cell malignancies, achieving partial responses in leukemias and indolent and aggressive lymphomas,25 although a follow‐up phase 2 study in DLBCL was less promising (overall response rate 3%).26 A series of phase 2 studies were conducted in patients with RA; statistically significant improvements in ACR50 scores were achieved in methotrexate‐resistant patients27, 28 but not in patients who had failed prior therapy with TNF inhibitors.29 Unfortunately, phase 3 studies for fostamatinib in RA did not meet their primary endpoints for clinical benefit.30 Fostamatinib is currently being studied in a phase 3 trial in immune thrombocytopenia31 (reference 31 and NCT02077192). Although it is difficult to clearly interpret these data as clinical validation of targeting SYK,32 the clinical results are suggestive and consistent with the known etiology of these diseases. In comparison to fostamatinib, PRT062607 is more selective as an SYK inhibitor10, 33 and more potent when tested head‐to‐head in whole blood BCR/SYK and FcεR1/SYK signaling and functional assays (data not shown). Braselmann et al32 reported an IC50 of 1.06 μM with fostamatinib in the basotest assay following oral administration to healthy volunteers; we report here an IC50 of 0.2 μM and an IC90 of 0.7 μM in healthy volunteers following the same route of dosing. Given the specificity and potency differences, it is possible that PRT062607 will elicit a different safety and efficacy profile relative to fostamatinib. Of note, a dual SYK/ZAP70 inhibitor (MK‐8457) recently demonstrated efficacy in patients with rheumatoid arthritis but ultimately was not tolerated due to serious infections, and the program was discontinued.34
Another SYK inhibitor in clinical development, GS9973, appears to have a better selectivity profile relative to fostamatinib.35 Kinases inhibited within a 10‐fold potency range by this compound were CSNK2A2, JAK2, SRC, and TNK1,36 with JAK2 and SRC being relevant to B‐cell and tumor biology. Efficacy in relapsed CLL patients was reported from a phase 2 study, demonstrating that 94.5% of patients achieved decreased adenopathy, with 61.5% of patients having experienced a partial response. Duration of response was greater than 6 months in the majority of responding patients.37 It is difficult to assess the exact level of SYK inhibition in this study, as only changes in serum cytokines/chemokines were reported. Estimating from reduction in serum levels of CCL3 and CCL4 (∼50% to 75%) as measures of BCR signaling, it appears that the BCR pathway was not completely suppressed since more potent inhibition of these markers has been reported (>90%) by the covalent BTK modifier ibrutinib.38 Combining GS9973 with idelalisib in patients with B‐cell malignancies was not tolerated, and the study was discontinued.39 It is unclear if the combination of these 2 agents was not tolerated due to the combined SYK/PI3Kδ mechanism or due to overlapping off‐target toxicities of these 2 distinct kinase inhibitors derived from separate chemistry scaffolds.
The PK profiles of PRT062607 following multiple‐dose administration were comparable to that following single‐dose administration, and plasma exposures observed showed a greater than dose‐proportional increase as a function of dose in both SAD and MAD studies. Caco‐2‐cell data suggest that PRT062607 is highly effluxed by P‐gp (data not shown), which may be a contributing factor to the nonlinear increases in exposure observed up to 100 mg in the SAD study and up to 110 mg in the MAD study. Dose‐proportional increases, however, are seen beyond 200 mg in the SAD study, suggesting that the efflux mechanism of P‐gp in the gut may be saturated at these higher doses. Despite the nonlinear increases in exposure, intersubject variability was found to be low to moderate, suggesting that the PK behavior of PRT062607 is predictable. In addition, PD response appeared to be directly related to PK response, and the pharmacologic activity of PRT062607 was maintained on repeat dosing. There was little contribution via renal excretion to the overall elimination of PRT062607. Renal clearance of PRT062607 was determined to be nearly equivalent to the glomerular filtration rate when the protein binding of PRT062607 in human plasma (93.5%) is taken into account. Low renal clearance offers a benefit when this drug is administered to elderly patient populations, such as those with rheumatoid arthritis, where compromised renal function is prevalent.
One consistent observation made with PRT062607 both preclinically and following oral dosing in humans is a potent inhibition of FcεR1 signaling. We do not have a clear mechanistic understanding of this observation, but the combined SAD/MAD human data indicate an approximately 2‐fold increase in potency of suppressing FcεR1‐mediated basophil degranulation relative to BCR‐mediated signaling or activation. Based on the clinical studies, we have estimated that inhibition of peripheral blood basophil degranulation by 50% can be achieved at a concentration of ∼0.2 μM, which would be maintained at a dose of 51 mg, and by 90% at around 0.7 μM, which would require a projected dose of 130 mg. Preclinical models support targeting SYK for allergic disease. Wex et al40 demonstrated in a conditional knockout model that SYK was critical for the generation of mast cell–mediated allergic and inflammatory responses. Aerosolized antisense oligonucleotides targeting SYK effectively blocked inflammatory responses in a rat model of allergen‐induced asthma.41 Independent chemical scaffolds that target SYK have demonstrated efficacy in animal models of allergic disease (reviewed by Pamuk and Tsokos),42 and intranasal delivery of the SYK inhibitor R112 improved clinical symptoms of seasonal allergic rhinitis.43 The data suggest that PRT062607 could be utilized as a tool to explore the full range of FcεR1 signaling inhibition for therapy in type I hypersensitivity allergic reactions such as allergic asthma, atopic dermatitis, or even severe life‐threatening food or drug allergies.
As reported here, an average of 40% inhibition of rat peripheral blood BCR signaling was associated with resolution of inflammation and joint destruction in the animal model of RA. Most likely, this was a function of a mixed reaction of suppressing BCR signaling and autoantibody production as well as inhibiting FcγR‐mediated activation of effector myeloid cells. The combined effect of targeting multiple relevant immunological mechanisms may have contributed to the observation that low‐level inhibition of a single pathway was sufficient for disease resolution. Experimentally, SYK is required for FcγR‐mediated activation of macrophages and neutrophils,6, 17 and SYK expression in neutrophils is required for the development of inflammation and disease symptoms in the K/BxN serum transfer model of arthritis.44 Consistently, reconstitution of the hematopoietic system of irradiated mice with SYK knockout fetal liver cells protects against autoantibody‐mediated arthritis.45 To confirm the requirement of SYK in established mature immune cells for arthritis development, Ozaki et al46 have demonstrated that conditional SYK knockout mice were completely protected from collagen antibody–induced arthritis. The protection from inflammation was associated with defective signaling and functional responses following FcγR ligation in macrophages and neutrophils.
In conclusion, SYK remains an attractive target for certain autoimmune diseases and B‐cell malignancies, yet its potential has not been fully explored. Animal models predict that complete suppression of SYK activity may not be needed to elicit clinical response by PRT062607. Dose‐ranging clinical studies will be required with PRT062607 to determine the level of pathway suppression required for efficacy. Results from the healthy normal volunteer studies suggest that the full range of SYK inhibition by PRT062607 can be safely explored in humans and can enable identification of a pharmacological profile customized to individual target patient populations.
Disclosures
G.C., A.B., Y.P., H.D., A.P., S.H., D.G., and U.S. are current or former employees and shareholders of Portola Pharmaceuticals, Inc.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supplemental Figure S1: Relationship between Cmax, AUC, and Dose of PRT062607 following single and multiple oral administration of PRT062607.
Supplemental Figure S2: PK parameters following the first oral administration of PRT062607 in the MAD study.
Supporting Table
Supporting Information
References
- 1. Geisberger R, Crameri R, Achatz G. Models of signal transduction through the B‐cell antigen receptor. Immunology. 2003;110(4):401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cheng AM, Rowley B, Pao W, Hayday A, Bolen JB, Pawson T. Syk tyrosine kinase required for mouse viability and B‐cell development. Nature. 1995;378(6554):303–306. [DOI] [PubMed] [Google Scholar]
- 3. Turner M, Mee PJ, Costello PS, et al. Perinatal lethality and blocked B‐cell development in mice lacking the tyrosine kinase Syk. Nature. 1995;378(6554):298–302. [DOI] [PubMed] [Google Scholar]
- 4. Le Roux D, Lankar D, Yuseff MI, et al. Syk‐dependent actin dynamics regulate endocytic trafficking and processing of antigens internalized through the B‐cell receptor. Mol Biol Cell. 2007;18(9):3451–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Costello PS, Turner M, Walters AE, et al. Critical role for the tyrosine kinase Syk in signalling through the high affinity IgE receptor of mast cells. Oncogene. 1996;13(12):2595–2605. [PubMed] [Google Scholar]
- 6. Kiefer F, Brumell J, Al‐Alawi N, et al. The Syk protein tyrosine kinase is essential for Fcgamma receptor signaling in macrophages and neutrophils. Mol Cell Biol. 1998;18(7):4209–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Regnault A, Lankar D, Lacabanne V, et al. Fcgamma receptor‐mediated induction of dendritic cell maturation and major histocompatibility complex class I‐restricted antigen presentation after immune complex internalization. J Exp Med. 1999;189(2):371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mocsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA. Syk is required for integrin signaling in neutrophils. Immunity. 2002;16(4):547–558. [DOI] [PubMed] [Google Scholar]
- 9. Coffey G, DeGuzman F, Inagaki M, et al. Specific inhibition of spleen tyrosine kinase suppresses leukocyte immune function and inflammation in animal models of rheumatoid arthritis. J Pharmacol Exp Ther. 2011;340(2):350–359. [DOI] [PubMed] [Google Scholar]
- 10. Coffey G, Betz A, Graf J, et al. Methotrexate and a spleen tyrosine kinase inhibitor cooperate to inhibit responses to peripheral blood B cells in rheumatoid arthritis. Pharmacol Res Perspect. 2013;1(2):e00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheng S, Coffey G, Zhang XH, et al. SYK inhibition and response prediction in diffuse large B‐cell lymphoma. Blood. 2011;118(24):6342–6352. [DOI] [PubMed] [Google Scholar]
- 12. Hoellenriegel J, Coffey GP, Sinha U, et al. Selective, novel spleen tyrosine kinase (Syk) inhibitors suppress chronic lymphocytic leukemia B‐cell activation and migration. Leukemia. 2012;26(7):1576–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Spurgeon SE, Coffey G, Fletcher LB, et al. The selective SYK inhibitor P505‐15 (PRT062607) inhibits B cell signaling and function in vitro and in vivo and augments the activity of fludarabine in chronic lymphocytic leukemia. J Pharmacol Exp Ther. 2013;344(2):378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ritz C, Streibig JC. Bioassay analysis using R. J Stat Software. 2005;12:1–22. [Google Scholar]
- 15. Ritz C, Baty F, Streibig JC, Gerhard D. Dose‐response analysis using R. PloS One. 2015;10(12):e0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang H, Kadlecek TA, Au‐Yeung BB, et al. ZAP‐70: an essential kinase in T‐cell signaling. Cold Spring Harbor Perspect Biol. 2010;2(5):a002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Crowley MT, Costello PS, Fitzer‐Attas CJ, et al. A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J Exp Med. 1997;186(7):1027–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jakus Z, Simon E, Frommhold D, Sperandio M, Mocsai A. Critical role of phospholipase Cgamma2 in integrin and Fc receptor‐mediated neutrophil functions and the effector phase of autoimmune arthritis. J Exp Med. 2009;206(3):577–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. O'Brien S, Furman RR, Coutre SE, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: an open‐label, multicentre, phase 1b/2 trial. Lancet Oncol. 2014;15(1):48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenstrom's macroglobulinemia. N Engl J Med. 2015;372(15):1430–1440. [DOI] [PubMed] [Google Scholar]
- 22. Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle‐cell lymphoma. N Engl J Med. 2013;369(6):507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gopal AK, Kahl BS, de Vos S, et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370(11):1008–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Friedberg JW, Sharman J, Sweetenham J, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non‐Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115(13):2578–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Flinn IW, Bartlett NL, Blum KA, et al. A phase II trial to evaluate the efficacy of fostamatinib in patients with relapsed or refractory diffuse large B‐cell lymphoma (DLBCL). Eur J Cancer. 2016;54:11–17. [DOI] [PubMed] [Google Scholar]
- 27. Weinblatt ME, Kavanaugh A, Burgos‐Vargas R, et al. Treatment of rheumatoid arthritis with a Syk kinase inhibitor: a twelve‐week, randomized, placebo‐controlled trial. Arthritis Rheum. 2008;58(11):3309–3318. [DOI] [PubMed] [Google Scholar]
- 28. Weinblatt ME, Kavanaugh A, Genovese MC, Musser TK, Grossbard EB, Magilavy DB. An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. N Engl J Med. 2010;363(14):1303–1312. [DOI] [PubMed] [Google Scholar]
- 29. Genovese MC, Kavanaugh A, Weinblatt ME, et al. An oral Syk kinase inhibitor in the treatment of rheumatoid arthritis: a three‐month randomized, placebo‐controlled, phase II study in patients with active rheumatoid arthritis that did not respond to biologic agents. Arthritis Rheum. 2011;63(2):337–345. [DOI] [PubMed] [Google Scholar]
- 30. Weinblatt ME, Genovese MC, Ho M, et al. Effects of fostamatinib, an oral spleen tyrosine kinase inhibitor, in rheumatoid arthritis patients with an inadequate response to methotrexate: results from a phase III, multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group study. Arthritis Rheum. 2014;66(12):3255–3264. [DOI] [PubMed] [Google Scholar]
- 31. Podolanczuk A, Lazarus AH, Crow AR, Grossbard E, Bussel JB. Of mice and men: an open‐label pilot study for treatment of immune thrombocytopenic purpura by an inhibitor of Syk. Blood. 2009;113(14):3154–3160. [DOI] [PubMed] [Google Scholar]
- 32. Braselmann S, Taylor V, Zhao H, et al. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex‐mediated inflammation. J Pharmacol Exp Ther. 2006;319(3):998–1008. [DOI] [PubMed] [Google Scholar]
- 33. Coffey G, DeGuzman F, Inagaki M, et al. Specific inhibition of Syk suppresses leukocyte immune function and alleviates inflammation in rodent models of rheumatoid arthritis. Blood. 2010;116 (21):723 abstract # 1727. [Google Scholar]
- 34. Vollenhoven RCSB, Mease PJ, Peterfy CG, et al. Efficacy and safety of MK‐8457, a novel SYK inhibitor for the treatment of rheumatoid arthritis in two randomized, controlled, phase 2 studies. Presented at the American College of Rheumatology; November 14, 2014; Boston, MA.
- 35. Sharman JP, Klein L, Boxer M, et al. Phase 2 trial of GS‐9973, a selective Syk inhibitor, in chronic lymphocytic leukemia (CLL) and non‐Hodgkin lymphoma (NHL). Presented at the American Society of Hematology; December 7, 2013; New Orleans, LA.
- 36. Burke RT, Meadows S, Loriaux MM, et al. A potential therapeutic strategy for chronic lymphocytic leukemia by combining Idelalisib and GS‐9973, a novel spleen tyrosine kinase (Syk) inhibitor. Oncotarget. 2014;5(4):908–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sharman J, Klein LM, Boxer M, et al. Phase 2 trial of entospletinib (GS‐9973), a selective Syk inhibitor, in chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL). Presented at the American Society of Hematology; December 5, 2015; Orlando, FL.
- 38. Ponader S, Chen SS, Buggy JJ, et al. The Bruton tyrosine kinase inhibitor PCI‐32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119(5):1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Barr PM, Saylors GB, Spurgeon S, et al. Phase 2 trial of GS‐9973, a selective syk inhibitor, and idelalisib (idela) in chronic lymphocytic leukemia (CLL) and non‐Hodgkin lymphoma (NHL). Presented at the American Society of Clinical Oncology; May 30, 2014; Chicago, IL.
- 40. Wex E, Bouyssou T, Duechs MJ, et al. Induced Syk deletion leads to suppressed allergic responses but has no effect on neutrophil or monocyte migration in vivo. Eur J Immunol. 2011;41(11):3208–3218. [DOI] [PubMed] [Google Scholar]
- 41. Stenton GR, Ulanova M, Dery RE, et al. Inhibition of allergic inflammation in the airways using aerosolized antisense to Syk kinase. J Immunol. 2002;169(2):1028–1036. [DOI] [PubMed] [Google Scholar]
- 42. Pamuk ON, Tsokos GC. Spleen tyrosine kinase inhibition in the treatment of autoimmune, allergic and autoinflammatory diseases. Arthritis Res Ther. 2010;12(6):222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Meltzer EO, Berkowitz RB, Grossbard EB. An intranasal Syk‐kinase inhibitor (R112) improves the symptoms of seasonal allergic rhinitis in a park environment. J Allergy Clin Immunol. 2005;115(4):791–796. [DOI] [PubMed] [Google Scholar]
- 44. Elliott ER, Van Ziffle JA, Scapini P, Sullivan BM, Locksley RM, Lowell CA. Deletion of Syk in neutrophils prevents immune complex arthritis. J Immunol. 2011;187(8):4319–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jakus Z, Simon E, Balazs B, Mocsai A. Genetic deficiency of Syk protects mice from autoantibody‐induced arthritis. Arthritis Rheum. 2010;62(7):1899–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ozaki N, Suzuki S, Ishida M, et al. Syk‐dependent signaling pathways in neutrophils and macrophages are indispensable in the pathogenesis of anti‐collagen antibody‐induced arthritis. Int Immunol. 2012;24(9):539–550. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supplemental Figure S1: Relationship between Cmax, AUC, and Dose of PRT062607 following single and multiple oral administration of PRT062607.
Supplemental Figure S2: PK parameters following the first oral administration of PRT062607 in the MAD study.
Supporting Table
Supporting Information