

Summary
Introduction
Mutations in leucine‐rich repeat kinase 2 (LRRK2) are the most prevalent cause of familial and sporadic Parkinson's disease (PD). Because most pathogenic LRRK2 mutations result in enhanced kinase activity, it suggests that LRRK2 inhibitors may serve as a potential treatment for PD. To evaluate whether LRRK2 inhibitors are effective therapies for PD, it is crucial to know whether LRRK2 inhibitors will affect dopaminergic (DAergic) neurotransmission. However, to date, there is no study to investigate the impact of LRRK2 inhibitors on DAergic neurotransmission.
Aims
To address this gap in knowledge, we examined the effects of three types of LRRK2 inhibitors (LRRK2‐IN‐1, GSK2578215A, and GNE‐7915) on dopamine (DA) release in the dorsal striatum using fast‐scan cyclic voltammetry and DA neuron firing in the substantia nigra pars compacta (SNpc) using patch clamp in mouse brain slices.
Results
We found that LRRK2‐IN‐1 at a concentration higher than 1 μM causes off‐target effects and decreases DA release, whereas GSK2578215A and GNE‐7915 do not. All three inhibitors at 1 μM have no effect on DA release and DA neuron firing rate. We have further assessed the effects of the inhibitors in two preclinical LRRK2 mouse models (i.e., BAC transgenic hG2019S and hR1441G) and demonstrated that GNE‐7915 enhances DA release and synaptic vesicle mobilization/recycling.
Conclusion
GNE‐7915 can be validated for further therapeutic development for PD.
Keywords: LRRK2, Parkinson's disease, Dopamine, Fast‐scan cyclic voltammetry, Inhibitor
Introduction
Parkinson's disease (PD) is a progressive neurodegenerative movement disorder, affecting approximately 1–2% of the population over the age of 65 1. It is the second most common neurodegenerative disease. The neuropathological hallmark of PD is progressive loss of the dopamine (DA) neurons within the substantia nigra pars compacta (SNpc), associated with deficiency of the neurotransmitter DA in the striatum. Presently, despite extensive research, the etiology of PD remains unknown and there are no disease‐modifying therapeutic agents to slow the neuronal degeneration. Many factors such as age, genetic predisposition, and environmental factors have been involved to cause PD. Recent genetic studies have revealed an underlying genetic cause in at least 10% of all PD cases, which provides new opportunities for the discovery of molecularly targeted therapeutics that may ameliorate neurodegeneration. Among the genes associated with PD, leucine‐rich repeat kinases 2 (LRRK2/PARK8) is the most common cause of familial and sporadic late‐onset forms of PD 2, 3, 4, 5.
LRRK2 is a large gene whose transcript encodes 2527 amino acids. Several independent domains have been established or predicted for the LRRK2 protein, including an ankyrin‐like (ANK) domain, a leucine‐rich repeat (LRR) domain, a Ras‐of‐complex (Roc) GTPase domain followed by its associated C‐terminal of ROC (COR) domain, a serine–threonine kinase domain, and a C‐terminal WD40 domain 6. More than 40 LRRK2 mutations are identified, and six of them are considered definitely pathogenic (R1441C/G/H, Y1699C, G2019S, I2020T) 7, 8. The G2019S, R1441C, and R1441G mutations increase LRRK2 kinase activity 7, and both the kinase and the GTPase activities of LRRK2 are obligatory to induce cell death 9. To date, G2019S is the most common mutation and it is associated with increases in toxic putative kinase activity 3, 10, 11. These findings support the notion that G2019S may play a pathogenic role through a “gain‐of‐function” mechanism, suggesting that small molecule LRRK2 kinase inhibitors may be able to block aberrant LRRK2‐dependent signaling in PD 12, 13, 14, 15. Hence, LRRK2 kinase inhibitors begin to be considered as a potential PD therapy. Rapid progress has been made toward the development of LRRK2 kinase inhibitors 15, 16. LRRK2‐IN‐1 17 is the first selective and exceptionally potent LRRK2 inhibitor (WT‐LRRK2 IC50 = 13 nM, G2019S‐LRRK2 IC50 = 6 nM). However, it cannot pass the blood–brain barrier (BBB). Several LRRK2 inhibitors have recently been synthesized with the designation to cross the BBB, such as the potent and highly selective GSK2578215A 18 and GNE‐7915 19. Both of them are highly potent, selective, metabolically stable, and brain‐penetrant LRRK2 inhibitors (Table S1).
However, does the growing number of developed and patented LRRK2 kinase inhibitors mean that we are closer to a PD disease‐modifying therapy targeting LRRK2? To test whether LRRK2 inhibitors are effective therapies for PD, it is crucial to know, besides safety concerns, whether LRRK2 inhibitors will affect DA neurotransmission or not. To address this gap in knowledge, we investigated the effects of LRRK2 inhibitors on DA release in the dorsal striatum (dSTR) and DA neuron firing in the SNpc using three structurally distinct LRRK2 inhibitors, LRRK2‐IN‐1, GSK2578215A, and GNE‐7915. We included LRRK2‐IN‐1 in the study as it is now the standard/benchmark in cellular assays to investigate LRRK2 function 20, 21, 22, 23, 24.
Materials and Methods
Animals and Slice Preparation
The use of the animals followed the National Institutes of Health guidelines and was approved by the Institutional Animal Care and Use Committee at Thomas Jefferson University. All efforts were made to minimize the number of animals used. BAC LRRK2(hR1441G) transgenic (Tg) mice (stock #009604, The Jackson Laboratory) and BAC LRRK2(hG2019S) Tg mice (stock #009609, The Jackson Laboratory) were all obtained from Chenjian Li's laboratory at Weill Medical College of Cornell University and maintained on Taconic FVB/N background. Non‐Tg, G2019S‐LRRK2‐Tg, R1441G‐LRRK2‐Tg heterozygote mice were prepared by crossing male heterozygote Tg mice with female wild‐type (WT) FVB mice. LRRK2 knockout (KO) mice 25 were a kind gift from Dr. Jie Shen at Harvard Medical School.
Five‐ to 12‐month‐old male WT, LRRK2 KO, G2019S‐LRRK2‐Tg, R1441G‐LRRK2‐Tg mice and their non‐Tg littermates were used. For preparing striatal slices, mice were decapitated without anesthesia and brains were immediately dissected out. Coronal striatal brain slices at 300 μm were prepared on a vibratome (VT1200, Leica, Solms, Germany) for electrophysiological recording. The striatal slices were allowed to recover for at least 1 h at 36°C in a holding chamber containing oxygenated artificial CSF (ASCF) and then placed in a recording chamber superfused (1.5 mL/min) with ACSF (in mM: 125 NaCl, 2.5 KCl, 26 NaHCO3, 2.4 CaCl2, 1.3 MgSO4, 0.3 KH2PO4, and 10 glucose) at 36°C. For preparing midbrain slices, adult mice were anesthetized by intraperitoneal injection of ketamine and xylazine (100 and 10 mg/kg) and then transcardially perfused with 15–20 mL N‐methyl‐d‐glucamine (NMDG) ACSF 26 at room temperature. Coronal midbrain slices at 250 μm were prepared on a vibratome with NMDG ACSF, recovered in NMDG ACSF at 36°C for 10–12 min, and then transferred into a new holding chamber containing HEPES holding ACSF under constant carbogenation at room temperature. NMDG ACSF (in mM): 92 NMDG, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na‐ascorbate, 3 Na‐pyruvate, 0.5 CaCl2 and 10 MgSO4·7H2O. HEPES holding ACSF (in mM): 92 NaCl, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na‐ascorbate, 3 Na‐pyruvate, 0.1 CaCl2 and 4 MgSO4·7H2O. The pH of all ACSF solutions was adjusted to 7.3–7.4 with concentrated hydrochloric acid, and ACSF solutions were saturated with carbogen (95% O2/5% CO2) prior to use to ensure stable pH buffering and adequate oxygenation.
To examine the effects of the LRRK2 inhibitors, slices were incubated for 2 h or perfused for 30 min in ACSF containing LRRK2 inhibitors. For the incubation treatment, striatal slices were bisected, and one striatum was exposed to LRRK2 inhibitor (2 h), while the other was exposed to vehicle (DMSO) as the control.
LRRK2‐IN‐1 and GSK2578215A were purchased from Tocris Bioscience (Minneapolis, MN, USA). GNE‐7915 was a kind gift from Dr. Nathanael Gray at Harvard Medical School. All drugs at the highest concentration used did not alter the carbon fiber electrode sensitivity. The drugs were diluted from frozen aliquots immediately before use and were applied through bath application.
Fast‐scan Cyclic Voltammetry Recording (FSCV)
FSCV and amperometry were used to measure evoked DA release in the dSTR. Electrochemical recordings and electrical stimulation were performed as previously described 27. Briefly, freshly cut carbon fiber electrodes ~5 μm in diameter were inserted ~50 μm into the dSTR slice. For FSCV, a triangular voltage wave (−400 to 900 mV at 280 V/s versus Ag/AgCl) was applied to the electrode every 100 ms. For amperometry, a constant voltage of 500 mV was applied. Current was recorded with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA, USA), with a low‐pass Bessel filter set at 10 kHz, digitized at 25 kHz (ITC‐18 board; InstruTech, Great Neck, NY, USA). Triangular wave generation and data acquisition were controlled by a personal computer running a locally written (Dr. E. Mosharov, Columbia University, New York, NY, USA) IGOR program (WaveMetrics, Lake Oswego, OR, USA). Striatal slices were electrically stimulated (400 μA × 1 ms pulse duration) by an Iso‐Flex stimulus isolator triggered by a Master‐8 pulse generator (AMPI, Jerusalem, Israel) using a bipolar stimulating electrode placed at a distance of 100 μm from the recording electrode. Background‐subtracted cyclic voltammograms served for electrode calibration and to identify the released substance. DA oxidation current was converted to concentration based upon a calibration of 5 μM DA in ACSF after the experiment. In some cases, that is, for experiments measuring the ratio of 2p@100 Hz/1p ratio, after using FSCV to confirm the identity of DA, we switched to amperometry to increase temporal and detection sensitivity. The slices were stimulated every 2 min in the dSTR, and experiments were generally initiated after three stimuli to confirm a consistent and robust response.
Patch‐clamp Recording
In vitro electrophysiological patch‐clamp recordings were performed on SNpc DA neurons through an upright Olympus BX50WI (Olympus, Tokyo, Japan) differential interference contrast microscope with a 40× water immersion objective and an IR‐sensitive video camera. Whole‐cell or cell‐attached patch‐clamp recordings were carried out with a multiclamp 700B amplifier (Molecular Devices, Forster City, CA, USA) and digitized at 10 kHz with a Digidata 1440A (Molecular Devices). Data were acquired using Clampex 10.2 software (Molecular Devices) for subsequent analysis. DA neurons in the SNpc were identified by their characteristic morphologic and physiological features 28, 29. In brief, the hyperpolarization‐activated cation current Ih mediated by hyperpolarization‐activated cyclic nucleotide‐gated (HCN) ion channels is a reliable electrophysiological marker to identify DA neurons in the SNpc (Ih > 100pA). For cell‐attached recording, DA neurons were confirmed by Ih after the experiments.
All experiments were conducted at 35–36°C. For the recordings, slices were submerged in a perfusion of ACSF (1.5 mL/min). Pipettes (3–5 MΩ) were filled with (in mM): 115 K‐gluconate, 10 HEPES, 2 MgCl2, 20 KCl, 2 MgATP, 1 Na2‐ATP, 0.3 GTP, pH = 7.3; 285 ± 5 mOsm. Bridge balance was not compensated, but access resistance was periodically monitored and data were discarded if access resistance exceeded 25 MΩ or changed by more than 20% during an experiment.
Western Blot
Mouse brains slices were homogenized in sucrose buffer (20 mM HEPES (pH 7.4), 0.32 M sucrose, 1 mM NaHCO3, 2.5 mM CaCl2, 1 mM MgCl2) containing complete protease and phosphatase inhibitor cocktail (Fisher Scientific, Pittsburgh, PA, USA). Triton X‐100 was added to a final concentration of 1%, and the solution was incubated on ice for 30 min. Brain lysate was centrifuged at 2000 g, 4 degree for 10 min, and supernatant was transferred into a fresh tube. Protein concentration was measured with BCA assay (Pierce, Fisher Scientific). 35 μg protein was run using 4–12% Bis–Tris precasting Invitrogen gel and immunoblotted with LRRK2 (Neuromab, Davis, CA, USA 1:500 dilution) and pS935 (Abcam, Cambridge, MA, USA 1:500 dilution). The LI‐COR Odyssey system was used for detection and quantification.
Data Analysis and Statistics
Values given in the text and in the figures are mean ± SEM. Data obtained after administration of drugs were analyzed using a paired two‐tailed t‐test for individual comparisons of drug effect versus predrug (GraphPad Software, San Diego, CA, USA) unless specified. The difference was considered significant at levels of P < 0.05 (*).
For each experimental condition, at least six neurons or slices from at least three different mice were examined unless specified otherwise.
Results
No Alteration of DA Release and Recovery in LRRK2 KO Mice
To evaluate whether LRRK2 inhibitors are possible drug targets to treat patients with PD, we need to know whether loss of LRRK2 has any effects on DAergic neurotransmission. Therefore, we examined evoked DA release in striatal slices from LRRK2 KO mice and WT littermates at the age of 10‐12 months using FSCV 27. FSCV allows detection of synaptically released DA levels with subsecond resolution, providing insights into DA signaling dynamics 30. A bipolar stimulating electrode was placed in the dSTR ~150 μm from the recording microelectrode, and depolarizing currents were applied at 2‐min intervals to elicit the release of DA. Due to the heterogeneity of DAergic innervation, for single‐pulse (1p) stimulation, the apparent peak amplitudes of release from three sites in the dSTR of each slice were measured and averaged. Recordings from KO and WT slices at the similar coordinators were compared to further minimize the variation. No significant difference in DA release was detected (WT: 2.10 ± 0.29 μM, n = 11; KO: 2.16 ± 0.28 μM, n = 11, P > 0.05, Figure 1B). There was no alteration of DA release evoked by train stimulation of 4p at 20 Hz mimicking the phasic firing (WT: 2.43 ± 0.05 μM, n = 11; KO: 2.45 ± 0.06 μM, n = 11, P > 0.05, Figure 1B). The ratio of DA release by two pulses at 100 Hz to 1p is an indicator of release probability 31, 32, and it was not altered either (WT: 1.15 ± 0.06, n = 13; KO: 1.14 ± 0.04, n = 13, P > 0.05). The ratio of DA release by 4p at 20 Hz to 1p was not altered either (WT: 1.15 ± 0.02, n = 11; KO: 1.14 ± 0.02, n = 11, P > 0.05). To measure the rate of presynaptic recovery, we stimulated DA release with pairs of pulses separated by intervals at 5, 10, and 20 seconds and pair‐pulse ratio (PPR) was determined (release evoked by the 2nd stimulus/release evoked by the 1st stimulus 33, 34. PPR was not altered in KO mice (Figure 1C) indicating that DA synaptic vesicle replenishment/recycling is not affected by the deletion of KO either 34. Taken together, the results demonstrate that loss of LRRK2 has no effect on DA release and synaptic vesicle replenishment/recycling.
Figure 1.
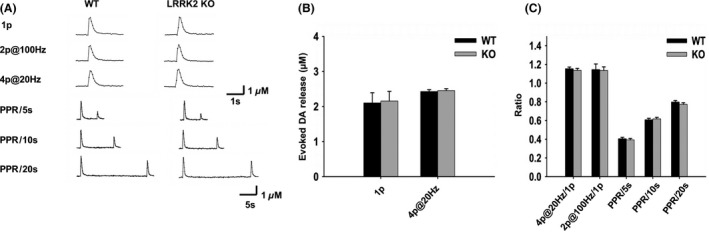
Loss of LRRK2 does not alter DA release and synaptic vesicle replenishment /recycling. (A) Representative voltammetric traces of evoked DA release with different stimulations (one pulse (1p), two pulses at 100 Hz (2p@100 Hz), four pulses at 20 Hz (4p@20 Hz), paired stimuli at variable interpulse intervals) in WT and LRRK2 KO mice. (B) Bar graphs showing no alteration of DA release evoked by 1p or 4p@20 Hz, n = 11. (C) Bar graphs showing no alteration of DA release and PPR by LRRK2 deletion. 4p@20 Hz/1p: 4p@20 Hz train stimuli evoked DA release normalized to 1‐p‐evoked DA release, n = 11; 2p@100 Hz/1p: 2p@100 Hz stimuli evoked DA release normalized to 1p‐evoked DA release, n = 13; PPR/5s, PPR/10s, and PPR/20s: paired‐pulse stimulation at 5‐, 10‐, and 20‐seconds interval, n = 19.
Effects of LRRK2 Inhibitors on DA Release and Recovery in WT Mice
We then examined the effects of these three LRRK2 inhibitors on evoked DA release and recovery in WT mice. Striatal slices were bisected, and one striatum was exposed to a LRRK2 inhibitor at various concentrations for 2 h at 36°C, while the other was exposed to vehicle (DMSO). As all three LRRK2 inhibitors are highly potent with low nanomolar biochemical and cellular activities, each LRRK2 inhibitor was tested at 0.1, 0.3, 1, 3 μM, respectively, to determine the dose response. LRRK2‐IN‐1 at concentrations of 0.1, 0.3, and 1 μM had no effect on DA release and PPR. However, LRRK2‐IN‐1 at 3 μM decreased single‐pulse‐evoked DA release by 28% (n = 7; P < 0.05) and 4p@20 Hz‐evoked DA release by 25% (n = 7; P < 0.05; Figure 2B). PPR at 5‐, 10‐, and 20‐seconds interval was also attenuated (Figure 2B). In contrast, GSK2578215A (Figure 2C) and GNE‐7915 (Figure 2D) at all concentrations had no effect on DA release and recovery. Western blot confirmed the dose–response inhibition by the LRRK2 inhibitions (Figure S1).
Figure 2.

Effects of different concentrations of LRRK2 inhibitors on DA release in WT mice. (A) Representative voltammetric traces of evoked DA release with different stimulations before and after LRRK2‐IN‐1 (3 μM, 2‐h incubation) treatment. (B) Effects of LRRK2‐IN‐1 (1 and 3 μM, 2‐h incubation) on evoked DA release and PPR. DMSO, n = 14; 1 μM, n = 10; 3 μM, n = 7. LRRK2‐IN‐1 at 1 μM had no effect on evoked DA release and PPR. However, LRRK2‐IN‐1 at a concentration of 3 μM decreased 1p‐evoked DA release (treated group: 1.57 ± 0.21 μM, n = 7; control group: 2.19 ± 0.08 μM, n = 7, P < 0.05) and 4p@20 Hz‐evoked DA release (treated group: 2.01 ± 0.13 μM, n = 7; control group: 2.67 ± 0.13 μM, n = 7, P < 0.05) and PPR with 5‐s interval (treated group: 0.40 ± 0.02, n = 7; control group: 0.46 ± 0.02, n = 7, P < 0.05), 10‐s interval (treated group: 0.54 ± 0.02, n = 7; control group: 0.64 ± 0.02, n = 7, P < 0.05), and 20‐s interval (treated group: 0.73 ± 0.02, n = 7; control group: 0.83 ± 0.03, n = 7, P < 0.05). (C) GSK2578215A (1 and 3 μM) had no effect on evoked DA release and PPR. DMSO, n = 14; 1 μM, n = 10; 3 μM, n = 10. (D) GNE‐7915 (1 and 3 μM) had no effect on evoked DA release and PPR. DMSO, n = 14; 1 μM, n = 11; 3 μM, n = 9. *P < 0.05, paired t‐test. All the three inhibitors at 0.1 or 0.3 μM had no effect on evoked DA release (data not shown here, but summarized in Figure 3B).
LRRK2‐IN‐1 at a Concentration of 3 μM Shows Off‐target Effects, Whereas GNE‐7915 and GSK2578215A Do Not
The inhibitory effects of 3 μM LRRK2‐IN‐1 on evoked DA release and recovery could be LRRK2 dependent, but could also be due to off‐target effects. To address concerns about off‐target pharmacology, we conducted the same experiments in LRRK2 KO mice with 1 or 3 μM LRRK2 inhibitors, the highest concentrations we used in this study. LRRK2‐IN‐1 at 3 μM decreased 1p‐evoked DA release by 39 ± 11% (n = 14; P < 0.01; Figure 3B, right panel). PPR at 5‐, 10‐, and 20‐seconds intervals was also attenuated (Figure 3A, bottom panel). We performed a comparison between WT and KO. There was no clear difference between genotypes at 3 μM (WT: 28 ± 7%, n = 7; KO: 39 ± 11%, n = 14, P > 0.05), suggesting that LRRK2‐IN‐1 mediates its inhibitory effect independently of LRRK2. GSK2578215A and GNE‐7915 at both concentrations had no effect on DA release and recovery, confirming on‐target effects. Therefore, we chose 1 μM as an appropriate concentration for these three LRRK2 inhibitors for short‐time treatments in the subsequent experiments.
Figure 3.
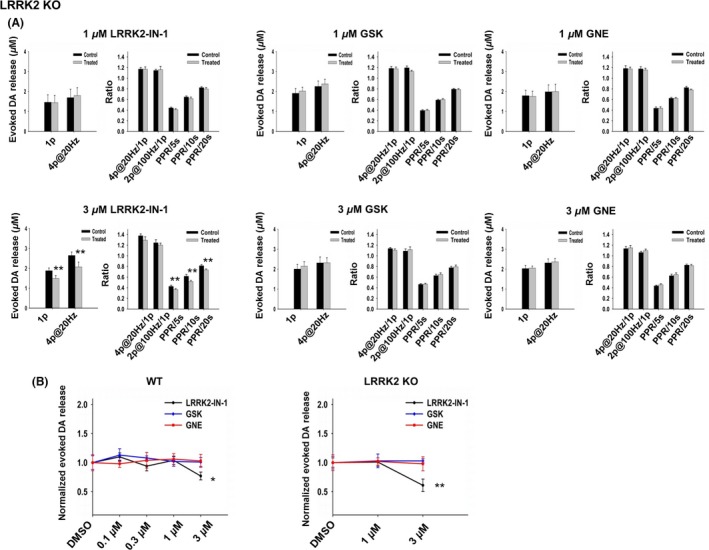
LRRK2‐IN‐1 at 3 μM shows off‐target effects on evoked DA release in KO mice, whereas GSK2578215A and GNE‐7915 do not. (A) Upper panels show no effect of the three LRRK2 inhibitors (1 μM, 2‐h incubation) on DA release and PPR. LRRK2‐IN‐1, n = 12 for each condition; GSK, n = 8 for each condition; GNE, n = 10 for each condition. Lower panels show that LRRK2‐IN‐1 at a concentration of 3 μM decreased 1‐p‐evoked DA release (treated group: 1.49 ± 0.22 μM, n = 14; control group: 1.88 ± 0.20 μM, n = 4, P < 0.01), 4p@20 Hz‐evoked DA release (treated group: 2.07 ± 0.26 μM, n = 14; control group: 2.65 ± 0.26 μM, n = 4, P < 0.01) and PPR with 5‐s interval (treated group: 0.37 ± 0.02, n = 9; control group: 0.43 ± 0.04, n = 9, P < 0.01), 10‐s interval (treated group: 0.52 ± 0.02, n = 9; control group: 0.61 ± 0.04, n = 9, P < 0.01), and 20‐s interval (treated group: 0.74 ± 0.02, n = 9; control group: 0.82 ± 0.02, n = 9, P < 0.01). GSK, n = 8 for each condition; GNE, n = 10 for each condition. *P < 0.05, **P < 0.01, paired t‐test. (B) Dose–response curves for each LRRK2 inhibitor, respectively, in WT mice (left panel) and KO mice (right panel). The amplitude of 1p‐evoked DA release in the treated group was normalized to the control group (DMSO‐treated group). 3 μM LRRK2‐IN‐1 decreased 1p‐evoked DA release in both WT and KO mice suggesting off‐target effects. *P < 0.05, **P < 0.01, One‐way ANOVA followed by Bonferroni test.
All Three LRRK2 Inhibitors Show No Inhibitory Effect on DA Release and Recovery in WT Mice with 30‐min Perfusion
Although these three LRRK2 inhibitors had no effects on DA release and recovery in WT mice (1 μM, 2‐h incubation), the experiments were conducted in different slices, that is, control slice versus treated slice. To make sure that these inhibitors at 1 μM have no acute effect on DA release and recovery in WT mice, we decided to examine the effects of LRRK2 inhibitors on the same recording site for a short period of time. We recorded the evoked DA releases in the same site before and after perfusing the slice with 1 μM LRRK2 inhibitor for 30 min. There was no alteration of DA release and PPR before and after 30‐min perfusion for LRRK2‐IN‐1 (Figure 4A), GSK2578215A (Figure 4B), and GNE‐7915 (Figure 4C). Thus, 30‐min treatment of LRRK2 inhibitor had no effect on DA release or synaptic vesicle replenishment/recycling, confirming that these three LRRK2 inhibitors at concentrations of 1 μM or lower have no acute effect on DA release or recovery.
Figure 4.

None of the three LRRK2 inhibitors have any significant effect on DA release or synaptic vesicle replenishment/recycling with 30‐min perfusion. Evoked DA release was recorded by FSCV at one site within a slice, and 1 μM LRRK2 inhibitor was perfused for 30 min. There was no significant difference of DA release and PPR before and after 30‐min perfusion. (A) LRRK2‐IN‐1, n = 6 for each condition. (B) GSK, n = 6 for each condition; (C) GNE, n = 6 for each condition.
None of the Three LRRK2 inhibitors Have Any Effects on DA Release and Recovery in G2019S Transgenic Mice
We then chose 1 μM as an appropriate concentration to examine whether LRRK2 inhibitors could enhance DA neurotransmission in G2019S Tg mice 35. G2019S Tg mice showed no alteration of DA release or recovery at the age of 10‐12 months old (Figure S2, five pairs of 10‐ to 12‐month‐old mice). Acute brain slices from ~10‐month‐old G2019S Tg mice were cut in half, one half as control group and the other half treated with a LRRK2 inhibitor for 2 h at 36°C. None of the three inhibitors had any effect on DA release or recovery (indicated by PPR) in G2019S Tg mice (Figure 5B). We then performed FSCV at the same site in one slice before and after 30‐min perfusion with LRRK2 inhibitor, and no significant difference in DA release or recovery was detected either (Figure 5C). As the lack of the effect on G2019S TG mice could be related to an increased LRRK2 kinase activity, for which 1 μM might be below the threshold, we performed a concentration–response curve and found that there was still no effect even at 3 μM (Figure S3). Western blot confirmed the dose–response inhibition by the LRRK2 inhibitions (Figure S4).
Figure 5.
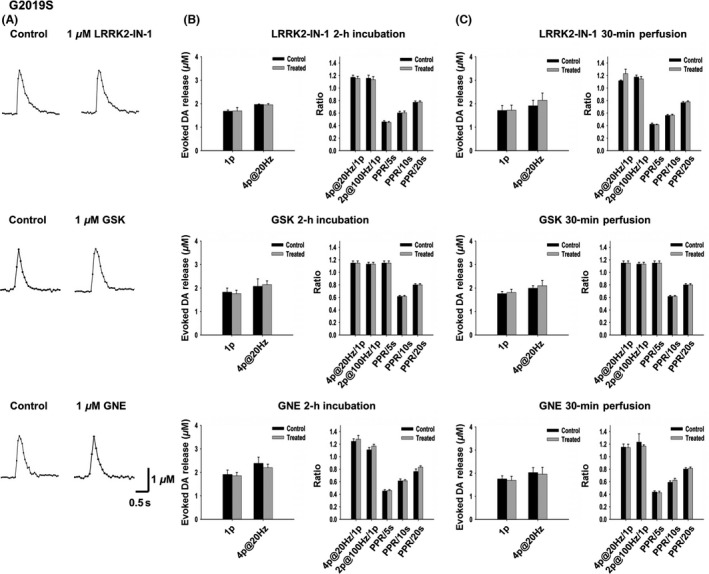
None of the three LRRK2 inhibitors have any significant effect on DA release or synaptic vesicle replenishment/recycling in the G2019S Tg mice with 2‐h incubation or 30‐min perfusion. (A) Representative voltammetric traces of 1‐p‐evoked DA release under control condition and with LRRK2 inhibitor (1 μM, 2 hr) treatment. (B) No inhibitor (1 μM, 2‐h incubation) produced a significant effect on DA release and recovery. LRRK2‐IN‐1, n = 10 for each condition; GSK, n = 11 for each condition; GNE, n = 11 for each condition. (C) No inhibitor (1 μM, 30‐min perfusion) produced a significant effect on DA release and recovery. LRRK2‐IN‐1, n = 7 for each condition; GSK, n = 7 for each condition; GNE, n = 7 for each condition.
GNE‐7915 Enhances DA Release and Recovery in R1441G Transgenic Mice
Next, we examined the effects of LRRK2 inhibitor on another preclinical mouse model, R1441G Tg mice, to see whether any of the inhibitors could alleviate DA transmission deficits. R1441G Tg mice recapitulate the motor behavioral, neurochemical, and histopathological features of PD 36. In addition, the Tg mice exhibit age‐dependent decrease of DA release and recovery, which starts at the age of 5 months. PPR was significantly decreased in Tg mice starting at the age of 5 months, and 1p‐evoked DA release was decreased by 25% at the age of 10 months (2014 SFN abstract, 512.08/S5, data now shown). When treated with 1 μM LRRK2‐IN‐1 or GSK2578215A for 30‐min perfusion or 2‐h incubation, DA release and recovery did not show any significant difference. However, when treated with 1 μM GNE‐7915 for 2 h, we observed a significant increase of PPR (PPR/5s, control: 0.45 ± 0.01, treated: 0.48 ± 0.01, n = 10, P < 0.05; PPR/10s, control: 0.61 ± 0.005, treated: 0.66 ± 0.008, n = 10, P < 0.01; PPR/20s, control: 0.80 ± 0.015, treated: 0.84 ± 0.002, n = 10, P < 0.001, Figure 6). Single‐pulse‐evoked DA release was also enhanced (control: 1.93 ± 0.15 μM, n = 10; treated: 2.17 ± 0.19 μM, n = 10, P < 0.05, Figure 6). Thirty‐minute perfusion also increased DA release and the PPR recorded from the same site.
Figure 6.
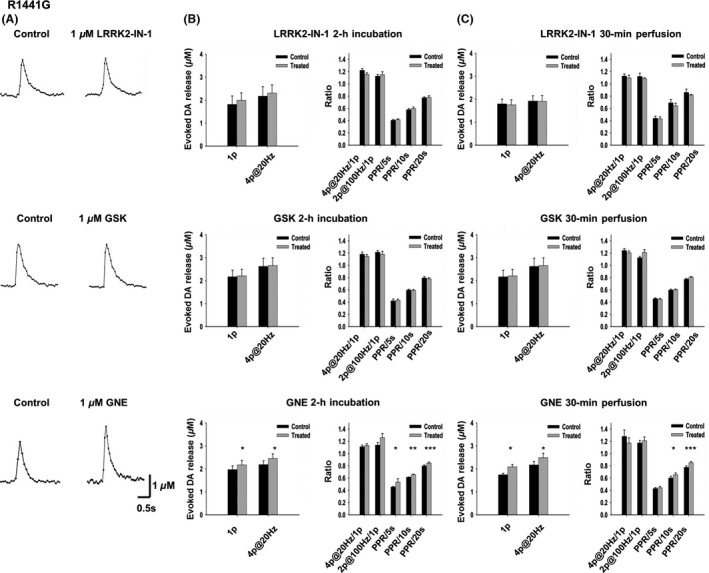
GNE‐7915 enhances DA release and synaptic vesicle replenishment/recycling in the R1441G Tg mice. (A) Representative voltammetric traces of 1‐p‐evoked DA release under control condition and with LRRK2 inhibitor (1 μM, 2‐h incubation) treatment. LRRK2‐IN‐1 and GSK had no significant effect on DA release, whereas GNE enhanced DA release. (B) Neither LRRK2‐IN‐1 (1 μM, 2‐h incubation, n = 10 for each condition) nor GSK (1 μM, 2‐h incubation, n = 10 for each condition) had any significant effect on DA release and recovery. In contrast, GNE‐7915 (1 μM, 2‐h incubation) increased 1‐p and 4p@20 Hz‐evoked DA release (n = 10, P < 0.05) and PPR at 5s (n = 10, P < 0.05), 10s (n = 10, P < 0.01) and PPR at 20s (n = 10, P < 0.001). 4p@20 Hz/1p, n = 9; 2p@100 Hz/1p, n = 9. (C) Neither LRRK2‐IN‐1 (1 μM, 30‐min perfusion, n = 7 for each condition) nor GSK (1 μM, 30‐min perfusion, n = 7 for each condition) had any effect on DA release and kinetics. In contrast, GNE‐7915 (1 μM, 30‐min perfusion) increased single‐pulse and 4p@20 Hz‐evoked DA release (n = 6, P < 0.05) and PPR at 10s (n = 9, P < 0.05) and PPR at 20s (n = 9, P < 0.001). 4p@20 Hz/1p, n = 6; 2p@100 Hz/1p, n = 6; PPR/5s, n = 9. *P < 0.05, ***P < 0.001, paired t‐test.
LRRK2 Inhibitors Have No Effects on the Firing Rates of DA Neurons from WT, G2019S, or R1441G TG Mice
LRRK2 inhibitors can exert their effects on DA neurotransmission either on the release of DA from the terminals, or on the firing of DA neurons. To investigate whether the firing properties of DA neuron are affected by the LRRK2 inhibitors, we used patch‐clamp recording to examine the tonic firing rates of SNpc DA neurons in acute midbrain slices from 5‐ to 6‐month‐old WT mice. DA neurons in the midbrain are vulnerable to various stresses, and it is very challenging to do patch‐clamp recording in adult slices (> 4 weeks). We have successfully conducted patch‐clamp experiments in up to 10‐month‐old mice using a modified cutting solution and holding solution 26. No difference of the firing rate was observed between the control group and the inhibitor‐treated group (LRRK2‐IN‐1: 2.07 ± 0.18 Hz versus 2.10 ± 0.22 Hz, n = 10; GSK: 2.27 ± 0.22 Hz versus 2.34 ± 0.20 Hz, n = 10; GNE: 2.22 ± 0.20 Hz versus 2.18 ± 0.19 Hz, n = 10, Figure 7A, middle panel). A 30‐min perfusion of the same neuron recorded with LRRK2 inhibitor did not have any detectable effects (Figure 7A, right panel). Thus, these three LRRK2 inhibitors had no effect on the firing rate of SNpc DA neurons from WT mice. We then used patch‐clamp recording to examine the effects of these three LRRK2 inhibitors on SNpc DA neurons from G2019S (Figure 7B) and R1441G (Figure 7C) Tg mice. No effect was observed for any of these three inhibitors. G2019S and R1441G mutations did not affect the tonic firing rate of the SNpc DA neurons either (WT: 2.19 ± 0.06, n = 66; G2019S: 2.24 ± 0.07, n = 63; R1441G: 2.23 ± 0.06, n = 54).
Figure 7.
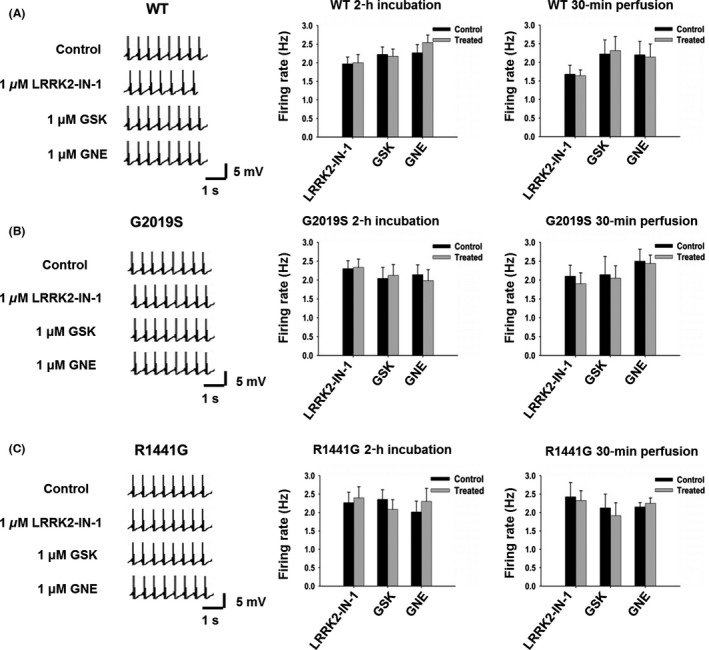
None of the three LRRK2 inhibitors have any significant effect on the tonic firing rates of SNpc DA neurons from WT, G2019S, and R1441G Tg mice. (A) No significant alteration of DA neuron firing rates in WT mice after 1 μM LRRK2‐IN‐1, GNE7915, and GSK2578215A treatment. Left panel, representative traces of patch recording before and after 30‐min perfusion of LRRK2 inhibitors; middle panel, bar graph showing no effects of the three inhibitors with 2‐h incubation, n = 10 for each inhibitor; right panel, bar graph showing no effects of the three inhibitors with 30‐min perfusion, n = 12 for each inhibitor. (B) No significant alteration of DA neuron firing rates in G2019S mice after 1 μM LRRK2‐IN‐1, GNE7915, and GSK2578215A treatment. Left panel, representative traces of patch recording before and after 30‐min perfusion of LRRK2 inhibitors; middle panel, bar graph showing no effects of the three inhibitors with 2‐h incubation, n = 11 for each inhibitor; right panel, bar graph showing no effects of the three inhibitors with 30‐min perfusion, n = 10 for each inhibitor. (C) No significant alteration of DA neuron firing rates in R1441G mice after 1 μM LRRK2‐IN‐1, GNE7915, and GSK2578215A treatment. Left panel, representative traces of patch recording before and after 30‐min perfusion of LRRK2 inhibitors; middle panel, bar graph showing no effects of the three inhibitors with 2‐h incubation, n = 10 for each inhibitor; right panel, bar graph showing no effects of the three inhibitors with 30‐min perfusion, n = 8 for each inhibitor.
Discussion
Our goal in this study was to examine the effects of various LRRK2 inhibitors on DA neurotransmission. To evaluate the potential of LRRK2 inhibitors as disease‐modifying therapies in PD, the first key issue is to assess the safety of kinase inhibitors. Therefore, we need to investigate whether loss of LRRK2 has any detrimental effects on DA neurotransmission. Consistent with a previous report 37, our study provides further evidence that loss of LRRK2 does not affect DA neurotransmission. LRRK2 KO mice show normal DA release and recovery compared to WT controls. Although loss of LRRK2 is not equivalent to the situation of LRRK2 kinase inhibition, the result suggests that inhibiting LRRK2 will not have side effects in terms of DA neurotransmission, which is critical to PD treatment. Our findings are consistent with the findings that LRRK2 deletion in mice does not cause neurodegeneration and motor deficits 38.
Using pharmacological approaches together with the LRRK2 KO mice, we investigated the effects of three structurally distinct LRRK2 inhibitors (LRRK2‐IN‐1, GSK2578215A, and GNE‐7915) on synaptically released DA in the dSTR using FSCV and DA neuron firing in the SNpc using patch‐clamp recording. We found that LRRK2‐IN‐1 at a concentration of 3 μM caused off‐target effects and decreased DA release, whereas GSK2578215A and GNE‐7915 did not with regard to DAergic transmission. We have further examined the effects in two preclinical LRRK2 mouse models (i.e., BAC transgenic hLRRK2‐G2019S and BAC transgenic hLRRK2‐R1441G) and demonstrate that GNE‐7915 enhances DA release and recovery (i.e., increasing synaptic vesicle mobilization/recycling) in R1441G mice. Thus, GNE‐7915 may be a better choice in the development of LRRK2 related therapeutics for PD. The lack of any effect of the LRRK2 inhibitors on G2019S mice is not surprising as the mice themselves have no DA neurotransmission deficits up to 12 months old.
Although all three inhibitors are selective, the selectivity of LRRK‐IN‐1 is not as good as GSK2578215A or GNE‐7915 15. Therefore, LRRK2‐IN‐1 at high concentrations may worsen DA neurotransmission by targeting other kinases. A recent study has also reported the off‐target effects of LRRK2‐IN‐1 39. Caution should be exerted when using LRRK2‐IN‐1 at high concentrations even for short periods of time or at low concentrations for long periods of time for biochemical or cellular studies. Genetic controls (KO or knockdown) should be used in conjunction to rule out off‐target effects. GSK2578215A or GNE‐7915 is highly selective as there is no observable off‐target effect in terms of DAergic neurotransmission even at 3 μM.
Whereas GNE‐7915 enhances DA release and recovery in R1441G mice, GSK2578215A has no effect. Although both inhibitors are BBB permeable, GSK2578215A fails to reduce the phosphorylation levels of LRRK2 in the CNS, although the mechanism is unclear 15. Our Western blot results from brain slices also showed less dephosphorylation by GSK2578215A compared to GNE‐7915. This may explain the different effects of GSK2578215A and GNE‐7915 in our study.
In addition to G2019S Tg, we chose R1441G Tg mice as this model has shown progressive motor deficits and DA release deficits. Human R1441C BAC Tg rats have also shown progressive motor deficits and DA release deficits 40. Although R1441G mutation is within the GTPase domain instead of the kinase domain, recent studies suggest that the two domains interact with each other 41, 42, 43, 44 and kinase activity is increased in R1441G Tg mice based on the Ser1292 autophosphorylation assay 7. GNE‐7915 enhanced DA release and recovery in R1441G mice, but not in G2019S mice. This is intriguing because kinase activity is more elevated in G2019S mice. One can speculate that decreased DA release and recovery in R1441G was a consequence of both altered GTPase and kinase activities and the rescue may depend on the interplay of GTPase and kinase domains. In our study, PPR is a measurement of the recovery of the rapid releasable pool of DA synaptic vesicles, which includes the mobilization, endocytosis, and refilling of DA into synaptic vesicles. The mechanism whereby GNE‐7915 enhances the recovery and release of DA vesicles warrants further studies.
Our work is the first study to examine whether LRRK2 inhibitors affect DAergic neurotransmission acutely and ameliorate DAergic neurotransmission deficits in preclinical models of PD; however, our study is limited in nature. First, our study was conducted in acute brain slices and we examined the acute effects (30‐min to 2‐h treatment). As PD is a chronic disorder, we need to investigate the effects of chronic treatment on DA neurotransmission. For example, mice will be orally administered with GNE‐7915 for months and we will then examine DA neurotransmission either in slice preparations or in vivo to know the effects following long‐term treatment with LRRK2 inhibitors. Second, DA neurons have two firing patterns in vivo, that is, tonic firing and phasic firing 45. We could only examine the effects of LRRK2 inhibitor on tonic firing using slice preparation as phasic firing depends on inputs from other brain areas to the SNpc. We cannot exclude the possibility that the inhibitor may affect DA neuron firing by exerting its effects on the inputs to SNpc. Finally, our results could not tell whether these LRRK2 inhibitors will slow down DA neuron degeneration, and therefore, the current data may not be good predictions of the therapeutic effects of the inhibitors.
Despite these limitations, our work here clearly demonstrates that LRRK2‐IN‐1, GSK2578215A, and GNE‐7915 at concentrations of 1 μM have no acute effects on synaptically released DA in the dSTR and the tonic firing rate of DA neurons in the SNpc in WT mice. GNE‐7915 could even enhance DA release and recovery in a R1441G mouse model. These findings are encouraging, as they not only fill in the gap in our knowledge on the safety in terms of DAergic neurotransmission, but also allow us to determine whether certain LRRK2 inhibitors can be validated for further therapeutic development for PD.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Figure S1. Western blot was used to measure the effects of different concentrations of LRRK2 inhibitors on s935 phosphorylation of LRRK2 in striatal slices from WT mice.
Figure S2. There is no alteration of DA release and synaptic vesicle replenishment /recycling in G2019S Tg mice.
Figure S3. FSCV was used to measure the effects of different concentrations of LRRK2 inhibitors on DA release and recovery in G2019S mice.
Figure S4. Western blot was used to measure the effects of different concentrations of LRRK2 inhibitors on s935 phosphorylation of LRRK2 in striatal slices from G2019S mice.
Table S1. Supplementary table.
Acknowledgments
We thank Dr. Jie Shen for providing the LRRK2 KO mice, and Dr. Nathanael S Gray for supplying GNE‐7915. This work was supported in part by a grant from the Michael J. Fox Foundation to HZ and by the Department of Neuroscience at Thomas Jefferson University Startup Funds to HZ.
Contributor Information
Guo‐Zhong Li, Email: hydlgz1962@163.com.
Hui Zhang, Email: Hui.X.Zhang@jefferson.edu.
References
- 1. Lees AJ, Hardy J, Revesz T. Parkinson's disease. Lancet 2009;373:2055–2066. [DOI] [PubMed] [Google Scholar]
- 2. Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal‐dominant parkinsonism with pleomorphic pathology. Neuron 2004;44:601–607. [DOI] [PubMed] [Google Scholar]
- 3. Gilks WP, Abou‐Sleiman PM, Gandhi S, et al. A common LRRK2 mutation in idiopathic Parkinson's disease. Lancet 2005;365:415–416. [DOI] [PubMed] [Google Scholar]
- 4. Paisan‐Ruiz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8‐linked Parkinson's disease. Neuron 2004;44:595–600. [DOI] [PubMed] [Google Scholar]
- 5. Di Fonzo A, Rohe CF, Ferreira J, et al. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson's disease. Lancet 2005;365:412–415. [DOI] [PubMed] [Google Scholar]
- 6. Cookson MR. The role of leucine‐rich repeat kinase 2 (LRRK2) in Parkinson's disease. Nat Rev Neurosci 2010;11:791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sheng Z, Zhang S, Bustos D, et al. Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med 2012;4:164ra161. [DOI] [PubMed] [Google Scholar]
- 8. Lee BD, Dawson VL, Dawson TM. Leucine‐rich repeat kinase 2 (LRRK2) as a potential therapeutic target in Parkinson's disease. Trends Pharmacol Sci 2012;33:365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kumar A, Cookson MR. Role of LRRK2 kinase dysfunction in Parkinson disease. Expert Rev Mol Med 2011;13:e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Greggio E, Jain S, Kingsbury A, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis 2006;23:329–341. [DOI] [PubMed] [Google Scholar]
- 11. West AB, Moore DJ, Biskup S, et al. Parkinson's disease‐associated mutations in leucine‐rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A 2005;102:16842–16847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dusonchet J, Kochubey O, Stafa K, et al. A rat model of progressive nigral neurodegeneration induced by the Parkinson's disease‐associated G2019S mutation in LRRK2. J Neurosci 2011;31:907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee BD, Shin JH, VanKampen J, et al. Inhibitors of leucine‐rich repeat kinase‐2 protect against models of Parkinson's disease. Nat Med 2010;16:998–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yao C, Johnson WM, Gao Y, et al. Kinase inhibitors arrest neurodegeneration in cell and C. elegans models of LRRK2 toxicity. Hum Mol Genet 2013;22:328–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kavanagh ME, Doddareddy MR, Kassiou M. The development of CNS‐active LRRK2 inhibitors using property‐directed optimisation. Bioorg Med Chem Lett 2013;23:3690–3696. [DOI] [PubMed] [Google Scholar]
- 16. Estrada AA, Sweeney ZK. Chemical biology of Leucine‐Rich repeat Kinase 2 (LRRK2) inhibitors. J Med Chem 2015;58:6733–6746. [DOI] [PubMed] [Google Scholar]
- 17. Deng X, Dzamko N, Prescott A, et al. Characterization of a selective inhibitor of the Parkinson's disease kinase LRRK2. Nat Chem Biol 2011;7:203–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reith AD, Bamborough P, Jandu K, et al. GSK2578215A; a potent and highly selective 2‐arylmethyloxy‐5‐substitutent‐N‐arylbenzamide LRRK2 kinase inhibitor. Bioorg Med Chem Lett 2012;22:5625–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Estrada AA, Liu X, Baker‐Glenn C, et al. Discovery of highly potent, selective, and brain‐penetrable leucine‐rich repeat kinase 2 (LRRK2) small molecule inhibitors. J Med Chem 2012;55:9416–9433. [DOI] [PubMed] [Google Scholar]
- 20. Marker DF, Puccini JM, Mockus TE, Barbieri J, Lu SM, Gelbard HA. LRRK2 kinase inhibition prevents pathological microglial phagocytosis in response to HIV‐1 Tat protein. J Neuroinflammation 2012;9:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matta S, Van Kolen K, da Cunha R, et al. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron 2012;75:1008–1021. [DOI] [PubMed] [Google Scholar]
- 22. Caesar M, Zach S, Carlson CB, Brockmann K, Gasser T, Gillardon F. Leucine‐rich repeat kinase 2 functionally interacts with microtubules and kinase‐dependently modulates cell migration. Neurobiol Dis 2013;54:280–288. [DOI] [PubMed] [Google Scholar]
- 23. Papkovskaia TD, Chau KY, Inesta‐Vaquera F, et al. G2019S leucine‐rich repeat kinase 2 causes uncoupling protein‐mediated mitochondrial depolarization. Hum Mol Genet 2012;21:4201–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Esteves AR, Fernandes MG, Santos D, Januario C, Cardoso SM. The upshot of LRRK2 inhibition to Parkinson's disease paradigm. Mol Neurobiol 2015;52:1804–1820. [DOI] [PubMed] [Google Scholar]
- 25. Tong Y, Yamaguchi H, Giaime E, et al. Loss of leucine‐rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha‐synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci U S A 2010;107:9879–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ting JT, Daigle TL, Chen Q, Feng G. Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol Biol 2014;1183:221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang H, Sulzer D. Glutamate spillover in the striatum depresses dopaminergic transmission by activating group I metabotropic glutamate receptors. J Neurosci 2003;23:10585–10592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ding S, Wei W, Zhou FM. Molecular and functional differences in voltage‐activated sodium currents between GABA projection neurons and dopamine neurons in the substantia nigra. J Neurophysiol 2011;106:3019–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Watts AE, Williams JT, Henderson G. Baclofen inhibition of the hyperpolarization‐activated cation current, Ih, in rat substantia nigra zona compacta neurons may be secondary to potassium current activation. J Neurophysiol 1996;76:2262–2270. [DOI] [PubMed] [Google Scholar]
- 30. Robinson DL, Venton BJ, Heien ML, Wightman RM. Detecting subsecond dopamine release with fast‐scan cyclic voltammetry in vivo. Clin Chem 2003;49:1763–1773. [DOI] [PubMed] [Google Scholar]
- 31. Rice ME, Cragg SJ. Nicotine amplifies reward‐related dopamine signals in striatum. Nat Neurosci 2004;7:583–584. [DOI] [PubMed] [Google Scholar]
- 32. Zhang H, Sulzer D. Frequency‐dependent modulation of dopamine release by nicotine. Nat Neurosci 2004;7:581–582. [DOI] [PubMed] [Google Scholar]
- 33. Abeliovich A, Schmitz Y, Farinas I, et al. Mice lacking alpha‐synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000;25:239–252. [DOI] [PubMed] [Google Scholar]
- 34. Hernandez D, Torres CA, Setlik W, et al. Regulation of presynaptic neurotransmission by macroautophagy. Neuron 2012;74:277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Choi I, Kim B, Byun JW, et al. LRRK2 G2019S mutation attenuates microglial motility by inhibiting focal adhesion kinase. Nat Commun 2015;6:8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li Y, Liu W, Oo TF, et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nat Neurosci 2009;12:826–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arranz AM, Delbroek L, Van Kolen K, et al. LRRK2 functions in synaptic vesicle endocytosis through a kinase‐dependent mechanism. J Cell Sci 2015;128:541–552. [DOI] [PubMed] [Google Scholar]
- 38. Tong Y, Shen J. Genetic analysis of Parkinson's disease‐linked leucine‐rich repeat kinase 2. Biochem Soc Trans 2012;40:1042–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luerman GC, Nguyen C, Samaroo H, et al. Phosphoproteomic evaluation of pharmacological inhibition of leucine‐rich repeat kinase 2 reveals significant off‐target effects of LRRK‐2‐IN‐1. J Neurochem 2014;128:561–576. [DOI] [PubMed] [Google Scholar]
- 40. Sloan M, Alegre‐Abarrategui J, Potgieter D, et al. LRRK2 BAC transgenic rats develop progressive, L‐DOPA‐responsive motor impairment, and deficits in dopamine circuit function. Hum Mol Genet 2016;25:951–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li T, Yang D, Zhong S, et al. Novel LRRK2 GTP‐binding inhibitors reduced degeneration in Parkinson's disease cell and mouse models. Hum Mol Genet 2014;23:6212–6222. [DOI] [PubMed] [Google Scholar]
- 42. Biosa A, Trancikova A, Civiero L, et al. GTPase activity regulates kinase activity and cellular phenotypes of Parkinson's disease‐associated LRRK2. Hum Mol Genet 2012;22:1140–1156. [DOI] [PubMed] [Google Scholar]
- 43. Rudenko IN, Chia R, Cookson MR. Is inhibition of kinase activity the only therapeutic strategy for LRRK2‐associated Parkinson's disease? BMC Med 2012;10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rudenko IN, Cookson MR. Heterogeneity of leucine‐rich repeat kinase 2 mutations: genetics, mechanisms and therapeutic implications. Neurotherapeutics 2014;11:738–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schultz W. Responses of midbrain dopamine neurons to behavioral trigger stimuli in the monkey. J Neurophysiol 1986;56:1439–1461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Western blot was used to measure the effects of different concentrations of LRRK2 inhibitors on s935 phosphorylation of LRRK2 in striatal slices from WT mice.
Figure S2. There is no alteration of DA release and synaptic vesicle replenishment /recycling in G2019S Tg mice.
Figure S3. FSCV was used to measure the effects of different concentrations of LRRK2 inhibitors on DA release and recovery in G2019S mice.
Figure S4. Western blot was used to measure the effects of different concentrations of LRRK2 inhibitors on s935 phosphorylation of LRRK2 in striatal slices from G2019S mice.
Table S1. Supplementary table.