

Abstract
1. Purpose
In the era of next‐generation sequencing, we are increasingly confronted with sequence variants of unknown significance. This phenomenon is also known for variations in Caveolin‐3 and can complicate the molecular diagnosis of the disease. Here, we aimed to study the ambiguous character of the G56S Caveolin‐3 variant.
2. Experimental design
A comprehensive approach combining genetic and morphological studies of muscle derived from carriers of the G56S Caveolin‐3 variant were carried out and linked to biochemical assays (including phosphoblot studies and proteome profiling) and morphological investigations of cultured myoblasts.
3. Results
Muscles showed moderate chronic myopathic changes in all carriers of the variant. Myogenic RCMH cells expressing the G56S Caveolin‐3 protein presented irregular Caveolin‐3 deposits within the Golgi in addition to a regular localization of the protein to the plasma membrane. This result was associated with abnormal findings on the ultra‐structural level. Phosphoblot studies revealed that G56S affects EGFR‐signaling. Proteomic profiling demonstrated alterations in levels of physiologically relevant proteins which are indicative for antagonization of G56S Caveolin‐3 expression. Remarkably, some proteomic alterations were enhanced by osmotic/mechanical stress.
4. Conclusions and clinical relevance
Our studies suggest that G56S might influence the manifestation of myopathic changes upon the presence of additional cellular stress burden. Results of our studies moreover improve the current understanding of (genetic) causes of myopathic disorders classified as caveolinopathies.
Keywords: Caveolin‐3, LGMD‐1C, Proteome profile of muscle cells, RCMH
Abbreviations
- EGFR
epidermal growth factor receptor
- LGMD‐1C
limb‐girdle muscular dystrophy type 1C
- PM
plasma membrane
- RMD
rippling muscle disease
- RT
room temperature
- wt
wild type
1. Introduction
In the era of next‐generation sequencing, geneticists, clinicians, and pathologists are increasingly confronted with sequence variants of unknown significance. Assigning pathogenicity to such variants solely based on genetic data and prediction programs might be misleading. Here, we characterize the ambiguous sequence of Caveolin‐3 G56S, which is located within the scaffolding domain of Caveolin‐3 1.
Clinical Relevance
This article represents a translational medical study with clinical impact toward the better understanding and characterization of sequence variants with unknown significance, which is a very important topic in the era of next‐generation sequencing as part of the diagnosis and management of (neuromuscular) patients as well as for drug treatment.
Caveolins are major structural components of caveolae that serve as signaling hubs and membrane reservoirs at the cell surface 2. They also function as scaffolding proteins that organize and enrich Caveolin‐interacting proteins and lipids 3. During muscle contraction, caveolae are passively flattened and compensate plasma membrane (PM) stretch by extending the cell surface 4. Caveolin‐3, a 21‐ to 24‐kDa protein mainly expressed in muscle fibers 5, 6, is localized at the sarcolemma within caveolae, associates with the dystroglycan complex 7, and binds to dysferlin. So far about 40 CAV3 mutations have been described in various autosomal dominant conditions affecting the striated muscle. The phenotypes range from asymptomatic HyperCKemia to Rippling Muscle Disease (RMD), Limb‐girdle muscular dystrophy type 1C (LGMD‐1C), or cardiomyopathy; the severity of the phenotype is highly variable 8, 9. Caveolin‐3 mutants are commonly associated with lowered sarcolemmal Caveolin‐3 levels, which are related to dissociation of the hetero‐oligomers at the PM, degradation by the ubiquitin‐proteasome pathway, and abnormal accumulation of mutated and wild‐type (wt) Caveolin‐3 in the Golgi causing activation of the unfolded protein response 1, 10, 11.
McNally et al. considered homozygosity for G56S as pathogenic in a single muscular dystrophy patient 12. The glycine at position 56 is conserved among many species, only in elephant an exchange to Serine in the Caveolin‐3 sequence at this position is described (UCSC: www.genome.ucsc.edu). Various DNA sequencing databases report frequencies between 1.07 and 25% for the G56S CAV3 allele 13, suggesting a benign character of this variant. However, biochemical characteristics and previous findings of cell biological investigations are not in line with a completely harmless nature of G56S: The nonpolar amino acid Glycine (G; MW = 57.05) does not have a side chain. It is often found at the surface of proteins, commonly within loops, providing high flexibility to these regions, whereas the polar amino acid Serine (S; MW = 87.08) might form so‐called side chain‐side chain or side chains‐main chain hydrogen bonds with polar amide carbonyl groups. Such interactions are likely to alter the 3D protein structure. In addition, Caveolin‐binding proteins such as signaling molecules are known to interact with the region of the protein where codon 56 is located 14.
Previously, we had reported that G56S Caveolin‐3 partially accumulates in the Golgi in transfected C2C12 and NIH3T3 cells, resulting in reduced sarcolemmal expression of both G56S and wt protein, similar to what is observed for Caveolin‐3 mutants known to be pathogenic 15. In order to address this discrepancy further, we performed comprehensive clinical, genetic, histopathological, and electron microscopic studies on three LGMD patients from unrelated families who carried the G56S Caveolin‐3 sequence variant. In addition, we performed cell culture experiments focusing on potential alterations induced or forced by the G56S amino acid exchange including pulse‐chase studies combined with immunoblotting, immunofluorescence, electron microscopy, and proteome profiling under both unstressed and stressed cellular conditions. Combined results of our investigations indicate that G56S might contribute to manifestation of myopathic changes for instance upon the presence of additional stress burden.
2. Materials and methods
Comprehensive clinical, genetic, histopathological, and electron microscopic studies on three LGMD patients from unrelated families who carried the G56S Caveolin‐3 sequence variant as well as cell culture experiments focusing on potential alterations induced or forced by the G56S amino acid exchange were carried out. Paradigmatic proteomic findings were confirmed in muscle tissue derived from two of these patients. Human material was analyzed following the guidelines of the Ethics Committee of RWTH Aachen University hospital.
2.1. Histology, immunoblotting, and electron microscopy
Histology of paraffin and semithin sections and electron microscopy and immunoblotting (patient 3) of the patients’ tissue were performed using standard methods as described previously 15, 16, 17. The following proteins were investigated: Lamin A/C (Vector Laboratories, Burlingame, CA, USA), beta‐Spectrin, Calpain‐3, Myotilin, alpha‐Sarcoglycan, gamma‐Sarcoglycan, beta‐Dystroglycan, and Emerin (all Leica Biosystems, Nussloch, Germany). Transmission electron microscopic studies on the RCMH cell lines were performed as described previously 18.
2.2. Genetic analysis
EDTA blood samples were collected with informed patients’ and parental consent. Using the QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany), DNA was isolated from the blood of the patients and their family members as well as 100 European controls. Fifty nanograms of the isolated DNA were used to amplify the coding sequence of CAV3. Primer sequences for the amplification are listed in the Supporting Information Document 1. The CAV3 PCR products were purified using Centri‐SepTM columns (Applied Biosystems, Darmstadt, Germany). Sequencing PCRs were performed using the BigDye Terminator version 1.1 Cycle Sequencing Kit (Applied Biosystems). Products were separated on an ABI Prism 310 (Applied Biosystems). For the detection of the sequence variant, the reference sequence NM_033337.2 (NCBI: http://www.ncbi.nlm.nih.gov/pubmed) was used. Protein references were as follows: NP203123 (NCBI) and P56539 (UniProtKB: http://www.uniprot.org). The analysis of the CAV3 sequence in 100 individual control samples derived from propositi without any muscular disease was performed as described above. For FSHD diagnostics, the DNA of patient 1 and of his family members was cleaved via the restriction enzymes EcoRI and EcoRI + BlnI (double digestion). Southern blot analysis and hybridization via probe p13E‐11 were performed to detect the fragments of locus D4Z4 as described by Lemmers et al. 19.
For the analysis of possible sequence variations in LMNA, FKRP (Exon 4), and CAPN3 in all three patients, the primers listed in the Supporting Information Document 1 were used. Moreover, whole exome sequencing (Exome Sequencing FAQ by Oxford Gene Technology, OGT, UK) was carried out using the DNA of patient 3 to exclude mutations in further known myopathy‐related genes. As human genome reference, the company uses the 1000 genomes version of GRCh37 (hg19) in alignment. They use dbSNP release 132 to provide information on whether variants are known or novel. The evaluation was designed to cover severe heterozygous and homozygous variants in myopathy‐related genes, alterations in essential splice sites, frameshift coding, gains or losses of stop codons, and nonsynonymous coding exchanges. Whole exome sequencing could not be performed in the other two patients due to limited amounts of DNA.
2.3. Cell culture, plasmids, and transfection
The murine Swiss 3T3 fibroblast cell line NR6W, which is stably expressing wt epidermal growth factor receptor (EGFR; kindly provided by Dr. Darell Bigner, Duke University, NC, USA) 20, was cultured in DMEM (Gibco, Darmstadt, Germany) with 10% FCS (Biowest, Nuaillé, France). Serum starvation of NR6W cells was performed by preincubation of the cells with DMEM containing 0.5% BSA (Roche, Mannheim, Germany) for 4 h to reduce background tyrosine phosphorylation caused by serum‐containing medium. The human RCMH cells (made by Drs. Caviedes and Freeman, Universidad de Chile, Santiago, Chile; further information could be inquired from Dr. Pablo Caviedes; pablo.caviedes@cicef.cl) were cultured in DMEM F‐12‐Ham containing 0.1% sodium bicarbonate (Sigma‐Aldrich, Taufkirchen, Germany) and 12.5% FCS (Biowest). These cells are classified as suitable to in vitro study myopathological changes including ER‐function and mechanical stress burden on both, the morphological and the biochemical levels 21.
Cells were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer´s protocol. The wt Caveolin‐3‐HA construct was kindly provided by Drs. S. Roy and R. Parton (University of Queensland Medical School, Australia). Plasmids carrying the Caveolin‐3 sequence variations R26Q and G56S were created by using the QuikChange II Site‐Directed Mutagenesis Kit (Stratagene, Cedar Creek, TX, USA). The vector pEGFP‐N1 (Clontech Laboratories, Mountain View, CA, USA) was used as a control of transfection efficiency. The primer sequences to create the mutated plasmids were described previously 15.
2.4. Immunofluorescence of RCMH cells
RCMH cells were seeded on coverslips coated with Poly‐l‐Lysine (Sigma‐Aldrich). At a density of 60–70%, cells were transfected as described in Section 2.3. The next day cells were washed once with 1× PBS (Gibco) and subsequently fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA, USA) for 10 min. After the fixation step, cells were permeabilized with 0.1% Triton X‐100 (Sigma‐Aldrich) and blocked with 10% goat serum (Sigma‐Aldrich) diluted in 1× PBS with 1% BSA. Then cells were washed with 1× PBS and incubated with a polyclonal rabbit anti‐HA.11‐antibody (Covance, Berkeley, CA, USA) and a monoclonal mouse anti‐gm130‐antibody (BD Transduction Laboratories, Franklin Lakes, NJ, USA) diluted 1:100 in 1× PBS with 1% BSA overnight at 4°C. The next day the cells were washed with 1× PBS and incubated with the appropriate secondary antibody for 30 min at room temperature (RT; labeling: AlexaFluor488 (green) against Caveolin‐3‐HA or AlexaFluor555 (red) against gm130 (Molecular Probes, Carlsbad, CA, USA)). Nuclei were stained using the Hoechst 33342 dye (Invitrogen). After washing with 1× PBS, the cells were mounted with Immumount (Thermo, Pittsburgh, PA, USA) and analyzed with an Axiovert Apotome fluorescence microscope (Zeiss, Jena, Germany). All immunostainings were performed at least three times with similar results.
2.5. Studies of EGFR signaling
In order to study the potential influence of the G56S sequence variant on EGFR signaling, which is important for the integrity of muscle cells 15, 22, NR6W cells endogenously expressing EGFR 20 were transiently transfected either with the wt Caveolin‐3‐HA plasmid or constructs carrying the amino acid changes R26Q or G56S. As a control of transfection efficiency, one fraction of the cells was transiently transfected with pEGFP‐N1 plasmids. Twenty‐four hours later, the NR6W cells were prestarved for 4 h to minimize background phosphorylation 23. After stimulation with EGF (0.1 ng/μL) (Sigma‐Aldrich) for 10 min, the transfected NR6W cells were lysed as described previously 23. Lysates of unstimulated NR6W cells, which were transfected with the same constructs, were used as negative controls for this experiment. For the immunoblot studies, lysates were stained with antibodies against EGFR and pEGFR, Akt, pAkt, ERK1/2, and pERK1/2 (Cell Signaling Technology, Cambridge, UK). Loading control lysates were stained with antibodies against Tubulin (Sigma‐Aldrich) and the HA‐tag (of the Caveolin‐3 constructs) (Covance). All assays were performed as previously described 15 for at least three times with similar results. Densitometric analyses of the immunoblots were performed using the ImageJ program (Open source: Wayne Rasband, NIH, Bethesda, MD, USA, v. 1,45e, 4‐2‐2011; http://imagej.nih.gov/ij/download.html).
2.6. Statistics
Statistical significance of immunoblot results was determined by the Kruskal–Wallis test followed by the Dunn's comparison test. A p‐value ≤ 0.05 was considered statistically significant. Data are shown as mean values ± SEM.
2.7. Comparative proteomic analysis
In order to analyze effects of the G56S Caveolin‐3 variant on cellular protein composition in cells exposed to mechanical stress and untreated controls, human RCMH myoblast cells were transfected with wt or G56S Caveolin‐3, seeded on 150 × 25 mm culture plates and exposed to hypoosmotic stress for 30 min or were left unstressed (three biological replicates, respectively). The stress medium contained 30% DMEM (Gibco) and 70% Aqua dest. (modified from Sinha et al. 4). After treatment, the medium was replaced by normal DMEM containing 10% FCS (Biowest). Myoblast swelling causes caveolae extension on the cell surface by passive flattening, and mimics mechanical stress 4. Presence of osmotic/mechanical stress was documented by light microscopy. Two hours after incubation, cells were harvested and washed with ice‐cold 1× PBS (Gibco); then cell pellets were frozen in liquid nitrogen.
2.7.1. Cell lysis and carbamidomethylation
Approximately, 1 mg of cells was lysed in 0.3 mL of 50 mM Tris‐HCl (Applichem Biochemica, Darmstadt, Germany), pH 7.8 buffer containing 150 mM NaCl (Merck, Darmstadt, Germany), 1% SDS (Roth, Karlsruhe, Germany), protease (cOmplete mini, Roche, Mannheim, Germany), and phosphatase inhibitor (PhosSTOP, Roche) tablets. After addition of 5 μL benzonase (25 U/μL; Novagen, Pretoria, South Africa) and 2 mM MgCl2 (Sigma‐Aldrich), lysates were incubated at 37°C for 30 min. Samples were centrifuged at 4°C and 18 000 × g for 20 min. Protein concentration of the supernatant was determined by BCA (Pierce, Schwerte, Germany) assay according to the manufacturer's protocol. Cysteines were reduced by addition of 10 mM DTT (Roche) at 56°C for 30 min followed by alkylation of free thiol groups with 30 mM IAA (Sigma‐Aldrich) at RT in the dark for 30 min.
2.7.2. Sample preparation and trypsin digestion
Sample preparation and proteolysis were performed using filter aided sample preparation (FASP) 24, 25 with minor changes. Briefly, 70 μl of each sample lysate corresponding to 100 μg of protein were diluted up to 400 μl with freshly prepared 8 M urea (Sigma‐Aldrich) /100 mM Tris‐HCl (pH 8.5) buffer 26 and loaded on a Nanosep centrifugal device (PALL, 30 kDa cutoff). The devices were centrifuged at 13,500 g at RT for 20 min. All the following centrifugation steps were performed under similar conditions. To eliminate residual SDS, three washing steps were carried out using 100 μL of 8.0 M urea buffer and finally for the buffer exchange, the devices were washed thrice with 100 μL of 50 mM NH4HCO3, pH 7.8 (Sigma‐Aldrich). To the concentrated proteins, 100 μL of proteolysis buffer comprising of trypsin (Sigma Aldrich; 1:40 w/w, enzyme to protein), 0.2 M GuHCl (Sigma‐Aldrich), 2 mM CaCl2 (Merck) in 50 mM NH4HCO3, pH 7.8, were added and incubated at 37°C for 14 h. The generated tryptic peptides were recovered by centrifugation with 50 μL of 50 mM NH4HCO3 followed by 50 μL of ultrapure water. Finally, the peptides were acidified to pH <3 using 10% TFA (v/v; Biosolve, Valkenswaard, the Netherlands) and the digests were quality controlled by monolithic column HPLC as described previously 27.
2.7.3. LC–MS/MS analysis
All 12 samples (three biological replicates, each 1 μg) were analyzed by nano‐LC–MS/MS as described in the Supporting Information Document 2.
2.7.4. Label‐free data analysis
Label‐free quantification was conducted using the Progenesis LC‐MS software (NonLinear Dynamics), in combination with peptide shaker 28, as described in the Supporting Information Document 2. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 29 partner repository with the dataset identifier PXD003719.
2.7.5. Functional classification of proteome alterations
Proteins were categorized manually according to the information found on the Uniprot website http://www.uniprot.org. As most of the proteins have multiple roles and/or localizations, for manual categorization only their main function and localization were considered per protein. The significance level for classifying a protein as significantly regulated was set at p ≤ 0.05.
2.7.6. Verification of proteome results via immunoblotting
Immunoblots were performed as previously described 4. Lysates were stained with antibodies against HSP75 (GeneTex, Irvine, CA, USA), SNX1 (Abcam, Tel Aviv, Israel), CSTF2 (Abcam), SEC63 (GeneTex), HNRA3 (GeneTex), RAB1A (Cell Signaling Technology), and tubulin (Sigma‐Aldrich).
3. Results
3.1. Case 1
This male patient of Caucasian descent presented with delayed motor development. At the age of 13, he complained about slowly progressive loss of muscle strength but had no myalgias or muscle cramps. Clinical examination revealed generalized muscle weakness pronounced in limb girdle muscles. Muscle tendon reflexes were preserved; bilateral pes cavus, slight calf pseudohypertrophy, and a positive Gower´s sign were present. There were no fasciculations, muscular atrophy, sensory abnormalities, or clinical signs of RMD. The patient could walk on tiptoe but not on his heels. Electromyography of the quadriceps femoris and vastus medialis muscles showed a myopathic pattern. Blood levels of creatine kinase were elevated at 624 U/L. Histopathological analysis of a vastus lateralis muscle biopsy revealed marked chronic myopathy with an extended spectrum of muscle fiber calibers including atrophic, rounded, and hypertrophic muscle fibers (Fig. 1A I). There were numerous muscle fibers with nonsubsarcolemmal nuclei and few necrotic muscle fibers. Electron microscopy revealed reduced numbers of caveolae; some of the remaining caveolar structures appeared enlarged (Fig. 1B I), similar to what has been described by Fischer et al. 9 and Woodman et al. 30 in cases of clear‐cut caveolinopathy.
Figure 1.
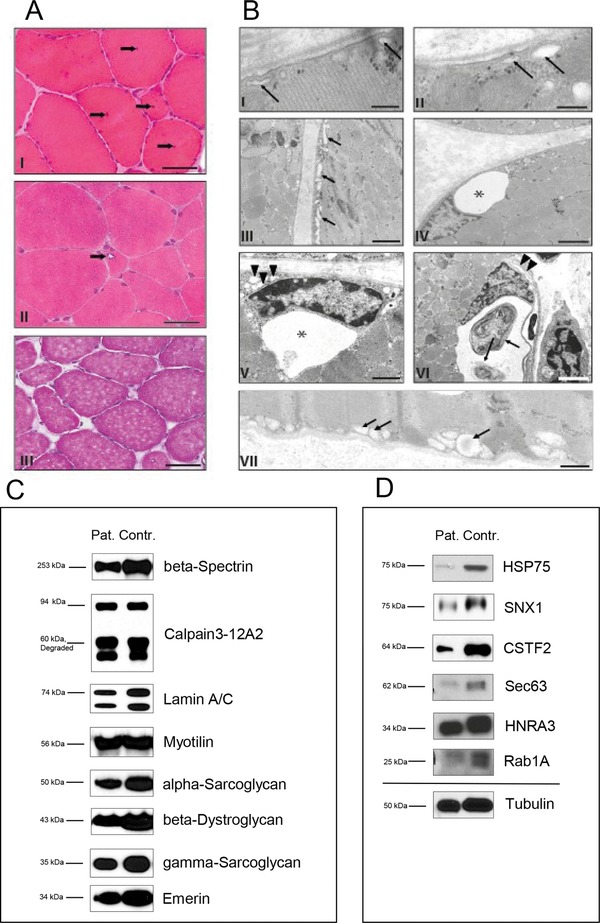
Analysis of patient material. Microscopical investigations (1A): (I) M. vastus lateralis biopsy of patient 1 showing a myopathic pattern with minor muscle fiber atrophy and hypertrophy as well as frequent nonsubsarcolemmal nuclei (arrows). Cryostat section, H&E. Scale bar: 80 μm. (II) Low‐grade muscle fiber atrophy and solitary rimmed vacuole (arrow) in cryostat section of the rectus femoris muscle biopsy of patient 2. Cryostat section, H&E. Scale bar: 50 μm. (III) M. quadriceps femoris biopsy of patient 3 showing minor atrophic as well as hypertrophic muscle fibers. Cryostat section, H&E. Scale bar: 80 μm Electron microscopic studies (1B): (I) Subsarcolemmal membrane‐bound vacuoles, presumably corresponding to enlarged caveolae (arrows) in the muscle biopsy of patient 1. Scale bar: 250 nm. (II–VI) Patient 2; (II) subsarcolemmal membrane‐bound vacuoles (arrows) similar to the one depicted in Fig. 1A. Scale bar: 200 nm. (III) Further abnormal membrane bound subsarcolemmal vacuoles as well as tubular structures (arrows). Scale bar: 2 μm. (IV) Large vacuole (asterisk) associated with a myonucleus. Scale bar: 4 μm. (V) Large (asterisk) and small (arrowheads) vacuoles in the vicinity of a myonucleus. Scale bar: 2 μm. (VI) Large vacuole containing autophagic material (arrows) associated with a deformed, possibly degenerating myonucleus. Arrowheads: Membranous material, presumably corresponding to a focal outfolding of the sarcoplasmic reticulum. Scale bar: 4 μm. (VII) Patient 3: subsarcolemmal membrane‐bound vacuoles (arrows). Scale bar: 800 nm. Immunoblots of myopathy‐associated proteins (1C): comparison of beta‐Spectrin, Calpain‐3, Lamin A/C, Myotilin, alpha‐ and gamma‐Sarcoglycan, beta‐Dystroglycan and Emerin in G56S Caveolin‐3 patient and control muscle revealed no significant changes in protein abundances. Confirmation of proteome data via immunoblotting (1D): immunoblot analyses of HSP75, SNX1, CSTF2, Sec63, HNRA3, and Rab1A revealed same changes in protein abundances upon G56S Caveolin‐3 expression like in the G56S Caveolin‐3 in vitro model. Tubulin was used as loading control.
Sequencing of the CAV3 gene revealed the heterozygous c.163G>A (G56S) sequence variant in the patient's DNA. No other affected family members were known. However, the G56S sequence variant was also found in the patient's mother and one sister (Supporting Information Table 1). Cursory clinical examinations of these individuals revealed no abnormalities. They refused further neurological, laboratory, and electrophysiological studies. Additional molecular genetic studies excluded alterations of the D4Z4 locus (D4Z4 repeat) as well as mutations within the CAPN3, FKRP, and LMNA genes.
3.2. Case 2
This 38‐year‐old woman of African descent first noticed weakness of the proximal limb girdle musculature at the age of 26. The symptoms had been slowly progressive with some relapses. Her mother and her maternal grandfather were reported to have shown similar symptoms but had died years before initiation of this study; no DNA or tissue was available for analysis. The patient's 3‐year‐old daughter did not show any symptoms or signs of neuromuscular disease. Symmetric weakness of the proximal muscles was found by clinical examination. Deep tendon reflexes were normal. MRI of the limb‐girdle musculature showed mild atrophy. No abnormalities were found in the sensory or extrapyramidal systems. The ischemic exercise test yielded normal results. Electromyography revealed myopathic alterations. Serum creatine kinase was slightly elevated up to 315 U/L. Biopsy of the rectus femoris muscle revealed signs of a moderate chronic myopathy with increased variability of muscle fiber calibers and elevated numbers of nonsubsarcolemmal muscle fiber nuclei. Autophagic vacuoles were encountered sporadically (Fig. 1A II). The partially atrophic muscle fibers were both rounded and angular; there was incipient fiber‐type grouping, indicating a minor neurogenic component (not shown). Electron microscopy revealed membrane‐bound vacuolar and tubular structures of variable size mostly in the subsarcolemmal compartment and the perinuclear region, but also between myofibrils (Fig. 1B II–VI). These structures for the most part did not contain electron dense material and probably correspond to enlarged caveolae and/or enlarged components of the ER/Golgi system. They were repeatedly associated with signs of altered autophagy such as cytoplasmic bodies and/or extensive focal proliferations of membranes.
Sequencing of the CAV3 gene revealed the heterozygous c.163G>A (G56S) sequence variant in the patient's DNA; both Caveolin‐3 alleles of the clinically unaffected daughter were normal (Supporting Information Table 1). Sequencing of the CAPN3, FKRP (Exon 4), and LMNA genes revealed no mutations.
3.3. Case 3
This 40‐year‐old male patient of Portuguese descent with ancestors from Cape Verde had normal motor development. He suffered from mild, slowly progressive generalized muscle weakness starting at age 38 combined with moderate serum CK elevations up to 800 U/L. The electromyographic examination did not reveal a characteristic pattern. There was no family history of neuromuscular disease; no relatives were available for neurological or genetic examination. Histopathological analysis of a quadriceps muscle biopsy revealed moderate chronic myopathy with few atrophic and several hypertrophic muscle fibers and a moderately elevated number of nonsubsarcolemmal muscle fiber nuclei (Fig. 1A III). There was no fiber‐type grouping. As in the above‐described cases, electron microscopy revealed subsarcolemmal and perinuclear vacuolar and tubular structures up to 0.5 μm in size (Fig. 1B VII). In addition to the neuromuscular condition the patient also had chronic ulcerative colitis, which was treated with Mesalazine with good effects.
Sequencing of the CAV3 gene revealed the heterozygous c.163G>A (G56S) variant (Supporting Information Table 1). No alterations were detected in the FKRP (Exon 4), CAPN3, and LMNA genes. Whole exome sequencing confirmed the c.166G>A sequence variant. No clearly pathogenic sequence variation in any gene known to be related to neuromuscular disorders was found (Supporting Information Table 2). Immunoblots for beta‐Spectrin, Calpain‐3, Lamin A/C, Myotilin, alpha‐ and gamma‐Sarcoglycan, beta‐Dystroglycqan, and Emerin gave normal results (Fig. 1C).
3.4. Occurrence of the Caveolin‐3 G56S variant in genetic resources
The Exome Aggregation Consortium (ExAC, containing exome sequencing data from a wide variety of large‐scale sequencing projects; http://exac.broadinstitute.org/) reports the heterozygous G56S CAV3 variant in 1130 and the homozygous variant in 84 out of 60495 individuals (allele frequency 1.07%). Another resource, the Exome Variant Server (http://evs.gs.washington.edu/EVS/), provides frequency information for distinct ethnic groups. When only individuals of African origin are considered, the heterozygous variant is observed in 421 and the homozygous variant in 30 out of 2203 individuals (allele frequency 10.7%). One study even found a prevalence of 25% of the G56S allele in the African population 13.
3.5. G56S Caveolin‐3 accumulates in the Golgi of transfected RCMH cells
To determine the localization of the G56S Caveolin‐3 in muscle cells, human RCMH myoblastic cells were transiently transfected either with the wt Caveolin‐3‐HA or the constructs carrying the amino acid changes R26Q or G56S. Immunofluorescence with antibodies to detect the HA‐tag (indirect detection of Caveolin‐3) and the Golgi marker gm130 revealed the normal association of wt Caveolin‐3 with the PM (Fig. 2). R26Q Caveolin‐3, on the other hand, was for the most part associated with the Golgi apparatus; only minor amounts of the mutated protein were found at the sarcolemma, confirming our previously reported results obtained in mouse C2C12 cells 15. After transfection with the G56S Caveolin‐3‐HA‐construct, immunoreactivity was found both in the Golgi and associated with the PM (Fig. 2), again confirming our previous results in C2C12 cells 15.
Figure 2.
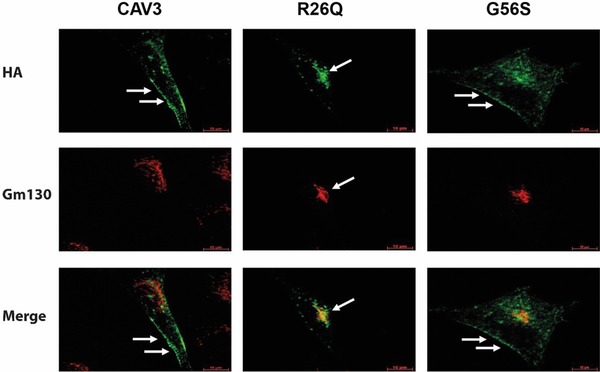
Analysis of human RCMH cells transfected with wild‐type Caveolin‐3, R26Q or G56S Caveolin‐3 constructs. After transient transfection of RCMH cells with the wt Caveolin‐3‐HA constructs, the HA antibody mainly detects Caveolin‐3 protein at the sarcolemma (green fluorescence, arrows). After transfection of the R26Q Caveolin‐3‐HA plasmid Caveolin‐3 is mainly detectable in the Golgi (arrow). The transfection of the G56S Caveolin‐3‐HA construct, in contrast, led to a mislocalization of the mutated protein to the Golgi (gm130: red fluorescence) while some of the protein is still located at the sarcolemma (arrows). Green: Caveolin‐3‐HA, red: Golgi. White arrows: localization of Caveolin‐3. Scale bar: 10 μm. The assays were performed at least for three times with similar results.
3.6. The G56S sequence variant causes buildup of vacuoles and aberrant Golgi and ER structures in RCMH myoblasts
To obtain insights into the cellular reactions upon expression of the G56S Caveolin‐3 sequence variant, transfected RCMH myogenic cells were analyzed by electron microscopy (Fig. 3). Our studies revealed a dilatation of ER structures compared to RCMH cells transfected with wt Caveolin‐3. These structures were often filled with osmiophilic membranous material (Fig. 3). Frequently, autophagic vacuoles filled with osmiophilic membranous material were found in the G56S Caveolin‐3 transfected cells (Fig. 3). Golgi structures of cells transfected with the variant form of Caveolin‐3 protein appeared more widened compared to cells overexpressing the wt protein (Fig. 3).
Figure 3.
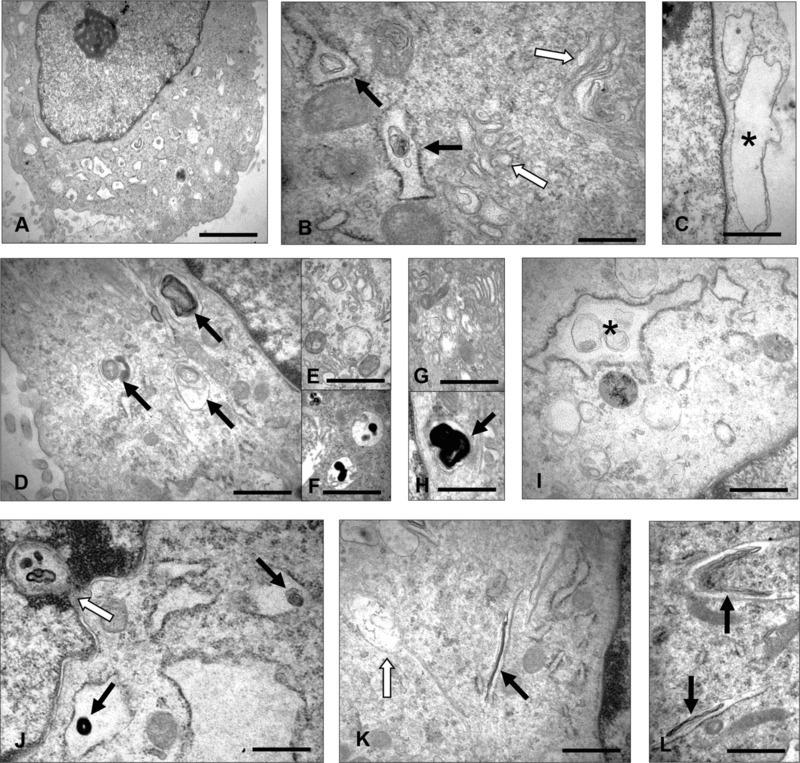
Electron microscopic findings in human RCMH myoblasts transfected with wild‐type or G56S Caveolin‐3 constructs. EM of RCMH cells transfected with wt Caveolin 3 (A–B) or the G56S mutant form (C–L): (A) A representative RCMH cell overexpressing wt Caveolin‐3 presents with proliferated, dilated endoplasmic reticulum (ER) partially filled with osmiophilic material. Scale bar: 2.0 μm. (B) ER structures at a higher magnification within another myoblast (black arrows). Golgi cisternae and vesicles are marked by white arrows. Scale bar: 0.5 μm. (C) RCMH myoblast transfected with G56S Caveolin‐3: Large subplasmalemmal vacuoles, one of them marked by an asterisk. Scale bar: 0.5 μm. (D) Osmiophilic, often membranous material localized within enlarged ER structures and autophagic vacuoles (arrows). Scale bar: 0.5 μm. (E) Proliferated and widened Golgi structures within the same RCMH cell. Scale bar: 0.5 μm. (F) Autophagic vacuoles also within the same cell. Scale bar: 0.5 μm (G) Widened Golgi structures within a different RCMH cell. Scale bar: 0.5 μm. (H) Autophagic vacuole (arrow) filled with membranous material adjacent to widened ER. Scale bar: 0.5 μm. (I) Widened ER containing membranous material (asterisk). Scale bar: 0.5 μm. (J) Osmiophilic material (black arrows) localized within enlarged ER structures and directly adjacent to a nuclear invagination (white arrow). Scale bar: 0.5 μm. (K–L) Tubular, presumably ER‐derived structures filled with elongated osmiophilic membranous material (black arrows). White arrow: early phase of accumulation of electron dense material. Scale bar: 0.5 μm.
3.7. The sequence variant G56S alters EGFR signaling
G56S is located within the scaffolding domain of Caveolin‐3, which is important for the association of the protein with membranes 31, 32. This domain is also involved in the self‐assembly of Caveolin proteins into homo‐oligomers and in the interaction with signaling molecules 33. Therefore, we studied the phosphorylation of EGFR upon expression of wt or mutant Caveolin‐3 in NR6W cells and stimulation with EGF 20. Immunoblots showed that both the R26Q (Fig. 4A, lane 3) and G56S (Fig. 4A, lane 5) amino acid exchange result in reduced phosphorylation levels of EGFR (Fig. 4A, lane 1). As expected, the immunoblots for the detection of total EGFR levels showed equal amounts of the protein (Fig. 4A). Lysates of unstimulated NR6W cells, which were transfected with the same constructs, were used as negative controls. As expected they showed minimal levels of phosphorylated EGFR (Fig. 4A; lanes 2, 4, 6, 8).
Figure 4.
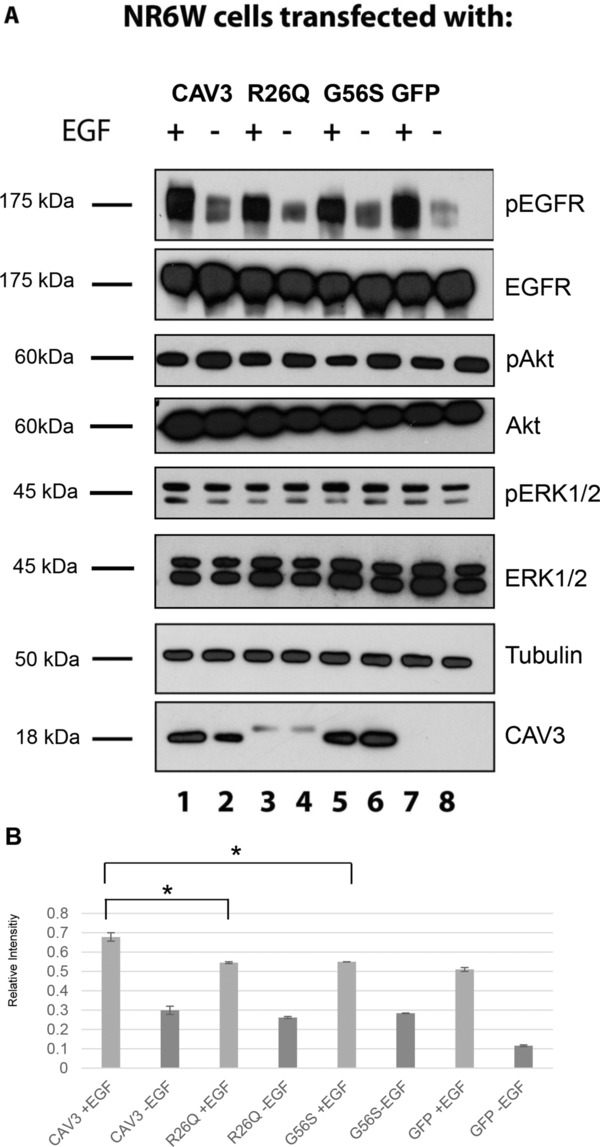
Analysis of EGFR‐ and downstream signaling after transfection of mutated Caveolin‐3 constructs into NR6W cells. (A) Immunoblots detecting phosphorylated EGFR (pEGFR), phosphorylated Akt (pAkt), and phosphorylated ERK1/2 (pERK1/2). Transfection of the R26Q and G56S constructs leads to lower phosphorylation levels of EGFR. Lysates of unstimulated NR6W cells transfected with the same constructs were used as negative controls. As expected, these cells show minimal levels of pEGFR. Transfection of R26Q and G56S has no influence on phosphorylation levels of Akt and ERK1/2. Loading control: Caveolin‐3 using the antibody against HA and tubulin. (B) Quantitative assessment of three Immunoblots. Statistical significance was determined by the Kruskal–Wallis test followed by the Dunn's comparison test. A p‐value ≤ 0.05 was considered statistically significant. Data were shown as mean values ± SEM. *Statistically significant (p‐value ≤ 0.05).
Next, we assessed levels of the MAP Kinases ERK1/2 (p42/p44) and of Akt after transient transfection of NR6W cells with the wt Caveolin‐3 and mutant plasmids carrying the amino acid changes R26Q and G56S in the same way as for the EGFR (see above). Figure 4A displays the ratios of phosphorylated ERK1/2 after transfection of wt Caveolin‐3 (lane 1), R26Q (lane 3), G56S (lane 5), and pEGFP‐N1 constructs (lane 7), respectively. Our data revealed that both sequence variants have no net effect on the phosphorylation of the downstream factor ERK1/2 (p42/p44). Similar results could be obtained for Akt (Fig. 4A): The banding pattern for wt Caveolin‐3 transfected cell lysates is the same as for lysates transfected either with the construct carrying the amino acid change R26Q or G56S. The immunoblots for the detection of total protein amount showed equal loading (Fig. 4A). The immunoblot for tubulin revealed equal protein loading for each lysate. The immunoblot for Caveolin‐3‐HA showed an upward shift for the protein bands of R26Q Caveolin‐3 HA‐transfected cells and a lower expression level, in accordance with previously reported results 15, 34. HA‐tagged G56S Caveolin‐3 showed the same banding pattern as the HA‐tagged wt protein. The densitometry results plotted in Fig. 4B represent the mean phosphorylation values of the EGF stimulated and unstimulated NR6W cells normalized to the total EGFR levels in the same cell lysates of three independent immunoblot experiments.
3.8. Expression of G56S causes alterations in the cellular proteome profile
In order to obtain an unbiased view on the cellular response upon G56S Caveolin‐3 expression, especially in terms of potential compensatory strategies, we performed quantitative LC–MS/MS studies. Using label‐free quantification, we found that levels of 97 out of 1803 quantified proteins (5.4%) were significantly altered due to the expression of the sequence variant. Proteins localized in the Golgi‐ER network (including proteins of the secretory pathway), in the PM, in mitochondria, in the cytoplasm (cytoskeleton), the nucleus and those that shuttle between the two latter compartments were altered. Most protein functions are assigned according to the information listed in the UniProtKB website (http://www.uniprot.org) as of July 2015. Data that are not derived from this site are marked by their references. Potential roles of these proteins in the context of G56S‐pathophysiology are listed in Supporting Information Table 3 and discussed below.
3.9. Influence of hypoosmotic stress on G56S Caveolin‐3 RCMH myoblasts
In vivo muscle fibers are continuously exposed to mechanical stress as a result of muscle exertion. Caveolae are mechanically deformable invaginations of the sarcolemma 35 and buffer mechanical stress caused by cell stretch or osmotic swelling 4. Therefore, we studied the effects of the G56S Caveolin‐3 variant in this context. We treated transfected RCMH cells carrying wt or G56S Caveolin‐3 with highly hypoosmotic medium for 30 min. During this time, the cells expressing wt Caveolin‐3 as well as those expressing G56S Caveolin‐3 started to swell (Fig. 5A). After the shift to normal culture medium, wt cells remained distended for up to 120 min. The RCMH cells carrying G56S Caveolin‐3 showed PM ruptures and leakage of organelles after the shift to normal culture medium (Fig. 5A). For proteomic analysis, cells were harvested before this period of rupture to avoid loss of informative proteins into the medium. Comparative proteome profiling revealed that levels of 25 out of the 97 proteins (25.8%) already altered by transfection with G56S were further influenced by osmotic stress (Fig. 5B). Proteins of all subcellular localizations mentioned above were affected. Four proteins (4.1%) that had already been found to be reduced in G56S transfected RCMH cells were further decreased after application of hypoosmotic stress. Further increases of protein levels in G56S myoblasts upon application of hypoosmotic stress were detectable for eight proteins (8.2%; Fig. 5B, Supporting Information Table 3). Upon hypoosmotic stress, a reduction of levels that had been increased initially after transfection with the G56S construct was detected for 13 proteins (13.4%; Fig. 5B, Supporting Information Table 3).
Figure 5.
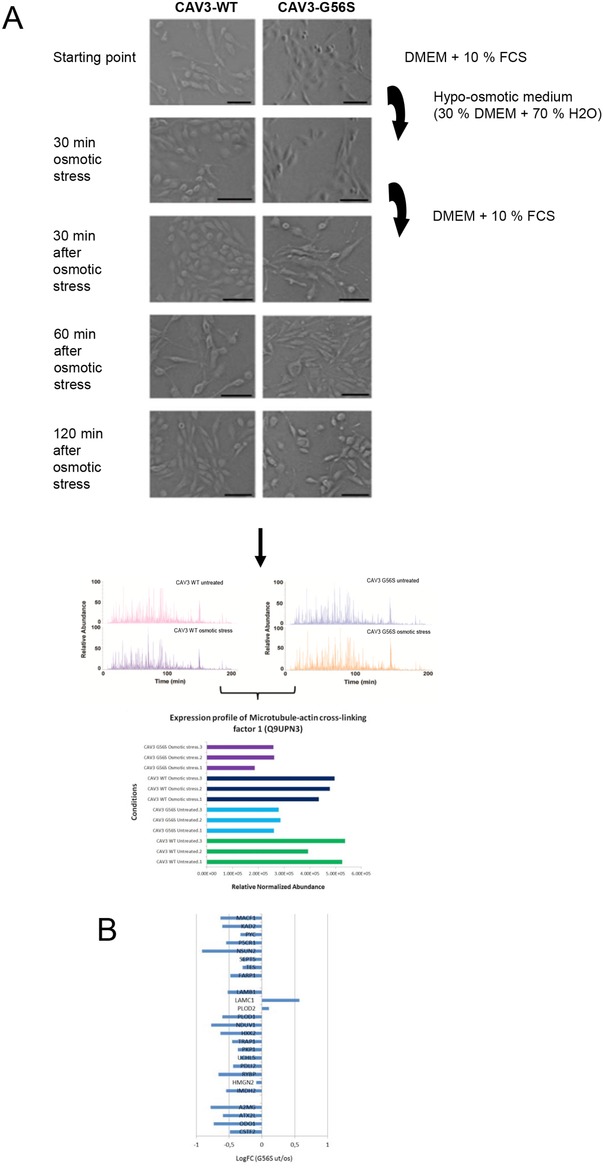
Analysis of RCMH cells transfected with wild‐type or G56S Caveolin‐3 and treated with osmotic stress. (A) RCMH myoblast cells were transfected with either the wt or G56S Caveolin‐3 and were treated with 30% hypoosmotic stress medium for 30 min or were left unstressed. The medium was replaced by normal DMEM containing 10% FCS. Both wt and G56S Caveolin‐3 cells started swelling during the 30 min of stress. After the shift to normal culture medium, wt cells remained distended within the analyzed 120 min. Shortly after the shift to normal culture medium, the G56S Caveolin‐3 cells started to rupture. One hundred twenty minutes after osmotic stress, cells were prepared for quantitative label‐free MS. Each condition was measured in triplicate in randomized order; four representative base peak chromatograms are shown. Only proteins that were quantified with at least two unique peptides (at 1% false discovery rate) between all samples were considered to represent proteins that show significant regulation. Among these, microtubule‐actin cross‐linking factor 1 (Q9UPN3) showed a significant regulation upon both, G56S Caveolin‐3 overexpression and osmotic stress burden. (B) logFC fold changes of protein expression. ut, untreated; os, osmotic stress.
4. Discussion
In the era of next‐generation sequencing, we are confronted with increasing numbers of sequence variants with unknown significance that remain of unclear relevance even after careful consideration of segregation data and prediction programs. Genotype–phenotype correlations have been notoriously difficult in caveolinopathies, because one and the same mutation may lead to different clinical manifestations of the disease even within the same family ranging from asymptomatic hyperCKemia to severe RMD or LGMD‐1C 36. Prompted by histological and ultrastructural features indicative of a caveolinopathy in muscle biopsies of three patients suffering from mild or moderate chronic myopathy, we sequenced the CAV3 gene and detected the heterozygous c.163G>A (G56S) CAV3 sequence alteration in all three patients. Two clinically normal relatives of patient 1 were also found to carry this variant. Autosomal dominant inheritance seemed likely in the family of patient 2, but no relatives were available for clinical examination and DNA analysis. In case of patient 3, no relatives could be examined, and no family history was available.
The frequency of the G56S variant amounts to 11% in the African‐American population in large‐scale exome sequencing projects. Even higher prevalence rates among healthy black individuals have been described in the literature 13. Notably, our cases 2 and 3 have African ancestry suggesting that discovery of the G56S variant is an incidental finding and that they suffer from another genetic or nongenetic neuromuscular condition. The high prevalence of the G56S variant in the general population strongly argues against this variant being a cause of a monogenic disease.
4.1. G56S causes partial mislocalization of Caveolin‐3
In previous studies, Caveolin‐3 immunohistochemistry was described to be normal in biopsies of patient harboring the G56S sequence variant 12, 37. The authors concluded that G56S does not alter the subcellular localization of the protein but may change the functional activity of Caveolin‐3 12, 37. Expression of recombinant G56S Caveolin‐3 in muscle cell lines led to localization at the sarcolemma and in the Golgi apparatus (Fig. 2). G56S Caveolin‐3 did not significantly sequester wt Caveolin‐3 to the Golgi in the RCMH muscle cell line, confirming our previous results using C2C12 cells 15.
4.2. Ultrastructural alterations in G56S‐transfected RCMH myoblasts reflect patients’ muscle biopsy findings
In all three biopsies, electron microscopy revealed abnormal subsarcolemmal vacuolar and tubular structures (Fig. 1B) corresponding to those that have been described previously in cases of caveolinopathy 9, 15, 30. The vacuolar structures were often located in the perinuclear region and were especially prominent in the biopsy of patient 2 (Fig. 1B IV–VI). Such perinuclear vacuoles were also found in our G56S Caveolin‐3 transfected RCMH cells (Fig. 3C), but not in control transfected cells. Several vacuoles in muscle fibers of patient 2 contained autophagic, myelin‐like material indicative of altered autophagy. Autophagic vacuoles containing myelin‐like material were also increased in RCMH myoblasts expressing G56S Caveolin‐3 (Fig. 3D, F, and H) compared to cells expressing wt Caveolin‐3 (Fig. 3A–B). This finding suggests altered proteolytic clearance in G56S Caveolin‐3 expressing muscle cells. The alterations of Golgi structures detected by EM in the G56S Caveolin‐3 transfected cells is in line with the mislocalization of the G56S Caveolin‐3 protein within this compartment found by immunofluorescence (Fig. 2). Taking together, these results suggest that mislocalization of G56S Caveolin‐3 within the Golgi alters protein processing, which leads to an increase of unfolded proteins and initiation of protein clearance as a cellular stress defense mechanism.
4.3. EGFR signaling is altered by G56S Caveolin‐3
Caveolae are known to be involved in intracellular signaling and endocytosis 38. They harbor numerous signaling molecules including EGFR, which is important for proliferation and regeneration processes in myoblasts 39. EGF signaling is not only important for muscle cell differentiation but also for regeneration: impaired EGFR phosphorylation rendered myotubes sensitive to apoptotic cell death induced by hypoxic stress, suggesting that activation of EGFR in skeletal muscle has a prosurvival function 40. Immunoblots using antibodies against phosphorylated EGFR revealed that the transfection of the G56S Caveolin‐3 construct—similar to the R26Q Caveolin‐3 plasmid 15—leads to a small, but significant decrease in phosphorylation of the receptor in comparison to the wt transfected cells (Fig. 4A). The well‐described pathogenic mutation R26Q was used as a control. Sotgia et al. could show that the sequence variant leads to RMD, hyperCKemia, or distal myopathy 34, 39. The mutation leads to a retention of Caveolin‐3 in the Golgi complex. Interestingly, when coexpressed with the wt‐protein R26Q does not behave in a dominant negative way, which may explain the different phenotypes of the disease caused by the mutation 34.
Our investigation of the downstream signaling and endocytosis of the EGFR interestingly revealed that G56S does not significantly affect the phosphorylation of ERK1/2 and Akt. The banding pattern of the phosphoblots after transfection of the cells either with the wt Caveolin‐3, R26Q Caveolin‐3, G56S Caveolin‐3 or GFP construct is unchanged (Fig. 4A). We conclude that the sequence variant only affects the interaction of Caveolin‐3 and the EGFR while the downstream factors are potentially regulated independently. This is in line with our findings in HEK293 cells transfected with constructs carrying the mutation R26Q or P28L 15
4.4. The G56S sequence variant alters the proteome profile in RCMH myoblasts
In order to obtain insights into potential compensatory mechanisms, we used a fourth, unbiased approach, proteome profiling of RCMH cells transfected with G56S Caveolin‐3. LC–MS/MS revealed altered levels of 5.4% out of a total of 1803 proteins compared to wt‐transfected cells (Supporting Information Table 3). Verification of protein alterations in muscle biopsy specimen derived from G56S Caveolin‐3 patients (Fig. 1, Supporting Information Fig. 1) not only provides a confirmation of our data across from in vitro to in vivo but also links the protein profiles to the physiological phenotypes of patients. Meaning of the altered abundance of paradigmatic proteins is discussed according to their respective functions listed on the Uniprot website (http://www.uniprot.org). The decrease of several ER‐Golgi network proteins (GLU2B, KCC2D, NUDC3, PRAF2, RAB1A, SCAM1, SCFD1, SEC11A, SEC63) is indicative of protein processing/translocation arrest compatible with the abnormal morphology of the ER‐Golgi network and the signs of altered autophagy found both in patient muscle fibers and in G56S transfected cells. C‐terminal‐binding protein 1 (CTBP1) is involved in controlling the equilibrium between tubular and stacked structures in the Golgi complex and its decrease suggests morphological alterations of the Golgi structure. Decrease of ER‐associated Ataxin‐2‐like protein ATXL2 reflects involvement of stress‐related RNA translation. By the same token, RNA‐binding protein EWS functions as a transcriptional repressor and decreased level could be in line with activation of stress‐related protein synthesis. Altered RNA processing is also indicated by decrease of HNRH3 and of CSTF2. Altered levels of proteins involved in ubiquitination processes (CSN4, ephrin type‐A receptor 2 (EPHA2), HUWE1, NEDD4, UBE1, and UBP14; Supporting Information Table 3) suggest altered proteolysis. As VATH is required for the formation of endosomes and acidficates vacuoles, its increase in G56S Caveolin‐3 expressing RCMH is in accordance with the occurrence of autophagic vacuoles detected on the electron microscopic level (Fig. 3). However, these alterations might reflect compensatory mechanisms, as activation of protein clearance mechanisms rescues muscular dystrophy phenotypes 41 and is considered to be a therapeutic target in these disorders 42.
In the context of the above‐described alterations in EGFR activation and signaling (Fig. 4A), the decreased expression of SNX1 is of special interest: SNX1 retrieves lysosomal enzyme receptors from endosomes to the trans‐Golgi network and targets ligand‐activated EGFR to the lysosomes for degradation after endocytosis from the cell surface and release from the Golgi 43. As a decrease of this protein stimulates the ligand‐induced endocytosis of EGFR and increases EGFR phosphorylation 44, its downregulation in G56S Caveolin‐3 expressing RCMH together with the reduced phosphorylation of EGFR (Fig. 4A) is indicative of a compensative strategy. The increased expression of proteins involved in the regulation of cellular survival (A2MG, CSRP1, ENOG, G6P, NSUN2, PARP3, RALB, SEPT5) can be interpreted accordingly (Supporting Information Table 3). As in their function as protective factors, ENOG and SEPT5 translocate to PM, these findings along with the concomitant upregulated proteins FERM and PKP1 are indicative for PM alterations likewise suggested by decreased factors (see below).
Microtubules and actin filaments control lipid raft/caveolae localization. A major role of Caveolin‐3 in the processing and assembly of these cytoskeletal proteins has been demonstrated 45. Our proteomic findings revealed several indications for enhanced PM vulnerability in RCMH cells overexpressing G56S Caveolin‐3: decreased 14‐3‐3 protein eta belongs to a group of major adaptor molecules also binding to cell surface receptors 46. Concomitantly, the ephrin type‐A receptor 2 (EPHA2), the peripheral membrane proteins UBP14 and NEDD4 as well as GNAI3, which acts as a modulator or transducer in various transmembrane signaling systems, are decreased. In addition, our findings indicate an influence of changed PM protein composition on cytoskeleton: spectrin alpha and beta chains (SPTAN1 and SPTBN1, fodrins) that are involved in calcium‐dependent movement of the cytoskeleton at the PM and are concomitantly decreased with further factors involved in cytoskeletal organization such as T‐complex protein 1 subunits alpha, delta, and epsilon; tubulin‐specific chaperone A; AFAP; ARPC2 and ARPC3; CKAP5; and DBNL. At high concentrations, PFN2 prevents the polymerization of actin, so that PFN2 decrease might reflect an attempt to antagonize cytoskeleton disorganization. However, involvement of intermediate filaments is given by decrease of PRPH. Disassembly of cytoskeletal proteins leads to fusion of intracellular vesicular membranes with the PM 47. The reduced expression of these proteins in G56S Caveolin‐3 transfected cells might thus contribute to the decrease in sarcolemmal Caveolin‐3 protein (Fig. 2). However, besides the above‐mentioned downregulation of proteins involved in cytoskeleton, several are also elevated. This effect presumably prevents total breakdown of cellular skeleton: MAP4 promotes microtubule assembly, MRP, TES, ABP620 as well as PKP1 regulate actin cytoskeleton homeostasis. Fold of alteration of protective or compensatory factors might contribute to clinical manifestation of the G56S Caveolin‐3 variant and thus an important aspect in the case of additional cellular stress burden.
4.5. The G56S sequence alteration increases vulnerability against osmotic stress in RCMH myoblasts
Caveolae protect the sarcolemma from mechanical stress. Increased expression of Caveolin‐3, as observed in several muscular dystrophies, may thus be a compensatory phenomenon 35. The reduced number of caveolae associated with the Caveolin‐3 missense mutation P28L has been shown to predispose cells to enhanced vulnerability against mechanical stress 4. In addition, accumulation of mutated and wt Caveolin‐3 in the Golgi is thought to affect the synthesis of PM proteins 48. We found that G56S Caveolin‐3 is partially mislocalized to the Golgi in transfected cells, and that Golgi ultrastructure is altered in G56S Caveolin‐3 transfected RCMH cells (Fig. 3). This finding is supported by our results of our proteomic comparison of RCMH cells overexpressing wt and G56S Caveolin‐3, respectively (Supporting Information Table 3). Treatment of G56S Caveolin‐3 RCMH myoblasts with 30% hypoosmotic medium elicited further alterations in the proteome profile (Supporting Information Table 3). Moreover, the proteomic analysis provided unbiased information on the cellular pathomechanisms involved. For example, ABP620 (microtubule‐actin cross‐linking factor 1) expression was increased due to G56S Caveolin‐3 transfection and was additionally enhanced after application of osmotic stress. ABP620 is involved in the guidance of microtubule growth along actin fibers 49 and is a sensor of stress to the cytoskeleton 49. Disruption of ABP620 affects the transport of proteins along the microtubule network resulting in decreased Caveolin‐3 levels at the PM 49. Thus, the upregulation of ABP620 after induction of osmotic stress should prevent the breakdown of trafficking processes along the microtubule network. Taken together, these results suggest that G56S Caveolin‐3 transfected cells show increased vulnerability to osmotic stress.
5. Conclusion
Taken together, our results suggest that the G56S variant is not a clearly pathogenic mutation, but may influence cellular functions and morphologies resulting in an increased cellular vulnerability in terms of a modifying factor. The latter assumption is supported by the proteomic results of our mechanical/osmotic stress experiment. Interestingly, two patients listed with the G56S variant in the database of the Leiden Muscular dystrophy Pages (http://www.dmd.nl) are reported to present with “double trouble”: they additionally carry mutations in the Calpain 3 (CAPN3) and the Fukutin‐related protein (FKRP) gene, respectively. Thus, modifier genes might determine the pathogenic effects of G56S. Our genetic analysis did not yield evidence for double trouble caused by alterations of additional myopathy genes in our patients thus far. However, future comprehensive next‐generation sequencing studies of larger cohorts of G56S carriers might be able to detect genetic alterations that promote phenotypical manifestation. In this context, it is interesting to note that patient 3 was treated chronically with mesalazine for ulcerative colitis. Sulfasalazine, a derivative of mesalazine, has been linked to the induction of myopathy in rare cases 50. Thus, the mesalazine treatment might have triggered myopathy in this patient. No such confounding factors were found in patient 1 or 2.
The authors have declared no conflict of interest.
Supporting information
Supplementary Material
Acknowledgments
We thank the German Federal Ministry for Education and Research (BMBF) for funding (MD‐Net P4, 01GM0887 to JW). LK and RPZ gratefully acknowledge the financial support by the Ministerium für Innovation, Wissenschaft und Forschung des Landes Nordrhein‐Westfalen, and by the German Research Foundation (DFG ZA 639/1‐1). This work has also been supported by a grant from START program of RWTH Aachen University (to AR; grant no. 41/12). We highly appreciate the financial support of the Deutsche Gesellschaft für Muskelkranke e.V.
We thank H. Mader, E. Beck, and E. Pascual for expert technical assistance. The wt‐Caveolin‐3 plasmid was kindly provided by Drs. Sandrine Roy and Robert G. Parton (University of Queensland Medical School, Australia). The NR6W cells were kindly provided by Dr. Darell Bigner, Duke University, NC, USA). The RCMH cells were kindly provided by Drs. Caviedes and Freeman (Universidad de Chile, Santiago, Chile).
Colour Online: See the article online to view Figs. 1, 2 and 5 in colour.
Contributor Information
Eva Brauers, Email: ebrauers@ukaachen.de.
Andreas Roos, Email: andreas.roos@isas.de.
6 References
- 1. Galbiati, F. , Volonte, D. , Minetti, C. , Chu, J. B. , Lisanti, M. P. , Phenotypic behavior of caveolin‐3 mutations that cause autosomal dominant limb girdle muscular dystrophy (LGMD‐1C). Retention of LGMD‐1C caveolin‐3 mutants within the golgi complex. J. Biol. Chem. 1999, 274, 25632–25641. [DOI] [PubMed] [Google Scholar]
- 2. Herrmann, R. , Straub, V. , Blank, M. , Kutzick, C. et al., Dissociation of the dystroglycan complex in caveolin‐3‐deficient limb girdle muscular dystrophy. Hum. Mol. Genet. 2000, 9, 2335–2340. [DOI] [PubMed] [Google Scholar]
- 3. Couet, J. , Li, S. , Okamoto, T. , Ikezu, T. , Lisanti, M. P. , Identification of peptide and protein ligands for the caveolin‐scaffolding domain. Implications for the interaction of caveolin with caveolae‐associated proteins. J. Biol. Chem. 1997, 272, 6525–6533. [DOI] [PubMed] [Google Scholar]
- 4. Sinha, B. , Koster, D. , Ruez, R. , Gonnord, P. et al., Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 2011, 144, 402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aboumousa, A. , Hoogendijk, J. , Charlton, R. , Barresi, R. et al., Caveolinopathy—new mutations and additional symptoms. Neuromuscul. Disord. 2008, 18, 572–578. [DOI] [PubMed] [Google Scholar]
- 6. Tang, Z. , Scherer, P. E. , Okamoto, T. , Song, K. et al., Molecular cloning of caveolin‐3, a novel member of the caveolin gene family expressed predominantly in muscle. J. Biol. Chem. 1996, 271, 2255–2261. [DOI] [PubMed] [Google Scholar]
- 7. Song, K. S. , Scherer, P. E. , Tang, Z. , Okamoto, T. et al., Expression of caveolin‐3 in skeletal, cardiac, and smooth muscle cells. Caveolin‐3 is a component of the sarcolemma and co‐fractionates with dystrophin and dystrophin‐associated glycoproteins. J. Biol. Chem. 1996, 271, 15160–15165. [DOI] [PubMed] [Google Scholar]
- 8. Traverso, M. , Gazzerro, E. , Assereto, S. , Sotgia, F. et al., Caveolin‐3 T78M and T78K missense mutations lead to different phenotypes in vivo and in vitro. Lab. Invest. 2008, 88, 275–283. [DOI] [PubMed] [Google Scholar]
- 9. Fischer, D. , Schroers, A. , Blumcke, I. , Urbach, H. et al., Consequences of a novel caveolin‐3 mutation in a large German family. Ann. Neurol. 2003, 53, 233–241. [DOI] [PubMed] [Google Scholar]
- 10. Galbiati, F. , Volonte, D. , Minetti, C. , Bregman, D. B. , Lisanti, M. P. , Limb‐girdle muscular dystrophy (LGMD‐1C) mutants of caveolin‐3 undergo ubiquitination and proteasomal degradation. Treatment with proteasomal inhibitors blocks the dominant negative effect of LGMD‐1C mutanta and rescues wild‐type caveolin‐3. J. Biol. Chem. 2000, 275, 37702–37711. [DOI] [PubMed] [Google Scholar]
- 11. Schroder, M. , Kaufman, R. J. , ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [DOI] [PubMed] [Google Scholar]
- 12. McNally, E. M. , de Sa Moreira, E. , Duggan, D. J. , Bonnemann, C. G. et al., Caveolin‐3 in muscular dystrophy. Hum. Mol. Genet. 1998, 7, 871–877. [DOI] [PubMed] [Google Scholar]
- 13. Vatta, M. , Ackerman, M. J. , Ye, B. , Makielski, J. C. et al., Mutant caveolin‐3 induces persistent late sodium current and is associated with long‐QT syndrome. Circulation 2006, 114, 2104–2112. [DOI] [PubMed] [Google Scholar]
- 14. Couet, J. , Sargiacomo, M. , Lisanti, M. P. , Interaction of a receptor tyrosine kinase, EGF‐R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J. Biol. Chem. 1997, 272, 30429–30438. [DOI] [PubMed] [Google Scholar]
- 15. Brauers, E. , Dreier, A. , Roos, A. , Wormland, B. et al., Differential effects of myopathy‐associated caveolin‐3 mutants on growth factor signaling. Am. J. Pathol. 2010, 177, 261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weis, J. , Schroder, J. M. , Adult polyglucosan body myopathy with subclinical peripheral neuropathy: case report and review of diseases associated with polyglucosan body accumulation. Clin. Neuropathol. 1988, 7, 271–279. [PubMed] [Google Scholar]
- 17. Weis, J. , Kaussen, M. , Calvo, S. , Buonanno, A. , Denervation induces a rapid nuclear accumulation of MRF4 in mature myofibers. Dev. Dyn. 2000, 218, 438–451. [DOI] [PubMed] [Google Scholar]
- 18. Prause, J. , Goswami, A. , Katona, I. , Roos, A. et al., Altered localization, abnormal modification and loss of function of Sigma receptor‐1 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 1581–1600. [DOI] [PubMed] [Google Scholar]
- 19. Lemmers, R. J. , van der Maarel, S. M. , van Deutekom, J. C. , van der Wielen, M. J. et al., Inter‐ and intrachromosomal sub‐telomeric rearrangements on 4q35: implications for facioscapulohumeral muscular dystrophy (FSHD) aetiology and diagnosis. Hum. Mol. Genet. 1998, 7, 1207–1214. [DOI] [PubMed] [Google Scholar]
- 20. Batra, S. K. , Castelino‐Prabhu, S. , Wikstrand, C. J. , Zhu, X. et al., Epidermal growth factor ligand‐independent, unregulated, cell‐transforming potential of a naturally occurring human mutant EGFRvIII gene. Cell Growth Differ. 1995, 6, 1251–1259. [PubMed] [Google Scholar]
- 21. Kollipara, L. , Buchkremer, S. , Weis, J. , Brauers, E. et al., Proteome profiling and ultrastructural characterization of the human RCMH cell line: myoblastic properties and suitability for myopathological studies. J. Proteome Res. 2016, 15, 945–955. [DOI] [PubMed] [Google Scholar]
- 22. Leroy, M. C. , Perroud, J. , Darbellay, B. , Bernheim, L. , Konig, S. , Epidermal growth factor receptor down‐regulation triggers human myoblast differentiation. PLoS One 2013, 8, e71770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kiel, C. , Serrano, L. , Cell type‐specific importance of ras‐c‐raf complex association rate constants for MAPK signaling. Sci. Signal. 2009, 2, ra38. [DOI] [PubMed] [Google Scholar]
- 24. Manza, L. L. , Stamer, S. L. , Ham, A. J. , Codreanu, S. G. , Liebler, D. C. , Sample preparation and digestion for proteomic analyses using spin filters. Proteomics 2005, 5, 1742–1745. [DOI] [PubMed] [Google Scholar]
- 25. Wisniewski, J. R. , Zougman, A. , Nagaraj, N. , Mann, M. , Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [DOI] [PubMed] [Google Scholar]
- 26. Kollipara, L. , Zahedi, R. P. , Protein carbamylation: in vivo modification or in vitro artefact? Proteomics 2013, 13, 941–944. [DOI] [PubMed] [Google Scholar]
- 27. Burkhart, J. M. , Schumbrutzki, C. , Wortelkamp, S. , Sickmann, A. , Zahedi, R. P. , Systematic and quantitative comparison of digest efficiency and specificity reveals the impact of trypsin quality on MS‐based proteomics. J. Proteomics 2012, 75, 1454–1462. [DOI] [PubMed] [Google Scholar]
- 28. Vaudel, M. , Burkhart, J. M. , Zahedi, R. P. , Oveland, E. et al., PeptideShaker enables reanalysis of MS‐derived proteomics data sets. Nat. Biotechnol. 2015, 33, 22–24. [DOI] [PubMed] [Google Scholar]
- 29. Vizcaíno, J. A. , Csordas, A. , del‐Toro, N. , Dianes, J. A. et al., 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Woodman, S. E. , Sotgia, F. , Galbiati, F. , Minetti, C. , Lisanti, M. P. , Caveolinopathies: mutations in caveolin‐3 cause four distinct autosomal dominant muscle diseases. Neurology 2004, 62, 538–543. [DOI] [PubMed] [Google Scholar]
- 31. Schlegel, A. , Schwab, R. B. , Scherer, P. E. , Lisanti, M. P. , A role for the caveolin scaffolding domain in mediating the membrane attachment of caveolin‐1. The caveolin scaffolding domain is both necessary and sufficient for membrane binding in vitro. J. Biol. Chem. 1999, 274, 22660–22667. [DOI] [PubMed] [Google Scholar]
- 32. Sowmya, B. L. , Jagannadham, M. V. , Nagaraj, R. , Interaction of synthetic peptides corresponding to the scaffolding domain of Caveolin‐3 with model membranes. Biopolymers 2006, 84, 615–624. [DOI] [PubMed] [Google Scholar]
- 33. Okamoto, T. , Schlegel, A. , Scherer, P. E. , Lisanti, M. P. , Caveolins, a family of scaffolding proteins for organizing "preassembled signaling complexes" at the plasma membrane. J. Biol. Chem. 1998, 273, 5419–5422. [DOI] [PubMed] [Google Scholar]
- 34. Sotgia, F. , Woodman, S. E. , Bonuccelli, G. , Capozza, F. et al., Phenotypic behavior of caveolin‐3 R26Q, a mutant associated with hyperCKemia, distal myopathy, and rippling muscle disease. Am. J. Physiol. Cell. Physiol. 2003, 285, C1150–1160. [DOI] [PubMed] [Google Scholar]
- 35. Huang, H. , Bae, C. , Sachs, F. , Suchyna, T. M. , Caveolae regulation of mechanosensitive channel function in myotubes. PLoS One 2013, 8, e72894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fulizio, L. , Nascimbeni, A. C. , Fanin, M. , Piluso, G. et al., Molecular and muscle pathology in a series of caveolinopathy patients. Hum. Mutat. 2005, 25, 82–89. [DOI] [PubMed] [Google Scholar]
- 37. de Paula, F. , Vainzof, M. , Bernardino, A. L. , McNally, E. et al., Mutations in the caveolin‐3 gene: When are they pathogenic? Am. J. Med. Genet. 2001, 99, 303–307. [DOI] [PubMed] [Google Scholar]
- 38. Liu, L. , Mohammadi, K. , Aynafshar, B. , Wang, H. et al., Role of caveolae in signal‐transducing function of cardiac Na+/K+‐ATPase. Am. J. Physiol. Cell. Physiol. 2003, 284, C1550–1560. [DOI] [PubMed] [Google Scholar]
- 39. Chen, X. , Raab, G. , Deutsch, U. , Zhang, J. et al., Induction of heparin‐binding EGF‐like growth factor expression during myogenesis. Activation of the gene by MyoD and localization of the transmembrane form of the protein on the myotube surface. J. Biol. Chem. 1995, 270, 18285–18294. [DOI] [PubMed] [Google Scholar]
- 40. Horikawa, M. , Higashiyama, S. , Nomura, S. , Kitamura, Y. et al., Upregulation of endogenous heparin‐binding EGF‐like growth factor and its role as a survival factor in skeletal myotubes. FEBS Lett. 1999, 459, 100–104. [DOI] [PubMed] [Google Scholar]
- 41. Grumati, P. , Coletto, L. , Sandri, M. , Bonaldo, P. , Autophagy induction rescues muscular dystrophy. Autophagy 2011, 7, 426–428. [DOI] [PubMed] [Google Scholar]
- 42. De Palma, C. , Morisi, F. , Cheli, S. , Pambianco, S. et al., Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. 2012, 3, e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pelkmans, L. , Helenius, A. , Endocytosis via caveolae. Traffic 2002, 3, 311–320. [DOI] [PubMed] [Google Scholar]
- 44. Nishimura, Y. , Takiguchi, S. , Yoshioka, K. , Nakabeppu, Y. , Itoh, K. , Silencing of SNX1 by siRNA stimulates the ligand‐induced endocytosis of EGFR and increases EGFR phosphorylation in gefitinib‐resistant human lung cancer cell lines. Int. J. Oncol. 2012, 41, 1520–1530. [DOI] [PubMed] [Google Scholar]
- 45. Head, B. P. , Patel, H. H. , Roth, D. M. , Murray, F. et al., Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components. J. Biol. Chem. 2006, 281, 26391–26399. [DOI] [PubMed] [Google Scholar]
- 46. Stetler, R. A. , Gan, Y. , Zhang, W. , Liou, A. K. et al., Heat shock proteins: cellular and molecular mechanisms in the central nervous system. Prog. Neurobiol. 2010, 92, 184–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mundy, D. I. , Machleidt, T. , Ying, Y. S. , Anderson, R. G. , Bloom, G. S. , Dual control of caveolar membrane traffic by microtubules and the actin cytoskeleton. J. Cell Sci. 2002, 115, 4327–4339. [DOI] [PubMed] [Google Scholar]
- 48. Lippincott‐Schwartz, J. , Yuan, L. C. , Bonifacino, J. S. , Klausner, R. D. , Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 1989, 56, 801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fassett, J. T. , Xu, X. , Kwak, D. , Wang, H. et al., Microtubule Actin Cross‐linking Factor 1 regulates cardiomyocyte microtubule distribution and adaptation to hemodynamic overload. PLoS One 2013, 8, e73887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Norden, D. K. , Lichtenstein, G. R. , Williams, W. V. , Sulfasalazine‐induced myopathy. Am. J. Gastroenterol. 1994, 89, 801–802. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material