

Abstract
BACKGROUND
Increased susceptibility to 5‐fluorouracil (5‐FU)/capecitabine can lead to rapidly occurring toxicity caused by impaired clearance, dihydropyrimidine dehydrogenase deficiency, and other genetic variations in the enzymes that metabolize 5‐FU. Life‐threatening 5‐FU overdoses occur because of infusion pump errors, dosage miscalculations, and accidental or suicidal ingestion of capecitabine. Uridine triacetate (Vistogard) was approved in 2015 for adult and pediatric patients who exhibit early‐onset severe or life‐threatening 5‐FU/capecitabine toxicities or present with an overdose. Uridine triacetate delivers high concentrations of uridine, which competes with toxic 5‐FU metabolites.
METHODS
In 2 open‐label clinical studies, patients who presented with a 5‐FU/capecitabine overdose or an early onset of severe toxicities were treated. Patients received uridine triacetate as soon as possible (most within the first 96 hours after 5‐FU/capecitabine). Outcomes included survival, resumption of chemotherapy, and safety. Their survival was compared with the survival of a historical cohort of overdose patients who received only supportive care.
RESULTS
A total of 137 of 142 overdose patients (96%) treated with uridine triacetate survived and had a rapid reversal of severe acute cardiotoxicity and neurotoxicity; in addition, mucositis and leukopenia were prevented, or the patients recovered from them. In the historical cohort, 21 of 25 patients (84%) died. Among the 141 uridine triacetate–treated overdose patients with a diagnosis of cancer (the noncancer patients included 6 intentional or accidental pediatric overdoses), 53 resumed chemotherapy in < 30 days (median time after 5‐FU, 19.6 days), and this indicated a rapid recovery from toxicity. Adverse reactions in patients receiving uridine triacetate included vomiting (8.1%), nausea (4.6%), and diarrhea (3.5%).
CONCLUSIONS
In these studies, uridine triacetate was a safe and effective lifesaving antidote for capecitabine and 5‐FU overexposure, and it facilitated the rapid resumption of chemotherapy. Cancer 2017;123:345–356. © 2016 American Cancer Society.
Keywords: capecitabine, fluoropyrimidines, 5‐fluorouracil, overdose, toxicity, uracil
Short abstract
An overdose of or excessive toxicity due to 5‐fluorouracil or capecitabine can be lethal. Uridine triacetate provides a treatment option for patients at risk for excessive 5‐fluorouracil/capecitabine toxicity.
INTRODUCTION
In 2007, the Institute for Safe Medication Practices (ISMP) released an alert that described a fatal 5‐fluorouracil (5‐FU) overdose, an error caused by a misprogrammed chemotherapy pump that resulted in an avoidable death. In this instance, a 43‐year‐old patient received a 5250‐mg dose of 5‐FU over 4 hours instead of the intended 4 days and died 22 days later from toxicity1 even after aggressive supportive care. Accidental overdoses with 5‐FU continue to be a significant problem.2, 3, 4, 5
Capecitabine is an orally bioavailable prodrug that is converted by 3 sequential enzymatic steps to yield 5‐FU.6 The most common toxicities caused by both agents are consequences of cytotoxic damage to rapidly dividing cells, and they include mucositis, diarrhea, and cytopenias. Cutaneous toxicities such as hand‐foot syndrome are more common with capecitabine than 5‐FU. Toxicities are generally cumulative over repeated treatment cycles. Capecitabine overdoses have occurred in suicide attempts and in young children accidentally ingesting a relative's tablets. Genetic enzyme variants, resulting in either enhanced conversion of capecitabine to 5‐FU or increased sensitivity to 5‐FU toxicity, can result in life‐threatening and fatal toxicities from planned dosages of capecitabine or 5‐FU.6, 7, 8, 9, 10, 11
The natural history of early‐onset severe toxicity from 5‐FU and capecitabine is well documented.6, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 Patients with an exaggerated sensitivity to 5‐FU or capecitabine may quickly develop severe, life‐threatening toxicities during or after administration, which can manifest as severe forms of common toxicities such as mucositis and cytopenias but can also include central neurotoxicity and acute cardiomyopathy. Early‐onset severe toxicities can occur during or after a patient's first course of capecitabine or 5‐FU treatment, and the virulence can rival or exceed cases of massive overdose. These early‐onset patients are more susceptible to toxicities due to the pharmacogenetics of 5‐FU metabolism, including variations in dihydropyrimidine dehydrogenase (DPD),17, 22, 23, 24 thymidylate synthase (TYMS),23, 25 and orotate phosphoribosyltransferase (OPRT),6, 17, 23 or due to renal impairment or other less well‐defined causes.6
It has been demonstrated clinically and in animals that uridine, through a simple mechanism of action, prevents or reduces 5‐FU–related mortality and toxicity to the gastrointestinal tract and hematopoietic system.26, 27, 28, 29, 30, 31, 32 Exogenous uridine, when it is administered after 5‐FU, competes with the toxic 5‐FU metabolite fluorouridine triphosphate (FUTP) for incorporation into RNA in normal tissues (Fig. 1).26, 29, 30, 31, 32, 33 Preclinical and clinical studies have shown that a sustained uridine concentration of 70 μmol/L provides protection of normal tissues from the toxic effects of FUTP.26, 27, 28, 29, 30, 31, 32, 33, 34, 35 However, the oral administration of adequate doses of uridine is not clinically feasible because of poor bioavailability, and parenteral infusion presents safety issues, including phlebitis and fever.27, 30, 31, 32, 33, 34, 35
Figure 1.
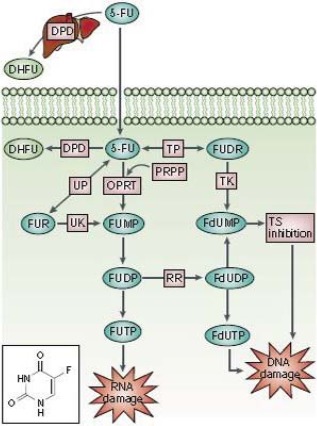
5‐FU (see structure) is converted into 3 main active metabolites: FdUMP, FdUTP, and FUTP. The main mechanism of 5‐FU activation is conversion to FUMP; this occurs either directly by OPRT with PRPP as the cofactor or indirectly via FUR through the sequential action of UP and UK. FUMP is then phosphorylated to FUDP, which can be either further phosphorylated to the active metabolite FUTP or converted to FdUDP by RR. In turn, FdUDP can be either phosphorylated or dephosphorylated to generate the active metabolites FdUTP and FdUMP, respectively. An alternative activation pathway involves the TP‐catalyzed conversion of 5‐FU to FUDR, which is then phosphorylated by TK to FdUMP. The DPD‐mediated conversion of 5‐FU to DHFU is the rate‐limiting step of 5‐FU catabolism in normal and tumor cells. Up to 80% of administered 5‐FU is broken down by DPD in the liver.56 Abbreviations: DHFU, dihydrofluorouracil; DPD, dihydropyrimidine dehydrogenase; FdUDP, fluorodeoxyuridine diphosphate; FdUMP, fluorodeoxyuridine monophosphate; FdUTP, fluorodeoxyuridine triphosphate; 5‐FU, 5‐fluorouracil; FUDP, fluorouridine diphosphate; FUDR, fluorodeoxyuridine; FUMP, fluorouridine monophosphate; FUR, fluorouridine; FUTP, fluorouridine triphosphate; OPRT, orotate phosphoribosyltransferase; PRPP, phosphoribosyl pyrophosphate; RR, ribonucleotide reductase; TK, thymidine kinase; TP, thymidine phosphorylase; TS, thymidylate synthase; UK, uridine kinase; UP, uridine phosphorylase. Longley et al.33 Used with permission.
Uridine triacetate (Vistogard; Wellstat Therapeutics Corporation, Gaithersburg, MD) is an oral pyrimidine analogue of uridine. Uridine triacetate is lipophilic, is quickly absorbed in the gut, and is rapidly deacetylated in the circulation to yield free uridine.34, 37, 38, 39, 40, 41 It provides 4‐ to 6‐fold more bioavailable uridine than equimolar doses of uridine itself.34, 37, 38, 39, 40, 41 The administration of uridine triacetate to mice, in models of either lethal 5‐FU overdosing or clearance defects, significantly improved survival, especially when it was administered within the first 24 hours after 5‐FU. Later administration provided a diminishing but still significant benefit.37, 41 The effectiveness of uridine triacetate as an antidote to 5‐FU has been studied previously37, 38, 39, 40, 41 in animals37, 40 and in clinical trials of intentional 5‐FU dose escalation.41 This report presents the final overall safety and efficacy of uridine triacetate in 2 compassionate‐use, open‐label clinical studies conducted in the United States and outside the United States, including Europe, Canada, and Australia, by Wellstat Therapeutics Corporation in adult and pediatric patients overdosed with 5‐FU or capecitabine or presenting with an early onset of severe toxicity.38, 39 The primary efficacy endpoint was patient survival 30 days after a 5‐FU/capecitabine overdose or after the appearance of early‐onset severe toxicity following the administration of a planned dose of 5‐FU or capecitabine. The standard treatment for a 5‐FU overdose before uridine triacetate was supportive care, including antiemetics, intravenous fluids, antibiotics, intensive care, and medications to treat myelosuppression. Therefore, in this report, the overall efficacy of treatment with uridine triacetate is compared with a historical cohort of cases in which documented overdose patients received best supportive care. The resumption of chemotherapy treatment within the 30‐day posttreatment period was also examined. Vistogard was approved by the Food and Drug Administration (FDA) in December 2015.42
MATERIALS AND METHODS
In both clinical trials, uridine triacetate was provided to patients because of a 5‐FU or capecitabine overdose and/or a rapid early onset of serious toxicity within the first 96 hours after the administration of planned doses of 5‐FU or capecitabine. Because there were no existing antidotal treatments for 5‐FU toxicity and the use of a placebo would have been unethical, a historical control cohort of patients who overdosed on 5‐FU and received only best supportive care was sourced from all available literature and used as a comparator.
Key inclusion criteria included consenting patients who were determined to be at excess risk for 5‐FU toxicity due to a frank overdose (the administration of 5‐FU at a dose or infusion rate greater than the intended dose or maximum tolerated dose for the patient's intended regimen) or who demonstrated a rapid onset of severe toxicity or were known or suspected to have increased susceptibility to 5‐FU (eg, a patient known to be DPD‐deficient). Early‐onset severe toxicities manifested during or within the 96 hours after the administration of 5‐FU or during a standard 14‐day course of capecitabine (days 3‐9) and included severe cytotoxic mucosal and/or hematologic toxicities as well as acute encephalopathy and/or cardiomyopathy. Most early‐onset toxicities in this study occurred after a patient's first exposure to 5‐FU or capecitabine, and this indicated an unusual susceptibility to the toxicity of these chemotherapy drugs.
Patients began emergency treatment with uridine triacetate (10 g orally every 6 hours for a total of 20 doses) as soon as possible after 5‐FU or capecitabine and the recognition of the need for antidotal treatment. The regimen was based on previous pharmacokinetic and clinical studies of uridine triacetate; data from these studies established that the proposed adult dose (fixed at 10 g) and pediatric dose (6.2 g/m2 of body surface area up to a maximum of 10 g) given every 6 hours for a total of 20 doses for 5 days would provide steady‐state plasma uridine concentrations of at least 70 μM, the threshold for protecting normal tissues from the toxic effects of 5‐FU.29, 30, 31, 32 This concentration was maintained for 5 days to ensure the clearance of 5‐FU metabolites from tissues and thus the optimum inhibition of the course of 5‐FU toxicities.25, 26, 27, 28, 29, 38, 39, 40, 41
Collected data included overdose details (dose, cause, infusion start and stop times, and symptoms associated with the overdose), the early onset of toxicities after 5‐FU (or capecitabine), or known or suspected impaired clearance and outcomes. Each patient's course and outcome, including survival, were assessed for 30 days after the initial event. Investigators supplied deidentified medical records, dosing logs, and toxicity and adverse event data to the sponsor, and independent study coordinators completed all case report forms with the investigator‐supplied documentation.
The overall primary endpoint was survival or the resumption of chemotherapy during the 30‐day monitoring period; all patients who resumed chemotherapy also survived beyond the 30‐day monitoring period. The primary safety endpoint was the monitoring of adverse events with Common Terminology Criteria for Adverse Events (version 4.03) within the same 30‐day period. Adverse events known to be associated with 5‐FU, other chemotherapeutic agents administered together with 5‐FU, other anticancer treatments received, and the patient's underlying diagnosis and medical history were documented.
All patients (or their legal representatives) provided informed consent before the initiation of any study‐related procedure. Uridine triacetate was provided by Wellstat Therapeutics Corporation after a review of eligibility upon a physician's request for the antidote under emergency single‐patient investigational new drug regulations (or the equivalent outside the United States) or to those meeting eligibility criteria for the expanded access protocol. Both protocols were approved by a central institutional review board, and any use of uridine triacetate was subject to institutional review board (or ethics committee) notification at the site. This study was approved and conducted in compliance with good clinical practice and is registered at ClinicalTrials.gov (NCT01432301).
No comparative analyses or inferential statistics were performed.
Historical Case Cohort
In lieu of an active or placebo control, to permit a comparison of active treatment with uridine triacetate and best supportive care, case records of patients who experienced 5‐FU overdoses were obtained from a review of the available literature, published medicolegal cases, the FDA's Manufacturers and User Facility Device Experience database (MAUDE), the FDA's Adverse Event Databases (FDAble), the FDA's Medical Product Safety Network (MedSun), and reports from the ISMP. This search yielded 47 cases. Key data points sought from this review included the total 5‐FU dose administered, the time and rate of administration, and the patient outcomes; 25 of the 47 cases included all 3 data elements.
Acute 5‐FU toxicity is proportional to the plasma 5‐FU exposure, which is a function of both the dose and the infusion rate. To provide a graphical method of comparison of the current uridine triacetate–treated cases (this study) and the historical cohort (best supportive care) and to allow the comparison of levels of toxicity of disparate 5‐FU doses/infusion rates in the historical cohort, a severity score was calculated for each uridine triacetate study case and for the 25 cases of 5‐FU overdose from the historical cohort as follows:
Square root of ([Log10 Dose]2 + [Log10 Infusion Rate]2
The units were total milligrams delivered (combined bolus and infusion) for the dose and milligrams per hour for the infusion rate. Severity scores were assigned to the historical cohort cases on the basis of the overdose dose and/or rate and were correlated with the outcomes of cases in the historical cohort. The same severity score calculation was then used for each of the cases in the study cohort to provide a method for comparing the historical cohort and the study cases. Historical cases with severity scores of 4.0 to 4.5 had high probabilities of severe, life‐threatening toxicities; severity scores > 4.5 indicated a high probability of death. Severity scores for maximum tolerated dosages of 5‐FU in a variety of standard regimens are below 4.0 and, in the graphical representation, delineate the boundary of the expected tolerated zone.
RESULTS
Patients
Details of the disposition of the patients are shown in Figure 2. For all 173 uridine triacetate–treated patients, the median number of doses taken per subject was 20 of 20 (range, 1‐23). Overall, more patients were male, and the mean patient age was 58.1 years. Overall demographics, including causes of overdoses, are listed in Table 1. The most common causes of overdoses were infusion pump programming errors and dose miscalculations. There were a total of 6 pediatric cases, including 1 patient with autoimmune disease who received 5‐FU instead of cyclophosphamide, 3 patients aged 1 to 2 years who accidentally ingested a family member's capecitabine, and 2 pediatric cancer patients who received a 5‐FU overdose. There were a total of 6 adult capecitabine suicide attempts with doses ranging from 7000 to 28,000 mg ingested at once. All pediatric and intentional‐overdose patients were treated with uridine triacetate. Fourteen patients were treated with uridine triacetate via a nasogastric or gastrostomy tube (1 via an orogastric tube) because they had severe mucositis, were unable to swallow, and/or were comatose or intubated.
Figure 2.
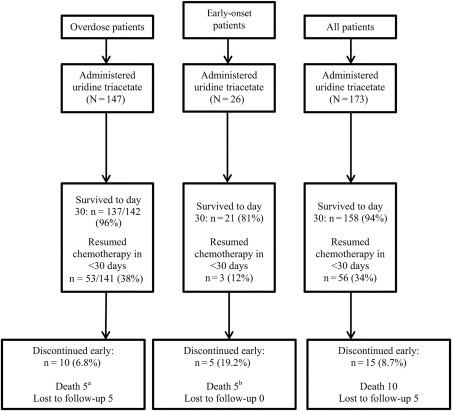
Disposition of the patients. aThree of the 5 deaths in this group were attributed to progression of the underlying cancer; 1 death was attributed to septic shock associated with acute ischemic enteritis and ileus, gram‐negative bacteremia, and respiratory failure; and 1 death was due to apparent tumor lysis syndrome. bIn all these patients, uridine triacetate was started more than 96 hours after 5‐fluorouracil or capecitabine was stopped. These deaths were attributed to sequelae of 5‐fluorouracil toxicities: acute respiratory distress syndrome (n = 1), multisystem organ failure secondary to sepsis (n = 1), and septic shock (n = 3).
Table 1.
Baseline Characteristics of Patients Treated With Uridine Triacetate
| Characteristic | Overdose (n = 147) | Early Onset (n = 26) | Overall (n = 173) |
|---|---|---|---|
| Age, mean (SD), y | 58.1 (15.10) | 56.1 (16.07) | 58.1 (15.14) |
| Female sex, No. (%) | 60 (40.8) | 15 (57.7) | 75 (43.4) |
| Cancer diagnosis, No. (%) | |||
| Pancreatic | 9 (6.1) | 0 | 9 (5.2) |
| Colorectal | 80 (54.4) | 11 (42.3) | 91 (52.6) |
| Head and neck | 22 (15.0) | 6 (23.1) | 28 (16.2) |
| Breast | 3 (2.0) | 2 (7.7) | 5 (2.9) |
| Gastric | 11 (7.5) | 1 (3.8) | 12 (6.9) |
| Unknown | 0 | 0 | 0 |
| Not applicablea | 6 (4.1) | 0 | 6 (3.5) |
| Other | 16 (10.9) | 6 (23.1) | 22 (12.7) |
| Cause of overdose, No. (%) | |||
| Pump programming error | 56 (38.1) | NA | — |
| Pump malfunction | 35 (23.8) | NA | — |
| Dose miscalculation | 12 (8.2) | NA | — |
| Wrong pump/filter used | 13 (8.8) | NA | — |
| Suicidal ingestion of capecitabine | 6 (4.1) | NA | — |
| Accidental ingestion of capecitabine (pediatric) | 3 (2.0) | NA | — |
| Unknown/other | 22 (15.0) | NA | — |
Abbreviation: NA, not applicable; SD, standard deviation.
Patients with an accidental or intentional (suicidal) overdose.
A tabulation of case characteristics for the historical case cohort is provided in Table 2.
Table 2.
Historical Case Data
| Case | 5‐FU | Time, h | Rate, mg/h | Severity Score | Outcome | Source |
|---|---|---|---|---|---|---|
| 1 | 10,400 mg | 2 | 5200 | 5.47 | Death | ISMP |
| 2 | 10,000 mg | 3 | 3333 | 5.33 | Death | ISMP |
| 3 | 4370 mg | 0.75 | 5827 | 5.24 | Death | Nevada Board of Pharmacy |
| 4 | 7500 mg | 2.5 | 3000 | 5.21 | Death | Legal document |
| 5 | 27,200 mg | 96 | 283 | 5.07 | Death | Nursing malpractice report |
| 6 | 4800 mg | 1.9 | 2526 | 5.01 | Death | ISMP |
| 7 | 3000 mg | 0.75 | 4000 | 5.01 | Death | ISMP |
| 8 | 4400 mg | 2 | 2200 | 4.94 | Death | ISMP |
| 9 | 6000 mg | 4 | 1500 | 4.94 | Death | ISMP |
| 10 | 6000 mg | 4 | 1500 | 4.94 | Death | FDA MAUDE |
| 11 | 5250 mg | 4 | 1313 | 4.85 | Death | ISMP |
| 12 | 4500 mg | 4 | 1125 | 4.76 | Death | Physician report |
| 13 | 10,000 mg | 36 | 278 | 4.69 | Survived | Hospital report |
| 14 | 4800 mg | 24 | 200 | 4.34 | Survived | Physician report |
| 15 | 1500 mg | 2 | 750 | 4.28 | Death | News report |
| 16 | 2000 mg | 24 | 83 | 3.82 | Survived | FDA MAUDE |
| 17 | 5000 mg | 26 | 192 | 4.35 | Death | Physician report |
| 18 | 5040 mg | 5 | 1008 | 4.77 | Death | Physician report |
| 19 | 1000 mg/m2/d × 5 | — | Bolus | >5.31 | Death | Reference 43 |
| 20 | 1000 mg/m2/d × 5 | — | Bolus | >5.31 | Death | Reference 43 |
| 21 | 1000 mg/m2/d × 5 | — | Bolus | >5.31 | Death | Reference 43 |
| 22 | 1000 mg/m2/d × 5 | — | Bolus | >5.31 | Death | Reference 43 |
| 23 | 1000 mg/m2/d × 5 | — | Bolus | >5.31 | Death | Reference 43 |
| 24 | 1000 mg/m2/d × 5 | — | Bolus | >5.31 | Death | Reference 43 |
| 25 | 1000 mg/m2/d × 5 | — | Bolus | >5.31 | Survived | Reference 43 |
Abbreviations: FDA, Food and Drug Administration; 5‐FU, 5‐fluorouracil; ISMP, Institute for Safe Medication Practices; MAUDE, Manufacturers and User Facility Device Experience.
Patients 19 to 25 were supposed to have received a 5‐day continuous infusion of 1000 mg/m2/d but instead were given 5 daily bolus doses of 1000 mg/kg/d. These patients were not included in the nomogram in Figure 3 because the nomogram is based on a single infusion or bolus/infusion in order to compare the relative severity of the most common overdose situations.
Outcomes: Early‐Onset Patients
Treatment was initiated within 96 hours for 18 of the 26 patients with early‐onset severe toxicity (the early‐onset group). Notably, all surviving early‐onset patients started uridine triacetate within the protocol‐specified 96 hours after the termination of 5‐FU or capecitabine (Fig. 2).
Three of the 8 early‐onset patients (38%) who initiated uridine triacetate beyond 96 hours survived. The 5 deaths occurred in early‐onset patients who started uridine triacetate beyond 96 hours. These deaths were attributed to sequelae of 5‐FU toxicities: acute respiratory distress syndrome (n = 1), multisystem organ failure secondary to sepsis (n = 1), and septic shock (n = 3).
Outcomes: Overdose Patients
All overdose patients (n = 147) received 1.9 to 576 times the planned infusion rate of 5‐FU or doses up to 10 times higher than intended. Of the 147 patients who received uridine triacetate, 5 were lost to follow‐up, leaving 142 evaluable patients in the 30 day survival evaluation. A total of 137 of these patients (96%) survived; in comparison, only 4 of the 25 historical (supportive‐care) controls (16%) survived. To aid in comparing the study patients with the historical control patients, a nomogram showing survival as a function of overdose severity is shown in Figure 3. Survival data for the study patients treated with uridine triacetate were plotted as a function of severity scores and are shown in Figure 4.
Figure 3.
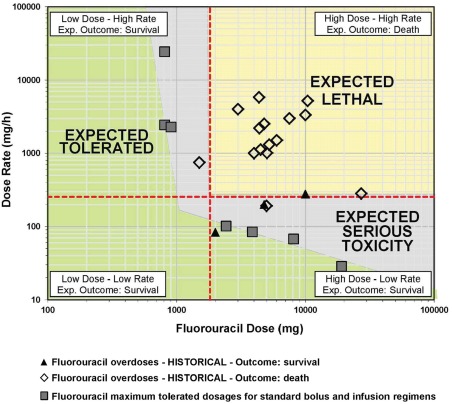
Historical case outcomes as a function of the 5‐fluorouracil infusion rate and dose. The expected tolerated zone is defined by the maximum tolerated doses of a variety of 5‐fluorouracil regimens. Patients in the expected lethal zone would be expected to die on the basis of the infusion rate and dose; patients in the expected tolerated zone as well as those in the expected serious toxicity zone would be expected to survive. Patients 19 to 25 in Table 2 were supposed to have received a 5‐day continuous infusion of 1000 mg/m2/d but instead were given 5 daily bolus doses of 1000 mg/kg/d. These patients are not included in the nomogram in this figure because the nomogram is based on a single infusion or bolus/infusion in order to compare the relative severity of the most common overdose situations.
Figure 4.
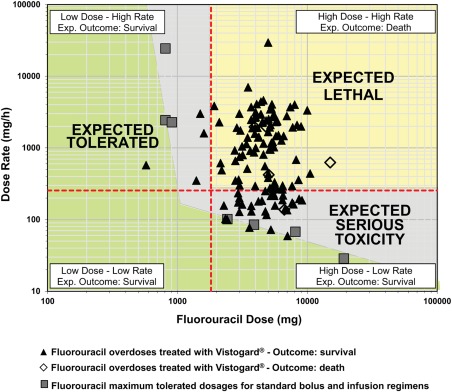
5‐Fluorouracil overdose case outcomes as a function of the 5‐fluorouracil infusion rate and dose. The expected tolerated zone is defined by the maximum tolerated doses of a variety of 5‐fluorouracil regimens. Patients in the expected lethal zone would be expected to die on the basis of the infusion rate and dose; patients in the expected tolerated zone as well as those in the expected serious toxicity zone would be expected to survive.
Resumption of chemotherapy
Of the 147 patients who received uridine triacetate, 6 were accidental or intentional overdoses. Fifty‐three of the 141 evaluable overdose patients (38%) treated with uridine triacetate resumed chemotherapy within the 30‐day observation period; the majority of these patients resumed treatment in less than 3 weeks (mean time to resumption of chemotherapy, 19.6 days). All of these patients also survived beyond the 30‐day monitoring period.
Safety
The adverse reactions reported at a frequency ≥ 2% were vomiting (n = 14 or 8.1%), nausea (n = 8 or 4.6%) and diarrhea (n = 6 or 3.5%). One head and neck cancer patient with a history of serious nausea and vomiting experienced grade 3 nausea and vomiting while receiving uridine triacetate. These events are also consistent with those observed generally in cancer patients receiving chemotherapy.
DISCUSSION
Overdoses due to a variety of clinical mishaps, most often the excessively rapid administration of 5‐FU, can lead to excessive 5‐FU exposures.3 The frequency of overdoses is unknown, but with at least 275,000 patients in the United States receiving multiple cycles of 5‐FU annually (most adjusted for a patient's body surface area), a substantial number of unreported overdoses probably occur.44
Severe toxicity generally develops faster in predisposed patients than patients who receive overdoses.7, 8, 22, 23, 24 The best elucidated predisposing genetic factor is a DPD deficiency (3%‐5% of 5‐FU patients).9, 10 The National Institutes of Health has estimated the number of deaths due to a DPD deficiency to be approximately 1300 patients per year (0.5% of 5‐FU patients).2, 44 Approximately 80% of a standard dose of 5‐FU is degraded by DPD, with toxic overexposure occurring if DPD activity is depressed. An overdose alone can saturate the capacity of DPD to degrade 5‐FU. Mutations in OPRT can increase 5‐FU anabolism to form toxic intracellular 5‐fluorouridine nucleotides and are associated with an allele (Gly213Ala) that has an incidence of 16% to 17% in the United States,9 with homozygotes (2.5% of the US population) at high risk for a rapid onset of serious toxicities.9 Particular polymorphisms in the genes encoding TYMS and methylene tetrahydrofolate reductase are also associated with increased 5‐FU toxicity.10, 11, 17, 23, 45, 46, 47, 48 Life‐threatening capecitabine toxicity can be caused by the same gene variants that exacerbate 5‐FU toxicity6 and also by overactivity of cytidine deaminase, which mediates the conversion of capecitabine to 5‐FU.17 In all of these forms of genetic susceptibility to 5‐FU, the proximal cause of death or life‐threatening toxicity is tissue damage caused by 5‐FU metabolites; this tissue damage is often ulcerative mucositis with neutropenia leading to sepsis, shock, and organ failure. Genetic testing for a DPD deficiency before the initiation of 5‐FU or capecitabine chemotherapy has not been widely adopted, perhaps because of low penetrance, and it does not detect other consequential mutations (or the low expression of genetically normal DPD) that may cause serious toxicity after planned dosages of 5‐FU. DPD mutations are found in 50% or fewer of severe 5‐FU toxicity cases.17 Other major risk factors for excess fluoropyrimidine toxicity include mutations in OPRT and TYMS, compromised renal function, older age, elevated pretreatment uracil levels, and increased body surface area.6
Rare but potentially catastrophic side effects of 5‐FU include acute central neurotoxicity and cardiotoxicity, which occur in patients with no identifiable predisposing factors.49, 50, 51 In this study, 10 patients presented with serious central neurotoxicity, and 2 presented with acute, life‐threatening cardiotoxicity. Potential mediators of these toxicities may be 5‐FU catabolites such as fluoroacetate. Central neurologic toxicities related to 5‐FU and capecitabine treatment can range from confusion and altered mental status to encephalopathy and coma. Reported cardiotoxic effects include arrhythmias, transient vasospasm, takotsubo syndrome, heart failure, cardiogenic shock, and cardiac arrest49, 50, 51; these neurologic and cardiac toxicities can rapidly become fatal.
The absence of a comparator antidote and obvious ethical prohibitions against a placebo control in the study made direct quantitative efficacy comparisons difficult. The use of severity scores allowed a comparison of overdoses and outcomes between cases in the historic cohort and the clinical study cases. Although there was a possible selection bias introduced by the overdose cases that were publicly available (perhaps these reported cases were more severe), the natural history of a severe 5‐FU overdose typically ends with death, especially when the dosage exceeds that planned by 3‐fold or more. A countervailing bias is based on medical liability issues that can keep these cases from being publicly disseminated. During the review of the new drug application for uridine triacetate, members of the FDA staff performed their own analysis of fluorouracil toxicity cases from the FDA's Adverse Event Reporting System. The Adverse Event Reporting System was searched for voluntary postmarketing reports of fatalities in patients who experienced early‐onset severe or life‐threatening toxicity after capecitabine or 5‐FU. Fifty‐eight cases for 5‐FU and 145 cases for capecitabine were uncovered, and many of the cases had a spectrum of signs and symptoms consistent with our description of early‐onset severe or life‐threatening toxicity associated with 5‐FU or capecitabine. All cases were treated with supportive care and had uniformly fatal outcomes.52 This case analysis further bolsters the poor survival data observed for the historical case patients who received only supportive care.
In the historical case cohort, 16% of the patients who overdosed with 5‐FU (and did not receive uridine triacetate) survived. In contrast, 96% of similar patients who received uridine triacetate survived. A large fraction of the patients treated with uridine triacetate had received 5‐FU overdoses at least comparable in severity to overdoses that were uniformly lethal in the historical group (Figs. 3 and 4). Importantly, the timing of the administration of uridine triacetate proved to be a significant survival factor. Patients who received uridine triacetate within the first 96 hours after the end of 5‐FU or capecitabine had increased survival. All 18 of the early‐onset patients (100%) who were started on uridine triacetate treatment within the 96 hours after the termination of 5‐FU or capecitabine survived and recovered; 5 of the 8 patients who initiated treatment beyond 96 hours died. This illustrates both the potential severity of early‐onset toxicities and the need for prompt recognition and treatment.
Although direct statistical comparisons (beyond survival) of patients treated with uridine triacetate and patients treated with best supportive care are difficult, individual case descriptions and comparisons further illustrate the agent's efficacy. In one case that parallels the aforementioned 43‐year‐old patient who died from a 5‐FU overdose (see the introduction),1 this study included a 43‐year‐old female patient who received an overdose of 5‐FU of a very similar magnitude (5000 mg over 0.17 hours instead of the intended 96 hours). Unlike the patient in the ISMP report, this patient received uridine triacetate 8 hours after the overdose, and she survived after experiencing only grade 1 and 2 anemia and thrombocytopenia. She was discharged from the hospital 6 days after the overdose. In a separate case, a 46‐year‐old male patient, subsequently discovered to be severely DPD‐deficient, received an intended low 5‐FU dose of 1710 mg over 96 hours but experienced grade 4 mucositis, pancytopenia, and hand‐foot syndrome as well as grade 3 fever and diarrhea, but he survived a 15‐day hospital stay after receiving intensive supportive care. Months later, the same patient was overdosed with a 1‐minute bolus of 1000 mg of 5‐FU instead of the intended 100 mg dose of 5‐FU. He began treatment with uridine triacetate 8 hours after the overdose and experienced only mild (grade 1/2) cytopenia that was present before the 5‐FU administration, and he did not require a change in his daily routines. These individual cases illustrate well the profound therapeutic benefit of the early administration of uridine triacetate.
All patients treated with uridine triacetate after suicidal overdoses of capecitabine (acute ingestion of up to 28,000 mg) survived with minimal toxicities, as did young children who accidentally ingested a relative's capecitabine tablets. All of these patients received uridine triacetate within the first 96 hours after the ingestion of capecitabine.
Recovery from severe 5‐FU and capecitabine toxicity generally causes a delay in subsequent treatment, and this affects the patient's cancer therapy.17, 53 In these clinical studies, a significant proportion (38%) of overdose patients treated with uridine triacetate resumed chemotherapy within the 30‐day observation period; most did so in less than 3 weeks.
Generally, early‐onset patients presented with severe toxicity within hours or several days of the termination of 5‐FU or capecitabine; many did so after or during their very first cycle. In contrast, standard 5‐FU toxicities typically develop over a period of weeks and multiple cycles. Accordingly, many overdose patients did not show immediate signs and symptoms upon the recognition of their overdose, and they began treatment with uridine triacetate emergently before symptoms appeared. The incidence of grade 3 or 4 mucositis was extremely low in overdose patients, and it was resolved completely in almost all patients (overdose and early‐onset patients) within the 30‐day observation period. This course parallels published clinical experience with intentional 5‐FU dose escalation enabled by planned posttreatment uridine triacetate, in which low frequencies of serious hematologic and gastrointestinal toxicities, including mucositis, were noted.10, 37, 54
Uridine triacetate competitively inhibits components of cytotoxicity attributable to 5‐FU incorporation into RNA; it does not interfere with thymidylate synthase (TS) inhibition, which is established quickly and irreversibly.55 Uridine triacetate acts by augmenting intracellular uridine triphosphate to compete with FUTP, a toxic RNA precursor. Uridine catabolites derived from uridine triacetate, including uracil and its breakdown products, also dilute and compete with some 5‐FU byproducts such as fluoroacetate that have been implicated in neurotoxicity and cardiotoxicity. RNA‐directed cytotoxicities account for a major proportion of dose‐limiting and life‐threatening 5‐FU toxicities after either an overdose or early onset (whether due to pharmacogenetic susceptibility, impaired clearance, or other underlying causes).
Treatment‐emergent adverse events were consistent with the setting of a 5‐FU or capecitabine overdose. Of the 10 deaths that occurred among the 173 patients in these studies, 5 were among patients who received the antidote more than 96 hours after 5‐FU or capecitabine; this agrees with a time window of approximately 96 hours for effective treatment, as found in preclinical studies.37, 40 Three deaths among overdose patients during the 30‐day monitoring period were attributed to progression of the underlying cancer; 1 was attributed to septic shock associated with acute ischemic enteritis and ileus, gram‐negative bacteremia, and respiratory failure; and 1 was due to apparent tumor lysis syndrome.
Uridine triacetate had a high rate of treatment compliance. Most patients who took uridine triacetate started treatment in the hospital, but because it is an oral agent with general ease of use and administration, some of these patients were able to complete their therapy at home. Some patients who were unable to swallow or were unconscious when treatment with uridine triacetate began received the medication via nasogastric or gastrostomy tubes; the administration of uridine triacetate in this manner did not appear to affect its efficacy or safety in comparison with oral ingestion.
In this patient set, attempts to assign causality for toxicity in specific patients resulted in the identification of mutations in enzymes involved in 5‐FU clearance (DPD), anabolism (OPRT), or the conversion of capecitabine to 5‐FU (cytidine deaminase), but often an associated defect was not identified. The recognition of early‐onset severe 5‐FU or capecitabine toxicity is complicated by a lack of diagnostic tools beyond the observation of signs and symptoms contrasting with the expected degrees and types of toxicities for individual patients. A grade 3/4 toxicity is reported for approximately 30% of patients treated with systemic 5‐FU (much of it is cumulative, but in some cases it develops rapidly during and after a patient's first dose), and toxic death is reported for >0.5% of patients.56 A recent publication by Mazzuca et al57 described a technique for measuring the 5‐FU degradation (or anabolism) rate in peripheral mononuclear blood cells. This technique potentially allows better identification of patients at increased risk for early‐onset severe toxicity in comparison with an assessment of the DPD mutation status. According to this method, there is a high risk of serious toxicity in patients with either ultrahigh 5‐FU metabolism (excessive anabolism) or poor 5‐FU metabolism (impaired degradation), and on the basis of the data, the method indicates that up to 10% of the population overall is at high risk for developing severe toxicity.57 In the current report, the number of patients with an overdose exceeded the number of patients with an early‐onset severe toxicity, in part because the identification of overdose cases is straightforward. If we assume that the overall incidence for the risk of developing severe toxicity is >10%,9 it is not unrealistic to conclude that the frequency of early‐onset patients in reality is probably much higher than the frequency of patients with an overdose. This reinforces the need for physicians, nurses, and patients to remain vigilant for signs and symptoms of early‐onset severe toxicity, especially during the initial course or courses of 5‐FU and capecitabine, and to be prepared to treat it urgently. The identification of a specific genetic cause for already severe toxicities of 5‐FU or capecitabine is not a prerequisite or even recommended before the initiation of antidotal treatment with uridine triacetate because its mode of action (dilution of and competition with toxic 5‐FU metabolites) applies, regardless of the specific details underlying the exaggerated toxicity. An early onset of serious symptoms, especially during or after the first cycle of treatment, often leads to rapid worsening of toxicities or a fatal outcome that may occur before the identification of an underlying molecular or metabolic defect is possible.
The safety of uridine triacetate has been established. No safety issues were observed in any preclinical studies, and the adverse events experienced by patients in the current study were common in patients undergoing chemotherapy, especially after overexposure to toxic agents. The adverse reactions at least possibly related to oral uridine triacetate were almost entirely mild to moderate gastrointestinal events. Those experienced by ≥ 2% of patients were vomiting (8.1%), nausea (4.6%) and diarrhea (3.5%), which occurred in the context of the emergency treatment of patients, many of whom had mucositis or other gastrointestinal distress, with an oral agent every 6 hours. The results from this study demonstrate that uridine triacetate is a safe and highly effective antidote. In comparison with historical cases, uridine triacetate significantly improved the survival of patients overdosed with 5‐FU or capecitabine. Uridine triacetate (Vistogard; https://www.vistogard.com/) was approved by the FDA in December 2015 and is available commercially.
FUNDING SUPPORT
Wellstat Therapeutics Corporation is the sponsor of uridine triacetate, the subject of this article, and provided the funding source for the studies upon which this work is based.
CONFLICT OF INTEREST DISCLOSURES
Thomas R. King is a paid employee of BTG International, Inc, which is a partner of Wellstat Therapeutics Corporation. Reid W. von Borstel and Michael K. Bamat are both paid employees of Wellstat Therapeutics Corporation (the sponsor and developer of uridine triacetate).
AUTHOR CONTRIBUTIONS
Wen Wee Ma: Clinical input, writing, editing, and data interpretation. Muhammad Wasif Saif: Clinical input, writing, editing, and data interpretation. Bassel F. El‐Rayes: Clinical input, writing, editing, and data interpretation. Marwan G. Fakih: Clinical input, writing, editing, and data interpretation. Thomas H. Cartwright: Clinical input, writing, editing, and data interpretation. James A. Posey: Clinical input, writing, editing, and data interpretation. Thomas R. King: Writing, editing, and data interpretation. Reid W. von Borstel: Writing, editing, data interpretation, study planning, conduct management, and guarantors of data and content of this article. Michael K. Bamat: Writing, editing, data interpretation, study planning, conduct management, and guarantor of data and content of this article.
Portions of these data were presented at the American Society of Clinical Oncology Gastrointestinal Cancers Symposium; January 21‐23, 2016; San Francisco, CA.
We acknowledge Julie Searle Vanas, BS, for her assistance with the clinical trial and data.
REFERENCES
- 1. ISMP Canada . Fluorouracil incident root cause analysis, 2007. http://www.ismp-canada.org/download/reports/FluorouracilIncidentMay2007.pdf. Accessed on June 15, 2016.
- 2. Andreica I, Pfeifer E, Rozov M, et al. Fluorouracil overdose: clinical manifestations and comprehensive management during and after hospitalization. J Hematol Oncol Pharm. 2015;5:43–47. [Google Scholar]
- 3. ISMP Canada . Fluorouracil error ends tragically, but application of lessons learned will save lives. ISMP Med Saf Alert. 2007;12:1806–1809. [Google Scholar]
- 4. Fyhr A, Akselsson R. Characteristics of medication errors with parenteral cytotoxic drugs. Eur J Cancer Care. 2012;21:606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cohen MR, Smetzer JL. Alteplase and tenecteplase confusion; lack of E‐prescribing interoperability leads to double dosing; accidental overdoses involving fluorouracil infusions. Hosp Pharm. 2015;50:849–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meulendijks D, van Hasselt JG, Huitema AD, et al. Renal function, body surface area, and age are associated with risk of early‐onset fluoropyrimidine‐associated toxicity in patients treated with capecitabine‐based anticancer regimens in daily clinical care. Eur J Cancer. 2016;54:120–130. [DOI] [PubMed] [Google Scholar]
- 7. Amstutz U, Froehlich TK, Largiader CR. Dihydropyrimidine dehydrogenase gene as a major predictor of severe 5‐fluorouracil toxicity. Pharmacogenomics. 2011;12:1321–1336. [DOI] [PubMed] [Google Scholar]
- 8. Van Kuilenburg AB, Haasjes J, Van Lenthe H, et al. Dihydropyrimidine dehydrogenase deficiency and 5‐fluorouracil associated toxicity In: Zoref‐Shani E, Sperling O, eds. Purine and Pyrimidine Metabolism in Man X. New York, NY: Springer; 2002:251–255. [DOI] [PubMed] [Google Scholar]
- 9. Ichikawa W, Takahashi T, Suto K, et al. Orotate phosphoribosyltransferase gene polymorphism predicts toxicity in patients treated with bolus 5‐fluorouracil regimen. Clin Cancer Res. 2006;12:3928–3934. [DOI] [PubMed] [Google Scholar]
- 10. Saif MW, Seller S, Diasio RB. Atypical toxicity associated with 5‐fluororacil in a DPD‐deficient patient with pancreatic cancer. Is ethnicity a risk factor? JOP. 2008;9:226–229. [PubMed] [Google Scholar]
- 11. van Kuilenburg AB, Muller EW, Haasjes J, et al. Lethal outcome of a patient with a complete dihydropyrimidine dehydrogenase (DPD) deficiency after administration of 5‐fluorouracil frequency of the common IVS14 + 1G > A mutation causing DPD deficiency. Clin Cancer Res. 2001;7:1149–1153. [PubMed] [Google Scholar]
- 12. Tsalic M, Bar‐Sela G, Beny A, et al. Severe toxicity related to the 5‐fluorouracil/leucovorin combination (the Mayo Clinic regimen): a prospective study in colorectal cancer patients. Am J Clin Oncol. 2003;26:103–106. [DOI] [PubMed] [Google Scholar]
- 13. Andre T, Boni C, Mounedji‐Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med. 2004;350:2343–2351. [DOI] [PubMed] [Google Scholar]
- 14. Cordier PY, Nau A, Ciccolini J, et al. 5‐FU‐induced neurotoxicity in cancer patients with profound DPD deficiency syndrome: a report of two cases. Cancer Chemother Pharmacol. 2011;68:823–826. [DOI] [PubMed] [Google Scholar]
- 15. Ducreux M, Bennouna J, Hebbar M, et al. Capecitabine plus oxaliplatin (XELOX) versus 5‐fluorouracil/leucovorin plus oxaliplatin (FOLFOX‐6) as first‐line treatment for metastatic colorectal cancer. Int J Cancer. 2011;128:682–690. [DOI] [PubMed] [Google Scholar]
- 16. Kuehr T, Ruff P, Rapoport BL, et al. Phase I/II study of first‐line irinotecan combined with 5‐fluorouracil and folinic acid Mayo Clinic schedule in patients with advanced colorectal cancer. BMC Cancer. 2004;4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mercier C, Ciccolini J. Severe or lethal toxicities upon capecitabine intake: is DPYD genetic polymorphism the ideal culprit? Trends Pharmacol Sci. 2007;28:597–598. [DOI] [PubMed] [Google Scholar]
- 18. Rothenberg ML, Meropol NJ, Poplin EA, et al. Mortality associated with irinotecan plus bolus fluorouracil/leucovorin: summary findings of an independent panel. J Clin Oncol. 2001;19:3801–3807. [DOI] [PubMed] [Google Scholar]
- 19. Sharif S, O'Connell MJ, Yothers G, et al. FOLFOX and FLOX regimens for the adjuvant treatment of resected stage II and III colon cancer. Cancer Invest. 2008;26:956–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tournigand C, Andre T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol. 2004;22:229–237. [DOI] [PubMed] [Google Scholar]
- 21. Schmoll HJ, Cunningham D, Sobrero A, et al. Cediranib with mFOLFOX6 versus bevacizumab with mFOLFOX6 as first‐line treatment for patients with advanced colorectal cancer: a double‐blind, randomized phase III study (HORIZON III). J Clin Oncol. 2012;30:3588–3595. [DOI] [PubMed] [Google Scholar]
- 22. Ezzeldin H, Diasio R. Dihydropyrimidine dehydrogenase deficiency, a pharmacogenetic syndrome associated with potentially life‐threatening toxicity following 5‐fluorouracil administration. Clin Colorectal Cancer. 2004;4:181–189. [DOI] [PubMed] [Google Scholar]
- 23. Johnson MR, Diasio R. Importance of dihydropyrimidine dehydrogenase (DPYD) deficiency in patients exhibiting toxicity following 5‐fluorouracil administration. Adv Enzyme Regul. 2001;41:151–157. [DOI] [PubMed] [Google Scholar]
- 24. van Kuilenburg AB, Haasjes J, Meinsma R, et al. Dihydropyrimidine dehydrogenase (DPD) deficiency: novel mutations in the DPD gene. Adv Exp Med Biol. 2000;486:247–250. [DOI] [PubMed] [Google Scholar]
- 25. Peters GJ, van Groeningen CJ, van der Wilt CL, et al. Time course of inhibition of thymidylate synthase in patients treated with fluorouracil and leucovorin. Semin Oncol. 1992;19:26–35. [PubMed] [Google Scholar]
- 26. Seiter K, Kemeny N, Martin D, et al. Uridine allows dose escalation of 5‐fluorouracil when given with N‐phosphonacetyl‐L‐aspartate, methotrexate, and leucovorin. Cancer. 1993;71:1875–1881. [DOI] [PubMed] [Google Scholar]
- 27. Klubes P, Cerna I, Meldon MA. Uridine rescue from the lethal toxicity of 5‐fluorouracil in mice. Cancer Chemother Pharmacol. 1982;8:17–21. [DOI] [PubMed] [Google Scholar]
- 28. Martin DS, Stolfi RL, Sawyer RC, Spiegelman S, Young CW. High‐dose 5‐fluorouracil with delayed “rescue” in mice. Cancer Res. 1982;42:3964–3970. [PubMed] [Google Scholar]
- 29. Peters G, Van Dijk J, Laurensse E, et al. In vitro biochemical and in vivo biological studies of the uridine ‘rescue’ of 5‐fluorouracil. Br J Cancer. 1988;57:259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van Groeningen C, Peters G, Pinedo H. Reversal of 5‐fluorouracil‐induced toxicity by oral administration of uridine. Ann Oncol. 1993;4:317–320. [DOI] [PubMed] [Google Scholar]
- 31. van Groeningen CJ, Peters GJ, Pinedo HM. Modulation of fluorouracil toxicity with uridine. Semin Oncol. 1992;19(suppl 3):148–154. [PubMed] [Google Scholar]
- 32. van Groeningen CJ, Pinedo HM, Heddes J, et al. Pharmacokinetics of 5‐fluorouracil assessed with a sensitive mass spectrometric method in patients on a dose escalation schedule. Cancer Res. 1988;48:6956–6961. [PubMed] [Google Scholar]
- 33. Longley D, Harkin DP, Johnston PG. 5‐Fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. [DOI] [PubMed] [Google Scholar]
- 34. Ashour OM, Naguib FN, Panzica RP, et al. Modulation of 5‐fluorouracil host toxicity by 5‐(benzyloxybenzyl) barbituric acid acyclonucleoside, a uridine phosphorylase inhibitor, and 2′, 3′, 5′‐tri‐O‐acetyluridine, a prodrug of uridine. Biochem Pharmacol. 2000;60:427–431. [DOI] [PubMed] [Google Scholar]
- 35. Martin DS, Stolfi RL, Sawyer RC. Use of oral uridine as a substitute for parenteral rescue of 5‐fluorouracil therapy, with and without the uridine phosphorylase inhibitor 5‐benzylacyclouridine. Cancer Chemother Pharmacol. 1989;24:9–14. [DOI] [PubMed] [Google Scholar]
- 36. Weinberg ME, Roman MC, Jacob P, et al. Enhanced uridine bioavailability following administration of a triacetyluridine‐rich nutritional supplement. PLoS One. 2011;6:e14709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saif MW, von Borstel R. 5‐Fluorouracil dose escalation enabled with PN401 (triacetyluridine): toxicity reduction and increased antitumor activity in mice. Cancer Chemother Pharmacol. 2006;58:136–142. [DOI] [PubMed] [Google Scholar]
- 38. Bamat M, Tremmel R, Helton J, et al. Clinical experience with uridine triacetate for 5‐fluorouracil overexposure: an update. Ann Oncol. 2013;24:iv71 [Google Scholar]
- 39. Bamat M, Tremmel R, O'Neil J, et al. Uridine triacetate: an orally administered, life‐saving antidote for 5‐FU overdose [abstract 9084]. J Clin Oncol. 2010;28(suppl):15s. [Google Scholar]
- 40. von Borstel R, O'Neil J, Saydoff J, et al. Uridine triacetate for lethal 5‐FU toxicity due to dihydropyrimidine dehydrogenase (DPD) deficiency [abstract]. J Clin Oncol. 2010;28(suppl):e13505. [Google Scholar]
- 41. Kelsen DP, Martin D, O'Neil J, et al. Phase I trial of PN401, an oral prodrug of uridine, to prevent toxicity from fluorouracil in patients with advanced cancer. J Clin Oncol. 1997;15:1511–1517. [DOI] [PubMed] [Google Scholar]
- 42. Wellstat Therapeutics Corporation . Vistogard approved prescribing information 2015. https://www.vistogard.com/Vistogard/media/Main-Media/final-labeling-text-vistogard.pdf.
- 43. JA Ajani, JR Hecht, L Ho, et al. An open‐label, multinational, multi‐center study of G17DT vaccination combined with cisplatin and 5‐fluorouracil in patients with untreated, advanced gastric or gastroesophageal cancer: the GC4 study. Cancer. 2006;106:1908–1916. [DOI] [PubMed] [Google Scholar]
- 44. Rodriguez RU. Public teleconference regarding licensing and collaborative research opportunities for: methods and compositions relating to dihydropyrimidine dehydrogenase (DPD). Fed Reg. 2008;73:38233. [Google Scholar]
- 45. Bajetta E, Floriani I, Di Bartolomeo M, et al. Randomized trial on adjuvant treatment with FOLFIRI followed by docetaxel and cisplatin versus 5‐fluorouracil and folinic acid for radically resected gastric cancer. Ann Oncol. 2014;25:1373–1378. [DOI] [PubMed] [Google Scholar]
- 46. Meulendijks D, Henricks LM, Sonke GS, et al. Clinical relevance of DPYD variants c. 1679T > G, c. 1236G > A/HapB3, and c. 1601G > A as predictors of severe fluoropyrimidine‐associated toxicity: a systematic review and meta‐analysis of individual patient data. Lancet Oncol. 2015;16:1639–1650. [DOI] [PubMed] [Google Scholar]
- 47. Gusella M, Bertolaso L, Bolzonella C, et al. Frequency of uridine monophosphate synthase Gly213Ala polymorphism in Caucasian gastrointestinal cancer patients and healthy subjects, investigated by means of new, rapid genotyping assays. Genet Test Mol Biomarkers. 2011;15:691–695. [DOI] [PubMed] [Google Scholar]
- 48. Schwab M, Zanger UM, Marx C, et al. Role of genetic and nongenetic factors for fluorouracil treatment–related severe toxicity: a prospective clinical trial by the German 5‐FU Toxicity Study Group. J Clin Oncol. 2008;26:2131–2138. [DOI] [PubMed] [Google Scholar]
- 49. Tsibiribi P, Descotes J, Lombard‐Bohas C, et al. Cardiotoxicity of 5‐fluorouracil in 1350 patients with no prior history of heart disease. Bull Cancer. 2006;93:10027–10030. [PubMed] [Google Scholar]
- 50. Polk A, Vaage‐Nilsen M, Vistisen K, et al. Cardiotoxicity in cancer patients treated with 5‐fluorouracil or capecitabine: a systematic review of incidence, manifestations and predisposing factors. Cancer Treat Rev. 2013;39:974–984. [DOI] [PubMed] [Google Scholar]
- 51. Sorrentino MF, Kim J, Foderaro AE, et al. 5‐Fluorouracil induced cardiotoxicity: review of the literature. Cardiol J. 2012;19:453–458. [DOI] [PubMed] [Google Scholar]
- 52. Ison G, Beaver JA, McGuinn D, et al. FDA approval: uridine triacetate for the treatment of patients following fluorouracil or capecitabine overdose or exhibiting early‐onset severe toxicities following administration of these drugs. Clin Cancer Res. Epub ahead of print. July 2016. [DOI] [PubMed] [Google Scholar]
- 53. Mercier C, Ciccolini J. Profiling dihydropyrimidine dehydrogenase deficiency in patients with cancer undergoing 5‐fluorouracil/capecitabine therapy. Clin Colorectal Cancer. 2006;6:288–296. [DOI] [PubMed] [Google Scholar]
- 54. Doroshow JH, McCoy S, Macdonald JS, et al. Phase II trial of PN401, 5‐FU, and leucovorin in unresectable or metastatic adenocarcinoma of the stomach: a Southwest Oncology Group study. Invest New Drugs. 2006;24:537–542. [DOI] [PubMed] [Google Scholar]
- 55. Codacci‐Pisanelli G, Van der Wilt C, Pinedo H, et al. Antitumour activity, toxicity and inhibition of thymidylate synthase of prolonged administration of 5‐fluorouracil in mice. Eur J Cancer. 1995;31:1517–1525. [DOI] [PubMed] [Google Scholar]
- 56. Levy E, Piedbois P, Buyse M, et al. Toxicity of fluorouracil in patients with advanced colorectal cancer: effect of administration schedule and prognostic factors. J Clin Oncol. 1998;16:3537–3541. [DOI] [PubMed] [Google Scholar]
- 57. Mazzuca F, Borro M, Botticelli A, et al. Pre‐treatment evaluation of 5‐fluorouracil degradation rate: association of poor and ultra‐rapid metabolism with severe toxicity in a colorectal cancer patients cohort. Oncotarget. 2016;7:20612–20620. [DOI] [PMC free article] [PubMed] [Google Scholar]