

Summary
The Chinese Taihu pig breeds are an invaluable component of the world's pig genetic resources, and they are the most prolific breeds of swine in the world. In this study, the genomes of 252 pigs of the six indigenous breeds in the Taihu Lake region were sequenced using the genotyping by genome reducing and sequencing approach. A total of 950 million good reads were obtained using an Illumina Hiseq2000 at an average depth of 13× (for SNP calling) and an average coverage of 2.3%. In total, 122 632 indels, 31 444 insertions, 44 056 deletions and 455 CNVs (copy number variants) were identified in the genomes of the pigs. Approximately 2.3% of these genetic markers were mapped to gene exon regions, and 25% were in QTL regions related to economically important traits. The KEGG pathway or GO enrichment analyses revealed that genetic variants assumed to be large‐effect mutations were significantly overrepresented in 22 SNP, 56 indel, 26 insertion, 28 deletion and three CNV gene sets. A total of 343 breed‐specific SNPs were also identified in the six Chinese indigenous pigs. The findings from this study can contribute to future investigations of the genetic diversity, population structure, positive selection signals and molecular evolutionary history of these pigs at the genome level and can serve as a valuable reference for improving the breeding and cultivation of these pigs.
Keywords: breed‐specific SNPs, Chinese indigenous pigs, genome sequencing, GGRS
Introduction
The Chinese Taihu pig breeds are the most prolific breeds of swine in the world. They are distributed mainly in a narrow region with a mild sub‐tropical climate in the Taihu Lake region in the lower Yangtze River Valley of China. They are currently classified into six breeds (Meishan, Fengjing, Shawutou, Erhualian, Jiaxing Black and Mizhu) according to the most recently reported classification of Chinese indigenous swine breeds (China National Commission of Animal Genetic Resources 2011), and the Meishan breed can also be subdivided into two types (Small Meishan and Middle Meishan) (Zhang 1991). Studies, such as genome‐wide association studies (GWAS) (Ai et al. 2014; Jung et al. 2014; Zhang et al. 2014; Xiong et al. 2015) and selection signature studies (Rubin et al. 2012; Li et al. 2014; Wang et al. 2014; Moon et al. 2015), are now being widely performed on pigs to investigate the genetic mechanisms of complex traits at the genome level. However, the prerequisite for conducting those studies is to determine genome‐wide genetic markers. Additionally, surveying for genetic variation across genomes can be quite valuable for investigating genetic diversity and population structure (Ai et al. 2013). Therefore, surveying for genetic variation can contribute to further research on the molecular mechanisms of pig evolution and domestication, and the findings of such a survey can serve as a valuable reference for improving the breeding and cultivation of Chinese Taihu pig breeds (Li et al. 2013; Ai et al. 2015).
In our previous study, we identified genome‐wide SNPs and investigated the genetic diversity and population structure (such as PCA, NJ‐tree, structure, FST etc.) of Taihu pig breeds (Wang et al. 2015). However, an original genome‐wide survey of genetic variants other than SNPs (such as indels, insertions, deletions and CNVs) in Chinese Taihu pig breeds has not yet been conducted. For example, indels are one of the main forms of genomic variation and have received increasing attention; with the rapid advancements in sequencing technology, considerable progress has been made in the identification of indels (Mullaney et al. 2010; Li et al. 2014; Yan et al. 2014). Indels have been reported to cause many common human diseases such as cystic fibrosis (Collins et al. 1987) and Huntington's disease (Ashley & Warren 1995). Indels have also been found to be responsible for a number of traits and diseases of domestic animals such as plumage colour (Kerje et al. 2004) and sex‐linked dwarfism (Agarwal et al. 1994) in chicken and carcass traits (He et al. 2010) and the double muscle trait (Grobet et al. 1997) in cattle. Studies in pigs have shown that an intronic insertion in KPL2 results in the immotile short‐tail sperm defect (Sironen et al. 2006) and that a 51‐bp insertion in the TEX14 gene causes an infertility defect. Those studies indicated that other types of genomic variations, such as indels, have important effects on traits. Therefore, genetic variants other than SNPs, such as indels, should be studied in conjunction with SNPs to reveal the associations between genes and traits and to accelerate the identification of causative mutations. Furthermore, the characteristics of genetic variations (including SNPs) in Chinese Taihu pig breeds are still unknown.
Therefore, the objective of this study was to identify and characterize genome‐wide genetic variations (including SNPs, indels, insertions, deletions and CNVs) in Chinese Taihu pig breeds using next generation sequencing (NGS) technology (i.e. GGRS: genotyping by genome reducing and sequencing).
Materials and methods
DNA sample collection and sequencing data preparation
A total of 252 samples from six Chinese pig breeds (Meishan, Fengjing, Shawutou, Erhualian, Jiaxing Black and Mizhu) were obtained. Detailed information about the samples as well as the methods of acquiring the raw Illumina DNA sequence data were reported in our previous study (Wang et al. 2015). We aligned the reads to the pig reference genome (SGSC Sscrofa10.2, http://hgdownload.soe.ucsc.edu/goldenPath/susScr3/bigZips/) using burrows‐wheeler aligner (bwa ver 0.7.5) (Li & Durbin 2009) with the default settings and the steps of the GGRS approach.
Genetic variation discovery
A total of 105 550 SNPs were detected in our previous study; thus, in this report we describe only the criteria of the other four types of genetic variants: indels, insertions, deletions and CNVs. The alignment results with mapping quality scores of <20 were filtered before the variants were called. The samtools (ver 0.1.19) (Li et al. 2009) mpileup algorithm was used to call indels (short deletions and insertions), and only those with calling quality scores of more than 20 and lengths more than 100 bp that were identified in more than five samples were retained. The cn.MOPS algorithm (ver 1.12.0, with mode = unpaired and normType = mean) was used to detect CNVs (Klambauer et al. 2012), and only those with a length of more than 1 kb that were identified in at least one sample and were not of the amphiploid form were retained for further analysis. To identify long insertions and deletions, we performed delly (ver 0.5.9) (Rausch et al. 2012) using paired mapping data and the following criteria: more than three paired‐end supports, a length between 300 bp and 1000 bp, and a MAPQ (mapping quality) equal to or greater than 30.
Identification of breed‐specific SNPs
Each SNP that passed the applied filtering criteria was analysed according to the information about the breed. A SNP was labelled as breed specific (Ramos et al. 2011) when it was detected in only one of the seven populations.
Function annotation
The Ensembl pig gene annotation set (Ensembl release 78) was downloaded from the Ensembl website (ftp://ftp.ensembl.org/pub/release-78/gtf/sus_scrofa/) (Flicek et al. 2013). Variants located in start and stop codons and variants that caused frameshifts in exons were defined as large‐effect mutations. The Gene Ontology (GO) categories and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichments for genes containing large‐effect mutations were determined using the DAVID tool (ver 6.7) (Dennis et al. 2003; Huang da et al. 2009). Because the GO annotation of the pig is incomplete, corresponding human orthologous Ensembl IDs were retrieved to determine enrichment function categories using a PERL script. The enriched GO terms and pathways with P‐values < 0.05 after correction for multiple testing [with a false discovery rate (FDR) of <25%] were considered to be statistically significant. Additionally, the pig QTL database (Animal QTL release 25) was downloaded from the Animal QTL database website (http://www.animalgenome.org/cgi-bin/QTLdb/GG/index, updated Dec 29, 2014) (Hu et al. 2013). Although the current release of the pig QTL database contains 12 618 QTLs, not all of them are suitable for analysis because the length of some are too large for efficient post‐processing. We removed QTLs with lengths >1 Mb and, when two or more QTLs overlapped by more than 50%, we merged them into one larger QTL. The merged QTLs were considered to be related to a trait if the origin trait type occurred one time (thus, the newly defined QTL might be related to several types of traits). The PERL script was used to conduct the above QTL‐based annotations.
Data availability
All BAM data were deposited in the NCBI Sequence Read Achieve (SRA) under the Bioproject number PRJNA281578. The experiment number for the 252 pigs is SRX999959. Indel data have been submitted to dbSNP of NCBI (NCBI_ss1966414102 to ss1966531374) and insertions and deletions to dbVar of NCBI. Detailed information about the variants is provided in Table S1. Phenotypic records related to reproduction and pedigree information have been uploaded to our website (http://klab.sjtu.edu.cn/pigbreeds/).
Results
Sequencing and mapping summary
A total of 950 million good reads were generated by the Illumina HiSeq2000. After removing the primer/adaptor contaminated reads, the average phred score for each position was >20 (Figs S1a and S1b provide an example of the sequencing quality of a library). On average, approximately 3.8 million good reads were generated for each individual (Table S2), and the number of average reads for individuals within a population ranged from 2.8 million (Jiaxing Black) to 4.4 million (Shawutou, Fig. S1c). Information about the coverage and depth of the raw good reads for each individual are shown in Table S2. The average depth of each population for SNP calling ranged from 8.8× to 23.5×, with an average of 13× (Table 1). The average coverage of each population ranged from 1.2% to 2.9%, with an average of 2.3% (Table 1).
Table 1.
Summary statistics for the sequencing data
| Breed | Meishan | Erhualian | Mi | Fengjing | Shawutou | Jiaxing Black | Overall | Average | |
|---|---|---|---|---|---|---|---|---|---|
| Middle | Small | ||||||||
| Number | 50 | 69 | 31 | 36 | 16 | 21 | 29 | 252 | – |
| Average genome coverage (%) | 2.0 | 2.9 | 1.7 | 2.7 | 1.2 | 1.8 | 2.9 | – | 2.3 |
| Average sequencing deptha | 11.1 | 10.5 | 23.5 | 8.8 | 21.2 | 20.4 | 9.4 | – | 13.0 |
| Breed‐specific SNPsb | 38 | 54 | 7 | 88 | 4 | 1 | 151 | 343 | – |
Sequencing data used for ultimate SNP identification.
Number of identified bred‐specific SNPs of the 105 550 overall SNPs.
Discovery of variants and their genomic distributions
A total of 122 632 indels, 31 444 insertions, 44 056 deletions and 455 CNVs were identified in Chinese indigenous pigs in the Taihu Lake region according to the above criteria (see Materials and methods). The distances between all pairs of adjacent SNPs are shown in Fig. 1a. The lengths of the indels detected in this study ranged from 1 to 5 bp, and the dominant indel length was a single base pair, accounting for 86.56% of all detected indels. The number of gain events detected was greater than the number of loss events detected (Fig. 1b).
Figure 1.
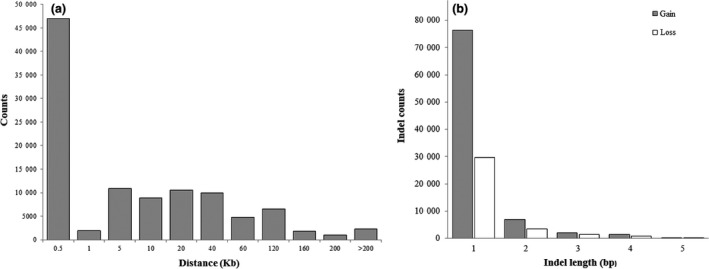
(a) Distance distribution of adjacent SNPs and (b) distribution of indels of different lengths.
The results of the density distribution analysis for each chromosome showed that the SNPs were distributed in a non‐uniform fashion (P < 0.001, chi‐square test), whereas the indels, insertions, deletions and CNVs were distributed in a uniform fashion (P > 0.998, chi‐square test). Chromosomes 18, 12 and 11 had the highest SNP densities, and chromosome 12 had the highest indel density (Fig. 2). The two sex chromosomes had the lowest SNP and indel densities (Fig. 2).
Figure 2.
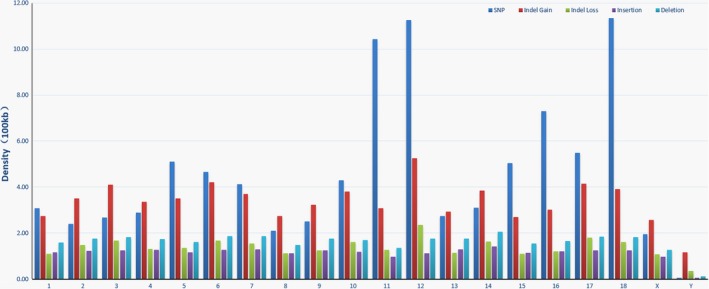
Distribution of the density of different types of genetic variants across chromosomes calculated as the number of variants per 100 kb.
To further explore the distributions of variants in genic regions, we annotated all detected variants using the Ensembl gene set (containing 25 332 genes). The genomic location (intergenic, start or stop codon, exonic, intronic, or untranslated region) and functional role (frameshifting or non‐frameshifting) for each variant were determined. In total, 37 484 (35.51%) SNPs (related to 7133 genes), 47 764 (38.95%) indels (related to 10 414 genes), 12 436 (39.54%) insertions (related to 4818 genes), 8645 (19.62%) deletions (related to 6071 genes) and 168 (36.92%) CNVs (related to 189 genes) were mapped to genic regions (Table 2). The distributions of the variants in each type of genomic location are shown in Table 3. Regarding the potential roles of indels in exons, the majority of them (3276, 98.85%) were non‐triplet; thus, they were predicted to cause frameshift mutations. The remaining 38 indels were triplet (non‐frameshifting). The indels in exons were located in numerous functional genes (2743, 10.83%), many of which (736, 26.83%) contained two or more exonic indels (Table S3).
Table 2.
Number distribution of variants detected in each chromosome
| Chr | SNP | Indel | Insertion | Deletion | CNV | Genes | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n a | Genes | Ratio (%) | Gain | Loss | Genes | Ratio (%) | n a | Genes | Ratio (%) | n a | Genes | Ratio (%) | NO. | Genes | Ratio (%) | n b | |
| 1 | 3632 | 662 | 30.00 | 3323 | 1321 | 1023 | 46.30 | 1420 | 516 | 23.36 | 1013 | 639 | 28.93 | 20 | 22 | 1.00 | 2209 |
| 2 | 2857 | 640 | 29.80 | 2284 | 922 | 904 | 42.10 | 859 | 361 | 16.81 | 575 | 491 | 22.87 | 10 | 11 | 0.51 | 2147 |
| 3 | 2065 | 482 | 33.40 | 2072 | 789 | 727 | 50.30 | 709 | 295 | 20.43 | 471 | 385 | 26.66 | 7 | 8 | 0.55 | 1444 |
| 4 | 1427 | 389 | 31.00 | 1430 | 550 | 547 | 43.60 | 637 | 266 | 21.21 | 456 | 335 | 26.71 | 9 | 10 | 0.80 | 1254 |
| 5 | 4764 | 369 | 31.50 | 3907 | 1507 | 540 | 46.10 | 576 | 239 | 20.39 | 411 | 290 | 24.74 | 8 | 9 | 0.77 | 1172 |
| 6 | 2490 | 635 | 32.70 | 2342 | 897 | 944 | 48.60 | 884 | 349 | 17.98 | 591 | 480 | 24.73 | 13 | 13 | 0.67 | 1941 |
| 7 | 2191 | 487 | 29.50 | 1785 | 769 | 632 | 38.30 | 771 | 280 | 16.96 | 526 | 358 | 21.68 | 11 | 12 | 0.73 | 1651 |
| 8 | 918 | 247 | 29.20 | 1161 | 442 | 422 | 49.80 | 518 | 227 | 26.80 | 390 | 269 | 31.76 | 8 | 9 | 1.06 | 847 |
| 9 | 2053 | 397 | 27.80 | 1701 | 668 | 560 | 39.20 | 738 | 293 | 20.53 | 540 | 353 | 24.74 | 6 | 6 | 0.42 | 1427 |
| 10 | 898 | 185 | 35.30 | 778 | 308 | 254 | 48.50 | 324 | 130 | 24.81 | 225 | 152 | 29.01 | 8 | 9 | 1.72 | 524 |
| 11 | 695 | 156 | 37.00 | 533 | 266 | 208 | 49.30 | 222 | 99 | 23.46 | 149 | 128 | 30.33 | 2 | 2 | 0.47 | 422 |
| 12 | 1408 | 392 | 34.20 | 1337 | 555 | 559 | 48.80 | 409 | 183 | 15.98 | 266 | 252 | 22.01 | 6 | 10 | 0.87 | 1145 |
| 13 | 1718 | 479 | 31.20 | 2045 | 804 | 717 | 46.70 | 1051 | 396 | 25.81 | 733 | 487 | 31.75 | 12 | 12 | 0.78 | 1534 |
| 14 | 2627 | 514 | 37.50 | 2369 | 963 | 717 | 52.30 | 1103 | 380 | 27.74 | 746 | 471 | 34.38 | 10 | 11 | 0.80 | 1370 |
| 15 | 1519 | 296 | 30.80 | 1228 | 532 | 445 | 46.40 | 562 | 221 | 23.02 | 398 | 266 | 27.71 | 10 | 11 | 1.15 | 960 |
| 16 | 642 | 154 | 34.70 | 642 | 285 | 225 | 50.70 | 339 | 122 | 27.48 | 234 | 146 | 32.88 | 4 | 4 | 0.90 | 444 |
| 17 | 3817 | 265 | 38.50 | 2892 | 1250 | 334 | 48.50 | 390 | 149 | 21.66 | 251 | 196 | 28.49 | 5 | 5 | 0.73 | 688 |
| 18 | 1023 | 195 | 38.10 | 817 | 315 | 248 | 48.40 | 352 | 121 | 23.63 | 241 | 147 | 28.71 | 2 | 3 | 0.59 | 512 |
| X | 739 | 169 | 15.10 | 1366 | 584 | 405 | 36.20 | 572 | 191 | 17.05 | 429 | 226 | 20.18 | 17 | 22 | 1.96 | 1120 |
| Y | 1 | 0 | 0.00 | 19 | 6 | 3 | 23.10 | 0 | 0 | 0.00 | 0 | 0 | 0.00 | 0 | 0 | 0.00 | 13 |
| Total | 37 484 | 7113 | 31.20 | 34 031 | 13 733 | 10 414 | 45.60 | 12 436 | 4818 | 21.11 | 8645 | 6071 | 26.60 | 168 | 189 | 0.83 | 22 824 |
Number of identified variants within genic regions.
Number of total genes contained in the Ensembl gene database of pig.
Table 3.
Statistics of variants in functional regions
| Category | SNP (%) | Indel (%) | Insertion (%) | Deletion (%) |
|---|---|---|---|---|
| Intergenic | 68 066 (64.5) | 74 868 (61.1) | 22 799 (72.5) | 31 620 (71.8) |
| Start/End codon | 4 (0.0) | 7 (0.0) | 64 (0.2) | 99 (0.2) |
| Exonic | 2400 (2.3) | 3314 (2.7) | 672 (2.1) | 1007 (2.3) |
| Intronic | 20 249 (19.2) | 25 398 (20.7) | 3344 (10.6) | 4870 (11.1) |
| Untranslated region | 14 831 (14.1) | 19 045 (15.5) | 4565 (14.5) | 6460 (14.7) |
| Total | 105 550 | 122 632 | 31 444 | 44 056 |
Breed‐specific SNPs and population structure
Among the 105 550 SNPs identified, a total of 343 SNPs were found to be putatively breed specific (Table 1), meaning that one of the alleles was present in only one of the six populations studied. SNPs specific to Jiaxing Black were the most abundant, whereas Shawutou had the fewest number of breed‐specific SNPs. We also investigated the genomic location distributions of the breed‐specific SNPs and found that the majority of them (n = 210, 61.2%) were located in intergenic regions and that four (1.2%) were located in exons, 92 (26.8%) in introns and 37 (10.8%) in untranslated regions.
We also investigated the differences in the breed‐specific SNP distributions between all breed pairs. The numbers of putatively breed‐specific SNPs identified when comparing two breeds ranged from 15 407 (Mi and Erhualian) to 32 586 (Fengjing and Small Meishan) (Table S4). The numbers of putatively breed‐specific SNPs identified were the highest when comparing the Fengjing breed to any of the other breeds. This finding indicates that the Fengjing breed has genetic distances from all other breeds greater than the genetic distances between any two of the other six breeds.
To further explore the population structure based on the SNPs in different genic regions, we conducted principal components analysis (PCA) using gcta (ver 1.24) (Yang et al. 2011). PCA revealed that, with the exception of the Erhualian and Mi breeds, the first two PC axes could clearly distinguish the pigs from each breed (including the two subdivisions of the Meishan, i.e. the Small Meishan and Middle Meishan) (Figs. 3a,b). This finding indicates that the Mi and Erhualian pigs are closely related. Interestingly, the SNPs in the genic regions (Fig. 3a, including 37 484 SNPs) are more likely to distinguish the Jiaxing Black and Fengjing breeds in comparing the SNPs in the intergenic regions (Fig. 3b, including 68 066 SNPs).
Figure 3.
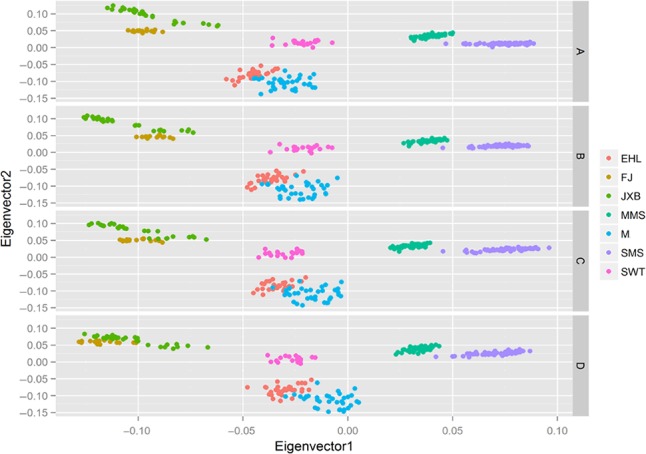
Population structures of Chinese indigenous pigs in the Taihu Lake region revealed by principal components analysis based on (a) the dataset of the SNPs in genic regions, (b) the dataset of the SNPs in the intergenic regions, (c) the dataset of the SNPs in the QTL regions related to meat and carcass quality traits, (d) the dataset of the SNPs in the QTL regions related to reproduction traits. MMS, Middle Meishan; SWT, Shawutou; EHL, Erhualian; M, Mi; FJ, Fengjing; JXB, Jiaxing Black; SMS, Small Meishan.
Minor allele frequency distribution
First, we investigated the minor allele frequency (MAF) distribution of SNPs in the entire population of Taihu pig breeds. The distribution of the observed SNPs was skewed toward an intermediate frequency (MAF = 0.05–0.20, 67.04%, Fig. S2). Then, we surveyed the MAF distributions in each population and found that they were skewed toward lower frequencies (MAF < 0.05, range: 28.3–41.4%, Fig. S3).
Gene enrichment and function annotation
The KEGG pathway and GO analyses were performed on 1381 genes whose exons contained SNPs, 2727 genes whose exons contained frameshift indels, 715 genes whose exons contained insertions, 1046 genes whose exons contained deletions and 189 genes that overlapped with CNVs, and we assumed these genetic variants to be large‐effect mutations. The KEGG pathway analysis revealed that the genes were significantly overrepresented in four, nine, three, two and zero pathways as related to SNPs, indels, insertions, deletions and CNVs respectively after FDR correction and that one pathway (hsa04510: focal adhesion) was significant in SNP and indel mutations (Table S5). The GO analysis results showed that 18, 47, 23, 26 and three GO terms were significant as related to SNPs, indels, insertions, deletions and CNVs respectively. The genes with SNPs were significantly enriched in the molecular functions of molecule binding (such as ion, tetrapyrrole, oxygen and carbohydrate binding) and as extracellular matrix structural constituents, whereas genes with indels were significantly enriched in a greater variety of molecular functions (such as molecule binding, transcription cofactor activity, enzyme activator activity, transcription activator activity and transmembrane transporter activity) than were genes with SNPs. Genes with insertions or deletions were significantly enriched in more types of molecular functions related to binding and enzyme activity.
We also investigated the distribution of variants in quantitative trait loci (QTL) regions (Table S6). Based on our filtering criteria, a total of 2031 non‐overlapping QTL regions were included in the analysis. In total, 25 414 (24.08%) SNPs, 30 890 (25.19%) indels, 7977 (25.37%) insertions, 11 308 (25.67%) deletions and 41 (9.01%) CNVs were located in the newly defined QTL regions. The number distribution of the variants and the related genes involved in five types of traits are shown in Table 4. Furthermore, we conducted PCA based on the SNPs in the QTL regions related to meat and carcass quality and to reproduction traits to explore the population structure. PCA revealed that the Jiaxing Black and Fengjing breeds are more closely clustered in the term of reproduction traits (Fig. 3d; including 8077 SNPs) than they are for meat and carcass quality traits (Fig. 3c; including 15 428 SNPs). This finding indicates that the relationship between the Jiaxing Black and Fengjing breeds are closer in terms of reproduction performance than in meat and carcass quality performance.
Table 4.
Distribution of SNPs and indels among different traits
| Trait | SNP | Indel | Insertion | Deletion | CNV | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n a | n b | Genesc | n a | n b | Genesc | n a | n b | Genesc | n a | n b | Genesc | n a | n b | Genesc | |
| Meat and carcass quality | 15 428 | 4309 | 1214 | 18 883 | 5856 | 1733 | 4950 | 1319 | 779 | 7044 | 1922 | 997 | 30 | 24 | 30 |
| Health | 2987 | 851 | 257 | 3807 | 1140 | 360 | 1023 | 280 | 165 | 1416 | 396 | 216 | 4 | 3 | 3 |
| Production | 658 | 1684 | 420 | 7675 | 2260 | 608 | 2012 | 542 | 300 | 2852 | 768 | 367 | 7 | 7 | 7 |
| Reproduction | 8077 | 357 | 78 | 9531 | 2876 | 795 | 2427 | 634 | 387 | 3451 | 943 | 492 | 13 | 11 | 13 |
| Exterior | 3876 | 946 | 263 | 4696 | 1348 | 383 | 1188 | 300 | 178 | 1718 | 152 | 220 | 12 | 11 | 11 |
Variants located in QTL regions.
Variants located in both gene and QTL regions.
Number of genes related to variants.
Discussion
In this study, we performed NGS on 252 Chinese Taihu pigs of six breeds to discover genetic variations and gain a comprehensive understanding of SNPs, indels, insertions, deletions and CNVs in their genomes. We surveyed the molecular and functional characteristics of five types of genetic variants, but we performed more analyses of SNPs than the other types of genetic variants.
Genetic variants discovery and NGS
To our knowledge, this is the first study to apply NGS (using the GGRS method with a design for sequencing a reduced representation of a genome) to the above‐mentioned pig breeds. The merits of the strategy of sequencing a reduced representation of a genome include discovering and genotyping hundreds of thousands of markers across the genome at a greatly reduced cost compared to whole‐genome resequencing and finding novel markers in the population being studied. Furthermore, this study was focused not only on identifying two common types of genetic variants (SNPs/indels) using NGS data but also on detecting other types of genetic variants (such as insertions, deletions, copy number variants and inversions). This helps to comprehensively reveal the genetic variation characteristics of the population being studied and to enrich the genetic variation database. NGS approaches (similar to GGRS approaches) are currently widely used with agricultural animals, especially in large sample studies (such as GWAS) because they obtain a sufficient number of markers at a reduced cost compared to whole‐genome resequencing.
Genotyping by genome reducing and sequencing confirmed that, although there are a few regional variations, in general, assigned reads for each individual are uniformly distributed across chromosomes (Chen et al. 2013). Therefore, when approximately 2% of an individual's genome is sequenced, the results reflect the genetic variation characteristics of the population being studied. The good reads (removing primer/adaptor contaminated reads) used for detecting variants had an average phred score of >20 (at each position), and the average depth for SNP calling was 13×, which allowed us to call variants with high confidence. Moreover, the accuracy of the detected structural variation can also reach a high confidence level by setting the least number of samples in which the same variant was detected (such as indel calling, for which the variant was detected in at least five samples) and the length of variants that can be reliably called by the used algorithm (such as the delly algorithm). For the pig population in this study, we identified and genotyped 105 550 SNPs, approximately 28% of which were newly discovered SNPs. This indicates that the identification of SNPs in pigs, or at least in Chinese indigenous pigs, is far from complete.
Molecular characteristics
To determine the genetic molecular characteristics of Chinese Taihu pig breeds, we surveyed the number and density distributions of variants across chromosomes, the MAF distributions of SNPs and putatively breed‐specific SNPs, and we found the following molecular characteristics. Firstly, the SNP density distributions were significantly non‐uniform across chromosomes, but those of the other types of genetic variants studied were uniform across chromosomes. Chromosome 12 was found to have higher densities of both SNPs and indels than the other chromosomes, which indicates that relatively high genetic variation exists in chromosome 12. Additionally, investigating known genes (Ensembl release 78) and QTLs (Animal QTL release 25) showed that chromosome 12 has the highest gene density (18/Mb) and a relatively higher QTL density (7/Mb, Top 3) than do the other chromosomes. These results suggest that other researchers should pay more attention to chromosome 12 when studying pigs in the future. Secondly, the MAF of SNPs was skewed toward intermediate frequencies in the Chinese Taihu pig breeds. Because the MAF can affect the statistical power of some methods, such as GWAS (Park et al. 2011), attention should be paid to selecting the proper method of MAF analysis based on allele frequency for future studies of Chinese Taihu pig breeds. Thirdly, the number of breed‐specific SNPs was small when all Taihu pig breeds were included in the analysis, but the numbers of breed‐specific SNPs identified when comparing two breeds to each other was large. These breed‐specific SNPs will help to classify pigs with unclear identities into breeds. Moreover, population structures of Chinese indigenous pigs in the Taihu Lake region revealed by PCA using the SNPs in different genic or QTL regions exhibited similar relationships among the breeds. The Erhualian and Mi, Fengjing and Jiaxing Black breeds were closely clustered. These observations are reasonable considering the assumption that the breeds with geographically close origins likely shared common ancestors and crossbred with each other.
Functional characteristics
We performed gene and QTL mapping as well as gene enrichment and functional annotation analyses to better understand the functions of these variants. Firstly, the proportions of each type of variant located in genic and QTL regions were similar. For example, most variants (64.5–72.5%) were located in intergenic regions. For all types of variants, approximately 2.3% were located in exons and nearly 25% were located in QTL regions. The results help us to understand that the proportions of each type of variants are evenly distributed in QTL regions and indicated that the proportions of each type of variants that might have a function in affecting the traits were similar, though they appeared with different forms in the genome.
Secondly, a large number of genes were affected by frameshifting indels. Some genes were associated with pig reproductive traits. For instance, RGS12 is a regulator of G‐proteins [important signalling molecules involved in a wide range of cell regulation activities, such as hormone signalling (Chatterjee & Fisher 2000)] and is thought to affect ovulation rate in swine (Campbell et al. 2003; Gladney et al. 2004). Dall'Olio et al. (2010) reported that the polymorphisms in the CXCL10 gene, which has a possible role in embryonic development and implantation (Kim et al. 2004), are suitable markers for association studies of litter size in Italian Large White pigs. We suggest that indels identified in the CXCL10 gene might be litter size markers. We also found many genes involved in immunity. For example, the IRF7 gene, which belongs to the interferon regulatory factors family, is a crucial regulator of type I interferons and is involved in resistance to pathogenic infections (Ning et al. 2011). Another example is the IFIT1 gene, encoding a molecule that functions both as a sensor and effector to inhibit the disease pathogenesis of several virus families (Diamond 2014). Interestingly, the results of the KEGG pathway analysis showed that genes containing indels were significantly overrepresented in pathways related to disease (such as hsa05200:pathways in cancer and hsa05222:pathways in small cell lung cancer), indicating that indels are likely associated with disease susceptibility. Previous studies have shown that Meishan pigs have higher immunity and disease resistance than do pigs of other breeds (Duchet‐Suchaux et al. 1991; Chen et al. 2010). These findings indicate that indels may have an effect on reproductive and immune response traits in these pigs. Therefore, indels are another common type of genetic variation and should be incorporated with SNPs and CNVs to reveal the associations between genes and traits to accelerate the identification of causative mutations.
Thirdly, the different types of variants may have different functions in the genome. The enrichment analysis for genes containing large‐effect mutations for each type of variant suggested that they are enriched in different GOs and pathways. SNPs were related mainly to adipose tissue. Indels were more related to disease. Both insertions and deletions were related to endocytosis, and CNVs were more related to nerve function. These findings might be the reason for the many outstanding economic traits of Chinese Taihu pig breeds, such as their delicious taste (which may be related to adipose tissue), high disease resistance and docile temperament (related to the nervous system).
In summary, we performed NGS on six Chinese indigenous pig breeds and discovered and identified a large number of variants across their genomes. The molecular and functional characteristics of these genetic variants were investigated. Many of the indels located in previously reported genes were related to reproductive and immune traits. Our results can be further explored in several other contexts, including investigations of genetic diversity, population structure, positive selection signals, molecular evolutionary history and the development of a national plan for the conservation and utilization of these breeds.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Authors′ contributions
Y. P. designed the study. Y. P. and Q. W. supervised the study. Z. W. analysed the data. Z. W. wrote the manuscript. Z. Z. implemented the method in the iblup software package. Q. C. developed the GGRS approach for outbred populations with the help of R. L, and X. Z. M. X., H. Y. and Y. Z. assisted with pig sample collection. All authors have read and edited the manuscript.
Supporting information
Figure S1 Sequencing data summary: (a) sequencing quality of the raw and filtered reads from R1 (5′), (b) sequencing quality of raw and filtered reads from R2 (3′), (c) distribution of the average good reads for individuals within a population.
Figure S2 Probability density of MAF.
Figure S3 Distribution of MAF across each breed.
Table S1 Detailed information of the detected variants including SNPs, indels, insertions, deletions and CNVs and their lengths and locations on chromosomes as well as the frequencies of alleles.
Table S2 Summary of number of good reads, depth and coverage for each sample.
Table S3 Indels in exon regions, their locations on chromosomes and the corresponding Ensembl gene ID and name.
Table S4 Breed‐specific SNP distributions when comparing two populations.
Table S5 Functional enrichment of genes with large‐effect mutation variants.
Table S6 Distribution of variants in QTL regions. Densities of four types of variants (including SNPs, indels, insertions and deletions) in each new defined QTL and the information of the newly defined QTL locations, raw QTL traits and variant counts and densities in the new QTLs.
Acknowledgements
This study was supported by the 2011–2015 Animal Germplasm Resources Conservation project of the Ministry of Agriculture of China, The National Natural Science Foundation of China (grant nos. 31472069, U1402266, 31370043, 31272414) and The National 948 Project of China (2012‐Z26, 2011‐G2A).
Contributor Information
Q. Wang, Email: wangqishan@sjtu.edu.cn
Y. Pan, Email: panyuchun1963@aliyun.com
References
- Agarwal S.K., Cogburn L.A. & Burnside J. (1994) Dysfunctional growth hormone receptor in a strain of sex‐linked dwarf chicken: evidence for a mutation in the intracellular domain. Journal of Endocrinology 142, 427–34. [DOI] [PubMed] [Google Scholar]
- Ai H., Huang L. & Ren J. (2013) Genetic diversity, linkage disequilibrium and selection signatures in Chinese and Western pigs revealed by genome‐wide SNP markers. PLoS One 8, e56001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai H., Xiao S., Zhang Z., Yang B., Li L., Guo Y., Lin G., Ren J. & Huang L. (2014) Three novel quantitative trait loci for skin thickness in swine identified by linkage and genome‐wide association studies. Animal Genetics 45, 524–33. [DOI] [PubMed] [Google Scholar]
- Ai H., Fang X., Yang B. et al (2015) Adaptation and possible ancient interspecies introgression in pigs identified by whole‐genome sequencing. Nature Genetics 47, 217–25. [DOI] [PubMed] [Google Scholar]
- Ashley C.T. Jr & Warren S.T. (1995) Trinucleotide repeat expansion and human disease. Annual Review of Genetics 29, 703–28. [DOI] [PubMed] [Google Scholar]
- Campbell E.M., Nonneman D. & Rohrer G.A. (2003) Fine mapping a quantitative trait locus affecting ovulation rate in swine on chromosome 8. Journal of Animal Science 81, 1706–14. [DOI] [PubMed] [Google Scholar]
- Chatterjee T.K. & Fisher R.A. (2000) Novel alternative splicing and nuclear localization of human RGS12 gene products. Journal of Biological Chemistry 275, 29660–71. [DOI] [PubMed] [Google Scholar]
- Chen J., Qi S., Guo R., Yu B. & Chen D. (2010) Different messenger RNA expression for the antimicrobial peptides beta‐defensins between Meishan and crossbred pigs. Molecular Biology Reports 37, 1633–9. [DOI] [PubMed] [Google Scholar]
- Chen Q., Ma Y., Yang Y. et al (2013) Genotyping by genome reducing and sequencing for outbred animals. PLoS One 8, e67500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- China National Commission of Animal Genetic Resources . (2011) Animal Genetic Resources in China Pigs. China Agriculture Press, Beijing. [Google Scholar]
- Collins F.S., Drumm M.L., Cole J.L., Lockwood W.K., Vande Woude G.F. & Iannuzzi M.C. (1987) Construction of a general human chromosome jumping library, with application to cystic fibrosis. Science 235, 1046–9. [DOI] [PubMed] [Google Scholar]
- Dall'Olio S., Fontanesi L., Tognazzi L. & Russo V. (2010) Genetic structure of candidate genes for litter size in Italian Large White pigs. Veterinary Research Communications 34(Suppl 1), S203–6. [DOI] [PubMed] [Google Scholar]
- Dennis G. Jr, Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C. & Lempicki R.A. (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biology 4, P3. [PubMed] [Google Scholar]
- Diamond M.S. (2014) IFIT1: a dual sensor and effector molecule that detects non‐2′‐O methylated viral RNA and inhibits its translation. Cytokine & Growth Factor Reviews 25, 543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchet‐Suchaux M.F., Bertin A.M. & Menanteau P.S. (1991) Susceptibility of Chinese Meishan and European Large White pigs to enterotoxigenic Escherichia coli strains bearing colonization factor K88, 987P, K99, or F41. American Journal of Veterinary Research 52, 40–4. [PubMed] [Google Scholar]
- Flicek P., Ahmed I., Amode M.R. et al (2013) Ensembl 2013. Nucleic Acids Research 41, D48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladney C.D., Bertani G.R., Johnson R.K. & Pomp D. (2004) Evaluation of gene expression in pigs selected for enhanced reproduction using differential display PCR and human microarrays: I. Ovarian follicles. Journal of Animal Science 82, 17–31. [DOI] [PubMed] [Google Scholar]
- Grobet L., Martin L.J., Poncelet D. et al (1997) A deletion in the bovine myostatin gene causes the double‐muscled phenotype in cattle. Nature Genetics 17, 71–4. [DOI] [PubMed] [Google Scholar]
- He H., Liu X., Gu Y. & Liu Y. (2010) A novel 18‐bp deletion mutation of the AMPD1 gene affects carcass traits in Qinchuan cattle. Molecular Biology Reports 37, 3945–9. [DOI] [PubMed] [Google Scholar]
- Hu Z.L., Park C.A., Wu X.L. & Reecy J.M. (2013) Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post‐genome era. Nucleic Acids Research 41, D871–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T. & Lempicki R.A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Jung E.J., Park H.B., Lee J.B., Yoo C.K., Kim B.M., Kim H.I., Cho I.C. & Lim H.T. (2014) Genome‐wide association study identifies quantitative trait loci affecting hematological traits in an F2 intercross between Landrace and Korean native pigs. Animal Genetics 45, 534–41. [DOI] [PubMed] [Google Scholar]
- Kerje S., Sharma P., Gunnarsson U. et al (2004) The dominant white, dun and smoky color variants in chicken are associated with insertion/deletion polymorphisms in the PMEL17 gene. Genetics, 168, 1507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.G., Rohrer G.A., Vallet J.L., Christenson R.K. & Nonneman D. (2004) Addition of 14 anchored loci to the porcine chromosome 8 comparative map. Animal Genetics 35, 474–6. [DOI] [PubMed] [Google Scholar]
- Klambauer G., Schwarzbauer K., Mayr A., Clevert D.A., Mitterecker A., Bodenhofer U. & Hochreiter S. (2012) cn.MOPS: mixture of Poissons for discovering copy number variations in next‐generation sequencing data with a low false discovery rate. Nucleic Acids Research 40, e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. & Durbin R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25, 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. & Genome Project Data Processing S (2009) The Sequence Alignment/Map format and samtools . Bioinformatics 25, 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Tian S., Jin L. et al (2013) Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nature Genetics 45, 1431–8. [DOI] [PubMed] [Google Scholar]
- Li M., Tian S., Yeung C.K. et al (2014) Whole‐genome sequencing of Berkshire (European native pig) provides insights into its origin and domestication. Scientific Reports 4, 4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon S., Kim T.H., Lee K.T. et al (2015) A genome‐wide scan for signatures of directional selection in domesticated pigs. BMC Genomics 16, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullaney J.M., Mills R.E., Pittard W.S. & Devine S.E. (2010) Small insertions and deletions (INDELs) in human genomes. Hum Mol Genet 19, R131‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning S., Pagano J.S. & Barber G.N. (2011) IRF7: activation, regulation, modification and function. Genes and Immunity 12, 399–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.H., Gail M.H., Weinberg C.R., Carroll R.J., Chung C.C., Wang Z., Chanock S.J., Fraumeni J.F. Jr & Chatterjee N. (2011) Distribution of allele frequencies and effect sizes and their interrelationships for common genetic susceptibility variants. Proceedings of the National Academy of Sciences of the United States of America 108, 18026–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos A.M., Megens H.J., Crooijmans R.P., Schook L.B. & Groenen M.A. (2011) Identification of high utility SNPs for population assignment and traceability purposes in the pig using high‐throughput sequencing. Animal Genetics 42, 613–20. [DOI] [PubMed] [Google Scholar]
- Rausch T., Zichner T., Schlattl A., Stutz A.M., Benes V. & Korbel J.O. (2012) delly: structural variant discovery by integrated paired‐end and split‐read analysis. Bioinformatics 28, i333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin C.J., Megens H.J., Martinez Barrio A. et al (2012) Strong signatures of selection in the domestic pig genome. Proceedings of the National Academy of Sciences of the United States of America 109, 19529–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sironen A., Thomsen B., Andersson M., Ahola V. & Vilkki J. (2006) An intronic insertion in KPL2 results in aberrant splicing and causes the immotile short‐tail sperm defect in the pig. Proceedings of the National Academy of Sciences of the United States of America 103, 5006–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Chen Q., Yang Y. et al (2014) A genome‐wide scan for selection signatures in Yorkshire and Landrace pigs based on sequencing data. Animal Genetics 45, 808–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Chen Q., Yang Y. et al (2015) Genetic diversity and population structure of six Chinese indigenous pig breeds in the Taihu Lake region revealed by sequencing data. Animal Genetics 46, 697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X., Liu X., Zhou L. et al (2015) Genome‐wide association analysis reveals genetic loci and candidate genes for meat quality traits in Chinese Laiwu pigs. Mammalian Genome 26, 181–90. [DOI] [PubMed] [Google Scholar]
- Yan Y., Yi G., Sun C., Qu L. & Yang N. (2014) Genome‐wide characterization of insertion and deletion variation in chicken using next generation sequencing. PLoS One 9, e104652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Lee S.H., Goddard M.E. & Visscher P.M. (2011) gcta: a tool for genome‐wide complex trait analysis. American Journal of Human Genetics 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. (1991) Chinese Taihu Pig. Shanghai Scientific and Technical Publishers, Shanghai. [Google Scholar]
- Zhang L.C., Li N., Liu X. et al (2014) A genome‐wide association study of limb bone length using a Large White × Minzhu intercross population. Genetics Selection Evolution 46, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Sequencing data summary: (a) sequencing quality of the raw and filtered reads from R1 (5′), (b) sequencing quality of raw and filtered reads from R2 (3′), (c) distribution of the average good reads for individuals within a population.
Figure S2 Probability density of MAF.
Figure S3 Distribution of MAF across each breed.
Table S1 Detailed information of the detected variants including SNPs, indels, insertions, deletions and CNVs and their lengths and locations on chromosomes as well as the frequencies of alleles.
Table S2 Summary of number of good reads, depth and coverage for each sample.
Table S3 Indels in exon regions, their locations on chromosomes and the corresponding Ensembl gene ID and name.
Table S4 Breed‐specific SNP distributions when comparing two populations.
Table S5 Functional enrichment of genes with large‐effect mutation variants.
Table S6 Distribution of variants in QTL regions. Densities of four types of variants (including SNPs, indels, insertions and deletions) in each new defined QTL and the information of the newly defined QTL locations, raw QTL traits and variant counts and densities in the new QTLs.
Data Availability Statement
All BAM data were deposited in the NCBI Sequence Read Achieve (SRA) under the Bioproject number PRJNA281578. The experiment number for the 252 pigs is SRX999959. Indel data have been submitted to dbSNP of NCBI (NCBI_ss1966414102 to ss1966531374) and insertions and deletions to dbVar of NCBI. Detailed information about the variants is provided in Table S1. Phenotypic records related to reproduction and pedigree information have been uploaded to our website (http://klab.sjtu.edu.cn/pigbreeds/).