

Abstract
Background
We postulated that Cerebral Amyloid Angiopathy (CAA), characterized by cortical vascular amyloid deposits, is associated with cortical tissue loss independent of parenchymal Alzheimer pathology. Patients with hereditary cerebral hemorrhage with amyloidosis–Dutch type (HCHWA-D), a monogenetic model of essentially pure CAA with minimal or no concomitant Alzheimer pathology, constitute a unique disease model to test this hypothesis.
Methods
In this observational case-control study, the primary outcome measure, the cortical thickness of prospectively enrolled patients diagnosed with HCHWA-D using genetic testing and non-demented patients with probable CAA according to Boston criteria was compared to relevant age-matched contrast groups including healthy controls (HC) and Alzheimer's Disease (AD), the latter diagnosed with National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association criteria. The data were collected at Massachusetts General Hospital in Boston MA, at Leiden University in Netherlands, and sites contributing to Alzheimer's Disease Neuroimaging Initiative (ADNI). The correlations between cortical thickness and structural lesions as well as Blood-Oxygen-Level-Dependent Time-to-Peak (BOLD-TTP, a physiologic measure of vascular dysfunction) were sought to understand the potential mechanistic link.
Results
Data were collected from March 15, 2006 to December 1, 2014. Patients with HCHWA-D (n=26) had thinner cortices (2.31mm± 0.18) compared to their HC (n=28) group (mean difference [MD]= −0.112, 95% confidence interval [CI] −0.190, −0.034, p=0.006). Non-demented sporadic CAA patients (n=63) had globally thinner cortices (2.17±0.11) compared to the two HC (n=63 and n=126) cohorts (MD=−0.14, 95% CI −0.17, −0.10 and MD=−0.10, 95% CI −0.13, −0.06, p<0.001 for both). All differences remained independent in multivariable analyses. As expected, patients with AD dementia (n=63) displayed more severe atrophy than CAA (2.1±0.14mm, p=0.005). There were strong associations between cortical thickness and vascular dysfunction in the HCHWA-D (rho=−0.58, p=0.003) and sporadic CAA (r= −0.4, p = 0.015) subjects but not in controls. Vascular dysfunction was identified as a mediator of the influence of hereditary CAA on cortical atrophy, accounting for 63% of the total effect.
Funding
The main funding for this study came from grants from National Institute of Health (NINDS K23-NS083711, NINDS R01-NS070834, NIA P01-AG036694, NIH U01-AG024904).
Conclusions
The appearance of cortical thinning in an essentially pure form of CAA indicates that vascular amyloid is an independent contributor to cortical atrophy. These results were reproduced in the more common sporadic CAA. Our findings also suggest that this effect is at least partly mediated by vascular dysfunction.
Introduction
Loss of cortical gray matter is a marker of many neurodegenerative diseases such as Alzheimer's Disease (AD) and a key mediator of associated cognitive impairment.1, 2 Cerebral small vessel diseases, conversely, are generally recognized as a cause of subcortical white matter injury.3, 4 Although cortical atrophy has been observed in small vessel disease, studies have not determined whether the observed atrophy represents true loss of cortical gray matter rather than widening of cortical sulci due to loss of the underlying subcortical white matter.5 It is important to understand the specific contributors to cortical atrophy in order to test targeted therapeutic approaches.
Cerebral amyloid angiopathy (CAA) is characterized by the gradual deposition of Aβ peptides in the media and adventitia of small leptomeningeal and cortical vessels.6 CAA has been identified as a major cause of lobar intracerebral hemorrhage (ICH), microbleeds and superficial siderosis.7, 8 In recent years, multiple studies have shown that CAA also causes ischemic subcortical tissue injury, an effect probably mediated by vascular dysfunction.9–12 As a result of such ischemic damage, CAA has emerged as an independent contributor to age-related cognitive decline, even in patients who have not had symptomatic ICH.13, 14
Although CAA, a cortical vascular pathology, has been linked to a plethora of subcortical lesions, its association with cortical structural changes has not been studied. The major impediment for such a study is the frequent coexistence of CAA and parenchymal AD pathologies, making it difficult to tease out the independent effects of vascular amyloid on cerebral cortex. To overcome such confounding by parenchymal AD, we analyzed a unique cohort of patients with hereditary cerebral hemorrhage with amyloidosis–Dutch type (HCHWA-D), a condition characterized by severe CAA with minimal or no neurofibrillary pathology.15 To assess the generalizability of findings from this select cohort, we further compared cortical thickness in the more common sporadic CAA to age matched healthy control (HC) and AD groups. We also examined the correlation between cortical thickness and markers of structural cerebral injury (lobar microbleeds and leukoaraiosis) to understand whether these widespread lesions might affect cortical loss or its measurement. Finally, we analyzed the relationship between cortical thickness and a functional MRI marker of vascular reactivity to explore the possibility that CAA-related vascular dysfunction mediates cortical damage.
Methods
Study design and participants
This prospective parallel case control study was conducted from March 15, 2006 through December 1, 2014 at two tertiary referral centers, namely Leiden University in Netherlands and Massachusetts General Hospital in Boston, MA. The cohort of patients with HCHWA-D and their age-matched HC were enrolled at the Leiden University Medical Center, Leiden, The Netherlands.16 The diagnosis of HCHWA-D was based on DNA analysis for confirmation of the codon 693 mutation in the amyloid-β precursor protein gene.17 A total of 26 DNA-proven HCHWA-D mutation carriers and 28 age-matched (±5 years) controls were prospectively enrolled and demographics, risk factors, and clinical features prospectively recorded.16 These healthy controls from the same communities in Leiden all had genetic testing to rule out HCHWA-D. Sporadic CAA subjects were prospectively enrolled in an ongoing longitudinal cohort study of CAA at Massachusetts General Hospital (MGH), Boston, MA. Detailed information including demographics, risk factors, and clinical characteristics were prospectively recorded at the time of enrollment.9, 11 None of the CAA subjects had dementia and all were free of symptoms suggestive of new stroke for 6 months prior to MRI acquisition. All sporadic CAA subjects had a diagnosis of probable CAA according to the pathologically validated Boston criteria.18 The Institutional Review Boards of the participating sites have approved this study. All participants completed written informed consent.
Sporadic CAA subjects were compared to two age-matched HC groups with complementary strengths: one comprised of control subjects in the Harvard Aging Brain (HAB) study19 drawn from the same population as the CAA subjects but imaged on a different scanner type, and a second obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database20 imaged using the same scanner type/strength as the CAA subjects. As a positive control group to demonstrate cortical atrophy, we also selected age-matched AD subjects from the ADNI database. Age-matching was done based on the closest aged individual to the 1/10th decimal for age, from the available HAB cohort as well as ADNI HC and AD cohorts. Matching was performed without knowledge of radiologic or other subject characteristics. Patient enrollment and data collection for these large prospective studies have been extensively described.19, 20 Specifically, part of the data used in the preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu, last accessed on December 14, 2015). The ADNI was launched in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer's disease (AD).
Overall, 63 non-demented patients with probable sporadic CAA were age-matched to 63 HC from the ADNI database, 126 HC from HAB study, and 63 patients with AD from ADNI.
MRI Acquisition
All patients with HCHWA-D and their respective controls had the same structural and functional MRI sequences obtained in a 3T GE scanner. The FLAIR (TR/TE: 11.0s/125ms, flip angle 90°, slices 25, FOV 252 × 179.76 × 250mm), 3D T1-weighted images (TR/TE 9/4.6ms, flip angle 8°, FOV = 224 × 177 × 168mm), and SWI-weighted images (TR = 45 ms, TE = 31 ms, FA = 13°, FOV = 250 × 175 × 112 mm) were obtained. All patients with sporadic CAA underwent structural MRI using a Siemens Avanto 1.5T scanner (12-channel head coil) and 37 of them consecutively underwent functional MRI (fMRI) after this latter protocol was implemented. Specifically, T1-weighted (Multi-echo MPRAGE: 1×1×1 mm voxel size), high-resolution Susceptibility Weighted Imaging (SWI: 0.75×0.75×1.30 mm voxel size), and fluid attenuated inversion recovery (FLAIR: 1×1×1 mm voxel size) scans were acquired.11 The Blood-Oxygen-Level-Dependent (BOLD) fMRI protocol that uses a block design with a visual stimulus has been previously described.9 The subjects obtained from the ADNI database underwent similar structural MRI sequences using Siemens 1.5T scanners at multiple sites, whereas the HC subjects from the HAB study underwent MRI using a Siemens TrioTim 3T scanner (12-channel head coil) within the same research facility as the sporadic CAA cohort.19, 20 Extensive details of MRI acquisition parameters for these studies were presented in previous publications.9,11,19,20
Image Processing
Three-dimensional cortical surface reconstruction was performed using the FreeSurfer software suite (https://surfer.nmr.mgh.harvard.edu, last accessed on November 12, 2015), version 5.3.21–23 The average cortical thickness of each hemisphere is calculated from representations of the gray/white and pial surfaces by taking into account the spatial and intensity singularities of the examined three-dimensional MPRAGE volume.24 This pipeline allowed the measurement of cortical thickness to sub-millimeter accuracy in previous validation studies.24, 25 All the results of the cortical surface reconstruction were visually examined, and manual interventions performed mainly by placing control points at regions distorted by gross pathology. This step improves the quality of segmentation and regional parcellations of the entire brain, when needed. In all patients who had ICH, we used results from the unaffected hemisphere for all cortical and white matter measures, similar to our previous work.9–11 Besides cortical thickness, the hippocampal volume and estimated total intracranial volumes (eTIV) were calculated again from FreeSurfer parcellations.26, 27 The eTIV represents the volume within the cranium and it allows correcting for head size variation within the population examined. Hippocampal volumes (HV) were expressed as a percent of eTIV.
Two other measures of CAA-related structural injury were examined: the total number of lobar microbleeds (LMB) and the white matter hyperintensity (WMH) volume. The number of LMBs was counted on a high resolution SWI-MRI, based on previously published guidelines.28 WMH volume was calculated with a homemade semi-automated algorithm that produced maps of WMH.11 All the maps generated by this algorithm were visually checked to verify their accuracy, manually corrected when needed and the WMH volume was expressed as a percentage of eTIV.
The BOLD Time-to-Peak (BOLD-TTP, in seconds), a physiologic marker of vascular reactivity was calculated from the fMRI acquisitions as described.9
Statistical Methods
The primary objective of the study was to compare cortical thickness between CAA and HCs, the overarching hypothesis was that CAA causes cortical atrophy. Bivariate analyses were performed using chi-square test for ratios,and t-test for continuous variables. Bivariate correlation analyses were performed to test the association between continuous variables, Spearman's rho for variables that did not have a normal distribution. Multivariate linear regression models were built to test the association between diagnosis of CAA and cortical thinning, after adjustment for covariates. These covariates were identified based on the results of bivariate analyses in this study and previous reports, and included age, gender, WMH volume, and finally HV as a surrogate for parenchymal amyloid related damage. Separate multivariate models were built and reported under Results for each group comparison, i.e. sporadic CAA and its contrast cohorts, HCHWA-D and respective controls. Additional multivariable models were performed omitting sporadic CAA patients with ICH to exclude confounding from these lesions. As LMBs are part of the presentation and diagnostic criteria for CAA, we performed multivariate models restricted to the respective CAA cohorts to test for their associations with cortical thickness. When regression models incorporating all covariates risked overfitting a model with a relatively small number of observations or false negatives due to multicollinearity, we applied backward elimination (p < 0.10 for retention) to remove non-significant terms. Mediation analyses were performed using methods of Baron and Kenny as well as the Sobel test.29, 30 For the mediation analysis, the diagnosis (CAA vs HC) was used as the independent variable (potential effector), TTP of BOLD response as the potential mediator and cortical thickness as the dependent variable. The Sobel test is useful in estimating the percentage of the total effect that is mediated and the results of both approaches were reported. All statistical analyses were performed using SPSS software. A two-tailed threshold for significance of p<0.05 was used.
Regional Comparisons of Cortical Thickness
A General Linear Model was computed in order to schematically explore the regional differences in cortical thickness between patients with CAA and Healthy Controls, after adjusting for age and gender. Topographic surface maps were generated using a threshold of p < 0.01 (with false discovery rate correction for multiple comparisons). The resulting maps (Figure 1, Figure 2) show the statistically significant regional differences in cortical thickness between the CAA and their age-matched HC cohorts.
Figure 1.
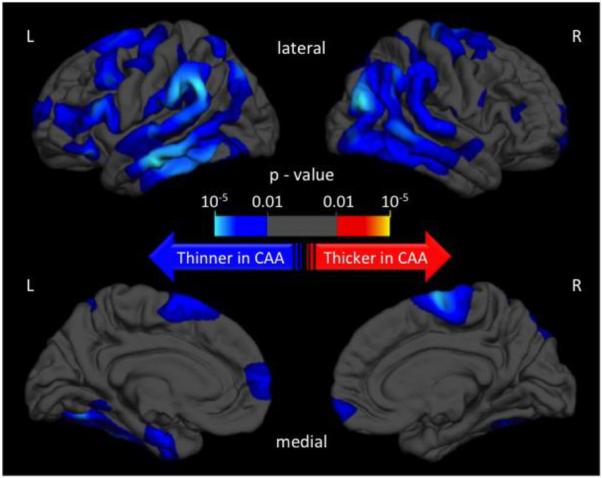
Topographic surface maps showing differences in cortical thickness between patients with hereditary cerebral hemorrhage with amyloidosis–Dutch type and their age-matched controls
Figure 2.
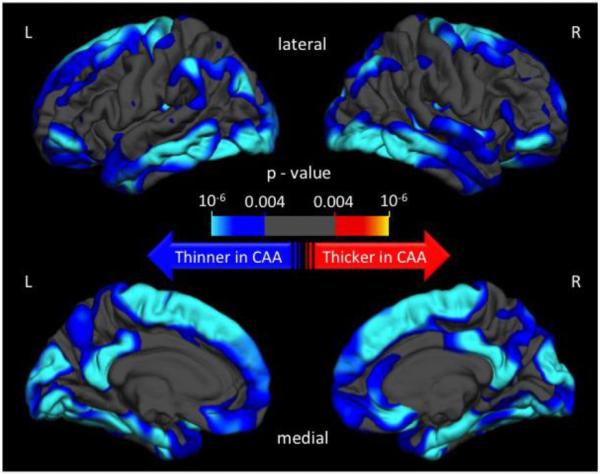
Topographic surface maps showing differences in cortical thickness between patients with sporadic cerebral amyloid angiopathy and their age-matched controls
Role of the Funding Source
No funding source had any role in the performance of this study, analyses, interpretation of results or manuscript preparation. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Cortical Atrophy in Hereditary CAA and Controls
The mean age (±SD) of patients with HCHWA-D (n=26) and their respective controls (n=28) was similar (HCHWA-D 45.8±14 vs controls 47.5±12, p=0.624). The comparisons of the demographic characteristics and imaging measures of interest are given in Table 1. Patients with HCHWA-D had thinner cortices (2.31mm± 0.18, mean difference [MD]= −0.112, 95% confidence interval [CI] −0.190, −0.034, p=0.006) than controls (Figure 1). Interestingly, HV was not significantly different between HCHWA-D and controls (Table 1). A multivariate linear regression model was built to test the association between diagnosis of HCHWA-D and cortical thinning, after adjustment for covariates that included age, gender, WMH volume, and finally HV as a surrogate for parenchymal amyloid related damage. The diagnosis of HCHWA-D was independently associated with lower cortical thickness (p<0.001). Other variables independently associated with cortical thickness were age and HV, but not WMH and gender. Cortical thickness did not correlate with WMH or LMBs in a multivariate model that also included age within the HCHWA-D cohort.
Table 1.
Comparisons between hereditary CAA and healthy controls
| Hereditary CAA Patients (n = 26) | Healthy Controls (n = 28) | |
|---|---|---|
| Demographics | ||
| Age (mean ± SD) | 45.8 ± 14 | 47.5 ± 12 |
| # Female patients (%) | 16 (61.5%) | 17 (60.7%) |
| Outcomes | ||
| Cortical Thickness in mm, (mean ± SD) | 2.31 ± 0.18 | 2.42 ± 0.1 p=0.006 |
| HV % of ICV (mean ± SD) | 0.54 ± 0.13 | 0.58 ± 0.07 p=0.113 |
The p values result from comparison to the same measure in the Hereditary CAA cohort
CAA: cerebral amyloid angiopathy
HV % of ICV: Hippocampal volume percent of intracranial volume
Cortical Atrophy in Sporadic CAA, Controls and AD
We tested the generalizability of the above findings from a pure CAA cohort to the more common situation of sporadic CAA. The groups of sporadic CAA patients (n=63), 2 HC cohorts (HAB n=126, ADNI n=63), and subjects with AD (n=63) were age matched with mean age (±SD) 72±5 years. The results of comparisons between CAA and these contrast groups are given in Table 2. A smaller proportion of sporadic CAA patients was female (Table 2) than both HC groups. Gender was therefore entered in all multivariate models. Patients with CAA presented significantly lower global cortical thickness (2.17±0.11 mm) when compared to both HC cohorts (ADNI: MD=−0.14, 95% CI −0.17, −0.10, p<0.001; HAB: MD=−0.10, 95% CI −0.13, −0.06, p<0.001). These differences were most pronounced in the occipital, temporal, posterior parietal, and medial frontal regions [Figure 2]. We repeated these comparisons after excluding 31 patients with CAA who had ICH (and their matched HCs), in order to overcome any potential confounding effect of these lesions. The remaining 32 patients with CAA without ICH again had significantly lower cortical thickness (2.16±0.12 mm) versus both HC groups (32 ADNI control subjects: MD=−0.13, 95% CI −0.18, −0.08, p<0.001; 64 HAB control subjects: MD=−0.11, 95% CI −0.15, −0.06, p<0.001). The diagnosis of CAA was independently associated with cortical thinning compared to either HC group after adjustment for the covariates above (p=0.007). Similar results were obtained when the multivariate models were limited to CAA patients without ICH and corresponding HC subjects (p=0.010). There was again no association between cortical thickness and total number of LMBs or WMH volume after controlling for age within the sporadic CAA cohort.
Table 2.
Comparisons between sporadic CAA and contrast groups
| Sporadic CAA Patients (n = 63) | ADNI Healthy Controls (n = 63) | HAB Healthy Controls (n = 126) | ADNI AD Patients (n = 63) | |
|---|---|---|---|---|
| Demographics | ||||
| Age (mean ± SD) | 72.2 ± 5 | 72.3 ± 5 | 72.4 ± 5 | 72.2 ± 5 |
| # Female patients (%) | 14 (22.2%) | 27 (42.9%) | 54 (42.9%) | 20 (31.7%) |
| Outcomes | ||||
| Cortical Thickness in mm (mean ± SD) | 2.17 ± 0.11 | 2.31 ± 0.07 p<0.001 |
2.27 ± 0.09 p<0.001 |
2.10 ± 0.14 p=0.005 |
| HV % of ICV (mean ± SD) | 0.41 ± 0.07 | 0.49 ± 0.07 p<0.001 |
0.51 ± 0.07 p<0.001 |
0.36 ± 0.08 p=0.001 |
The p values result from comparison to the same measure in the sporadic CAA cohort
CAA: cerebral amyloid angiopathy
HV % of ICV: Hippocampal volume percent of intracranial volume
The patients with AD had relatively early disease: 36 (57%) had a clinical dementia rating (CDR) of 0.5 and 27 (43%) had a CDR 1. Patients with AD showed significantly thinner cortices (Table 2) when compared to both CAA (MD=−0.07, 95% CI −0.11, −0.02, p=0.005) and HC (ADNI: MD=−0.20, 95% CI −0.24, −0.16, p<0.001; HAB: MD=−0.16, 95% CI −0.20, −0.13, p<0.001). HV was significantly reduced in CAA (Table 2) versus both HC cohorts (ADNI: MD=−0.08, 95% CI −0.11, −0.06, p<0.001; HAB: MD=−0.10, 95% CI −0.12, −0.08, p<0.001) and smaller in AD (Table 2) than CAA (MD=−0.05, 95% CI −0.07, −0.02, p=0.001).
Cortical Thickness and Vascular Reactivity
BOLD-TTP, a functional marker of vascular reactivity thought to mediate ischemic injury in CAA based on prior work,9,12 was prolonged in HCHWA-D compared to controls (10.6sec±8.5, MD=7.9, 95% CI 4.4, 11.4, p<0.001). Prolonged BOLD-TTP correlated with decreased cortical thickness within the HCHWA-D cohort (rho=−0.58, p=0.003) but not in the age-matched HC group (rho=0.05). Within the HCHWA-D cohort, BOLD-TTP remained independently correlated with cortical thickness after adjustment for age, gender, WMH volume, microbleed count, HV, and presence of ICH. In patients with sporadic CAA, cortical thickness was also negatively correlated with BOLD-TTP (r= −0.4, p = 0.015) and remained significant after adjusting for the above covariates (p = 0.007). We performed mediation analyses29,30 to address whether some of the effect of HCHWA-D on cortical thickness is mediated by vascular dysfunction as measured by prolonged BOLD-TTP. There was a significant initial relationship between the diagnosis of HCHWA-D and cortical thickness (β = 0.22, p = 0.01) that was non-significant (β = 0.12, p = 0.2) after controlling for BOLD-TTP, indicating that BOLD-TTP mediates the relationship between HCHWA-D diagnosis and cortical thickness. The Sobel test also found BOLD-TTP (t=2.4, p=0.018) to be a significant mediator of the influence of HCHWA-D on cortical thickness, explaining 63% of the total effect.
Discussion
To identify the contribution of cerebrovascular amyloid pathology to cortical atrophy, we evaluated the cortical thickness of a monogenic form of CAA largely devoid of parenchymal Alzheimer pathology as well as non-demented patients with sporadic CAA. Both hereditary and sporadic CAA patients showed significant global decreases in cortical thickness (5–6%) when compared to age-matched healthy controls, whereas demented patients diagnosed with AD exhibited still greater cortical thinning (3%) than age-matched sporadic CAA.
Cortical atrophy in CAA could be related to several factors: 1) confounding from concomitant parenchymal AD pathology, 2) cortical effects of CAA-related structural lesions, or 3) CAA-related vascular dysfunction. We addressed the first possibility through the unique opportunity afforded in HCHWA-D of minimizing the confounding of AD pathology, finding essentially the same extent and pattern of cortical thinning in these subjects as the sporadic CAA subjects. Our results further found no major effects on cortical thickness of structural lesions such as LMB or WMH and suggest instead that vascular dysfunction may at least in part mediate CAA-related cortical damage.
A strength of our study was inclusion of HCHWA-D patients and age-matched controls identified and scanned at a single site. This amyloid precursor protein codon 693 mutation in these patients15 causes extensive amyloid-deposition in the walls of meningocortical arterioles, little parenchymal Aβ deposition in the form of `diffuse' plaques and essentially no dense core plaque or tau deposition.31 We found decreased cortical thickness in this essentially pure human model of CAA in relatively young patients (mean age 46) and no association between cortical thickness and structural lesions. We have replicated these results in the more prevalent condition of sporadic CAA. Adding HV, a commonly used AD biomarker, as a covariate in multivariate regression models did not change the independent association between CAA and global cortical thinning, further supporting our hypothesis that vascular amyloid directly contributes to cortical atrophy.
Cortical atrophy could be related to any of the various CAA-related lesions: ICH, microbleeds, cortical microinfarcts and even white matter lesions through local or “dying back” mechanisms. As in previous studies,9, 11 we used the hemisphere that was not affected by ICH and repeated all analyses in the subgroup of patients without ICH without affecting the results. LMB counts and WMH volumes did not correlate with cortical thickness in either CAA cohort in multivariate models, similar to reported negative findings for another small vessel disease model, Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL).32 Future studies designed to detect cortical microinfarcts and measure the remote effects of small deep lesions such as lacunes will further increase our understanding of the mechanisms of cortical atrophy in CAA.
Cortical thinning in CAA was noted in the occipital, temporal, posterior parietal, and medial frontal regions, areas previously reported to be particularly susceptible to higher vascular amyloid load by pathology and in vivo amyloid imaging.6, 33, 34 Amyloid burden in the medial frontal region has also been identified as an independent predictor of future hemorrhages in CAA.35 The association between regional cortical atrophy patterns from this work and amyloid distribution in previous studies further support a link between vascular amyloid and cortical thinning. Although the distribution of cortical thinning in the CAA subjects resembles the distribution of vascular amyloid, it also overlaps with the pattern of cortical thinning observed in AD, suggesting that these patterns may not be entirely disease specific.36 We did not observe major qualitative differences in the patterns of cortical thinning between the hereditary and sporadic CAA subjects (Figure 1 and 2). Because of substantial age differences between the two CAA cohorts (approximately 25 years with no age overlap) and the large effect of age on cortical thickness, we could not quantitatively compare the pattern of cortical thinning between sporadic and hereditary CAA.
BOLD-TTP appears to be a marker of vascular dysfunction in sporadic CAA patients and, based on the current data, in the younger HCHWA-D patients as well.9, 12 There was a strong and independent negative correlation between BOLD-TTP and cortical thickness. Consequently, vascular dysfunction--expressed as delay in hemodynamic response--might contribute to cortical thinning in patients in CAA. Cortical thinning did not correlate with BOLD-TTP in the respective control subjects, supporting the conclusion that vascular amyloid, but not the cortical thickness itself, drives this association. Furthermore, both mediation analyses found that vascular dysfunction mediates a significant part of HCHWA-D's effect on cortical thinning. Some recent evidence also suggests reduced blood flow and possibly cortical atrophy in the setting of CAA. Two recent studies showed gray matter loss and hypometabolism in the temporal lobe of patients with AD with microbleeds when compared to AD without microbleeds, and widespread reductions in cortical perfusion in healthy octogenarians with cortical microbleeds when compared to age-similar HC without microbleeds.37, 38
The lack of a dedicated control group for sporadic CAA is a weakness of the study. We note, however, that the two complementary age-matched control groups used in this study, yielded essentially the same contrast from the sporadic CAA subjects, supporting the robustness of the finding. Other notable weaknesses were the lack of fMRI in the first 26 sporadic CAA patients enrolled early in the study and in the older healthy controls. We cannot exclude the possibility that the parenchymal Aß that has been observed as diffuse plaques in HCHWA-D might have some contribution to cortical thinning even in hereditary CAA.39 We also again note that in the absence of a validated molecular tracer specific to CAA or AD pathology, it is impossible to exclude some degree of confounding by concurrent parenchymal AD pathology on cortical thickness. Efforts to produce a specific marker for CAA are underway. Short of such a specific marker, we have analyzed a series of neuroimaging markers that capture aspects of CAA severity—LMBs, WMH volume, vascular reactivity—and find correlation only with vascular reactivity, raising the possibility that this aspect of CAA severity contributes to the CAA-related reduction in cortical thickness. In all other respects, the findings from the sporadic CAA analyses replicated those from the less confounded HCHWA-D analyses, arguing that the observed cortical thinning and association with vascular dysfunction are indeed driven by advanced CAA.
In conclusion, our findings show that cerebrovascular amyloid pathology is an independent contributor to cortical atrophy and suggest that this effect is at least partly mediated by CAA-related vascular dysfunction. These results indicate that a relatively common cerebral small disease results in cortical changes independent of its known profound subcortical effects. The current results also add further support to the view that the important contributory role of CAA and small vessel diseases should be considered in efforts to reduce age-associated cognitive impairment.
PANEL: Research in context.
Evidence before this study
We searched PubMed for articles on cerebral amyloid angiopathy (CAA) and cortical atrophy published between Jan 1, 1990, and Nov 1, 2015, with different combinations of the terms “cerebral amyloid angiopathy”, “cortical atrophy”, “cortical thickness”, ”cortical damage”. We have not found any article that addressed the association between CAA and cortical atrophy. We have then repeated PubMed searches within the same time interval using combinations of the term “cerebral amyloid angiopathy” and each one of the following terms: “leukoaraiosis”, “white matter”, “structural networks”, and “subcortical injury”. We have found multiple articles that established associations between CAA and subcortical tissue damage but the association of CAA and cortical atrophy was unknown. A major obstacle in clarifying this latter association has been the relatively common coexistence of CAA and parenchymal Alzheimer's Disease (AD) pathology, the latter being a well-established cause of cortical atrophy.
Added value of this study
This prospective study shows that patients with hereditary cerebral hemorrhage with amyloidosis–Dutch type, a monogenetic model of essentially pure CAA with minimal or no concomitant parenchymal AD pathology, have decreased cortical thickness when compared to age-matched controls. These findings were confirmed when comparing a well-defined cohort of common sporadic CAA patients to age-matched elderly controls. Mediation analyses also showed that vascular dysfunction explained at least part of the effect of CAA on cortical atrophy.
Implications of all the available evidence
Autopsy and imaging based studies show that vascular amyloid pathology and lobar microbleeds, the hallmarks of CAA, are common findings in otherwise healthy older adults as well as patients with mild cognitive symptoms. Multiple lines of evidence suggest that CAA is an important contributor to subcortical tissue injury through vascular dysfunction and ischemia. Cerebral amyloid angiopathy is also a well-established contributor to cognitive impairment in elderly, independent of intracerebral hemorrhages that constitute the other important consequences of the vascular amyloid. Our study for the first time suggests that CAA might also cause global cortical atrophy, an effect that might at least in part be mediated by vascular dysfunction. These new findings are important in better understanding the mechanisms of cerebral damage in CAA as they can potentially mediate disabling clinical effects and even confound therapeutic efforts. For example, some of the immunotherapies studied for Alzheimer's Disease resulted in superficial cerebral microbleeds typical of CAA and accelerated atrophy was observed in multiple antibody trials. Based on all available evidence, CAA will need to be taken into consideration in future efforts to develop therapies for AD and cognitive impairment.
Acknowledgments
Study Funding M. Edip Gurol: NINDS K23 NS083711
Steven M. Greenberg: NINDS R01 NS070834
Keith A. Johnson, Reisa A. Sperling (the HAB study) National Institutes of Health/National Institute on Aging (P01 AG036694, R01 AG046396, R01AG027435, P50 AG00513421), Fidelity Biosciences, Harvard Neurodiscovery Center, and the Alzheimer's Association.
Trey Hedden: K01AG040197
Jonathan Polimeni: Supported by NIH NIBIB K01-EB011498, P41-EB015896, and R01-EB019437 and the Athinoula A. Martinos Center for Biomedical Imaging; NIH NCRR Shared Instrumentation Grants S10-RR023401 and S10-RR020948.
Part of the data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors' Contributions: Literature search: Panagiotis Fotiadis, Eitan Auriel, Yael Reijmer, M. Edip Gurol
Figures: Panagiotis Fotiadis, M. Edip Gurol
Study design: Panagiotis Fotiadis, Keith A. Johnson, Mark A. van Buchem, Steven M. Greenberg, M. Edip Gurol
Data collection: Panagiotis Fotiadis, Sanneke van Rooden, Jeroen van der Grond, Aaron Schultz, Anna M. van Opstal, Alison Ayres, Kristin M. Schwab, the Alzheimer's Disease Neuroimaging Initiative, Trey Hedden, Anand Viswanathan, Jonathan Rosand, Marieke Wermer, Gisela Terwindt, Reisa A. Sperling, Jonathan R. Polimeni, Keith A. Johnson, Mark A. van Buchem, Steven M. Greenberg, M. Edip Gurol
Imaging Data analysis: Panagiotis Fotiadis, Sanneke van Rooden, Sergi Martinez-Ramirez, Eitan Auriel, MD, Yael Reijmer, Jonathan R. Polimeni, M. Edip Gurol
Statistical Analysis: Panagiotis Fotiadis, Steven M. Greenberg, M. Edip Gurol
Data interpretation: Sanneke van Rooden, Jonathan Rosand, Reisa A. Sperling, Jonathan R. Polimeni, Keith A. Johnson, Mark A. van Buchem, Steven M. Greenberg, M. Edip Gurol
Manuscript Writing: Steven M. Greenberg, M. Edip Gurol
Critical Review of the Manuscript: Panagiotis Fotiadis, Sanneke van Rooden, Jeroen van der Grond, Aaron Schultz, Sergi Martinez-Ramirez, Eitan Auriel, MD, Yael Reijmer, Anna M. van Opstal, Alison Ayres, Kristin M. Schwab, the Alzheimer's Disease Neuroimaging Initiative, Trey Hedden, Jonathan Rosand, Anand Viswanathan, Marieke Wermer, Gisela Terwindt, Reisa A. Sperling, Jonathan R. Polimeni, Keith A. Johnson, Mark A. van Buchem
Conflicts of Interest No other conflicts of interest by any other author related to this study.
References
- 1.Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harbor perspectives in medicine. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalaria RN, Erkinjuntti T. Small vessel disease and subcortical vascular dementia. Journal of clinical neurology. 2006;2:1–11. doi: 10.3988/jcn.2006.2.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9:689–701. doi: 10.1016/S1474-4422(10)70104-6. [DOI] [PubMed] [Google Scholar]
- 5.Nitkunan A, Lanfranconi S, Charlton RA, Barrick TR, Markus HS. Brain atrophy and cerebral small vessel disease: a prospective follow-up study. Stroke. 2011;42:133–138. doi: 10.1161/STROKEAHA.110.594267. [DOI] [PubMed] [Google Scholar]
- 6.Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke. 1987;18:311–324. doi: 10.1161/01.str.18.2.311. [DOI] [PubMed] [Google Scholar]
- 7.Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. Journal of neurology, neurosurgery, and psychiatry. 2012;83:124–137. doi: 10.1136/jnnp-2011-301308. [DOI] [PubMed] [Google Scholar]
- 8.Greenberg SM. Cerebral amyloid angiopathy: prospects for clinical diagnosis and treatment. Neurology. 1998;51:690–694. doi: 10.1212/wnl.51.3.690. [DOI] [PubMed] [Google Scholar]
- 9.Dumas A, Dierksen GA, Gurol ME, et al. Functional magnetic resonance imaging detection of vascular reactivity in cerebral amyloid angiopathy. Ann Neurol. 2012;72:76–81. doi: 10.1002/ana.23566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gurol ME, Irizarry MC, Smith EE, et al. Plasma beta-amyloid and white matter lesions in AD, MCI, and cerebral amyloid angiopathy. Neurology. 2006;66:23–29. doi: 10.1212/01.wnl.0000191403.95453.6a. [DOI] [PubMed] [Google Scholar]
- 11.Gurol ME, Viswanathan A, Gidicsin C, et al. Cerebral amyloid angiopathy burden associated with leukoaraiosis: A positron emission tomography/magnetic resonance imaging study. Ann Neurol. 2013;73:529–536. doi: 10.1002/ana.23830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peca S, McCreary CR, Donaldson E, et al. Neurovascular Decoupling is Associated with Severity of Cerebral Amyloid Angiopathy. Neurology. 2013 doi: 10.1212/01.wnl.0000435291.49598.54. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenberg SM, Gurol ME, Rosand J, Smith EE. Amyloid angiopathy-related vascular cognitive impairment. Stroke. 2004;35:2616–2619. doi: 10.1161/01.STR.0000143224.36527.44. [DOI] [PubMed] [Google Scholar]
- 14.Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ. Cerebral amyloid angiopathy and cognitive function: The HAAS autopsy study. Neurology. 2002;58:1629–1634. doi: 10.1212/wnl.58.11.1629. [DOI] [PubMed] [Google Scholar]
- 15.Zhang-Nunes SX, Maat-Schieman ML, van Duinen SG, Roos RA, Frosch MP, Greenberg SM. The cerebral beta-amyloid angiopathies: hereditary and sporadic. Brain Pathol. 2006;16:30–39. doi: 10.1111/j.1750-3639.2006.tb00559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Rooden S, van der Grond J, van den Boom R, et al. Descriptive analysis of the Boston criteria applied to a Dutch-type cerebral amyloid angiopathy population. Stroke. 2009;40:3022–3027. doi: 10.1161/STROKEAHA.109.554378. [DOI] [PubMed] [Google Scholar]
- 17.Watson DJ, Selkoe DJ, Teplow DB. Effects of the amyloid precursor protein Glu693-->Gln `Dutch' mutation on the production and stability of amyloid beta-protein. The Biochemical journal. 1999;340(Pt 3):703–709. [PMC free article] [PubMed] [Google Scholar]
- 18.Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology. 2001;56:537–539. doi: 10.1212/wnl.56.4.537. [DOI] [PubMed] [Google Scholar]
- 19.Dagley A, LaPoint M, Huijbers W, et al. Harvard Aging Brain Study: Dataset and accessibility. NeuroImage. 2015 doi: 10.1016/j.neuroimage.2015.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jack CR, Jr, Bernstein MA, Fox NC, et al. The Alzheimer's Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27:685–691. doi: 10.1002/jmri.21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis. I. Segmentation and surface reconstruction. NeuroImage. 1999;9:179–194. doi: 10.1006/nimg.1998.0395. [DOI] [PubMed] [Google Scholar]
- 22.Fischl B, Sereno MI, Dale AM. Cortical surface-based analysis. II: Inflation, flattening, and a surface-based coordinate system. NeuroImage. 1999;9:195–207. doi: 10.1006/nimg.1998.0396. [DOI] [PubMed] [Google Scholar]
- 23.Segonne F, Dale AM, Busa E, et al. A hybrid approach to the skull stripping problem in MRI. NeuroImage. 2004;22:1060–1075. doi: 10.1016/j.neuroimage.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 24.Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci U S A. 2000;97:11050–11055. doi: 10.1073/pnas.200033797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salat DH, Buckner RL, Snyder AZ, et al. Thinning of the cerebral cortex in aging. Cerebral cortex. 2004;14:721–730. doi: 10.1093/cercor/bhh032. [DOI] [PubMed] [Google Scholar]
- 26.Destrieux C, Fischl B, Dale A, Halgren E. Automatic parcellation of human cortical gyri and sulci using standard anatomical nomenclature. NeuroImage. 2010;53:1–15. doi: 10.1016/j.neuroimage.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- 28.Greenberg SM, Vernooij MW, Cordonnier C, et al. Cerebral microbleeds: a guide to detection and interpretation. The Lancet Neurology. 2009;8:165–174. doi: 10.1016/S1474-4422(09)70013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baron RM, Kenny DA. The moderator-mediator variable distinction in social psychological research: conceptual, strategic, and statistical considerations. Journal of personality and social psychology. 1986;51:1173–1182. doi: 10.1037//0022-3514.51.6.1173. [DOI] [PubMed] [Google Scholar]
- 30.Sobel ME. Asymptotic intervals for indirect effects in structural equations models. In: Leinhart S, editor. Sociological methodology. Jossey-Bass; San Francisco: 1982. pp. 290–312. [Google Scholar]
- 31.Bornebroek M, Haan J, Van Duinen SG, et al. Dutch hereditary cerebral amyloid angiopathy: structural lesions and apolipoprotein E genotype. Ann Neurol. 1997;41:695–698. doi: 10.1002/ana.410410523. [DOI] [PubMed] [Google Scholar]
- 32.Jouvent E, Mangin JF, Duchesnay E, et al. Longitudinal changes of cortical morphology in CADASIL. Neurobiol Aging. 2012;33:1002, e1029–1036. doi: 10.1016/j.neurobiolaging.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 33.Greenberg SM, Grabowski T, Gurol ME, et al. Detection of isolated cerebrovascular beta-amyloid with Pittsburgh compound B. Ann Neurol. 2008;64:587–591. doi: 10.1002/ana.21528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson KA, Gregas M, Becker JA, et al. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol. 2007;62:229–234. doi: 10.1002/ana.21164. [DOI] [PubMed] [Google Scholar]
- 35.Gurol ME, Dierksen G, Betensky R, et al. Predicting sites of new hemorrhage with amyloid imaging in cerebral amyloid angiopathy. Neurology. 2012;79:320–326. doi: 10.1212/WNL.0b013e31826043a9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dickerson BC, Bakkour A, Salat DH, et al. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cerebral cortex. 2009;19:497–510. doi: 10.1093/cercor/bhn113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gregg NM, Kim AE, Gurol ME, et al. Incidental Cerebral Microbleeds and Cerebral Blood Flow in Elderly Individuals. JAMA neurology. 2015;72:1021–1028. doi: 10.1001/jamaneurol.2015.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samuraki M, Matsunari I, Yoshita M, et al. Cerebral Amyloid Angiopathy-Related Microbleeds Correlate with Glucose Metabolism and Brain Volume in Alzheimer's Disease. Journal of Alzheimer's disease : JAD. 2015;48:517–528. doi: 10.3233/JAD-150274. [DOI] [PubMed] [Google Scholar]
- 39.Maat-Schieman M, Roos R, van Duinen S. Hereditary cerebral hemorrhage with amyloidosis-Dutch type. Neuropathology. 2005;25:288–297. doi: 10.1111/j.1440-1789.2005.00631.x. [DOI] [PubMed] [Google Scholar]