

Abstract
The final step in vitamin D activation is catalyzed by 1-alpha-hydroxylase (CYP27B1). Chronic kidney disease (CKD) is characterized by low levels of both 25(OH)D3 and 1,25(OH)2D3 provoking secondary hyperparathyroidism (2HPT). Therefore, treatments with active or native vitamin D compounds are common in CKD to restore 25(OH)D3 levels and also to decrease PTH. This study evaluates the dose of 25(OH)D3 that restores parathyroid hormone (PTH) and calcium levels in a model of CKD in CYP27B1-/- mice. Furthermore, we compare the safety and efficacy of the same dose in CYP27B1+/+ animals. The dose needed to decrease PTH levels in CYP27B1-/- mice with CKD was 50 ng/g. That dose restored blood calcium levels without modifying phosphate levels, and increased the expression of genes responsible for calcium absorption (TRPV5 and calbindinD- 28K in the kidney, TRPV6 and calbindinD-9k in the intestine). The same dose of 25(OH)D3 did not modify PTH in CYP27B1+/+ animals with CKD. Blood calcium remained normal, while phosphate increased significantly. Blood levels of 25(OH)D3 in CYP27B1-/- mice were extremely high compared to those in CYP27B1+/+ animals. CYP27B1+/+ animals with CKD showed increases in TRPV5, TRPV6, calbindinD-28K and calbindinD-9K, which were not further elevated with the treatment. Furthermore, CYP27B1+/+ animals displayed an increase in vascular calcification. We conclude that the dose of 25(OH)D3 effective in decreasing PTH levels in CYP27B1-/- mice with CKD, has a potentially toxic effect in CYP27B1+/+ animals with CKD.
Introduction
Vitamin D is a major regulator of Ca2+ and phosphate homeostasis and it is essential for proper development and maintenance of bones.[1] The active form of vitamin D, 1,25(OH)2D3, is synthesized from its precursor 25OHD3 by the 25-hydroxyvitamin-D-1α-hydroxylase (1-α-hydroxylase; CYP27B1).[2] Mutations in the CYP27B1 gene cause severe disorders of Ca2+ homeostasis, including vitamin D-dependent rickets type I (VDDRI).[3]
Chronic kidney disease- mineral and bone disorder (CKD-MBD) is a common problem in patients with kidney disease. It is characterized by abnormal levels of calcium (Ca) and phosphate (P) and biochemical alterations of mineral metabolism related hormones, alongside vascular calcification. Among those biochemical alterations, low levels of active vitamin D metabolites are of paramount importance.[4] To treat CKD-MBD related complications, vitamin D compounds have been widely used. However, the use of active vitamin D compounds such as 1,25(OH)2D3 has been accompanied by undesired side effects like hypercalcemia and hyperphosphatemia, which increase the risk of vascular calcification.[5] To avoid these effects, vitamin D analogs were developed in order to suppress PTH secretion with a minimal calcemic action. [6–8]
Active vitamin D compounds directly increase intestinal and renal Ca2+ reabsorption through up-regulation of Ca2+ transport proteins.[9] A distinct family of epithelial Ca2+ channels (TRPV6 and TRPV5) has been identified, which provides the molecular identity of the apical entry mechanism facilitating this active Ca2+ reabsorption process.[10, 11] Ca2+ entry via these Ca2+ channels is followed by cytosolic diffusion facilitated by Ca2+ binding proteins (calbindin-D28K and /or calbindin-D9k) and active extrusion of Ca2+ across the basolateral membrane by a high affinity plasma membrane Ca2+-ATPase (PMCA1b) and Na+-Ca2+ exchanger (NCX1). In this active process, TRPV5 and TRPV6 probably form the final target for hormonal control, suggesting that these channels could be the primary targets in the regulation of Ca2+ reabsorption.[9]
Not only 1,25(OH)2D3, but also 25OHD3 levels are low in CKD-MBD. Due to the existence of CYP27B1 in many tissues, international guidelines propose the correction of low 25OHD3 levels to maintain the pleiotropic beneficial effects of vitamin D.[12, 13] Thus, the use of cholecalciferol or even 25OHD3 to correct its own deficiency has been recommended.[14] Furthermore, the normalization of 25OHD3 levels has been reported to have an effect in decreasing PTH levels.[15] It has been also shown that 25OHD3 can mediate its effects independent of its conversion to 1,25(OH)2D3.[16] However, it is unknown whether the effect of 25OHD3 on PTH reduction is achieved directly by activation of VDR or by residual conversion into 1,25(OH)2D3.
The hypothesis of the present study is that the dose necessary to decrease PTH with 25OHD3 (without conversion into 1,25(OH)2D3) in CKD is very high, and could have toxic effects. Thus, the aim of the present study was to find the dose of 25OHD3 effective enough to restore Ca2+ and PTH levels in a CYP27B1 knockout (CYP27B1-/-) mouse model with CKD, therefore independently of conversion into 1,25(OH)2D3, and to test the same dose in CYP27B1+/+ mice to evaluate its safety and efficacy in decreasing PTH. We also investigated the effects of 25OHD3 treatment in regulating the expression of the main genes responsible for intracellular and paracellular calcium transport in the kidney and intestine of the same mice.
Methods
In Vivo study
All animal studies were approved by the University of Lleida Animal Ethics Committee in accordance with the guidelines of European Research Council for the care and use of laboratory animals. In all surgical procedures performed in animals, isoflurane was used as anesthetic and buprenorphine was used as analgesic after the surgery.
Generation and characterization of CYP27B1-/- mice
CYP27B1-/- mice were provided by Dr. David Goltzman (Montreal, Canada) and were generated by ablation of exon 6 to exon 9 (26). These mice were bred to C57/BL6 wild-type animals and the heterozygous offspring were crossed to produce CYP27B1-/- (KO) and CYP27B1+/+ (WT) animals. KO mice were fed a rescue diet (2% calcium, 1.25% phosphorus, 20% lactose and 2.2 IU/g vitamin D3; Harlan Teklad, TD.96348) during growth and maintenance. Before starting the experimental process the diet was changed to a standard mouse chow for KO animals (0.6% Ca2+, 0.8% phosphorus, and 0.6 IU/g vitaminD3; Harlan Teklad), while WT animals were maintained on a 0.9% phosphorus diet in order to induce an increase in PTH levels.
Model of CKD in mice
Subtotal nephrectomy was performed in 10 week-old mice after the two-step surgical procedure for 75% nephron reduction (NX), as previously described. [17] Briefly, the parenchyma of the left kidney was reduced 50%. The kidney was exposed, decapsulated and carefully cauterized, reducing the parenchyma of the upper and the lower pole. After 1-week of recovery period, right-sided total nephrectomy was performed. Treatments started two weeks after nephrectomy to facilitate recovery after the operation.
First, a dose response analysis was carried out in 10 week–old nephrectomized CYP27B1-/- mice (KO NX) using 25, 50, 100 ng/g of 25OHD3 (Sigma-Aldrich) administered intraperitoneally three times per week for 30 days (n = 8 per dose). Terminal blood samples were taken 24 hours after the last injection. In parallel, an additional group of KO NX mice (n = 8) was treated with a dose of 1,25(OH)2D3 (50 pg/g) three times per week for 30 days and a terminal blood sample was taken 24 hours after the last injection. The goal was to determine the dosage of 25OHD3 that induced changes in PTH levels similar to those achieved with 1,25(OH)2D3. After selecting the dosage of 50 ng/g for 25OHD3, nephrectomized CYP27B1+/+ mice (WT NX, n = 8) were treated with the same dose and route of administration for a 30 days period. A group of sham-operated mice were used as a control in the present study (CYP27B1+/+ (WT control) n = 6, CYP27B1-/- (KO control) n = 5).
Euthanasia was performed 24 hours after the last injection. Then, a terminal blood sample was collected and aortas were divided in two parts, one frozen in liquid nitrogen for calcium content determination and the other one fixed in formalin solution followed by processing and paraffin embedding. Kidney and duodenum were collected to study the expression of calcium transport proteins.
Serum biochemical analysis
Blood was collected by cardiac puncture and centrifuged at 2500 rpm for 10 min at 4°C to obtain serum.
Ca2 + and P were analyzed by a standard colorimetric assay analysis in the Biochemistry service of the Arnau de Vilanova Hospital (HUAV) in Lleida using a multichannel autoanalyzer (Roche/Hitachi Modular Analytics), using the following methods: 1) for calcium the o-cresolphthalein complexone method, 2) for phosphate the ammonium molybdate method. Blood urea nitrogen (BUN) was determined by colorimetric assay using the QuantiChrom Urea assay kit (DIUR-500, Gentaur, San Jose, CA, USA). Immunoassays were used to determine 25OHD3, 1,25 (OH)2D3 (IDS 25OHD3 EIA, Immunodiagnostic Systems, The Boldons, UK), and also PTH (PTH mouse ELISA kit, Immunotopics, San Clemente, CA, USA).
Quantitative analysis of aortic calcium
Aortic tissue was desiccated for 20–24 hours at 60°C, crushed to a powder with a pestle and mortar, and decalcified with HCl (1N) at 4°C, and then vortexed for 16 hours. After centrifugation, supernatant was collected and calcium content determined colorimetrically using the o-cresolphthalein complexone method, whereas total protein content was determined by the Lowry method (Bio-Rad, Hercules, CA, USA), as previously described.[7] Aortic calcium content was normalized by the protein amount in the sample and expressed as ng of Ca/ mg of protein.
Histology and immunohistochemistry
Immunostaining for CalbindinD28k and TRPV5 were carried out on 5-μm-thick kidney tissue sections. Sections were deparaffinized through xylene and rehydrated through graded ethanol concentrations into distilled water, as previously described.[18] Shortly, antigen retrieval was done by boiling the slides in 10mM citrate buffer (pH 6) for 10 minutes. Endogenous peroxidase quenching (30 min incubation in 0.66% (vol/vol) H2O2/PBS) was followed by blocking of nonspecific binding with normal horse blocking serum (Vector Laboratories) for 30 min at room temperature (RT). Anti-rabbit calbindinD28k (1:500) and anti-guinea pig TRPV5 antibody (1:1500) were incubated overnight at 4°C. After washing with PBS, slides were treated with goat anti-rabbit Alexa 488 (1:300) (for CalbindinD28k) and Cy2 AffiniPure donkey anti-guinea pig IgG (1:150) (for TRPV5) for 1 hour at RT. Sections were dehydrated in methanol and mounted with Mowiol. Negative controls were performed by incubation with nonimmune serum in place of a specific antibody, which resulted in complete absence of staining. Images were taken with Zeiss fluorescence microscope with a digital camera (Nikon DMX1200).
For calcium staining in aortic sections, samples were deparaffinized, rehydrated and stained in 2% Alizarin red solution (Sigma A3757, Sigma Aldrich, SL, MO, USA) at pH between 4,1–4,3 for 5 min. After staining, samples were rehydrated with acetone, acetone-xylene (1:1), xylene and mounted in synthetic mounting medium (DPX, Sigma Aldrich, SL, MO, USA).
Real time PCR
Total RNA was extracted from the kidney and duodenum samples using TRIzol reagent (Sigma Aldrich, SL, MO, USA) and following manufacturer’s instructions. The RNA concentrations were determined by nanodrop (ND-100) spectrophotometer. Reverse transcription was performed as previously described.[19] Real time PCR with gene-specific SYBR Green primers for mouse TRPV5 (5’CTGGAGCTTGTGGTTTCCTC3’), Calbindin-D28k (5’GACGGAAGTGGTTACCTGGA3’), Calbindin-D9k (5’CCTGCAGAAATGAAGAGCATTTT3’), PMCA1b (5’GTCACCGGCCTTACGTGTAT3’), NCX1 (5’GTGACTGCCGTTGTGTTTGT3’), TRPV6 (5’GGCCTCACAACCTCATTTAC3’) and GAPDH (5’TAACATCAAATGGGGTGAGG3’) was performed with a CFX Real-Time PCR detection system (Bio-Rad Laboratories, S.A., Madrid, Spain). Forty cycles at 95°C for 15 seconds and 60°C for 1 minute were performed. Duplicate readings were taken, and the average was calculated. The relative mRNA levels were calculated by standard formulae (ΔΔCt method) using mouse GAPDH as an endogenous control. The results referred to a randomly selected basal sample that was considered as value = 1.
Western blot analysis
Kidney tissue was homogenized in lysis buffer (50 mM HEPES, 250 mM NaCl, 5 mM EDTA, 0,1% Nonidet P-40) using polytron homogenizer. After centrifugation for 10 min at 10000 rpm, 4°C, supernatant was collected and protein concentration was determined using a BCA protein assay kit (Bio-Rad). 25μg of proteins were electrophoresed on 12% SDS-PAGE gels and transferred to PVDF membranes (Immobilon-P, Millipore), as previously described.[20] Membranes were probed with primary antibodies against CalbindinD28k (1:10000) and α-tubulin (1:5000) over night at 4°C. Appropriate horseradish peroxidase-conjugated secondary antibodies were used at 1:10000. The immunoreaction was visualized using chemiluminescent kits EZ ECL (Biological Industries) or ECL Advanced (Amersham Biosciences). Images were digitally acquired by Chemidoc (Bio Rad). Positive immunoreactive bands were quantified by densitometry and compared with the expression of adequate loading control (α-tubulin).
Statistical analysis
Differences between groups were assessed by one-way ANOVA and Bonferroni posthoc test. Differences between WT and KO mice were assessed by two-way ANOVA and Bonferroni posthoc test. A p<0.05 was considered statistically significant. All data examined are expressed as mean ± SEM.
Results
25OHD3 restores calcium and PTH levels in CYP27B1-/- mice with CKD
We first sought to determine the dose of 25OHD3 effective enough to maintain serum Ca2+ and PTH levels in CYP27B1-/- mouse model with CKD. CYP27B1-/- (KO) mice were subjected to subtotal nephrectomy (KO NX) and treated with 25, 50 and 100 ng/g of 25OHD3 three times per week for 30 days. The dose of 25OHD3 that was able to normalize serum Ca2+ and reduce PTH levels in KO NX animals similar to 1,25OH2D3 (50 pg/g) was 50 ng/g of 25OHD3 (S1 Fig). This dose was chosen for further investigation in CYP27B1+/+ (WT) and CYP27B1-/- (KO) mice. After subtotal nephrectomy, serum BUN levels increased in all nephrectomized groups of mice (Fig 1A), suggesting a similar degree of renal impairment. Calcium levels (Fig 1B), generally lower in KO mice (both in normal and NX conditions), increased with 25OHD3 treatment reaching similar levels as in WT animals. Neither nephrectomy nor 25OHD3 treatment did modify serum calcium levels of WT animals (Fig 1B). Phosphate levels, which were comparable in control and NX groups of mice, significantly increased only in WT NX animals treated with 25OHD3 (Fig 1C). Levels of PTH, which are very high in KO mice, increased even more after the nephrectomy and were partially corrected in KO NX mice after the treatment with 25OHD3 (Fig 1D). 25OHD3 had no effect on elevated PTH levels in WT NX mice (Fig 1D). Serum levels of 25OHD3 increased in both groups of mice treated with 25OHD3, but were higher in KO than in WT mice (Fig 1E). Circulating 1,25OH2D3 levels slightly increased in WT animals treated with 50 ng/g of 25OHD3 (WT NX: 69.49 ± 23.9; WT NX+25OH: 81,64 ± 15 pg/ml. p:0.6).
Fig 1. 25OHD3 restores calcium and PTH levels in CYP27B1-/- mice with CKD.

A: BUN B: Calcium C: Phosphate D: PTH and E: 25OHD3 serum levels from CYP27B1+/+ (WT) and CYP27B1-/- (KO) control mice, nephrectomized (nx) and nephrectomized treated with 50 ng/g of 25(OH)D3 for 1 month. Data are mean ± SEM * p< 0,001 vs. WT, # p< 0,05 vs. Control, † p< 0,05 vs. Nx, (n = 5 to 7).
25OHD3 does not induce vascular calcification in CYP27B1-/- mice with CKD
The effect of 25OHD3 on vascular calcification in WT and KO mice is shown in Fig 2. Treatment with 25OHD3 showed a tendency to increase calcium content in arteries of WT mice (Fig 2A), but did not modify calcium content in KO animals. Representative histochemical alizarin red staining from aortas showed an increase of the staining in the elastic laminas of WT mice treated with 25OHD3 (Fig 2B).
Fig 2. 25OHD3 does not induce vascular calcification in CYP27B1-/- mice with CKD.
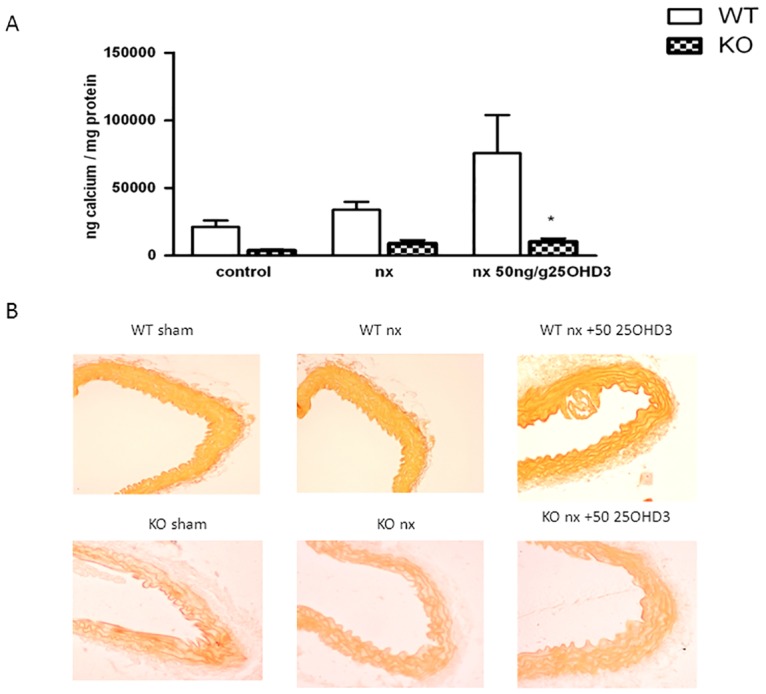
A: Calcium content in abdominal aorta normalized to protein in CYP27B1+/+ (WT) (white bars) and CYP27B1-/- (KO) control mice, nephrectomized (nx) and nephrectomized treated with 25OHD3 (50ng/g) for 1 month. B: Representative histochemical of alizarin red staining from aortas of mice from the same groups. Data are mean ± SEM (n = 5 to 7) * p< 0,001 vs WT mice. † p< 0,05 vs. nx.
25OHD3 increases expression of TRPV6 and Calbindin-D9k in duodenum of CYP27B1-/- mice with CKD
We further investigated whether treatment with 25OHD3 had influence on the expression of genes encoding Ca2+ transport proteins involved in duodenal transcellular Ca2+ absorption. Administration of 50 ng/g of 25OHD3 produced a 3-fold increase in TRPV6 mRNA in KO NX mice, while in WT animals the increase of TRPV6 mRNA was already evident after the nephrectomy and did not further increase with the treatment (Fig 3A). Calbindin-D9k mRNA levels showed a similar profile (Fig 3B). The expression of PMCa1b mRNA was not significantly modified by any of the conditions (Fig 3C).
Fig 3. 25OHD3 increases expression of TRPV6 and Calbindin-D9k in duodenum of CYP27B1-/- mice with CKD.

A: TRPV6 B: Calbindin-D9K and C: PMCA1b mRNA expression (qPCR) in CYP27B1+/+ (WT) and CYP27B1-/- (KO) control mice, nephrectomized (nx) and nephrectomized treated with 25OHD3 for 1 month. Data are mean ± SEM * p< 0,001 vs. WT, # p< 0,05 vs. Control, † p< 0,05 vs. Nx, (n = 5 to 7).
25OHD3 increases expression of Ca2+ transport genes and proteins in the kidney of CYP27B1-/- with CKD
The effect of 25OHD3 on mRNA expression of Ca2+ transport genes (TRPV5, Calbindin-D28k, NXC1 and PMCA1b) in the kidney was investigated by real-time quantitative PCR. The profile of Calbindin-D28k mRNA expression in the kidney was similar to the expression of Calbindin-D9k mRNA in the duodenum. Namely, nephrectomy (NX) increased Calbindin-D28k mRNA expression in the kidneys of WT mice, which was not further increased by 25OHD3 treatment (Fig 4A). On the contrary, nephron reduction in KO mice did not modify the expression of Calbindin-D28k mRNA, but the treatment with 25OHD3 managed to increase the expression of this gene in the kidney, although to the levels lower than in WT animals (Fig 4A). Analysis of protein expression, determined by Western blot (Fig 4B), corroborated results obtained by RT-PCR analysis. Furthermore, immunofluorescence analysis showed an increase of Calbindin-D28k staining in distal tubules of KO NX mice treated with 25OHD3 (Fig 4C).
Fig 4. 25OHD3 increases expression of CalbindinD-28K in the kidneys of CYP27B1-/- mice with CKD.

A: mRNA expression (qPCR) B: protein expression (western blot) and C: immunohistochemistry of CalbindinD-28K in CYP27B1+/+ (WT) and CYP27B1-/- (KO) control mice, nephrectomized (nx) and nephrectomized treated with 25OHD3 for 1 month. Data are mean ± SEM * p< 0,001 vs. WT, # p< 0,05 vs. Control, † p< 0,05 vs. Nx, (n = 5 to 7).
Expression of TRPV5 significantly increased in the WT group after the NX and did not further increase with the 25OHD3 treatment (Fig 5A). In the KO group, NX did not modify the expression of TRPV5 mRNA, but the treatment with 25OHD3 was able to increase the expression of this gene (Fig 5A). Immunofluorescence analysis showed an increase of TRPV5 staining in distal convoluted and connecting tubules of KO NX mice treated with 25OHD3 (Fig 5B). Expression of the basolateral extrusion genes (NCX1 and PMCA1b) was not affected by 25OHD3 (Fig 6A and 6B) in KO NX mice. Nevertheless, WT NX mice showed an increase of PMCA1b mRNA expression after administration of 25OHD3 treatment (Fig 6A), while the expression of NCX1 increased in the both WT NX groups (untreated and treated with 25OHD3) (Fig 6B).
Fig 5. 25OHD3 increases expression of TRPV5 in the kidneys of CYP27B1-/- mice with CKD.
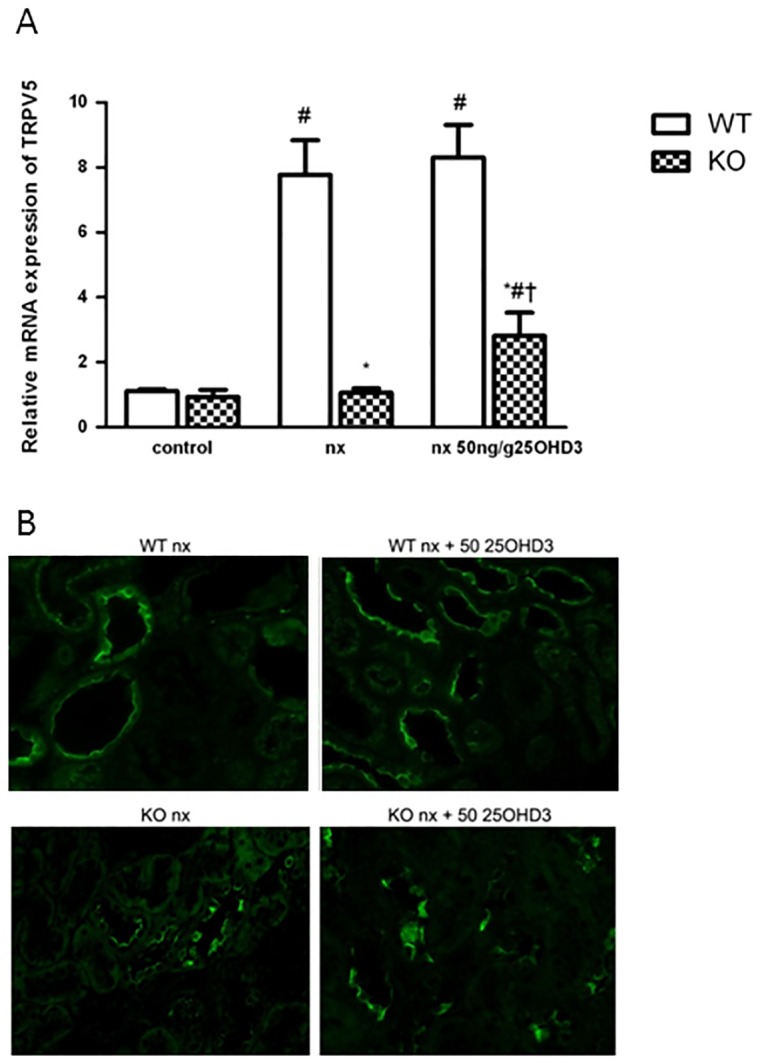
A: mRNA expression (qPCR) and B: immunohistochemistry of TRPV5 in CYP27B1+/+ (WT) and CYP27B1-/- (KO) control mice, nephrectomized (nx) and nephrectomized treated with 25OHD3 for 1 month. Data are mean ± SEM * p< 0,001 vs. WT, # p< 0,05 vs. Control, † p< 0,05 vs. Nx, (n = 5 to 7).
Fig 6. The effect of 25OHD3 on the expression of renal PMCA1b and NCX1 in CYP27B1+/+ and CYP27B1-/- mice.
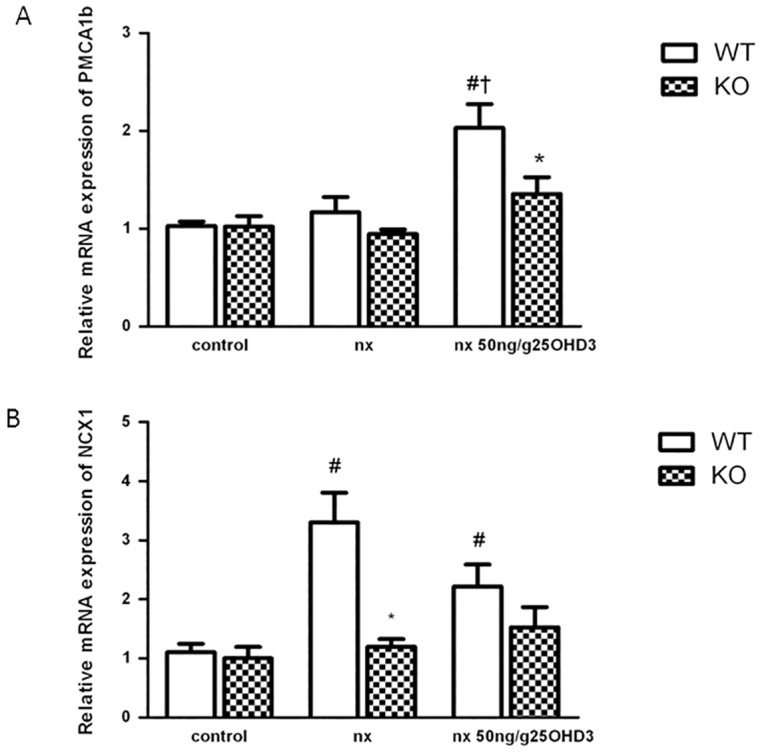
A: mRNA expression (qPCR) of PMCA1b and B: NCX1 in CYP27B1+/+ (WT) and CYP27B1-/- (KO) control mice, nephrectomized (nx) and nephrectomized treated with 25OHD3 for 1 month. Data are mean ± SEM * p< 0,001 vs. WT, # p< 0,05 vs. Control, † p< 0,05 vs. Nx, (n = 5 to 7).
Discussion
In the present paper we demonstrate that the suppression of PTH in an experimental model of CKD can be achieved directly by 25OHD3 without its conversion to 1,25(OH)2D3, but the blood levels necessary to attain the effect are extremely high. Furthermore, although the effect is undoubtedly due to an effect of 25OHD3, it is unclear whether this effect is directly stimulating VDR in the parathyroid gland or indirectly by increasing calcium absorption in the intestine. Thus, the administration of 50 ng/g of 25OHD3 in KO NX animals decreased serum PTH to levels below the ones observed in the same animals with normal renal function. This inhibition was achieved with blood levels of 25OHD3 around 7 times higher than the normal levels and in parallel to a normalization of blood calcium levels. Thus, the decrease of PTH could be attained by a combination of normalization of blood calcium levels and direct activation of VDR by 25OHD3 in the parathyroid gland.
The results of the present study demonstrate that 25OHD3 can activate VDR in uremic conditions, and agree with previous results showing similar results in animals with normal renal function.[21] It has been shown that uremic milieu contains toxins that block the binding of activated VDR with VDRE in the promoter of target genes.[22] Thus, inhibition of VDR target gene expression by uremia can partially explain the resistance to 1,25(OH)2D3 observed in CKD. Our results show that reaching levels of 25OHD3 in blood high enough can overcome the mentioned problem. We demonstrate that treatment with 25OHD3 increased the expression of calcium channels in the kidney and intestine, most likely by direct effect of activated VDR on the promoter of target genes.
Furthermore, the administration of the same dose of 25OHD3 to WT NX animals showed interesting results. Namely, the dose administered was unable to significantly reduce the increase of PTH levels induced by the nephrectomy and high phosphate feeding. Although the PTH levels obtained with the nephrectomy in WT NX animals were far lower than measured in KO NX animals, there are similar to the ones found in CKD patients.[23] In any case, this different behavior of the treatment could be due to two reasons. On the one hand, the levels of 25OHD3 achieved in blood of WT NX mice were much lower than the ones observed in the KO NX animals. This effect could be explained by a higher rate of degradation of 25OHD3 or by its conversion to 1,25(OH)2D3. The degradation of both, 1,25(OH)2D3 and 25OHD3 is mediated by the enzyme 24-hydroxylase, the levels of which are highly induced by active vitamin D compounds.[24] Thus the levels of 24-hydroxylase are expected to be very low in the KO animals, increasing the half-life of the 25OHD3 and its blood concentration. Furthermore, and in contrast to KO mice, part of the 25OHD3 administered to the WT animals can be converted to 1,25(OH)2D3, which will also decrease 25OHD3 levels. Although this represents a very small portion of 25OHD3, the circulating levels of 1,25(OH)2D3 did not significantly increase, it could also have an influence on 25OHD3 blood levels. On the other hand, the administration of 25OHD3 to the WT animals induced an increase in the levels of phosphate which have been shown to stimulate PTH synthesis and release.[25] This effect can also be explained by the partial conversion of 25OHD3 into 1,25(OH)2D3 and subsequent activation of intestinal VDR, which has been shown to increase phosphate absorption from the intestine, although this effect should minor since circulating levels of 1,25(OH)2D3 did not significantly increase.[26]
Furthermore, the administration of the same doses of 25OHD3, although ineffective to decrease PTH levels, showed a tendency to increase vascular calcification. Thus, calcium levels in arteries of WT NX animals treated with 25OHD3 were significantly increased compared to the treated KO NX mice. The Alizarin red staining, although not showing an evident increase in vascular calcification, showed an increase in the red staining of the elastic laminas, suggesting an increase in calcium deposition in that area. The involvement of elastic lamina in medial calcification in patients, although challenged in the past, seems to be accepted nowadays.[27, 28] This pattern is also found in some genetically modified mice affected by extensive medial vascular calcification.[29, 30] The increase of vascular calcification in WT animals can be explained by the increase in P levels. It has been shown that high P levels are of paramount importance in the genesis of vascular calcification. Thus, increases in blood P have been shown to be associated with vascular calcification both in patients and in experimental animals.[31] Furthermore, a recent paper from our laboratory has shown that the upregulation of local CYP27B1 in the vascular smooth muscle cells is of paramount importance in the genesis of uremic vascular calcification.[17] Thus, in KO animals the absence of CYP27B1 blocks the increase in vascular calcification. However, in WT animals the administration of 25(OH)D3 could exacerbate the pro-calcificant effects of uremia by increasing the substrate for local production of 1,25(OH)2D3 in the artery.
We have also shown an effect of the uremia and the treatment with 25OHD3 on the expression of the proteins related to calcium transport both in the duodenum and in the kidney. The effect of the 25OHD3 treatment on the expressions of TRPV5, TRPV6, calbindins D9K and 28K in KO NX animals was similar. Thus NX did not have an effect of the levels of any of the proteins, but the treatment with 25OHD3 significantly increased its expression. Several VDRE have been identified in both TRPV and calbindin gene promoters.[32–35] Thus, the extremely high levels of 25OHD3 reached in the treated KO NX animals could be able to activate the VDRE in the promoter of the genes. In the WT animals however, the NX already increased the expression levels of all four genes. The effect of CKD on renal calcium transporter has been recently described.[36] The increase of proteins related to calcium transport has been attributed to a possible effect of PTH, FGF23 or even vitamin D. Our results show that active vitamin D must be playing a central role in this increase, because the effect is absent in KO animals. Furthermore, this is the first report showing that experimental CKD also induces an increase in duodenal calcium transport mechanisms. In the WT NX animals, the increase of expression of all four genes was not further induced by the administration of 25OHD3, either because the expression was already submaximal or because the levels of 25OHD3 achieved in the WT NX animals were insufficient to further increase the promoter activity of those four genes. The effects of uremia and the treatments on NCX1 followed a similar pattern. However, the expression of PMCA1b on both tissues was not affected in the same way, suggesting a different regulation of this gene. Thus, and in agreement with previous results, renal levels were increased by treatment with vitamin D.[37] However, intestinal levels of PMCA1b were not affected in any of the conditions confirming that, as it has been suggested previously, vitamin D effects on PMCA1b gene could be tissue specific.[38]
As any study in genetically modified animals, the main strength of the study relays in the fact that we can be absolutely sure that all the effects observed in the treatments are due to a direct effect of 25OHD3, as conversion to the active metabolite is totally blocked. The main limitation is that we did not collect parathyroid glands in the treated animals, and we can not differentiate whether the PTH reduction effect is mediated by direct effects on VDR in the parathyroid gland or in intestinal cells.
In conclusion, 25OHD3 is able to reduce PTH in a CYP27B1-/- mouse model of CKD without direct conversion to active vitamin D, but the blood levels needed are extremely high. Although this effect on PTH is due to an increase of VDR signaling by 25OHD3, it could be a combination of a direct effect in the parathyroid gland and an induction of the calcium absorption machinery in the intestine, and the subsequent normalization of Ca blood levels.
Supporting Information
Levels of Ca (A), P (B) and PTH (C) in sham-operated KO animals, NX KO animals and NX KO animals treated with 50 pg/g of 1,25(OH)2D3 or 25, 50 and 100 ng/g of 25OHD3. *: p<0.01 vs KO sham. #: p<0.01 vs KO NX.
(TIF)
Acknowledgments
We thank Dr Joost Hoenderop and Dr Rene Bindels for their invaluable help in the elaboration of this manuscript.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the Instituto de Salud Carlos III PS12/01770, RD12/0021/0026. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Brown AJ, Dusso A, Slatopolsky E. Vitamin D. American Journal of Physiology-Renal Physiology. 1999;277(2):F157–F75. [DOI] [PubMed] [Google Scholar]
- 2.Fraser DR, Kodicek E. Unique Biosynthesis by Kidney of A Biologically Active Vitamin-D Metabolite. Nature. 1970;228(5273):764–6. [DOI] [PubMed] [Google Scholar]
- 3.Panda DK, Miao D, Tremblay ML, Sirois J, Farookhi R, Hendy GN, et al. Targeted ablation of the 25-hydroxyvitamin D 1 alpha-hydroxylase enzyme: Evidence for skeletal, reproductive, and immune dysfunction. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(13):7498–503. 10.1073/pnas.131029498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodman WG. Recent developments in the management of secondary hyperparathyroidism. Kidney International. 2001;59(3):1187–201. 10.1046/j.1523-1755.2001.0590031187.x [DOI] [PubMed] [Google Scholar]
- 5.Cozzolino M, Brancaccio D, Gallieni M, Slatopolsky E. Pathogenesis of vascular calcification in chronic kidney disease. Kidney International. 2005;68(2):429–36. 10.1111/j.1523-1755.2005.00421.x [DOI] [PubMed] [Google Scholar]
- 6.Brown AJ, Finch J, Slatopolsky E. Differential effects of 19-nor-1,25-dihydroxyvitamin D(2) and 1,25-dihydroxyvitamin D(3) on intestinal calcium and phosphate transport. JLab ClinMed. 2002;139(5):279–84. [DOI] [PubMed] [Google Scholar]
- 7.Cardus A, Panizo S, Parisi E, Fernandez E, Valdivielso JM. Differential effects of vitamin D analogs on vascular calcification. Journal of Bone and Mineral Research. 2007;22(6):860–6. 10.1359/jbmr.070305 [DOI] [PubMed] [Google Scholar]
- 8.Slatopolsky E, Cozzolino M, Finch JL. Differential effects of 19-nor-1,25-(OH)(2)D(2) and 1alpha-hydroxyvitamin D(2) on calcium and phosphorus in normal and uremic rats. Kidney Int. 2002;62(4):1277–84. 10.1111/j.1523-1755.2002.kid573.x [DOI] [PubMed] [Google Scholar]
- 9.Hoenderop JGJ, Nilius B, Bindels RJM. Molecular mechanism of active Ca2+ reabsorption in the distal nephron. Annual Review of Physiology. 2002;64:529–49. 10.1146/annurev.physiol.64.081501.155921 [DOI] [PubMed] [Google Scholar]
- 10.Hoenderop JGJ, van der Kemp AWCM, Hartog A, van de Graaf SFJ, Van Os CH, Willems PHGM, et al. Molecular identification of the apical Ca2+ channel in 1,25-dihydroxyvitamin D-3-responsive epithelia. Journal of Biological Chemistry. 1999;274(13):8375–8. [DOI] [PubMed] [Google Scholar]
- 11.Peng JB, Chen XZ, Berger UV, Vassilev PM, Tsukaguchi H, Brown EM, et al. Molecular cloning and characterization of a channel-like transporter mediating intestinal calcium absorption. Journal of Biological Chemistry. 1999;274(32):22739–46. [DOI] [PubMed] [Google Scholar]
- 12.KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney IntSuppl. 2009;(113):S1–130. [DOI] [PubMed] [Google Scholar]
- 13.K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis. 2003;42(4 Suppl 3):S1–201. [PubMed] [Google Scholar]
- 14.Mazzaferro S, Goldsmith D, Larsson TE, Massy ZA, Cozzolino M. Vitamin D Metabolites and/or Analogs: Which D for Which Patient? Current Vascular Pharmacology. 2014;12(2):339–49. [DOI] [PubMed] [Google Scholar]
- 15.Moslehi N, Shab-Bidar S, Mirmiran P, Hosseinpanah F, Azizi F. Determinants of parathyroid hormone response to vitamin D supplementation: a systematic review and meta-analysis of randomised controlled trials. British Journal of Nutrition. 2015;114(9):1360–74. 10.1017/S0007114515003189 [DOI] [PubMed] [Google Scholar]
- 16.Rowling MJ, Gliniak C, Welsh J, Fleet JC. High dietary vitamin D prevents hypocalcemia and osteomalacia in CYP27B1 knockout mice. Journal of Nutrition. 2007;137(12):2608–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torremadé N, Bozic M, Panizo S, Barrio-Vazquez S, Fernandez-Martín JL, Encinas M, et al. Vascular Calcification Induced by Chronic Kidney Disease Is Mediated by an Increase of 1α-Hydroxylase Expression in Vascular Smooth Muscle Cells. J Bone Miner Res. 2016;31(10):1865–76. 10.1002/jbmr.2852 [DOI] [PubMed] [Google Scholar]
- 18.Bozic M, de Rooij J, Parisi E, Ortega MR, Fernandez E, Valdivielso JM. Glutamatergic Signaling Maintains the Epithelial Phenotype of Proximal Tubular Cells. Journal of the American Society of Nephrology. 2011;22(6):1099–111. 10.1681/ASN.2010070701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bozic M, Alvarez A, de Pablo C, Sanchez-Nino M-D, Ortiz A, Dolcet X, et al. Impaired Vitamin D Signaling in Endothelial Cell Leads to an Enhanced Leukocyte-Endothelium Interplay: Implications for Atherosclerosis Development. Plos One. 2015;10(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bozic M, Panizo S, Sevilla MA, Riera M, Soler MJ, Pascual J, et al. High phosphate diet increases arterial blood pressure via a parathyroid hormone mediated increase of renin. Journal of Hypertension. 2014;32(9):1822–32. [DOI] [PubMed] [Google Scholar]
- 21.Deluca HF, Prahl JM, Plum LA. 1,25-Dihydroxyvitamin D is not responsible for toxicity caused by vitamin D or 25-hydroxyvitamin D. Archives of Biochemistry and Biophysics. 2011;505(2):226–30. 10.1016/j.abb.2010.10.012 [DOI] [PubMed] [Google Scholar]
- 22.Patel SR, Ke HQ, Vanholder R, Koenig RJ, Hsu CH. Inhibition of Calcitriol Receptor-Binding to Vitamin-D Response Elements by Uremic Toxins. Journal of Clinical Investigation. 1995;96(1):50–9. 10.1172/JCI118061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Craver L, Paz Marco M, Martinez I, Rue M, Borras M, Luisa Martin M, et al. Mineral metabolism parameters throughout chronic kidney disease stages 1–5—achievement of K/DOQI target ranges. Nephrology Dialysis Transplantation. 2007;22(4):1171–6. [DOI] [PubMed] [Google Scholar]
- 24.Tanaka Y, Deluca HF. Stimulation of 24,25-Dihydroxyvitamin-D3 Production by 1,25-Dihydroxyvitamin-D3. Science. 1974;183(4130):1198–200. [DOI] [PubMed] [Google Scholar]
- 25.Almaden Y, Hernandez A, Torregrosa V, Canalejo A, Sabate L, Cruz LF, et al. High phosphate level directly stimulates parathyroid hormone secretion and synthesis by human parathyroid tissue in vitro. Journal of the American Society of Nephrology. 1998;9(10):1845–52. [DOI] [PubMed] [Google Scholar]
- 26.Carlsson A. The Effect of Vitamin-D on the Absorption of Inorganic Phosphate. Acta Physiologica Scandinavica. 1954;31(4):301–7. [DOI] [PubMed] [Google Scholar]
- 27.Micheletti RG, Fishbein GA, Currier JS, Singer EJ, Fishbein MC. Calcification of the internal elastic lamina of coronary arteries. Modern Pathology. 2008;21(8):1019–28. 10.1038/modpathol.2008.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiao JH, Doherty TM, Fishbein MC, Salusky IB, Luthringer DL, Fitzpatrick LA, et al. Calcification of the coronary arteries in the absence of atherosclerotic plaque. Mayo Clinic Proceedings. 2005;80(6):807–9. 10.1016/S0025-6196(11)61536-X [DOI] [PubMed] [Google Scholar]
- 29.Murshed M, Schinke T, McKee MD, Karsenty G. Extracellular matrix mineralization is regulated locally; different roles of two gla-containing proteins. J Cell Biol. 2004;165(5):625–30. 10.1083/jcb.200402046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stubbs JR, Liu S, Tang W, Zhou J, Wang Y, Yao X, et al. Role of hyperphosphatemia and 1,25-dihydroxyvitamin D in vascular calcification and mortality in fibroblastic growth factor 23 null mice. Journal of the American Society of Nephrology. 2007;18(7):2116–24. 10.1681/ASN.2006121385 [DOI] [PubMed] [Google Scholar]
- 31.Kendrick J, Chonchol M. The Role of Phosphorus in the Development and Progression of Vascular Calcification. American Journal of Kidney Diseases. 2011;58(5):826–34. 10.1053/j.ajkd.2011.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gill RK, Christakos S. Identification of Sequence Elements in Mouse Calbindin-D(28K) Gene That Confer 1,25-Dihydroxyvitamin-D(3)-Inducible and Butyrate-Inducible Responses. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(7):2984–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H, Christakos S. Differential Regulation by 1,25-Dihydroxyvitamin-D3 of Calbindin-D9K and Calbindin-D28K Gene-Expression in Mouse Kidney. Endocrinology. 1991;128(6):2844–52. 10.1210/endo-128-6-2844 [DOI] [PubMed] [Google Scholar]
- 34.Meyer MB, Watanuki M, Kim S, Shevde NK, Pike JW. The human transient receptor potential vanilloid type 6 distal promoter contains multiple vitamin D receptor binding sites that mediate activation by 1,25-dihydroxyvitamin D-3 in intestinal cells. Molecular Endocrinology. 2006;20(6):1447–61. 10.1210/me.2006-0031 [DOI] [PubMed] [Google Scholar]
- 35.Nijenhuis T, Hoenderop JGJ, van der Kemp AWCM, Bindels RJM. Localization and regulation of the epithelial Ca2+ channel TRPV6 in the kidney. Journal of the American Society of Nephrology. 2003;14(11):2731–40. [DOI] [PubMed] [Google Scholar]
- 36.Pulskens WP, Verkaik M, Sheedfar F, van Loon EP, van de SB, Vervloet MG, et al. Deregulated Renal Calcium and Phosphate Transport during Experimental Kidney Failure. Plos One. 2015;10(11):e0142510 10.1371/journal.pone.0142510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glendenning P, Ratajczak T, Dick IM, Prince RL. Calcitriol upregulates expression and activity of the 1b isoform of the plasma membrane calcium pump in immortalized distal kidney tubular cells. Archives of Biochemistry and Biophysics. 2000;380(1):126–32. 10.1006/abbi.2000.1908 [DOI] [PubMed] [Google Scholar]
- 38.Glendenning P, Ratajczak T, Prince RL, Garamszegi N, Strehler EE. The promoter region of the human PMCA1 gene mediates transcriptional downregulation by 1,25-dihydroxyvitamin D-3. Biochemical and Biophysical Research Communications. 2000;277(3):722–8. 10.1006/bbrc.2000.3745 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Levels of Ca (A), P (B) and PTH (C) in sham-operated KO animals, NX KO animals and NX KO animals treated with 50 pg/g of 1,25(OH)2D3 or 25, 50 and 100 ng/g of 25OHD3. *: p<0.01 vs KO sham. #: p<0.01 vs KO NX.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.