

Asxl1 loss–induced myeloid malignancies are mediated partially through impaired ASXL1 cohesin interaction and cohesin functions.
Keywords: ASXL1, cohesin complex, hematopoiesis, gene regulation
Abstract
ASXL1 is frequently mutated in a spectrum of myeloid malignancies with poor prognosis. Loss of Asxl1 leads to myelodysplastic syndrome–like disease in mice; however, the underlying molecular mechanisms remain unclear. We report that ASXL1 interacts with the cohesin complex, which has been shown to guide sister chromatid segregation and regulate gene expression. Loss of Asxl1 impairs the cohesin function, as reflected by an impaired telophase chromatid disjunction in hematopoietic cells. Chromatin immunoprecipitation followed by DNA sequencing data revealed that ASXL1, RAD21, and SMC1A share 93% of genomic binding sites at promoter regions in Lin−cKit+ (LK) cells. We have shown that loss of Asxl1 reduces the genome binding of RAD21 and SMC1A and alters the expression of ASXL1/cohesin target genes in LK cells. Our study underscores the ASXL1-cohesin interaction as a novel means to maintain normal sister chromatid separation and regulate gene expression in hematopoietic cells.
INTRODUCTION
ASXL1 (additional sex combs like 1) gene mutations frequently occur in a spectrum of myeloid malignancies, including myelodysplastic syndrome (MDS), chronic myelomonocytic leukemia (CMML), myeloproliferative neoplasms (MPNs), and acute myeloid leukemia (AML) (1–4). ASXL1 mutation is a poor prognostic marker for MDS, CMML, and AML (5–7), suggesting an important role of ASXL1 mutations in disease initiation and progression. The ASXL1 gene encodes ASXL1, one of the polycomb group proteins. These proteins are necessary for the maintenance of stable repression of homeotic genes and other gene loci (8–10). We and others have reported that loss of Asxl1 leads to the development of MDS-like diseases in mice, which can progress to bone marrow (BM) failure or MPN (11, 12). ASXL1 has also been shown to regulate the self-renewal and differentiation of mesenchymal stromal cells and erythropoiesis (13, 14). In addition, loss of Asxl1 in hematopoietic stem cells (HSCs)/hematopoietic progenitor cells (HPCs) reduces global levels of histone H3 lysine 27 trimethylation (H3K27me3) and H3K4me3, and alters the expression of genes implicated in apoptosis (11).
Cohesin is a multiple-subunit protein complex that is highly conserved in mammalian cells (15). The cohesin complex consists of four major subunits: RAD21, SMC1A, SMC3, and STAG1/STAG2 (16, 17). The core cohesin proteins form a triangular ring, which embraces sister chromatids and prevents their premature separation (18, 19). Besides its major function in sister chromatid cohesion, the cohesin complex participates in many other cellular processes, such as transcriptional regulation through long-range cis interactions (20–26). Recently, clinical studies have discovered recurrent mutations or deletions in the cohesin genes in a variety of myeloid malignancies, including MDS, AML, CMML, and chronic myelogenous leukemia (16, 27, 28). Furthermore, cohesin mutations occur in a mutually exclusive manner (16, 29, 30). A more recent study by Merkenschlager and Odom (31) suggests that cohesin associates with enhancers, promoters, and sites defined by CTCF (CCCTC-binding factor) binding to form regulated networks of long-range interactions that can promote cell type–specific transcriptional programs.
Here, we report that ASXL1 interacts with the core proteins of the cohesin complex. Chromatin immunoprecipitation followed by DNA sequencing (ChIP-seq) analysis revealed a significant overlap between the ASXL1, RAD21, and SMC1A binding sites on the genome, mainly located at the promoter regions of genes. Loss of Asxl1 decreased RAD21 and SMC1A occupancy on the genome and altered expression of their target genes. Deletion of Asxl1 results in a significantly higher frequency of impaired telophase chromatid disjunction in hematopoietic cells, congruent with the previous finding by Díaz-Martínez et al. (32); the silencing of RAD21 leads to the nuclear bridging of HeLa cells. Collectively, these data demonstrate a novel biological function of ASXL1 in transcriptional regulation via interaction with cohesin complex proteins, in addition to the maintenance of normal chromatid separation. These findings provide strong evidence that ASXL1 is essential for the maintenance of gene expression and for preventing dysplastic morphology formation via cohesin complex in HSC/HPCs.
RESULTS
ASXL1 interacts with cohesin complex proteins
Located in the nucleus and capable of regulating gene expression, ASXL1 should exert its function in concert with its interacting partners. To identify functional ASXL1 interacting proteins, we performed protein affinity purification using an anti-FLAG antibody (fig. S1A) and nuclear extracts prepared from human embryonic kidney (HEK) 293T cells engineered to overexpress FLAG-tagged ASXL1 (fig. S1B). The ASXL1 interacting proteins were identified by liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis (table S1). We found that ASXL1 associates with all core members of the cohesin complex, including RAD21, SMC1A, SMC3, STAG1, and STAG2 (Fig. 1A). Co-immunoprecipitation (co-IP) and Western blotting analyses revealed that ASXL1 interacts with SMC1A, SMC3, and RAD21 in FLAG-ASXL1 overexpressing HEK293T cells (Fig. 1B). Furthermore, reciprocal IP confirmed the interaction of ASXL1 with SMC1A, SMC3, and RAD21 in FLAG-ASXL1 overexpressing HEK293T cells (Fig. 1, C to E). To study the endogenous interaction between ASXL1 and cohesin proteins, we carried out co-IP using primary BM cells of wild-type (WT) mice and showed that ASXL1 associates with endogenous SMC1A, SMC3, and RAD21 (Fig. 1F and fig. S1C).
Fig. 1. ASXL1 associates with cohesin complex proteins.
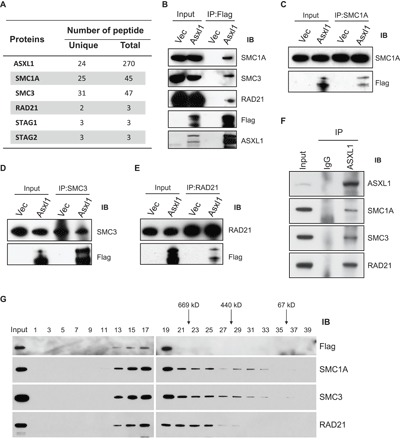
(A) Table of the most relevant proteins identified by LC-MS/MS in the affinity purification of ASXL1-associated proteins using FLAG-ASXL1 overexpressing HEK293T cells. Spectral counts (unique and total) for each interacting protein are shown. (B to E) Reciprocal IP and Western blotting confirmed interaction of ASXL1 with SMC1A, SMC3, and RAD21 in nuclear fraction derived from HEK293T cells transfected with pcDNA3.1+ (Vec) or FLAG-tagged ASXL1 (ASXL1). Nuclear extractions were subjected to IP using indicated antibodies against FLAG (B), SMC1A (C), SMC3 (D), or RAD21 (E). IB, immunoblot. (F) Western blot shows the endogenous interaction between ASXL1 and SMC1A, SMC3, and RAD21 in BM cells of WT mice. IgG, immunoglobulin G. (G) Gel filtration analysis of nuclear extracts from FLAG-ASXL1 overexpressing cells. ASXL1 and the cohesin complex were coeluted from a Superose 6 HR gel filtration column, as analyzed by Western blotting. The numbers over the lanes represent the eluted fraction numbers.
To determine whether ASXL1 and cohesin complex members coexist in a complex, we fractionated the nuclear extracts of ASXL1 overexpressing HEK293T cells on size exclusion chromatography. Western blot analysis showed that ASXL1 coeluted with SMC1A, SMC3, and RAD21 in high–molecular weight fractions (Fig. 1G), suggesting that ASXL1 is part of a large multiprotein complex that includes cohesin complex proteins.
To map the interacting region of ASXL1 with the cohesin complex, we generated a series of constructs that encode various FLAG-tagged ASXL1 truncations (Fig. 2A). We then overexpressed each of the ASXL1 truncates in HEK293T cells and performed Western blots on the anti-FLAG IP. ASXL1 full-length amino acids 1 to 1010 and 1 to 587, but not amino acids 1 to 420, successfully pulled down RAD21, SMC1A, and SMC3 (Fig. 2, B to D), indicating that the region spanning amino acids 420 to 587 is important for cohesin binding. Convincingly, amino acids 401 to 587 were capable of pulling down the cohesin complex (Fig. 2E). These data indicate that ASXL1 and cohesin form a complex in the nucleus, and the amino acid 401 to 587 region of ASXL1 mediates its interaction with the cohesin complex.
Fig. 2. Mapping the region of ASXL1 that mediates its binding to the cohesin complex.
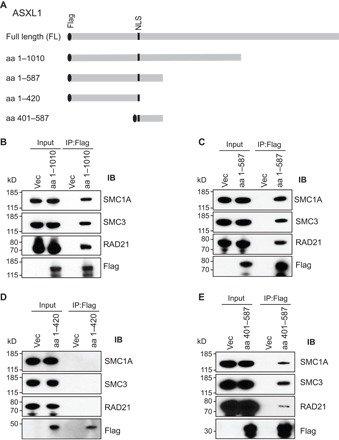
(A) Schematic diagram of the full-length (FL) ASXL1 and the truncated variants of Asxl1 [amino acids (aa) 1 to 1010, 1 to 420, 1 to 587, and 401 to 587]. Binding affinity was determined by the pull-down efficiency of IP with anti-FLAG and Western blotting with cohesin antibodies. NLS, nuclear localization signal. (B to E) Western blotting analysis of nuclear fractions and anti-FLAG immunoprecipitates from pcDNA3.1+, or each truncated ASXL1 transfected HEK293T cells using antibodies against FLAG, SMC1A, SMC3, or RAD21.
ASXL1 interacts with the cohesin complex to maintain the normal cell morphology and telophase chromatin disjunction
Cohesin complex proteins embrace sister chromatids by forming a ring-like structure; the defective function of any of the core cohesin proteins disrupts the sister chromatid separation (32, 33). Myeloid cells with depleted Asxl1 exhibit a specific dysplastic feature as a pseudo–Pelget-Hüet anomaly (11). Examination of the peripheral blood (PB) smear and BM of Asxl1+/− and Asxl1−/− mice revealed an increased frequency of cells with nuclear bridging and prominent disrupted sister chromatid separation in myeloid cells (Fig. 3, A and B, and fig. S2A). Consistently, significantly higher frequencies of cells with nuclear bridging and impaired telophase chromatid disjunction were observed, such as Asxl1+/− and Asxl1−/− cultures from Lin−cKit+ (LK) cells in the presence of a cocktail of growth factors including stem cell factor (SCF), interleukin-3 (IL-3), macrophage colony-stimulating factor, and thrombopoietin (Fig. 3, C and D).
Fig. 3. Loss of Asxl1 leads to premature sister chromatid separation in cells.
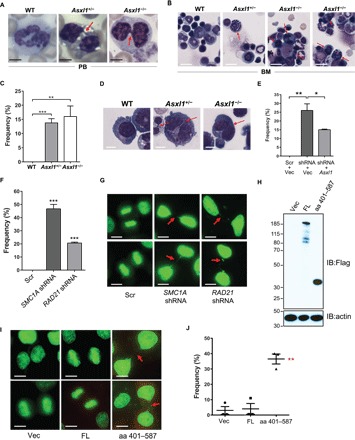
(A and B) The myeloid cells with premature sister chromatid separation are frequently seen in PB smears (A) and BM (B) of Asxl1+/− and Asxl1−/− mice with MDS. Red arrows indicate the abnormal nuclear bridging. Scale bars, 5 μm (A) and 10 μm (B). (C and D) Representative cells with premature sister chromatid separation in cultured WT, Asxl1+/−, and Asxl1−/− LK cells. Red arrows indicate the premature sister chromatid separation. The frequency of cells with premature sister chromatid separation is shown in (C). Y axis shows the percentage of cells with premature sister chromatid separation within all binucleated cells. Data are represented as means ± SEM from three independent experiments. ***P < 0.001 and **P < 0.01. Scale bars, 5 μm. (E) The frequency of cells with premature sister chromatid separation in the HeLaGFP-H2B cells with hASXL1 KD and hASXL1 KD plus mAsxl1 rescues. KD of ASXL1 leads to increased frequency of cells with premature sister chromatid separation in HeLaGFP-H2B cells. Reintroducing full-length mAsxl1 rescued the premature sister chromatid separation in HeLa cells with ASXL1 KD. Data are represented as means ± SEM from three independent experiments. ***P < 0.001 and **P < 0.01. (F and G) SMC1A or RAD21 KD leads to premature sister chromatid separation in HeLaGFP-H2B cells. Representative photomicrographs show the cells with premature sister chromatid separation, as indicated by red arrowheads (G). The frequency of cells with premature sister chromatid separation is shown in (F). Y axis shows the percentage of cells with premature sister chromatid separation within all binucleated cells. Data are represented as means ± SEM from three independent experiments. ***P < 0.001 and **P < 0.01. Scale bars, 5 μm. (H) Western blotting shows the expression of full-length ASXL1 and ASXL1 amino acids 401 to 587 in HeLaGFP-H2B cells transfected with vector only, full-length ASXL1, or ASXL1 amino acids 401 to 587. β-Actin serves as loading control. (I and J) ASXL1 amino acids 401 to 587 induce chromatin bridging in HeLaGFP-H2B cells. Quantification of the frequency of cells with premature sister chromatid separation in HeLaGFP-H2B cells transfected with pcDNA3.1+, full-length ASXL1, or ASXL1 amino acids 401 to 587 (J). Data are represented as means ± SEM from three independent experiments. **P < 0.01 for ASXL1 amino acids 401 to 587 fragment versus pcDNA3.1+ or full-length ASXL1. Red arrows indicate the premature sister chromatid separation. Scale bars, 5 μm.
To determine whether the increased frequency of cells with disrupted chromatin disjunction is a direct consequence of Asxl1 loss, we transfected human ASXL1-specific short hairpin RNA (shRNA-hASXL1) and/or mouse Asxl1 (mAsxl1) complementary DNA (cDNA) into HeLa cells stably expressing green fluorescent protein (GFP)–H2B (HeLaGFP-H2B). The mRNA levels of hASXL1 and mAsxl1 were determined by quantitative polymerase chain reaction (qPCR) with primers specific for hASXL1 and mAsxl1, respectively. Transduction of shRNA-hASXL1 successfully decreased the expression of hASXL1 mRNA by more than 40% but did not interfere with the expression of mAsxl1 (fig. S2B). Fluorescence microscopy was used to quantify the morphology of the cells with or without bridging in the nuclear. Knockdown (KD) of hASXL1 induced a markedly higher frequency of dysplastic nuclear bridging cells (Fig. 3E and fig. S2C), and reintroducing mASXL1 significantly reduced the frequency of cells with premature sister chromatid separation (Fig. 3E and fig. S2C).
A parallel study was carried out to knock down SMC1A or RAD21 in HeLaGFP-H2B cells. SMC1A KD resulted in a decreased expression of RAD21 and vice versa (fig. S2, D to F). This finding is consistent with those of previous studies by other groups using different cell systems that the protein levels of subunits of the cohesin complex are reduced upon depletion of other subunits of the complex, probably because of degradation (34–36). Consistently, both SMC1A and RAD21 KD led to higher frequency of cells with nuclear bridging in HeLa cells (Fig. 3, F and G). However, Western blot analysis showed that the levels of SMC1A, SMC3, and RAD21 proteins are comparable in WT and Asxl1−/− LK cells (fig. S2G). These results indicate that ASXL1, SMC1A, and RAD21 are required for the maintenance of normal cell morphology. Asxl1 loss–mediated cell bridging and premature chromatid separation are likely associated with an impaired cohesion function rather than dysregulating SMC1A or RAD21 expression.
To further determine whether ASXL1 maintains normal cell morphology through its interaction with the cohesin complex, we disrupted cohesin/ASXL1 interaction by expressing ASXL1 amino acids 401 to 587, the cohesin binding region of ASXL1 in HeLaGFP-H2B cells (Fig. 3H), and measured the frequency of cells with nuclear bridging. Expression of ASXL1 amino acids 401 to 587 markedly increased the frequency of cells containing premature sister chromatid separation compared to cells expressing full-length ASXL1 or vector control (Fig. 3I). This effect is presumably mediated by its disruption of the interaction between the endogenous WT ASXL1 and cohesin. These results indicate that ASXL1 is critical for the function of the cohesin complex to maintain normal cellular function during mitosis.
Asxl1 loss decreases the genomic occupancy of cohesin in LK cells
The cohesin complex binds to the genomic DNA sequence and regulates gene expression (37–39). To examine whether the genomic binding sites of ASXL1 and the cohesin proteins overlap, we next performed ChIP-seq using WT and Asxl1−/− BM LK cells, as well as antibodies against ASXL1, RAD21, and SMC1A. The genomic binding sites of ASXL1, SMC1A, and RAD21 significantly overlap (Fig. 4A). The percent binding cases at promoter regions for each of the three proteins are 93.45% (ASXL1), 42.61% (SMC1A), and 40.39% (RAD21), respectively. This result further confirmed the association of ASXL1 with the cohesin complex on the genome. Analysis of the genomic features showed that the ASXL1/SMC1A/RAD21 overlapping binding sites were enriched at promoter regions (93%), whereas 5 and 2% of these sites were located at the gene body and intergenic regions, respectively (Fig. 4B).
Fig. 4. Loss of Asxl1 leads to a decreased cohesin occupancy on the genome but does not affect their DNA recognition sequence.
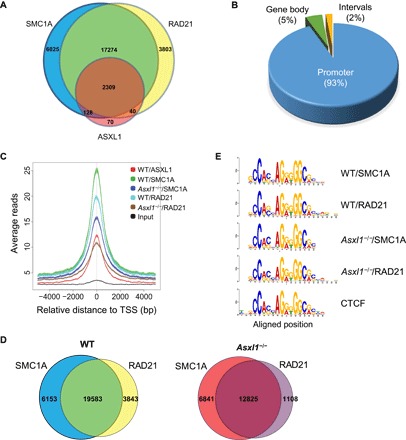
(A) Venn diagram showing overlapping peaks between ASXL1, SMC1A, and RAD21 ChIP-seq in WT LK cells. (B) Genomic distribution of ASXL1/SMC1A/RAD21 triple overlapping ChIP peaks in WT LK cells. (C) The overlap analysis shows the peak reads in WT and Asxl1−/− LK cells based on ASXL1/SMC1A/RAD21 overlapping peaks from WT LK cells. Zero base pair (bp) is defined as the peak of ASXL1 binding sites on the genome of WT LK cells. Decreased genomic cohesin complex occupancy is seen in Asxl1−/− LK cells. The overlap peaks of SMC1A and RAD21 represent the cohesin occupancy on the genome. Comparison of the SMC1A/RAD21 overlapping peaks between WT and Asxl1−/− LK cells identified 7833-peak loss and 1175-peak gain in the Asxl1−/− LK cells. TSS, transcription start site. (D) The pie chart represents the percentage of genes with no cohesin occupancy change (remain) or cohesin loss (SMC1A and/or RAD21 peak loss) in Asxl1−/− BM LK cells based on all ASXL1/SMC1A/RAD21 triple overlapping peaks of WT LK cells. (E) DNA recognition sequence of SMC1A and RAD21 in WT or Asxl1−/− LK cells. The SMC1A and RAD21 recognized identical DNA motif as CTCF.
Comparison of the ChIP-seq peaks in WT and Asxl1−/− cells showed that loss of Asxl1 markedly reduced cohesin occupancy on the genome (based on the SMC1A and RAD21 peaks; Fig. 4, C and D). Deletion of Asxl1 reduced SMC1A and RAD21 overlapping peaks by 35% (Fig. 4D). Note that the DNA sequence recognized by SMC1A or RAD21 (similar to the CTCF DNA binding sequence) remained unchanged regardless of the presence or absence of ASXL1 (Fig. 4E). These results suggest a role of ASXL1 in stabilizing, but not recruiting, RAD21 and SMC1A onto the genome.
ASXL1 regulates gene expression via the cohesin complex
It has been reported that loss of Asxl1 alters gene expression in LK cells (11, 12). To determine whether alterations in the genomic binding sites of ASXL1, SMC1A, and RAD21 are correlated with the changes of gene expression in Asxl1−/− LK cells, we performed an integrated analysis of ChIP-seq data with RNA sequencing (RNA-seq) data by focusing on the ~3000 genes with ASXL1/RAD21/SMC1A overlapping peaks in the promoter regions in WT LK cells. Among the ~1400 genes with RAD21/SMC1A overlapping peaks in both WT and Asxl1−/− LK cells, 9.3 and 4.4% were significantly up-regulated and down-regulated, respectively, in Asxl1−/− LK cells compared to WT cells (Fig. 5A). Of the ~1600 genes with loss of RAD21 and/or SMC1A occupancy in Asxl1−/− LK cells, 14.9 and 4.1% of genes were significantly up-regulated and down-regulated, respectively, in Asxl1−/− LK cells compared to WT cells (Fig. 5B). Gene ontology (GO) analysis of these up-regulated genes revealed associations with cell differentiation, regulation of programmed cell death, myeloid differentiation, RNA polymerase II activating transcription factor (TF) binding, and negative regulation of gene expression (Fig. 5C). In contrast, the down-regulated genes were associated with positive regulation of metabolic process, transcription from RNA polymerase II promoter, regulation of cell death, regulation of cell differentiation, and positive regulation of cell proliferation (Fig. 5D). An enrichment map was used for visualizing the networks of these GO terms enriched with up-regulated and down-regulated genes in Asxl1−/− LK cells (fig. S3). We identified a set of up-regulated genes that are enriched in the biological process termed “regulation of programmed cell death” between Asxl1−/− and WT cells, such as Atf4, Klf4, and Btg1. Up-regulated ATF4 has been reported to induce the transcriptional initiation of the apoptosis-related chop gene (40). In addition, among the down-regulated genes in Asxl1−/− relative to WT cells, we identified multiple genes that have a positive impact on cell proliferation, such as Myc, Nfya, and Slc. Patients with gene mutations of ASXL1 and/or the cohesin complex are found in all forms of myeloid malignancies, including MDS, MDS/MPN, and AML. These cellular phenotypes might be associated with changes of multiple genes. Further studies are warranted to validate these data in human primary cells. Two recent studies using Stag2 KD or Smc3 deletion demonstrated that cohesin proteins are required for the maintenance of normal gene expression in HSCs, and deletion of either Stag2 or Smc3 altered transcriptional output, leading to differentiation skewing of HSCs (35, 41).
Fig. 5. ASXL1 regulates gene expression via the cohesin complex in LK cells.
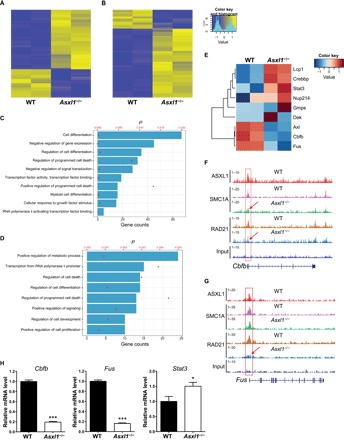
(A) The heatmap shows the differentially expressed genes associated with loci of no changes in SMC1A and RAD21 occupancy in Asxl1−/− BM LK cells. (B) The heatmap shows the differentially expressed genes associated with loss of SMC1A and/or RAD21 in Asxl1−/− BM LK cells. (C) The GO analysis of the 237 up-regulated genes (of the ~1600 genes with loss of RAD21 and/or SMC1A occupancy in Asxl1−/− LK cells) in Asxl1−/− LK cells compared to the WT LK cells. (D) GO analysis of the 65 down-regulated genes (of ~1600 genes with loss of RAD21 and/or SMC1A occupancy in Asxl1−/− LK cells) in Asxl1−/− LK cells compared to the WT LK cells. The P values of each GO term are represented by the red dots, and the gene set counts are represented by the bars. (E) Heatmap of differentially expressed genes of myeloid malignancy relevance within Asxl1/SMC1/RAD21 overlapping loci in Asxl1−/− LK cells. (F and G) Genome browser tracks of the Cbfb and Fus locus with overlapping ASXL1, SMC1A, or RAD21 peaks. (H) Relative RNA level of Cbfb, Fus, and Stat3 in LK cells as determined by qPCR. Data are represented as means ± SEM from three independent experiments. ***P < 0.001 and **P < 0.01.
A number of ASXL1/SMC1A/RAD21 target genes in Asxl1−/− LK cells are dysregulated, which are implicated in myeloid cell development and/or the pathogenesis of myeloid malignancies (Fig. 5E). For example, ASXL1, SMC1A, and RAD21 co-occupy Cbfb and Fus gene promoters in LK cells (Fig. 5, F and G). Asxl1 loss reduced both SMC1A and RAD21 occupancy at Cbfb and Fus promoters. qPCR confirmed the alteration of the gene expression levels, including Cbfb, Fus, and Stat3 (Fig. 5H). These data suggest that ASXL1 acts in concert with cohesin to regulate their target gene expression and that loss of Asxl1 decreases cohesin occupancy, leading to alterations in gene expression.
DISCUSSION
High frequencies of ASXL1 mutations occur in multiple forms of myeloid malignancies, and ASXL1 mutations are associated with poor prognosis, suggesting a driving role of ASXL1 mutations in the progression of disease (1, 3, 4, 42–45). However, the mechanisms by which ASXL1 loss leads to myeloid malignancy remain to be elucidated. ASXL1 has been shown to interact with various proteins to exert its regulatory function. ASXL1 recruits polycomb repressive complex 2 (PRC2) to exert its transcriptional repression through H3K27me3 (46, 47). ASXL1 has also been shown to interact with the retinoid acid receptor TF (48). Identification of the key ASXL1 binding partners would facilitate the investigation of mechanisms through which ASXL1 mediates its tumor-suppressive functions. Our protein purification and LC-MS/MS analyses identified cohesin proteins (including RAD21, SMC1A, SMC3, and STAG1/STAG2) as the major binding partners of ASXL1. Similar to ASXL1, cohesin genes are frequently mutated in multiple forms of myeloid malignancies (16, 30, 49). Although ASXL1 and cohesin form a big protein complex, the ASXL1 mutations and cohesin gene mutations are not strictly mutually exclusive in these patients (16, 28), indicating that ASXL1 and the cohesin complex have both overlapping and unique functions.
The cohesin complex has been shown to be critical for sister chromatid cohesion (23, 33) and gene expression regulation (37–39). Loss of any cohesin protein leads to premature sister chromatid separation (32, 50). Similar to cohesin loss, we show that Asxl1 loss also causes disruption of telophase chromatin disjunction, indicating that ASXL1 in concert with cohesin participates in this biological process. Furthermore, disruption of ASXL1 and the cohesin complex interaction by ASXL1aa401–587 expression, the region of ASXL1 mediating cohesin complex binding, impaired telophase chromatin separation. These results further demonstrate that ASXL1 is essential for the integral function of the cohesin complex in the maintenance of chromatin separation during cell division.
We and others have reported that Asxl1 loss leads to MDS-like disease in mice (11, 12). Here, we provide further evidence that Asxl1 loss increased frequencies of dysplastic myeloid cells with disrupted chromatin separation in the PB and BM. Recently, two independent groups reported that cohesin loss leads to the development of myeloid malignancies in mice (35, 41). Here, we show that ASXL1 binds to the cohesin complex and plays an essential role in maintaining normal chromatin separation during cell division, suggesting an overlapping molecular mechanism that underlies the pathogenesis of the myeloid disorders driven by alterations of ASXL1 or cohesin genes.
In addition to maintaining normal sister chromatid separation, the cohesin complex has also been shown to be important in multiple processes regulating transcription and gene expression (26, 51–54). We also explored whether the occupancy of ASXL1 and cohesin overlaps on the genome in WT LK cells and whether the deletion of Asxl1 alters the occupancies of cohesin on the genome. Analysis of the ChIP-seq data of ASXL1, RAD21, and SMC1A in WT and Asxl1−/− LK cells showed significant overlapping in ASXL1/SMC1A/RAD21 binding sites, which are enriched at promoter regions (93%). Furthermore, Asxl1 deletion reduced the occupancy of cohesin on the genome. The Database for Annotation, Visualization, and Integrated Discovery functional analysis of the up-regulated ASXL1 target genes revealed associations with cell differentiation, regulation of programmed cell death, myeloid differentiation, RNA polymerase II activating TF binding, and negative regulation of gene expression. Deletion of Asxl1 in LK cells dysregulated a number of ASXL1/SMC1A/RAD21 common target genes, including Stat3, Cbfb, and Fus, which are implicated in myeloid cell development and/or the pathogenesis of myeloid malignancies. These results suggest a role of ASXL1 in stabilizing, but not recruiting, RAD21 and SMC1A onto the genome. Cohesin proteins have been shown to be important for chromatin topology and facilitate enhancer-promoter looping to regulate gene transcription (55, 56). Future study is warranted to investigate whether ASXL1 mutations interfere with normal long-range chromosome interactions, altering gene expression and leading to the pathogenesis of myeloid malignancies.
In this study, we have identified a novel biological link that underlies the similar clinical features in MDS that are mediated by mutations in either ASXL1 or its major partners in cohesin genes. We also have shown that the ASXL1-cohesin interaction on the genome is important for regulating gene transcription in hematopoietic cells, establishing a novel mechanism of gene regulation by ASXL1 via the cohesin complex. Our data reinforce the view that ASXL1 has multifaceted functions in gene regulation by assembling epigenetic regulators and TFs to specific genomic loci.
MATERIALS AND METHODS
Mouse models
Asxl1+/− mice were generated as previously reported (11). Here, all mice were bred on a C57BL/6 genetic background. All animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals. All animal protocols were approved by the University of Miami Institutional Animal Care and Use Committee.
Plasmid constructs and shRNA
The cDNA of full-length mAsxl1 and its truncated variants (amino acids 1 to 1010, 1 to 587, 1 to 420, and 401 to 587) were tagged with 3×FLAG and engineered into pcDNA3.1+ (Invitrogen). The shRNA plasmids of ASXL1 (TG306527), SMC1A (TL513033), and RAD21 (TL501846) were purchased from OriGene.
Cell culture, retroviral transduction, and morphological analysis
HEK293T, HeLa, and HeLaGFP-H2B cells (57) were cultured and transfected with full-length Asxl1 and its truncation variant plasmids using Calcium Phosphate Transfection Kit (Invitrogen). The freshly isolated LK cells from fetal liver were cultured in RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum, SCF (100 ng/ml), and IL-3 (10 ng/ml) (PeproTech). For morphological analysis, PB was collected and smeared for May-Grünwald-Giemsa staining. Morphological analysis and cell differentiation of BM and fetal liver LK cells were performed on cytospins (5 × 105 cells per sample), followed by May-Grünwald-Giemsa staining. HeLaGFP-H2B cells were cultured on ultrathin glass coverslips. Images were acquired on a DeltaVision deconvolution microscope (Applied Precision) equipped with a 60× or 100× objective. All images were obtained and processed identically.
IP assay and LC-MS/MS
IP was performed using nuclear fraction buffer and washed with IP buffer [20 mM tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 5 mM EDTA, 2 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 2 mM NaF, and protease inhibitor cocktail (Roche)] for four times. For FLAG-tagged ASXL1, the IPs were eluted with 3×FLAG peptide (100 ng/ml; Sigma-Aldrich, F4799) in phosphate-buffered saline for 30 min at room temperature. All the IPs were performed with nuclear extraction.
FLAG-ASXL1 was immunoprecipitated from the nuclear extracts with anti-FLAG antibody–conjugated beads (Sigma-Aldrich), and the associated proteins were eluted from the beads by FLAG peptides. The eluates were then resolved on NuPAGE 4 to 12% Bis-Tris Gel (Invitrogen) followed by Coomassie brilliant blue staining, and lanes were excised for MS analysis by the Taplin Biological Mass Spectrometry Facility (Harvard Medical School).
qPCR analysis
Total RNA was isolated with TRIzol reagent (Invitrogen). The cDNA was synthesized using QuantiTect Reverse Transcription Kit (Qiagen). qPCR was performed using an ABI StepOnePlus with Fast SYBR Green Master Mix (Applied Biosystems). PCR amplifications were performed in triplicate for each gene of interest along with parallel measurements of β-actin. The primers used for the amplification of each gene are shown in table S2.
ChIP assays
BM LK cells were fixed with 1% formaldehyde for 15 min and quenched with 0.125 M glycine. Chromatin was isolated and sonicated to an average length of 300 to 500 bp. Genomic DNA regions of interest were isolated using antibodies against ASXL1 (Santa Cruz Biotechnology, sc-85283), SMC1A (Abcam, ab133643), and RAD21 (Abcam, ab154769). Illumina sequencing libraries were prepared and sequenced on a NextSeq 500.
Raw sequence reads from the FASTQ files of the six ChIP-seq samples were trimmed using sickle (https://github.com/najoshi/sickle) (58) with the Phred quality score threshold of 20 bases and the length threshold of 50 bases. Next, the trimmed reads were mapped against the mouse reference genome mm9 using bowtie 1.1.2 (59), allowing two mismatches. The promoter region was defined as the region 1000 bp upstream and 1000 bp downstream of the first transcription start site of a transcript cluster constructed using UCSC (University of California, Santa Cruz) known gene annotation of mm9. The uniquely mapped reads were counted in each promoter region. These counts of all samples were tabulated, and the differential bindings of ASXL1, SMC1A, and RADad21 in the promoter regions were tested using the Poisson test. The Benjamini-Hochberg procedure (60) was used to correct for multiple hypothesis testing. The differentially bound promoters were identified with a false discovery rate (FDR) cutoff of 0.05. To evaluate and identify statistical significance on the association between SMC1A, RAD21, and ASXL1 binding profiles in WT cells, we applied a hypergeometric test and calculated P values (table S3).
Specifically, the CTCF-like motifs were discovered on the basis of their high occurrences with significant E values, in the sequences where peaks were identified on each of the four ChIP-seq data sets: SMC1A in WT cells (3.3 × 10−61), RAD21 in WT cells (2 × 10−145), SMC1A in Asxl1−/− cells (7.1 × 10−146), and RAD21 in Asxl1−/− cells (2.9 × 10−183). The matches between these motifs and the known CTCF DNA binding motif were also highly significant, measured against the likelihood of the same matches by random sequences, with all significant FDR-adjusted P values for SMC1A in WT cells (4.12 × 10−13), RAD21 in WT cells (1.35 × 10−10), SMC1A in Asxl1−/− cells (2.05 × 10−13), and RAD21 in Asxl1−/− cells (2.64 × 10−9).
RNA-seq and analysis
Total RNA was isolated from BM LK cells of WT or Asxl1−/− mice (18- to 21-day-old mice) following standard protocol with TRIzol reagent (Invitrogen) followed by mRNA library preparation with the TruSeq strand-specific mRNA sample preparation system (Illumina). The library was sequenced (PE100bp) using the Illumina HiSeq 2500. Raw sequence reads from the FASTQ files of the four RNA-seq samples were mapped against the mouse reference genome mm9 using STAR2.3.1t with the default parameters (61). Only the uniquely mapped reads were used to count against the UCSC known gene annotation of mm9 to calculate the numbers of reads per gene. The counts of all samples were tabulated, then analyzed using DESeq (62) for normalization and identification of differentially expressed genes between the control and knockout samples using a standard workflow, as previously described (63, 64). The Benjamini-Hochberg procedure (61) was used to correct for multiple hypothesis testing. The differentially expressed genes were identified with an FDR cutoff of 0.05. GO analysis of differentially expressed genes of all comparisons was performed using Fisher’s exact test.
Statistical analysis
Differences between experimental groups were determined by Student’s t test or analysis of variance (ANOVA) followed by Newman-Keuls multiple comparison tests, as appropriate.
Supplementary Material
Acknowledgments
We thank C. Walczak (Indiana University, Bloomington, IN) for HeLaGFP-H2B. We also thank the histological processing and analysis services provided by the Satellite Histological Core of Sylvester Comprehensive Cancer Center Core Facility. Funding: This work was supported by the NIH (grant numbers 1R01CA172408-01 and 1R21CA185751-01). Author contributions: Z.L., P.Z., A.Y., Z.G., Y.B., J.L., S.C., H.Y., Y.H., J.L., Y.G., W.Z., E.H., H.A., and D.F. designed and performed the experiments and analyzed the data. M.X. and F.-C.Y. reviewed the blood smears and histopathologic sections. J.W.H., Y.R., S.D.N., P.Y., X.C., M.X., and F.-C.Y. designed and supervised the studies, performed the experiments, analyzed the data, wrote the manuscript, and were responsible for its final draft. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Raw and processed next-generation sequencing data have been deposited in the Gene Expression Omnibus GSE83962. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/1/e1601602/DC1
fig. S1. ASXL1 forms a complex with the cohesin complex.
fig. S2. Reintroducing mAsxl1 rescued the premature sister chromatid separation in HeLa cells with ASXL1 KD.
fig. S3. Enrichment map was used for visualizing the network of selected GO terms enriched with up-regulated and down-regulated genes in Asxl1−/− LK cells.
table S1. List of ASXL1 interaction proteins identified by MS in HEK293T cells transfected with FLAG-ASXL1.
table S2. qPCR primer sequences.
table S3. Statistical evidence for binding between SMC1A, RAD21, and ASXL1.
REFERENCES AND NOTES
- 1.Gelsi-Boyer V., Trouplin V., Adélaïde J., Bonansea J., Cervera N., Carbuccia N., Lagarde A., Prebet T., Nezri M., Sainty D., Olschwang S., Xerri L., Chaffanet M., Mozziconacci M.-J., Vey N., Birnbaum D., Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br. J. Haematol. 145, 788–800 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Brecqueville M., Rey J., Bertucci F., Coppin E., Finetti P., Carbuccia N., Cervera N., Gelsi-Boyer V., Arnoulet C., Gisserot O., Verrot D., Slama B., Vey N., Mozziconacci M.-J., Birnbaum D., Murati A., Mutation analysis of ASXL1, CBL, DNMT3A, IDH1, IDH2, JAK2, MPL, NF1, SF3B1, SUZ12, and TET2 in myeloproliferative neoplasms. Genes Chromosomes Cancer 51, 743–755 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Carbuccia N., Murati A., Trouplin V., Brecqueville M., Adélaïde J., Rey J., Vainchenker W., Bernard O. A., Chaffanet M., Vey N., Birnbaum D., Mozziconacci M. J., Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia 23, 2183–2186 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Boultwood J., Perry J., Pellagatti A., Fernandez-Mercado M., Fernandez-Santamaria C., Calasanz M. J., Larrayoz M. J., Garcia-Delgado M., Giagounidis A., Malcovati L., Della Porta M. G., Jädersten M., Killick S., Hellström-Lindberg E., Cazzola M., Wainscoat J. S., Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia 24, 1062–1065 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Chen T. C., Hou H.-A., Chou W.-C., Tang J.-L., Kuo Y.-Y., Chen C.-Y., Tseng M.-H., Huang C.-F., Lai Y.-J., Chiang Y.-C., Lee F.-Y., Liu M.-C., Liu C.-W., Liu C.-Y., Yao M., Huang S.-Y., Ko B.-S., Hsu S.-C., Wu S.-J., Tsay W., Chen Y.-C., Tien H.-F., Dynamics of ASXL1 mutation and other associated genetic alterations during disease progression in patients with primary myelodysplastic syndrome. Blood Cancer J. 4, e177 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Itzykson R., Kosmider O., Renneville A., Gelsi-Boyer V., Meggendorfer M., Morabito M., Berthon C., Adès L., Fenaux P., Beyne-Rauzy O., Vey N., Braun T., Haferlach T., Dreyfus F., Cross N. C. P., Preudhomme C., Bernard O. A., Fontenay M., Vainchenker W., Schnittger S., Birnbaum D., Droin N., Solary E., Prognostic score including gene mutations in chronic myelomonocytic leukemia. J. Clin. Oncol. 31, 2428–2436 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Pratcorona M., Abbas S., Sanders M. A., Koenders J. E., Kavelaars F. G., Erpelinck-Verschueren C. A. J., Zeilemakers A., Löwenberg B., Valk P. J. M., Acquired mutations in ASXL1 in acute myeloid leukemia: Prevalence and prognostic value. Haematologica 97, 388–392 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fisher C. L., Berger J., Randazzo F., Brock H. W., A human homolog of Additionalsexcombs, ADDITIONALSEXCOMBS-LIKE1, maps to chromosome 20q11. Gene 306, 115–126 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Milne T. A., Sinclair D. A. R., Brock H. W., The Additional sex combs gene of Drosophila is required for activation and repression of homeotic loci, and interacts specifically with Polycomb and super sex combs. Mol. Gen. Genet. 261, 753–761 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Fisher C. L., Randazzo F., Humphries R. K., Brock H. W., Characterization of Asxl1, a murine homolog of Additional sex combs, and analysis of the Asx-like gene family. Gene 369, 109–118 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Wang J., Li Z., He Y., Pan F., Chen S., Rhodes S., Nguyen L., Yuan J., Jiang L., Yang X., Weeks O., Liu Z., Zhou J., Ni H., Cai C.-L., Xu M., Yang F.-C., Loss of Asxl1 leads to myelodysplastic syndrome-like disease in mice. Blood 123, 541–553 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdel-Wahab O., Gao J., Adli M., Dey A., Trimarchi T., Chung Y. R., Kuscu C., Hricik T., Ndiaye-Lobry D., LaFave L. M., Koche R., Shih A. H., Guryanova O. A., Kim E., Li S., Pandey S., Shin J. Y., Telis L., Liu J., Bhatt P. K., Monette S., Zhao X., Mason C. E., Park C. Y., Bernstein B. E., Aifantis I., Levine R. L., Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 210, 2641–2659 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang P., Xing C., Rhodes S. D., He Y., Deng K., Li Z., He F., Zhu C., Nguyen L., Zhou Y., Chen S., Mohammad K. S., Guise T. A., Abdel-Wahab O., Xu M., Wang Q.-F., Yang F.-C., Loss of Asxl1 alters self-renewal and cell fate of bone marrow stromal cell, leading to Bohring-Opitz-like syndrome in mice. Stem Cell Reports 6, 914–925 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi H., Yamamoto S., Sheng M., Bai J., Zhang P., Chen R., Chen S., Shi L., Abdel-Wahab O., Xu M., Zhou Y., Yang F.-C., ASXL1 plays an important role in erythropoiesis. Sci. Rep. 6, 28789 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White G. E., Erickson H. P., The coiled coils of cohesin are conserved in animals, but not in yeast. PLOS ONE 4, e4674 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kon A., Shih L.-Y., Minamino M., Sanada M., Shiraishi Y., Nagata Y., Yoshida K., Okuno Y., Bando M., Nakato R., Ishikawa S., Sato-Otsubo A., Nagae G., Nishimoto A., Haferlach C., Nowak D., Sato Y., Alpermann T., Nagasaki M., Shimamura T., Tanaka H., Chiba K., Yamamoto R., Yamaguchi T., Otsu M., Obara N., Sakata-Yanagimoto M., Nakamaki T., Ishiyama K., Nolte F., Hofmann W.-K., Miyawaki S., Chiba S., Mori H., Nakauchi H., Koeffler H. P., Aburatani H., Haferlach T., Shirahige K., Miyano S., Ogawa S., Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. 45, 1232–1237 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Seitan V. C., Merkenschlager M., Cohesin and chromatin organisation. Curr. Opin. Genet. Dev. 22, 93–100 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Onn I., Heidinger-Pauli J. M., Guacci V., Ünal E., Koshland D. E., Sister chromatid cohesion: A simple concept with a complex reality. Annu. Rev. Cell Dev. Biol. 24, 105–129 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Losada A., The regulation of sister chromatid cohesion. Biochim. Biophys. Acta 1786, 41–48 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Nasmyth K., Haering C. H., Cohesin: Its roles and mechanisms. Annu. Rev. Genet. 43, 525–558 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Ström L., Karlsson C., Lindroos H. B., Wedahl S., Katou Y., Shirahige K., Sjögren C., Postreplicative formation of cohesion is required for repair and induced by a single DNA break. Science 317, 242–245 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Watrin E., Peters J.-M., The cohesin complex is required for the DNA damage-induced G2/M checkpoint in mammalian cells. EMBO J. 28, 2625–2635 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dorsett D., Cohesin, gene expression and development: Lessons from Drosophila. Chromosome Res. 17, 185–200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horsfield J. A., Anagnostou S. H., Hu J. K.-H., Cho K. H. Y., Geisler R., Lieschke G., Crosier K. E., Crosier P. S., Cohesin-dependent regulation of Runx genes. Development 134, 2639–2649 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Parelho V., Hadjur S., Spivakov M., Leleu M., Sauer S., Gregson H. C., Jarmuz A., Canzonetta C., Webster Z., Nesterova T., Cobb B. S., Yokomori K., Dillon N., Aragon L., Fisher A. G., Merkenschlager M., Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 132, 422–433 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Wendt K. S., Yoshida K., Itoh T., Bando M., Koch B., Schirghuber E., Tsutsumi S., Nagae G., Ishihara K., Mishiro T., Yahata K., Imamoto F., Aburatani H., Nakao M., Imamoto N., Maeshima K., Shirahige K., Peters J.-M., Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451, 796–801 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Matynia A. P., Szankasi P., Shen W., Kelley T. W., Molecular genetic biomarkers in myeloid malignancies. Arch. Pathol. Lab. Med. 139, 594–601 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Thota S., Viny A. D., Makishima H., Spitzer B., Radivoyevitch T., Przychodzen B., Sekeres M. A., Levine R. L., Maciejewski J. P., Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood 124, 1790–1798 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshida K., Toki T., Okuno Y., Kanezaki R., Shiraishi Y., Sato-Otsubo A., Sanada M., Park M.-j., Terui K., Suzuki H., Kon A., Nagata Y., Sato Y., Wang R. N., Shiba N., Chiba K., Tanaka H., Hama A., Muramatsu H., Hasegawa D., Nakamura K., Kanegane H., Tsukamoto K., Adachi S., Kawakami K., Kato K., Nishimura R., Izraeli S., Hayashi Y., Miyano S., Kojima S., Ito E., Ogawa S., The landscape of somatic mutations in Down syndrome–related myeloid disorders. Nat. Genet. 45, 1293–1299 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Thol F., Bollin R., Gehlhaar M., Walter C., Dugas M., Suchanek K. J., Kirchner A., Huang L., Chaturvedi A., Wichmann M., Wiehlmann L., Shahswar R., Damm F., Göhring G., Schlegelberger B., Schlenk R., Döhner K., Döhner H., Krauter J., Ganser A., Heuser M., Mutations in the cohesin complex in acute myeloid leukemia: Clinical and prognostic implications. Blood 123, 914–920 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Merkenschlager M., Odom D. T., CTCF and cohesin: Linking gene regulatory elements with their targets. Cell 152, 1285–1297 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Díaz-Martínez L. A., Giménez-Abián J. F., Clarke D. J., Cohesin is dispensable for centromere cohesion in human cells. PLOS ONE 2, e318 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michaelis C., Ciosk R., Nasmyth K., Cohesins: Chromosomal proteins that prevent premature separation of sister chromatids. Cell 91, 35–45 (1997). [DOI] [PubMed] [Google Scholar]
- 34.Kong X., Ball A. R. Jr, Pham H. X., Zeng W., Chen H.-Y., Schmiesing J. A., Kim J.-S., Berns M., Yokomori K., Distinct functions of human cohesin-SA1 and cohesin-SA2 in double-strand break repair. Mol. Cell. Biol. 34, 685–698 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mullenders J., Aranda-Orgilles B., Lhoumaud P., Keller M., Pae J., Wang K., Kayembe C., Rocha P. P., Raviram R., Gong Y., Premsrirut P. K., Tsirigos A., Bonneau R., Skok J. A., Cimmino L., Hoehn D., Aifantis I., Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J. Exp. Med. 212, 1833–1850 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laugsch M., Seebach J., Schnittler H., Jessberger R., Imbalance of SMC1 and SMC3 cohesins causes specific and distinct effects. PLOS ONE 8, e65149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lavagnolli T., Gupta P., Hörmanseder E., Mira-Bontenbal H., Dharmalingam G., Carroll T., Gurdon J. B., Fisher A. G., Merkenschlager M., Initiation and maintenance of pluripotency gene expression in the absence of cohesin. Genes Dev. 29, 23–38 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zuin J., Dixon J. R., van der Reijden M. I. J. A., Ye Z., Kolovos P., Brouwer R. W. W., van de Corput M. P. C., van de Werken H. J. G., Knoch T. A., van IJckenf W. F. J., Grosveld F. G., Ren B., Wendt K. S., Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. U.S.A. 111, 996–1001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan J., Enge M., Whitington T., Dave K., Liu J., Sur I., Schmierer B., Jolma A., Kivioja T., Taipale M., Taipale J., Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell 154, 801–813 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Jiang Q., Feng L., Kejian S., Pa W., An J., Yang Y., Caimin X., ATF4 activation by the p38MAPK-eIF4E axis mediates apoptosis and autophagy induced by selenite in Jurkat cells. FEBS Lett. 587, 2420–2429 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Viny A. D., Ott C. J., Spitzer B., Rivas M., Meydan C., Papalexi E., Yelin D., Shank K., Reyes J., Chiu A., Romin Y., Boyko V., Thota S., Maciejewski J. P., Melnick A., Bradner J. E., Levine R. L., Dose-dependent role of the cohesin complex in normal and malignant hematopoiesis. J. Exp. Med. 212, 1819–1832 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carbuccia N., Trouplin V., Gelsi-Boyer V., Murati A., Rocquain J., Adélaïde J., Olschwang S., Xerri L., Vey N., Chaffanet M., Birnbaum D., Mozziconacci M. J., Mutual exclusion of ASXL1 and NPM1 mutations in a series of acute myeloid leukemias. Leukemia 24, 469–473 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Boultwood J., Perry J., Zaman R., Fernandez-Santamaria C., Littlewood T., Kusec R., Pellagatti A., Wang L., Clark R. E., Wainscoat J. S., High-density single nucleotide polymorphism array analysis and ASXL1 gene mutation screening in chronic myeloid leukemia during disease progression. Leukemia 24, 1139–1145 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Sugimoto Y., Muramatsu H., Makishima H., Prince C., Jankowska A. M., Yoshida N., Xu Y., Nishio N., Hama A., Yagasaki H., Takahashi Y., Kato K., Manabe A., Kojima S., Maciejewski J. P., Spectrum of molecular defects in juvenile myelomonocytic leukaemia includes ASXL1 mutations. Br. J. Haematol. 150, 83–87 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Makishima H., Jankowska A. M., McDevitt M. A., O’Keefe C., Dujardin S., Cazzolli H., Przychodzen B., Prince C., Nicoll J., Siddaiah H., Shaik M., Szpurka H., Hsi E., Advani A., Paquette R., Maciejewski J. P., CBL, CBLB, TET2, ASXL1, and IDH1/2 mutations and additional chromosomal aberrations constitute molecular events in chronic myelogenous leukemia. Blood 117, e198–e206 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abdel-Wahab O., Adli M., LaFave L. M., Gao J., Hricik T., Shih A. H., Pandey S., Patel J. P., Rock Chung Y., Koche R., Perna F., Zhao X., Taylor J. E., Park C. Y., Carroll M., Melnick A., Nimer S. D., Jaffe J. D., Aifantis I., Bernstein B. E., Levine R. L., ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 22, 180–193 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmitges F. W., Prusty A. B., Faty M., Stützer A., Lingaraju G. M., Aiwazian J., Sack R., Hess D., Li L., Zhou S., Bunker R. D., Wirth U., Bouwmeester T., Bauer A., Ly-Hartig N., Zhao K., Chan H., Gu J., Gut H., Fischle W., Müller J., Thomä N. H., Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 42, 330–341 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Cho Y.-S., Kim E.-J., Park U.-H., Sin H.-S., Um S.-J., Additional sex comb-like 1 (ASXL1), in cooperation with SRC-1, acts as a ligand-dependent coactivator for retinoic acid receptor. J. Biol. Chem. 281, 17588–17598 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Leeke B., Marsman J., O’Sullivan J. M., Horsfield J. A., Cohesin mutations in myeloid malignancies: Underlying mechanisms. Exp. Hematol. Oncol. 3, 13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sonoda E., Matsusaka T., Morrison C., Vagnarelli P., Hoshi O., Ushiki T., Nojima K., Fukagawa T., Waizenegger I. C., Peters J.-M., Earnshaw W. C., Takeda S., Scc1/Rad21/Mcd1 is required for sister chromatid cohesion and kinetochore function in vertebrate cells. Dev. Cell 1, 759–770 (2001). [DOI] [PubMed] [Google Scholar]
- 51.Schaaf C. A., Misulovin Z., Gause M., Koenig A., Gohara D. W., Watson A., Dorsett D., Cohesin and polycomb proteins functionally interact to control transcription at silenced and active genes. PLOS Genet. 9, e1003560 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schaaf C. A., Kwak H., Koenig A., Misulovin Z., Gohara D. W., Watson A., Zhou Y., Lis J. T., Dorsett D., Genome-wide control of RNA polymerase II activity by cohesin. PLOS Genet. 9, e1003382 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schaaf C. A., Misulovin Z., Gause M., Koenig A., Dorsett D., The Drosophila Enhancer of split gene complex: Architecture and coordinate regulation by notch, cohesin, and polycomb group proteins. G3 (Bethesda) 3, 1785–1794 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kagey M. H., Newman J. J., Bilodeau S., Zhan Y., Orlando D. A., van Berkum N. L., Ebmeier C. C., Goossens J., Rahl P. B., Levine S. S., Taatjes D. J., Dekker J., Young R. A., Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mazumdar C., Shen Y., Xavy S., Zhao F., Reinisch A., Li R., Corces M. R., Flynn R. A., Buenrostro J. D., Chan S. M., Thomas D., Koenig J. L., Hong W.-J., Chang H. Y., Majeti R., Leukemia-associated cohesin mutants dominantly enforce stem cell programs and impair human hematopoietic progenitor differentiation. Cell Stem Cell 17, 675–688 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Faure A. J., Schmidt D., Watt S., Schwalie P. C., Wilson M. D., Xu H., Ramsay R. G., Odom D. T., Flicek P., Cohesin regulates tissue-specific expression by stabilizing highly occupied cis-regulatory modules. Genome Res. 22, 2163–2175 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cai S., Weaver L. N., Ems-McClung S. C., Walczak C. E., Proper organization of microtubule minus ends is needed for midzone stability and cytokinesis. Curr. Biol. 20, 880–885 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.N. Joshi, J. Fass, Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files [Software] (Version 1.33); https://github.com/najoshi/sickle (2011).
- 59.Langmead B., Trapnell C., Pop M., Salzberg S. L., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benjamini Y., Hochberg Y., Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 57, 289–300 (1995). [Google Scholar]
- 61.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anders S., Huber W., Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qian X., Li X., Ilori T. O., Klein J. D., Hughey R. P., Li C.-j., Alli A. A., Guo Z., Yu P., Song X., Chen G., RNA-seq analysis of glycosylation related gene expression in STZ-induced diabetic rat kidney inner medulla. Front. Physiol. 6, 274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guo Z., González J. F., Hernandez J. N., McNeilly T. N., Corripio-Miyar Y., Frew D., Morrison T., Yu P., Li R. W., Possible mechanisms of host resistance to Haemonchus contortus infection in sheep breeds native to the Canary Islands. Sci. Rep. 6, 26200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/1/e1601602/DC1
fig. S1. ASXL1 forms a complex with the cohesin complex.
fig. S2. Reintroducing mAsxl1 rescued the premature sister chromatid separation in HeLa cells with ASXL1 KD.
fig. S3. Enrichment map was used for visualizing the network of selected GO terms enriched with up-regulated and down-regulated genes in Asxl1−/− LK cells.
table S1. List of ASXL1 interaction proteins identified by MS in HEK293T cells transfected with FLAG-ASXL1.
table S2. qPCR primer sequences.
table S3. Statistical evidence for binding between SMC1A, RAD21, and ASXL1.