

Abstract
The Epstein-Barr virus (EBV) nuclear antigen 3C (EBNA3C) is a virus-encoded latent antigen essential for primary B-cell transformation. In this report we demonstrate that although the carboxy terminus of EBNA3C predominantly regulates cyclin A-dependent kinase activity, the region of greatest affinity for cyclin A lies within the EBNA3 amino-terminal homology domain of EBNA3C. Detailed mapping studies employing both in vitro binding assays and coimmunoprecipitation experiments implicated a small region of EBNA3C, amino acids 130 to 159 within the EBNA3 homology domain, as having the greatest affinity for cyclin A. The EBNA3 homology domain has the highest degree of amino acid similarity (approximately 30%) between the EBNA3 proteins, and, indeed, EBNA3B, but not EBNA3A, showed binding activity with cyclin A. We also show that EBNA3C binds to the α1 helix of the highly conserved mammalian cyclin box, with cyclin A amino acids 206 to 226 required for strong binding to EBNA3C amino acids 130 to 159. Interestingly, EBNA3C also bound human cyclins D1 and E in vitro, although the affinity was approximately 30% of that seen for cyclin A. Previously it was demonstrated that full-length EBNA3C rescues p27-mediated suppression of cyclin A-dependent kinase activity (J. S. Knight and E. S. Robertson, J. Virol. 78:1981-1991, 2004). It was also demonstrated that the carboxy terminus of EBNA3C recapitulates this phenotype. Surprisingly, the amino terminus of EBNA3C with the highest affinity for cyclin A was unable to rescue p27 suppression of kinase activity and actually downregulates cyclin A activity when introduced into EBV-infected cells. The data presented here suggests that the amino terminus of EBNA3C may play an important role in recruiting cyclin A complexes, while the carboxy terminus of EBNA3C is necessary for the functional modulation of cyclin A complex kinase activity.
Epstein-Barr virus (EBV) has been associated with human cancer since the early 1960s. EBV was discovered in 1962 by electron microscopy of cells cultured from Burkitt's lymphoma, an endemic and aggressive malignancy that is predominantly seen in equatorial Africa (7). EBV has since been implicated as the likely causative agent in other malignancies, including nasopharyngeal carcinoma, posttransplant lymphomas, and some forms of Hodgkins disease (11, 22). Further, EBV has been more tenuously linked to additional cancers such as gastric carcinoma and, somewhat controversially, metastatic breast carcinoma (11, 22, 28). Early studies of EBV noted its potent ability to transform resting primary B lymphocytes in cell culture. Subsequently, in vitro recombination studies identified five viral latency genes that are absolutely required for the transformation phenotype (11, 22). These include LMP1, EBNA1, EBNA2, EBNA3A, and EBNA3C (11, 22). Of these essential gene products, the EBNA3 proteins are clearly the least well understood in terms of their contribution to B-lymphocyte transformation.
EBNA3C of type 1 EBV is a 992-amino-acid protein that localizes to the nucleus in either a diffuse or punctate pattern depending on the cell type and expression level (12, 25, 28). EBNA3C has been shown to play a complex regulatory role in the transcription of viral and cellular genes. EBNA3C antagonizes EBNA2-mediated transactivation by competing with EBNA2 for RBP-Jκ binding (10, 19, 24), and, conversely, cooperates with EBNA2 in the upregulation of some promoters, like the EBV LMP1 promoter (2, 3, 33). Additionally, EBNA3C recruits both histone acetylase and deacetylase activities (12, 20, 29), in part through its association with the small acidic nuclear protein prothymosin-alpha (5). Further, EBNA3C associates with an antimetastatic factor, Nm23-H1, and may modulate the transcription of cellular genes involved in cell migration and invasion (28, 30).
In addition to this somewhat diffuse transcriptional regulatory picture, EBNA3C also modulates the mammalian cell cycle, presumably through direct protein-protein interactions, by targeting multiple checkpoint regulatory proteins (17, 18). To this end, EBNA3C has previously been shown to indirectly target pathways regulated by the retinoblastoma tumor suppressor (Rb) (17, 18). EBNA3C activates E2F-dependent promoters and induces foci formation similar to papillomavirus E7 in a colony formation assay (17). Additionally, EBNA3C overcomes the ability of the cyclin-dependent kinase inhibitor p16INK4A to block transformation and dramatically drives serum-starved cells through the G1/S restriction point (17, 18).
Despite this evidence, a clear molecular link between the Rb protein and EBNA3C has yet to be demonstrated in vivo. More recently, it was shown that EBNA3C may indirectly target Rb by associating with cyclin A complexes in EBV-transformed lymphoblastoid cell lines (13). Cell cycle progression is partially dependent on the activity of cyclin-dependent kinases (cdks). These kinases are allosterically activated by binding to cyclins, a family of proteins whose levels oscillate in synchrony with cell cycle progression (14). Cyclin A, synthesized beginning in G1 and maximally at the onset of S phase, is necessary for both progression through S phase and for entry into mitosis (15). Additionally, exogenous expression of cyclin A has been shown to accelerate exit from G1 (21). EBNA3C stimulates cyclin A-dependent kinase activity and rescues p27-mediated inhibition of cyclin A/Cdk2 kinase activity by decreasing the molecular association between cyclin A and p27 in cells (13). Cyclin A functionally interacts with a region of the carboxy terminus of EBNA3C shown to be important for both stimulation of cyclin A-dependent kinase activity and for cell cycle progression (13).
The molecular association between EBNA3C and cyclin A complexes is further examined in this study. Surprisingly, we have identified an additional and perhaps predominant cyclin A binding site at the amino terminus of EBNA3C not previously investigated in the initial report of Knight and Roberston (13) identifying cyclin A as an EBNA3C binding protein. An in-depth understanding of the different domains of EBNA3C that modulate cyclin A complexes will lead to a better understanding of the basic mechanism underlying the regulation of the mammalian cell cycle by EBV.
MATERIALS AND METHODS
Plasmids, antibodies, and cell lines.
pA3M-E3C constructs expressing either full-length EBNA3C or EBNA3C truncations 1-365, 366-620, and 621-992 with a carboxy-terminal myc tag have been described previously (5). pA3M-E3C constructs expressing amino acids 1 to 100 and 1 to 200 were prepared by cloning PCR-amplified cDNAs into the previously described pA3M vector (4). Glutathione S transferase (GST)-EBNA3C truncation mutants were prepared by cloning PCR-amplified cDNAs into pGEX-2TK. Point mutations in the EBNA3C gene were prepared by a standard PCR primer mutagenesis method. pSG5-EBNA3A, pSG5-EBNA3B, and pSG5-EBNA3C have been described previously (24). pA3M-cyclin A constructs expressing either full-length cyclin A or cyclin A truncations with a carboxy-terminal myc tag were prepared by cloning PCR-amplified cDNAs into the previously described pA3M vector (4). GST-cyclin A fusions were prepared by cloning PCR amplified fragments into the pGEX-2TK vector: the cyclin box clone represents amino acids 206 to 310, α1 represents amino acids 206 to 227, and α2-5 represents amino acids 228 to 310. pCMV-Cdk2, RC-cyclin A, and RC-cyclin E were kindly provided by Philip Hinds (8). The construct expressing GST-cyclin A was provided by Maria Mudryj (6). pCDNA3-p27 was provided by Michele Pagano (16). pCDNA3-cyclin D1 was provided by Alan Diehl (University of Pennsylvania). Rabbit polyclonal antibodies reactive to cyclin A, cyclin E, Cdk2, and p27Kip1 were purchased from Santa Cruz Biotechnology, Inc. A10 monoclonal and rabbit polyclonal antibodies reactive to EBNA3C have been previously described (5). HEK 293 cells are human embryonic kidney cells transformed by adenovirus type 5 DNA; HEK 293T cells stably express the simian virus 40 large T antigen (1). U2OS is a human osteosarcoma cell line. HEK 293T and U2OS cells were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco-BRL) with 10% fetal bovine serum unless otherwise indicated. For most experiments, 10 million 293T cells were transfected by electroporation at 210 V and 975 μF in a 0.4-μm cuvette.
GST pull-down assays.
GST fusion proteins were purified from bulk Escherichia coli cultures following induction with IPTG as described previously (5). For in vitro binding experiments, GST fusion proteins were incubated with 35S-labeled in vitro-translated protein in binding buffer (1× phosphate-buffered saline [PBS], 0.1% NP-40, 0.5 mM dithiothreitol [DTT], 10% glycerol, supplemented with protease inhibitors). In vitro translation was with the TNT T7 Quick Coupled Transcription/Translation System (Promega Inc., Madison, Wis.) according to the manufacturer's instructions.
Immunoprecipitation assays.
Immunoprecipitation was essentially performed as described previously (5). Proteins were fractionated by electrophoresis on a sodium dodecyl sulfate (SDS)-polyacrylamide gel and then transferred to a 0.45-μm nitrocellulose membrane. The membrane was blotted with appropriate rabbit polyclonal antibodies followed by horseradish peroxidase-linked Protein A (Amersham Biosciences, Piscataway, N.J.) at a 1:5,000 dilution in PBS.
In vitro kinase assay.
U2OS cells were seeded into 6-well plates and grown to confluence in 0.5% FBS for 48 h prior to transfection. Cells were transfected with Lipofectamine 2000 reagent (Invitrogen Corporation, Bethesda, Md.), harvested after 24 h with a cell scraper, washed with PBS, and lysed on ice in 500 μl of radioimmunoprecipitation assay (RIPA) buffer (0.5% NP-40, 10 mM Tris [pH 7.5], 2 mM EDTA, 150 mM NaCl, 1 mM EGTA, plus protease and phosphatase inhibitors). Lysates were precleared and then rotated with 1 μg of cyclin A antibody overnight at 4°C. Cyclin A complexes were captured by rotating with Protein A-Sepharose beads and were washed with RIPA buffer. Cyclin A complexes were then washed with histone wash buffer (25 mM Tris [pH 7.5], 70 mM NaCl, 10 mM MgCl2, 1 mM EGTA, 1 mM DTT, plus protease and phosphatase inhibitors). Complexes were incubated in 30 μl of histone wash buffer supplemented with 4 μg of histone H1 (Upstate Biotechnology, Inc., Lake Placid, N.Y.), 10 mM cold ATP, and 0.2 μCi of [γ-32P]-ATP/μl for 30 min at 37°C. The reaction was stopped by adding SDS-lysis buffer and heating to 95°C for 10 min. Labeled Histone H1 was resolved by SDS-12% polyacrylamide gel electrophoresis (PAGE). Quantitation was performed with ImageQuant software (Amersham Biosciences).
LCLs were transfected by electroporation with the Bio-Rad Gene Pulser II at 220 V and 975 μF, and transfection efficiency was monitored by using a cotransfected green fluorescent protein (GFP) expression plasmid. Prior to transfection, LCLs were grown exponentially and tested by trypan blue staining for live cells. All transfections were done when cells were greater than 90% positive for exclusion of trypan blue. Transfected cells were incubated in RPMI 1640 plus 10% FBS for 24 h and then were treated in the same manner as that for U2OS cells.
RESULTS
In a previous study, a truncated form of EBNA3C corresponding to amino acids 365 to 992 was fused in frame with the GAL4 DNA binding domain and was tested against an LCL-derived cDNA library in a yeast two-hybrid screen (5). Partial sequences from two positive cDNA clones identified coding sequences that were located within the cyclin A gene (13). Based on this initial screen, a region at the extreme carboxy terminus of EBNA3C was identified as functionally regulating cyclin A-dependent kinase activity while also possessing affinity for cyclin A, as determined by in vitro binding assays (13). Interestingly, in follow-up studies mutational analysis of the EBNA3C-cyclin A interaction revealed that another binding site with greater affinity for cyclin A lies at the amino terminus of EBNA3C. The following experiments dissect this interaction and attempt to understand the functional role of the amino terminus in modulation of cyclin A-dependent kinase activity.
Binding studies identify the amino terminus of EBNA3C as the predominant binding site for cyclin A.
To assess the association between different regions of EBNA3C and cyclin A in the context of cell lysates, 293T cells were transfected with expression constructs for cyclin A and either full-length EBNA3C, EBNA3C 1-365, EBNA3C 366-620, or EBNA3C 621-992. All EBNA3C expression constructs placed a myc tag in frame with the carboxy terminus of the protein. Cyclin A coimmunoprecipitated with relatively high affinity with both full-length EBNA3C and EBNA3C 1-365 (Fig. 1A). However, little to no coimmunoprecipiation of cyclin A above background was detected with EBNA3C 366-620. Moreover, and somewhat surprisingly, EBNA3C 621-992 showed significantly reduced coimmunoprecipitation in this assay. This suggests that the predominant region of interaction between cyclin A and EBNA3C lies in the amino terminus and that the interaction previously seen with the carboxy terminus using in vitro binding assays (13) may be somewhat transient or masked in transiently transfected cells expressing the truncated proteins (Fig. 1A).
FIG. 1.

The amino terminus of EBNA3C strongly binds cyclin A. (A) Ten million 293T cells were transfected with 10 μg of RC-cyclin A and 15 μg of pA3M-EBNA3C expression plasmids. Cells were harvested at 36 h and were immunoprecipitated (IP) with 1 μl of 9E10 myc-reactive ascites fluid. Samples were resolved by SDS-10% PAGE and probed by immunoblot. EBNA3C was detected with 9E10 myc hybridoma supernatant, and cyclin A was detected with rabbit polyclonal serum to cyclin A. (B) GST-cyclin A fusion protein was expressed in E. coli and purified with glutathione Sepharose beads. Full-length EBNA3C and EBNA3C truncations as indicated were labeled with [35S]methionine by in vitro translation and then were incubated with either GST control or GST-cyclin A beads normalized by Coomassie staining. In each case, 5% of in vitro translation (IVT) input was used for comparison. Precipitated proteins were resolved by SDS-PAGE, and bands were visualized with a phosphorimager screen. (C) GST-cyclin A fusion protein was expressed in E. coli and purified with glutathione Sepharose beads. Full-length EBNA3A, EBNA3B, or EBNA3C was labeled with [35S]methionine by in vitro translation and then was incubated with either GST control or GST-cyclin A beads normalized by Coomassie staining. In each case, 5% of in vitro translation input was used for normalization. Precipitated proteins were resolved by SDS-PAGE, and bands were visualized and quantified with a phosphorimager screen. The numerical data presented here represents the average of two representative experiments. WB, Western blot.
Bacterially expressed cyclin A binds the amino terminus of EBNA3C.
Coimmunoprecipitation experiments suggested that the amino terminus of EBNA3C is the predominant interacting site for cyclin A in cells. To corroborate this finding, the interaction with the amino-terminal domain was tested in the context of bacterially expressed human cyclin A and compared to that seen with the central and carboxy-terminal domains. GST-cyclin A beads strongly precipitated in vitro-translated, 35S-labeled EBNA3C, as has been shown previously (13). Interestingly, in vitro-translated EBNA3C 1-365 also precipitated strongly in this experiment, while both the central and carboxy-terminal domains of EBNA3C showed significantly less affinity for cyclin A than the amino terminus; however, the carboxy terminus did have slightly greater affinity than the central domain (Fig. 1B).
EBNA3A and EBNA3B interact weakly with bacterially expressed cyclin A.
The juxtaposition and colinear homology of the three EBNA3 family genes suggests gene duplication of a common ancestral gene (11, 22). While studies have revealed shared properties among the three EBNA3 gene products, such as association with the RBP-Jκ transcription factor (24), the proteins have clearly diverged functionally to some extent. This is emphasized by the fact that EBNA3A and EBNA3C are essential for primary B-cell transformation in vitro while EBNA3B appears to be dispensable (31). Previous sequencing and characterization of the EBNA3 family of proteins has identified a region known as the homology domain, located within amino acids 90 to 320 (23). As the EBNA3C-cyclin A interaction potentially falls within this region, we decided to test whether other EBNA3 family members might also interact with bacterially expressed cyclin A. As described above, GST-cyclin A strongly precipitated in vitro-translated EBNA3C (Fig. 1C). While EBNA3A, an essential latent protein for B-cell transformation, showed an association only slightly above background levels, EBNA3B was reproducibly precipitated with GST-cyclin A. This assay was repeated multiple times with independently prepared GST-cyclin A to ensure that the association with EBNA3B was significantly above that for either EBNA3A or GST control. The data presented in Fig. 1C is the quantification and average of two representative experiments. This finding suggests that the cyclin A binding site on EBNA3C may be conserved to some extent in EBNA3B and also that EBNA3B may have some limited role in regulating cyclin A complexes in virally infected cells.
Amino acids 130 to 159 of EBNA3C interact with high affinity with cyclin A.
To more finely map the region of EBNA3C responsible for cyclin A binding in the amino-terminal region of EBNA3C, additional truncation constructs of EBNA3C within the amino-terminal 365 amino acids were prepared. In vitro precipitation experiments with bacterially expressed GST-cyclin A showed that EBNA3C amino acids 1 to 200, but not 1 to 100, precipitated strongly with cyclin A (Fig. 2A). This result suggested that EBNA3C amino acids 101 to 200 contain the predominant mediator of cyclin A binding. Indeed, cyclin A coimmunoprecipitated strongly with EBNA3C 1-200 when expressed in cells (Fig. 2B). In repeated coimmunoprecipitation experiments, the association between cyclin A and EBNA3C 1-200 was actually stronger than the association between cyclin A and EBNA3C 1-365 (Fig. 2B). This result was in spite of the fact that 1-200 did not express as well as 1-365 in cells (compare lanes 5 and 6 in Fig. 2B). This suggests that EBNA3C sequences which lie outside of the primary binding domain play a potentially negative regulatory role in the functional association between EBNA3C and cyclin A.
FIG. 2.

Cyclin A (Cyc A) binding maps to EBNA3C amino acids 130 to 159. (A) GST-cyclin A fusion protein was expressed in E. coli and purified with glutathione Sepharose beads. EBNA3C truncations as indicated were labeled with [35S]methionine by in vitro translation (IVT) and then were incubated with either GST control or GST-cyclin A beads normalized by Coomassie staining. Precipitated proteins were resolved by SDS-PAGE, and bands were visualized with a phosphorimager screen. In each case, 5% of IVT input was used for comparison. (B) Ten million 293T cells were transfected with 10 μg of RC-cyclin A and 15 μg of pA3M-EBNA3C expression plasmids. Cells were harvested at 36 h and immunoprecipitated (IP) with 1 μl of 9E10 myc-reactive ascites fluid (for myc-tagged EBNA3C truncations). Samples were resolved by SDS-10% PAGE and probed by immunoblot. 1-200 and 1-365 represent the corresponding amino acids of EBNA3C. (C) GST-EBNA3C fusion proteins representing truncation mutants spanning amino acids 1 to 207 were expressed in E. coli and purified with glutathione Sepharose beads. Human cyclin A and p27 were labeled with [35S]methionine by in vitro transcription/translation and then were incubated with either GST control or GST-EBNA3C beads normalized by Coomassie staining. In each case, 5% of IVT input was used for comparison. (D) Ten million 293T cells were transfected with 10 μg of RC-cyclin A and 15 μg of pA3M-EBNA3C expression plasmids. Cells were harvested at 36 h and immunoprecipitated with 1 μl of 9E10 myc-reactive ascites fluid (for myc-tagged EBNA3C truncations). 1-200, 1-159, and 1-129 represent the corresponding amino acids of EBNA3C.
GST-EBNA3C truncation constructs were next used to further refine the association between cyclin A and EBNA3C. We first confirmed that, as expected, GST-EBNA3C fusion proteins corresponding to amino acids 1 to 207 and 90 to 190 were both able to strongly precipitate in vitro-translated cyclin A (Fig. 2C). This domain did not interact with in vitro-translated p27, a known cyclin A-interacting protein (Fig. 2C). We next tested EBNA3C fusion proteins corresponding to amino acids 90 to 129, 130 to 159, and 160 to 190. In this assay, amino acids 130 to 159 were the primary mediator of cyclin A binding, with amino acids 160 to 190 also having some affinity for cyclin A (Fig. 2C). To further confirm the importance of EBNA3C amino acids 130 to 159, cyclin A and EBNA3C truncations were expressed in cells for coimmunoprecipitation analysis. As described above, immunoprecipitation of EBNA3C amino acids 1 to 200 also strongly precipitated cyclin A (Fig. 2D). EBNA3C 1-159 also precipitated cyclin A, although at a level less than that for EBNA3C 1-200 (Fig. 2D, compare lanes 6 and 7). Importantly, EBNA3C 1-129 was unable to precipitate cyclin A (Fig. 2D, lane 8). These coimmunoprecipitation data suggest that, consistent with the above GST pull-down data, EBNA3C amino acids 160 to 200 as well as 130 to 159 play some role in binding cyclin A.
To identify individual residues mediating this association, we aligned EBV EBNA3C amino acids 130 to 159 with the known 3C homologues of Baboon and Rhesus lymphocryptovirus (LCV) (Fig. 3A). This region of EBNA3C was chosen because it demonstrated the strongest association with cyclin A in GST pull-down experiments (Fig. 2C). It has previously been demonstrated that transcriptional regulatory regions within EBNA3C are conserved in these EBNA3C homologues, and it is possible that cyclin A regulatory motifs might be similarly conserved (32). It is also possible that, given the association between cyclin A and EBNA3B, some of the residues important for the EBNA3C-cyclin A interaction would be conserved in EBNA3B but not EBNA3A (Fig. 3A). Amino acids 130 to 159 were about 70% conserved in Baboon and Rhesus LCV but were conserved less so in EBNA3A and EBNA3B. To address the role of individual residues, point mutations were constructed in the context of GST-EBNA3C 130-159. Mutated motifs are indicated by boxes in Fig. 3A. Mutation of phenylalanine 144 dramatically reduced cyclin A binding, suggesting that this residue is especially critical (Fig. 3B). Double mutation of arginine 149 and arginine 151 also significantly reduced cyclin A binding (Fig. 3B). These data confirm that specific conserved residues within EBNA3C 130-159 mediate cyclin A binding, with phenylalanine 144 playing an especially critical role in the EBNA3C-cyclin A association. However, additional mutational studies may be necessary to identify mutations in this domain capable of completely eliminating cyclin A binding to EBNA3C.
FIG. 3.

Point mutations of EBNA3C 130-159 more precisely map cyclin A (Cyc A) binding. (A) The schematic shows the alignment of EBNA3C 130-159 with Baboon (Ba-LCV) and Rhesus (Rh-LCV) lymphocryptovirus (32) (top panel) and EBNA3A and EBNA3B (bottom panel). Residues marked by an asterisk are functionally conserved. Boxed residues were assayed by point mutation below. (B) Single or double point mutations of EBNA3C were introduced into GST-EBNA3C 130-159 by PCR. GST-EBNA3C fusion proteins (amino acids 130 to 159) representing wild-type EBNA3C and the indicated mutations were expressed in E. coli and purified with glutathione Sepharose beads. Human cyclin A was labeled with [35S]methionine by in vitro transcription (IVT)/translation and then incubated with either GST control or GST-EBNA3C beads normalized by Coomassie staining. Precipitated proteins were resolved by SDS-PAGE, and bands were visualized and quantified with a phosphorimager screen. Five percent of cyclin A IVT input was used for comparison. RBU, relative binding units.
EBNA3C binds cyclin A at the conserved cyclin box.
While little is known about the tertiary and quaternary structure of EBNA3C, cyclin A is well understood structurally and, importantly, is understood in the context of structure-function relationships (9, 26). In an attempt to gain insights into the functionality of the association between cyclin A and EBNA3C, a number of cyclin A truncations were tested for their ability to bind GST-EBNA3C 130-159. Representative truncations are depicted here with cyclin A 1-205 showing no binding, cyclin A 171-399 showing strong binding similar to that of full-length cyclin A, and cyclin A 226-399 showing significantly weaker binding than full-length cyclin A (Fig. 4A). Other truncations, including cyclin A 1-170, showed no binding, and cyclin A 311-399 showed binding only slightly above background (data not shown). Given that cyclin A 1-205 shows no binding and deletion of cyclin A amino acids 171 to 225 significantly reduces binding affinity (Fig. 4A, compare right two panels), these data implicate cyclin A amino acids 206 to 225 as an important determinant of EBNA3C binding (Fig. 5B). This region of cyclin A is notable because it represents the α1 helix of the so-called cyclin box of cyclin A (9) (Fig. 4B). This helix contains a highly conserved MRAIL motif and has also been shown to be a primary determinant of both substrate recruitment and binding of the Cip/Kip family of inhibitors including p27 (26, 27).
FIG. 4.

The conserved cyclin box of cyclin A (Cyc A) is required for strong binding by EBNA3C amino acids 130 to 159. (A) GST-EBNA3C 130-159 was expressed in E. coli and purified with glutathione Sepharose beads. Full-length cyclin A and cyclin A truncation mutants as indicated were labeled with [35S]methionine by in vitro translation (IVT) and then were incubated with either GST control or GST-EBNA3C beads normalized by Coomassie staining. Precipitated proteins were resolved by SDS-PAGE, and bands were visualized and quantified with a phosphorimager screen. In each case, 5% of IVT input was used for comparison. RBU, relative binding units. (B) The schematic indicates known domains of cyclin A. For cyclin A truncation mutants, black bars indicate maximum binding, gray bars indicate minimal binding, and white bars indicate undetectable binding.
FIG. 5.

The cyclin box of cyclin A binds the amino terminus of EBNA3C. (A) GST-cyclin A cyclin box fusion protein (amino acids 206 to 310) was expressed in E. coli and purified with glutathione Sepharose beads. Full-length EBNA3C and EBNA3C truncations as indicated were labeled with [35S]methionine by in vitro translation (IVT) and then were incubated with either GST control or GST-cyclin A cyclin box (Cyc A Box) beads normalized by Coomassie staining. Precipitated proteins were resolved by SDS-PAGE, and bands were visualized with a phosphorimager screen. In each case, 5% of IVT input was used for comparison. (B) Ten million 293T cells were transfected with 15 μg of pCMV-Cdk2 or 15 μg of pA3M-EBNA3C 1-365 expression plasmid. Lysates were prepared at 36 h and incubated with either GST alone or GST-cyclin A cyclin box (Cyc A Box) fusion protein as described above. Samples were resolved by SDS-12% PAGE and probed by immunoblot. EBNA3C 1-365 was detected with 9E10 myc hybridoma supernatant, and Cdk2 was detected with rabbit polyclonal serum to Cdk2. (C) GST fusion proteins corresponding to the α1 helix of the cyclin A cyclin box (amino acids 206 to 225) or helices α2 to α5 (amino acids 226 to 310) were expressed in E. coli and were purified with glutathione Sepharose beads. EBNA3C amino acids 1-365 was labeled with [35S]methionine by in vitro translation (IVT) and then were incubated with GST control and either α1 (Cyc A Box α1) or α2 to α5 (Cyc A Box α2-5) beads normalized by Coomassie staining. In each case, 5% of IVT input was used for comparison.
To confirm the importance of the cyclin A cyclin box and specifically the α1 helix in EBNA3C binding, we constructed GST fusions containing the full-length cyclin A cyclin box (amino acids 206 to 310), the α1 helix (206 to 227), or helices α2 to α5 (228 to 310). As expected, in vitro-translated protein corresponding to both full-length EBNA3C and EBNA3C 1-365 bound strongly to a GST-cyclin A cyclin box fusion but not to GST control (Fig. 5A). As seen in Fig. 1B, EBNA3C 621-992 also showed limited binding slightly above background (Fig. 5A). In order to compare the affinity of EBNA3C 1-365 to a known functional interacting partner, the cyclin A cyclin box was used to precipitate proteins from the lysates of 293T cells transfected with expression plasmids for either Cdk2 or EBNA3C 1-365 (Fig. 5B). While Cdk2 precipitated more strongly with the cyclin A cyclin box, EBNA3C 1-365 precipitation was of a similar magnitude, hinting at the potential functionality of this interaction. Finally, we assessed the relative binding of the cyclin A α1 helix compared to that of helices α2 to α5 (Fig. 5C). In this in vitro binding assay, both elements of the cyclin A cyclin box confer binding to EBNA3C 1-365, consistent with the GST-EBNA3C 130-159 pull-downs of cyclin A truncation mutants (Fig. 4). In Fig. 5C, we show that the affinities of these two elements for the amino terminus of EBNA3C were approximately the same. This may suggest that, in the context of EBNA3C 130-159, the α1 helix is the predominant partner (Fig. 4) while the α2 to α5 helices may bind other regions of the amino terminus.
EBNA3C amino acids 130 to 159 bind other mammalian cyclins in addition to cyclin A.
The above truncation studies with cyclin A suggested that the amino terminus of EBNA3C targets the α1 helix of the cyclin box, a region of cyclin A conserved not only in cyclin A homologues but also in other mammalian cyclins. As such, we decided to test whether other mammalian cyclins can bind EBNA3C 130-159 in an in vitro binding assay. In this assay, we occasionally saw background precipitation of luciferase control when maximum amounts of GST-EBNA3C 130-159 protein were used (Fig. 6, lower right panel). However, this binding was always lower than that for the cyclins and was never seen when less fusion protein was used. Both cyclin E and cyclin D1 showed binding to EBNA3C 130-159 that was clearly above both GST control and the aforementioned luciferase background (Fig. 6). In repeated experiments, average binding was typically 30% of that seen with cyclin A, suggesting that residues unique to cyclin A confer some specificity for EBNA3C binding (Fig. 6). However, this data also suggests that EBNA3C may target other mammalian cyclins in addition to cyclin A. A previous study attempted rescue of p27-mediated suppression of cyclin E-dependent kinase activity similar to that seen for cyclin A (13). These attempts were unsuccessful, suggesting that the mechanism by which EBNA3C regulates other cyclins, such as cyclin E, is likely distinct from that seen for cyclin A.
FIG. 6.

EBNA3C amino acids 130 to 159 also binds other cyclins. GST-EBNA3C 130-159 was expressed in E. coli and purified with glutathione Sepharose beads. Full-length cyclin A (Cyc A), cyclin E, cyclin D1, and Luciferase (Luc) were labeled with [35S]methionine by in vitro transcription (IVT)/translation and then were incubated with either GST control or GST-EBNA3C beads normalized by Coomassie staining. Precipitated proteins were resolved by SDS-PAGE, and bands were visualized and quantified with a phosphorimager screen. In each case, 5% of IVT input was used for comparison. In lane 4, three times the amount of GST-EBNA3C fusion protein was used as in lane 3. RBU, relative binding units.
The amino-terminal 365 amino acids of EBNA3C are not sufficient to rescue p27-mediated inhibition of cyclin A-dependent kinase activity.
Previously it was demonstrated that expression of EBNA3C in either U2OS or BJAB cells rescues inhibition of cyclin A-dependent kinase activity by the kinase inhibitor p27 (13). This phenotype maps tightly to the carboxy terminus of EBNA3C, with mutants at the extreme carboxy terminus of EBNA3C eliminating the phenotype and the carboxy-terminal 372 amino acids (621 to 992) sufficient to recapitulate rescue of kinase activity similar to full-length EBNA3C (13). However, these experiments were performed without the present data demonstrating that the highest affinity for cyclin A lies within the amino-terminal 365 amino acids of EBNA3C. To test the ability of this domain to rescue p27-mediated kinase inhibition, U2OS cells were transfected with cyclin A, Cdk2, and p27 expression plasmids, along with either full-length EBNA3C or titrated amounts of EBNA3C 1-365 (Fig. 7), which binds strongly to cyclin A both in vitro and in cells (Fig. 1). In numerous experiments, including extreme titrations of EBNA3C 1-365 expression plasmid, we were unable to rescue p27 inhibition of cyclin A/Cdk2 kinase activity in spite of consistent rescue with the full-length molecule (Fig. 7). This implies that the amino terminus of EBNA3C is likely the primary domain for binding cyclin A and that other domains of EBNA3C, including the extreme carboxy terminus, are necessary for modulation or rescue of cyclin A-dependent kinase activity, at least in the context of p27 suppression.
FIG. 7.

EBNA3C 1-365 does not rescue p27-mediated suppression of cyclin A (Cyc A)-dependent kinase activity. U2OS cells were transfected with cyclin A, Cdk2, p27, and EBNA3C expression constructs as indicated. Specifically, cells were transfected with 2 μg of RC-cyclin A, 1 μg of CMV-Cdk2, 0 to 1 μg of pCDNA3-p27, 5 μg of pA3M-EBNA3C, and 0 to 15 μg of pA3M-EBNA3C 1-365 as indicated. After 24 h, cyclin A immunoprecipitates (IP) were captured and assayed for in vitro kinase activity toward histone H1.
The amino-terminal 365 amino acids of EBNA3C suppress cyclin A-dependent kinase activity when introduced into LCLs.
Previously it was demonstrated that the introduction of a stop codon at amino acid 365 in EBNA3C eliminates the ability of EBV to transform B lymphocytes (31). When a mixed population of wild-type and mutant virus was used to infect primary B cells, the resulting LCLs only harbored mutant EBNA3C when coinfected with wild-type (31). Further, in these coinfected LCLs the recombinant EBNA3C was lost when cultured continually (31). These studies hint that the amino-terminal domain of EBNA3C not only fails to mimic full-length EBNA3C but may also negatively regulate the effects of full-length EBNA3C in coinfected LCLs.
As EBNA3C amino acids 1 to 365 do not rescue p27 suppression of cyclin A-dependent kinase activity as full-length EBNA3C does, we tested whether 1-365 would actually suppress cyclin A kinase activity when introduced into LCLs. LCLs were transfected by electroporation with vector control, EBNA3C 1-365, or EBNA3C 1-129, which does not bind cyclin A. With this system, we achieved transfection efficiencies ranging from 10 to 20% in multiple experiments using GFP as a monitor. After 24 h, cyclin A complexes were immunoprecipitated and assayed for kinase activity toward histone H1. Expression of EBNA3C 1-365 reproducibly reduced cyclin A-dependent kinase activity, with representative data shown in Fig. 8. EBNA3C 1-365 suppressed the activity by 25 to 50% depending on the individual experiment, and, importantly, EBNA3C 1-129 (Fig. 8) and full-length EBNA3C (data not shown) did not reduce the kinase activity in this assay.
FIG. 8.
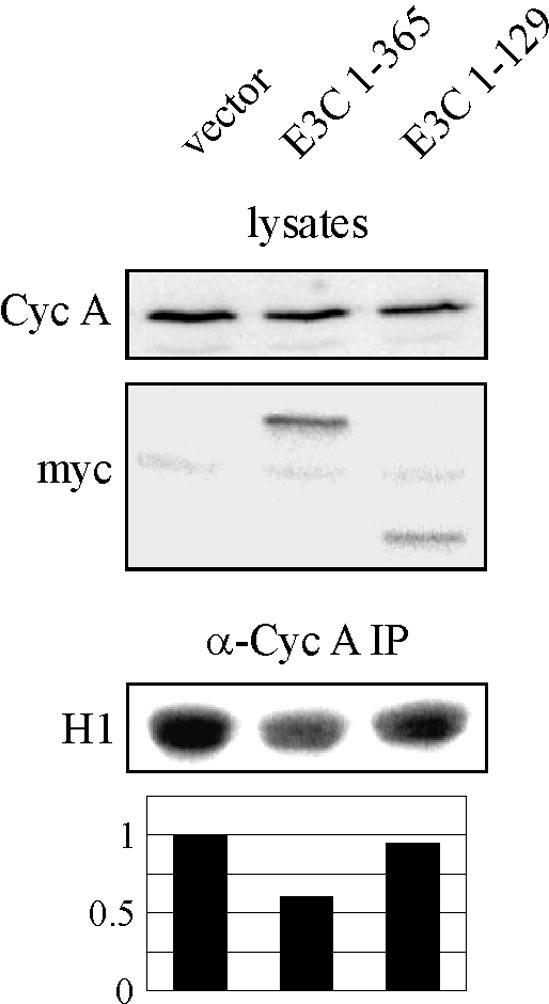
EBNA3C amino acids 1 to 365 downregulate cyclin A (Cyc A)-dependent kinase activity when expressed in LCLs. LCLs were transfected by electroporation with 10 μg of RC-cyclin A and 20 μg of pA3M vector, pA3M-EBNA3C 1-365, or pA3M-EBNA3C 1-129. After 24 h, cyclin A immunoprecipitates (IP) were captured and assayed for in vitro kinase activity toward histone H1.
DISCUSSION
In a previous study, a yeast two-hybrid screen with the carboxy terminus of EBNA3C as bait identified cyclin A as a potential EBNA3C-interacting protein. It was subsequently demonstrated that EBNA3C binds cyclin A in vitro and targets cyclin A kinase complexes in lymphoblastoid cell lines (13). Further, in the context of an experimental system, EBNA3C decreases the molecular association between cyclin A and the kinase inhibitor p27 and rescues p27-mediated inhibition of cyclin A-dependent kinase activity (13). This phenotype was at least partially mapped to the carboxy terminus of EBNA3C, which was consistent with the region of EBNA3C used as bait in the yeast two-hybrid screen (Fig. 9A). Subsequent to this study, we performed a more detailed dissection of the EBNA3C-cyclin A interaction in an attempt to better understand the functional relationship between these two molecules. Surprisingly, we found that the predominant interaction between EBNA3C and cyclin A actually occurred at the amino terminus of EBNA3C, a region of the molecule not included in the yeast two-hybrid screen (Fig. 9A). The cyclin A-interacting domain was mapped to amino acids 130 to 159 of EBNA3C, a region conserved in 3C homologues of other lymphocryptovirus species, Baboon lymphocryptovirus and Rhesus lymphocryptovirus (32). Specifically, 21 of 30 residues are either identical or demonstrate potential functional similarity, consistent with a conserved and potentially critical role for this region in the biology of EBV. This region of EBNA3C also falls within the so-called EBNA3 homology domain from amino acids 90 to 320, and, indeed, 10 of 30 residues show conservation between EBNA3A, 3B, and 3C. In multiple trials, EBNA3B, but not EBNA3A, showed some binding affinity for cyclin A, suggesting that the EBNA3C residues conserved in EBNA3B, but not EBNA3A, may be most important for cyclin A binding. We have yet to show a role for EBNA3B, individually, in modulating cyclin A function, although more thorough studies to evaluate potential synergism between EBNA3B and EBNA3C, both in vitro and in virally infected cells, are still needed.
FIG. 9.

EBNA3C schematics. (A) Schematic illustrating EBNA3C and known functional and interacting motifs. (B) Schematic illustrating potential cooperativity between the amino and carboxy termini of EBNA3C in regulation of cyclin A complexes in virally infected cells. A, cyclin A. N, EBNA3C amino terminus. C, EBNA3C carboxy terminus.
The strong interaction between EBNA3C 130-159 and cyclin A is at least partially dependent on the α1 helix of the conserved cyclin box of cyclin A. This helix has been shown to be critical for cyclin A function in at least two regards. First, the helix and especially its conserved MRAIL motif provide many of the critical residues required for binding of Cip/Kip family kinase inhibitors, such as p27 (26). Indeed, the crystal structure of cyclin A/Cdk2/p27 shows a hydrophobic groove in cyclin A that receives p27 (26). Among the residues contributing van der Waals contacts to p27 are Met 210, Ile 213, Leu 214, and Trp 217 (26). This observation is particularly noteworthy, as a previous study demonstrated that EBNA3C reduces the molecular association between cyclin A and p27 in cells. As such, an attractive model is that EBNA3C directly competes with p27 for cyclin A binding. However, we have yet to demonstrate such competition with either in vitro-translated or bacterially expressed proteins, suggesting that our observed competition in coimmunoprecipitation experiments may require other factors present in cell lysates, potentially recruited to cyclin A complexes by EBNA3C (Fig. 9B). We are presently seeking to identify such factors. In addition to mediating the binding of cyclin A inhibitors such as p27, the α1 helix also contributes residues to a hydrophobic patch on the surface of cyclin A that recruits substrates to Cdk2. This patch has been shown to be important for the binding of RXL-containing proteins, such as p107, E2F1, p27, and p21, and is necessary for Cdk2-mediated phosphorylation of some substrates (27). It is likely that cyclin A/Cdk2 can phosphorylate EBNA3C, although it is unclear what the functional significance of this modification would be in the context of virally infected cells.
Interestingly, previous second-site recombination experiments have demonstrated that a stop codon introduced at amino acid 365 eliminates the ability of EBV to transform B lymphocytes, suggesting that expression of the amino terminus alone is insufficient to mimic all functions of the full-length molecule (31). Further, in coinfected LCLs containing wild-type and recombinant EBNA3C (amino acids 1 to 365), the recombinant EBNA3C is lost when cultured continually in vitro, suggesting that the amino-terminal domain may negatively regulate the effects of the wild-type EBNA3C in the coinfected LCLs (31). Here we showed that introduction of EBNA3C amino acids 1 to 365 into LCLs negatively regulates cyclin A-dependent kinase activity, potentially providing selective pressure to lose this region in recombinant LCLs by sequestering cyclin A into nonfunctional complexes. However, it should be noted that the amino terminus of EBNA3C has also been shown to interact functionally with other cellular proteins. These include p300 and HDAC1, which are targeted by amino acids 1 to 207 (20, 29) and RBP-Jκ with interacting residues lying somewhere between amino acids 180 and 225 (24) (Fig. 9A). Importantly, the RBP-Jκ interaction appears to be conserved in the other EBNA3 family members, although it is not clear whether one EBNA3 protein may be the predominant functional interacting partner in EBV-transformed cells (32).
In combination with data from a previous study (13), our data suggests that multiple domains of EBNA3C contribute to viral modulation of cyclin A-dependent kinase activity. The amino terminus of EBNA3C appears to be the predominant recruitment domain, with both in vitro binding and coimmunoprecipitation studies suggesting that the amino terminus contains the strongest and predominant cyclin A binding domain. However, the previous work of Knight and Robertson demonstrated that the carboxy terminus of EBNA3C is the primary functional domain, with EBNA3C 621-992 able to recapitulate the ability of full-length EBNA3C to rescue p27-mediated inhibition of cyclin A-dependent kinase activity (13). It must be noted that these functional rescue experiments required exogenous expression of the EBNA3C polypeptides. With overexpression of the amino terminus of EBNA3C, cyclin A may be sequestered or recruited into complexes or compartments that are not particularly accessible to the carboxy terminus. It is possible that in the context of viral infection, where EBNA3C levels are undoubtedly significantly lower, strong cyclin A binding by the amino-terminal domain may be required to place the carboxy terminus in the proximity of cyclin A for functional modulation (Fig. 9B). We will continue to pursue the role of the carboxy terminus of EBNA3C in these complexes and also attempt to identify additional EBNA3C-recruited molecules in an attempt to better understand both EBNA3C cell cycle modulation and the ability of EBV to transform primary cells.
Acknowledgments
We thank Ed Harlow, Philip Hinds, Elliot Kieff, Maria Mudryj, Michele Pagano, Martin Rowe, and Alan Diehl for generously providing reagents.
J.S.K. is supported by the Lady Tata Memorial Trust. The project is supported by NIH grants NCI CA72150-07, NCI CA91792-01, and NIDCR DE14136-01 to E.S.R. E.S.R. is a scholar of the Leukemia and Lymphoma Society of America.
REFERENCES
- 1.Aiello, L., R. Guilfoyle, K. Huebner, and R. Weinmann. 1979. Adenovirus 5 DNA sequences present and RNA sequences transcribed in transformed human embryo kidney cells (HEK-Ad-5 or 293). Virology 94:460-469. [DOI] [PubMed] [Google Scholar]
- 2.Allday, M. J., D. H. Crawford, and J. A. Thomas. 1993. Epstein-Barr virus (EBV) nuclear antigen 6 induces expression of the EBV latent membrane protein and an activated phenotype in Raji cells. J. Gen. Virol. 74:361-369. [DOI] [PubMed] [Google Scholar]
- 3.Allday, M. J., and P. J. Farrell. 1994. Epstein-Barr virus nuclear antigen EBNA3C/6 expression maintains the level of latent membrane protein 1 in G1-arrested cells. J. Virol. 68:3491-3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aster, J. C., E. S. Robertson, R. P. Hasserjian, J. R. Turner, E. Kieff, and J. Sklar. 1997. Oncogenic forms of NOTCH1 lacking either the primary binding site for RBP-Jκ or nuclear localization sequences retain the ability to associate with RBP-Jκ and activate transcription. J. Biol. Chem. 272:11336-11343. [DOI] [PubMed] [Google Scholar]
- 5.Cotter, M. A., Jr., and E. S. Robertson. 2000. Modulation of histone acetyltransferase activity through interaction of Epstein-Barr nuclear antigen 3C with prothymosin alpha. Mol. Cell. Biol. 20:5722-5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Devoto, S. H., M. Mudryj, J. Pines, T. Hunter, and J. R. Nevins. 1992. A cyclin A-protein kinase complex possesses sequence-specific DNA binding activity: p33cdk2 is a component of the E2F-cyclin A complex. Cell 68:167-176. [DOI] [PubMed] [Google Scholar]
- 7.Epstein, M. A., B. G. Achong, and Y. M. Barr. 1964. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet i:702-703. [DOI] [PubMed]
- 8.Hinds, P. W., S. Mittnacht, V. Dulic, A. Arnold, S. I. Reed, and R. A. Weinberg. 1992. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 70:993-1006. [DOI] [PubMed] [Google Scholar]
- 9.Jeffrey, P. D., A. A. Russo, K. Polyak, E. Gibbs, J. Hurwitz, J. Massague, and N. P. Pavletich. 1995. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 376:313-320. [DOI] [PubMed] [Google Scholar]
- 10.Johannsen, E., C. L. Miller, S. R. Grossman, and E. Kieff. 1996. EBNA-2 and EBNA-3C extensively and mutually exclusively associate with RBPJkappa in Epstein-Barr virus-transformed B lymphocytes. J. Virol. 70:4179-4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kieff, E., and A. B. Rickinson. 2002. Epstein-Barr virus and its replication, p. 2511-2573. In D. Knipe and P. Howley (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 12.Knight, J. S., K. Lan, C. Subramanian, and E. S. Robertson. 2003. Epstein-Barr virus nuclear antigen 3C recruits histone deacetylase activity and associates with the corepressors mSin3A and NCoR in human B-cell lines. J. Virol. 77:4261-4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knight, J. S., and E. S. Robertson. 2004. Epstein-Barr virus nuclear antigen 3C regulates cyclin A/p27 complexes and enhances cyclin A-dependent kinase activity. J. Virol. 78:1981-1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgan, D. O. 1997. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 13:261-291. [DOI] [PubMed] [Google Scholar]
- 15.Pagano, M., R. Pepperkok, F. Verde, W. Ansorge, and G. Draetta. 1992. Cyclin A is required at two points in the human cell cycle. EMBO J. 11:961-971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pagano, M., S. W. Tam, A. M. Theodoras, P. Beer-Romero, G. Del Sal, V. Chau, P. R. Yew, G. F. Draetta, and M. Rolfe. 1995. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science 269:682-685. [DOI] [PubMed] [Google Scholar]
- 17.Parker, G. A., T. Crook, M. Bain, E. A. Sara, P. J. Farrell, and M. J. Allday. 1996. Epstein-Barr virus nuclear antigen (EBNA)3C is an immortalizing oncoprotein with similar properties to adenovirus E1A and papillomavirus E7. Oncogene 13:2541-2549. [PubMed] [Google Scholar]
- 18.Parker, G. A., R. Touitou, and M. J. Allday. 2000. Epstein-Barr virus EBNA3C can disrupt multiple cell cycle checkpoints and induce nuclear division divorced from cytokinesis. Oncogene 19:700-709. [DOI] [PubMed] [Google Scholar]
- 19.Radkov, S. A., M. Bain, P. J. Farrell, M. West, M. Rowe, and M. J. Allday. 1997. Epstein-Barr virus EBNA3C represses Cp, the major promoter for EBNA expression, but has no effect on the promoter of the cell gene CD21. J. Virol. 71:8552-8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Radkov, S. A., R. Touitou, A. Brehm, M. Rowe, M. West, T. Kouzarides, and M. J. Allday. 1999. Epstein-Barr virus nuclear antigen 3C interacts with histone deacetylase to repress transcription. J. Virol. 73:5688-5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Resnitzky, D., L. Hengst, and S. I. Reed. 1995. Cyclin A-associated kinase activity is rate limiting for entrance into S phase and is negatively regulated in G1 by p27Kip1. Mol. Cell. Biol. 15:4347-4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rickinson, A. B., and E. Kieff. 2002. Epstein-Barr Virus, p. 2575-2627. In D. Knipe and P. Howley (ed.), Fields Virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 23.Robertson, E. S. 1997. The Epstein-Barr Virus EBNA3 protein family as regulators of transcription. Epstein-Barr Vir. Rep. 4:143-150. [Google Scholar]
- 24.Robertson, E. S., J. Lin, and E. Kieff. 1996. The amino-terminal domains of Epstein-Barr virus nuclear proteins 3A, 3B, and 3C interact with RBPJ(κ). J. Virol. 70:3068-3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosendorff, A., D. Illanes, G. David, J. Lin, E. Kieff, and E. Johannsen. 2004. EBNA3C coactivation with EBNA2 requires a SUMO homology domain. J. Virol. 78:367-377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Russo, A. A., P. D. Jeffrey, A. K. Patten, J. Massague, and N. P. Pavletich. 1996. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature 382:325-331. [DOI] [PubMed] [Google Scholar]
- 27.Schulman, B. A., D. L. Lindstrom, and E. Harlow. 1998. Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc. Natl. Acad. Sci. USA 95:10453-10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramanian, C., M. A. Cotter, Jr., and E. S. Robertson. 2001. Epstein-Barr virus nuclear protein EBNA-3C interacts with the human metastatic suppressor Nm23-H1: a molecular link to cancer metastasis. Nat. Med. 7:350-355. [DOI] [PubMed] [Google Scholar]
- 29.Subramanian, C., S. Hasan, M. Rowe, M. Hottiger, R. Orre, and E. S. Robertson. 2002. Epstein-Barr virus nuclear antigen 3C and prothymosin alpha interact with the p300 transcriptional coactivator at the CH1 and CH3/HAT domains and cooperate in regulation of transcription and histone acetylation. J. Virol. 76:4699-4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian, C., J. S. Knight, and E. S. Robertson. 2002. The Epstein Barr nuclear antigen EBNA3C regulates transcription, cell transformation and cell migration. Front. Biosci. 7:704-716. [DOI] [PubMed] [Google Scholar]
- 31.Tomkinson, B., E. Robertson, and E. Kieff. 1993. Epstein-Barr virus nuclear proteins EBNA-3A and EBNA-3C are essential for B-lymphocyte growth transformation. J. Virol. 67:2014-2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao, B., R. Dalbies-Tran, H. Jiang, I. K. Ruf, J. T. Sample, F. Wang, and C. E. Sample. 2003. Transcriptional regulatory properties of Epstein-Barr virus nuclear antigen 3C are conserved in simian lymphocryptoviruses. J. Virol. 77:5639-5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao, B., and C. E. Sample. 2000. Epstein-barr virus nuclear antigen 3C activates the latent membrane protein 1 promoter in the presence of Epstein-Barr virus nuclear antigen 2 through sequences encompassing an Spi-1/Spi-B binding site. J. Virol. 74:5151-5160. [DOI] [PMC free article] [PubMed] [Google Scholar]